

Abstract
Peptide-based analogues of the gut-derived incretin hormone, glucagon-like peptide 1 (GLP1), stimulate insulin secretion in a glucose-dependent manner. Currently marketed GLP1 receptor (GLP1R) agonists are safe and effective in the management of Type 2 diabetes but often offer only modest weight loss. This has prompted the search for safe and effective alternatives to enhance the weight loss component of these treatments. We have demonstrated that concomitant activation GLP1R and the glucagon receptor (GCGR) can improve glucose metabolism and provide superior weight loss when compared to selective GLP1R agonism in preclinical species. This paper will highlight chemistry structure–activity relationship optimization and summarize in vivo efficacy studies toward the discovery of a once daily balanced dual agonist 12 (MK-1462), which was advanced into clinical trials.
Keywords: Glucagon-like peptide-1 (GLP1), glucagon, GLP1R/GCGR dual agonist, type 2 diabetes (T2DM), nonhuman primates, weight loss
Over the past two decades, the prevalence of diabetes has increased dramatically and is associated with a similarly dramatic increase in obesity and the associated comorbidities of metabolic syndrome.1,2 Several agents are available for treatment of elevated glycemia, including metformin, the sulfonylureas, DPP-IV, and SGLT2 inhibitors.2 While all of these mechanisms address the problem of elevated fasting glucose, as they only modestly impact the underlying issue of excess body weight, typically disease progression is only slowed and many type 2 diabetics ultimately progress to insulin therapy. One class of drugs that has shown promise in delivering modest reductions in appetite and body weight in addition to robust glucose lowering is the glucagon-like peptide-1 receptor (GLP1R) agonists, such as liraglutide (Lira) and semaglutide.3 However, high doses of GLP1R agonists only reduce body weight by about 4–6% and are associated with high incidences of nausea and GI intolerability.4
Leveraging additional incretin receptor agonism to maximize glycemic control and body weight reduction turns out to be quite successful. The first dual glucose-dependent insulinotropic polypeptide (GIP) and GLP1 receptor agonist tirzepatide has recently been approved by the U.S. Food and Drug Administration.5,6 In addition, GLP1R/glucagon receptor (GCGR) dual agonist injectable agents have been developed that are thought to improve glycemic control in patients with T2DM and provide superior body weight loss relative to GLP-1 monoagonists.7,8 It is anticipated that the dual agonist mechanism will deliver equal or better glucose lowering and superior weight loss versus GLP1R-selective agonists, dosed at a minimum once-daily or once-weekly with anticipated associated improvements in the overall metabolic profile.9,10
The effectiveness of GLP1R/GCGR dual agonist mechanism has been demonstrated in rodents showing dramatic weight loss and glucose lowering concomitantly with increases in FGF21, enhanced energy expenditure and improvements in lipid metabolism.11 We and others have conducted studies in obese, dysmetabolic nonhuman primates demonstrating superior weight loss and glucose lowering with a GLP1R/GCGR dual agonist relative to the benchmark liraglutide.11,12 Furthermore, oxyntomodulin (OXM), a balanced endogenous GLP1R/GCGR dual agonist, has been reported to induce weight loss in obese subjects after 4 weeks of subcutaneous TID (three times a day) dosing.13 In another study, OXM was compared to a matched analogue with a single point mutation at position 3, OXMQ3E, having equipotent activity on GLP1R but no activity on GCGR. In this study, OXM effected superior weight loss and similar glucose lowering activity compared to OXNQ3E in mice and establishing with pharmacological and genetic approaches that the GCGR pathway is required for the weight-lowering action of exogenous OXM in vivo.14
In addition to glucose and body weight lowering, efficacy studies in rodents have indicated that GLP1R/GCGR dual agonists have the potential to provide significant improvement in several markers associated with metabolic syndrome. These markers include markers for lipid metabolism and fatty acid oxidation such as FGF21 and ketones. In addition, rodent studies suggest that GLP1R/GCGR co-agonism increases energy expenditure and reduces hepatic steatosis.7
Based on this exciting and interesting biology, we initiated medicinal chemistry efforts toward the discovery of novel GLP1R/GCGR dual agonists. Our main goal here was to identify a molecule with a balanced receptor selectivity ratio and once-daily dosing for the treatment of diabetes. Such a dual agonist molecule was expected to provide glucose lowering efficacy comparable to that of GLP1R agonists but also to maximize differentiated weight loss.
At the time, structural data for GCGR-GCG or GLP1R-GLP1 complex were not available.15 In order to enable structure-guided design and optimization, a homology model of the GCGR-GCG complex was devised following the reported method.16 Glucagon (GCG) was chosen over GLP1 as our starting point for structure–activity relationship (SAR) development because (1) GCG’s endogenous precursor OXM is a well-studied GLP1R/GCGR dual agonist and (2) it is well documented that one can tune in GLP1 activity into the GCG sequence by chemical modifications. We used the reported GCGR 7TM structure (PDB: 4L6R),16 GCGR ECD domain structure (PDB: 4ERS),17 and GCG structure (PDB: 1GCN)18 to build up and assemble the corresponding domains. The complex model was further refined by manual adjustment to avoid steric clash among domains followed by iterative energy optimizations in MOE (Figure 1).
Figure 1.
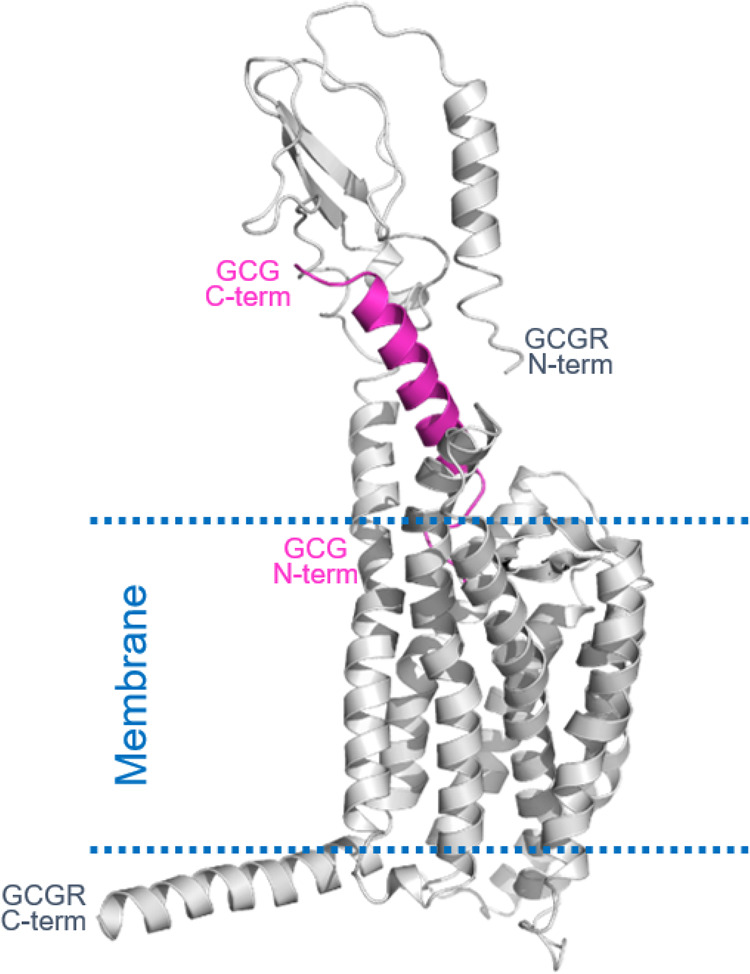
Homology model of the GCGR-GCG complex.
After careful mapping evaluation of the homology differences between native GLP1 and GCG sequences, we classified the GCG sequence amino acids into three categories (Figure 2). Group 1 (highlighted in green) amino acids are directly engaged in binding and not conserved between GCG and GLP1. This suggests that we can vary these positions to adjust the GCGR/GLP1R ratio (defined as tone) in a dual agonist. The group 2 class of amino acids (highlighted in blue) is conserved and directly involved in binding, suggesting that modification to these amino acids would potentially result in loss of activity. Lastly, the group 3 class of amino acids (highlighted in red) is hydrophilic and solvent-exposed, not directly involved in binding to both receptors. We envisioned that modification of these C-terminal amino acids could manipulate the physical properties and tune in GLP1 activity when needed as this region is only found in the GLP1 sequence.
Figure 2.

GLP1 and glucagon natural sequence and homology differences.
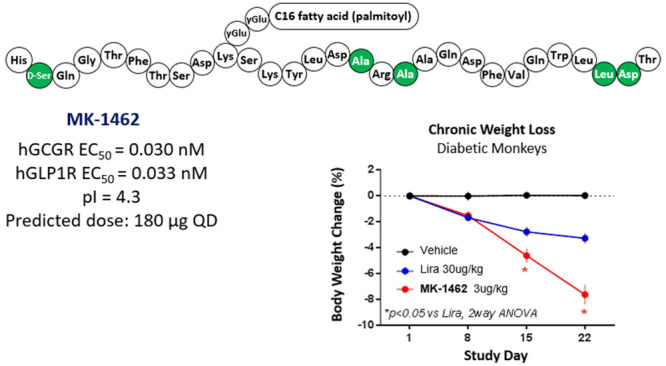
To accelerate the discovery of novel GLP1R/GCGR dual agonists for the treatment of T2DM, we took inspiration from the reported fatty acid acylated GCG analogues that exhibited promising dual agonism and pharmacological efficacy.19 Interestingly, fatty acid acylation alone at position 10 of GCG with Tyr to Lys(γGlu-γGlu-C16) substitution yielded a dual agonist 1 that is slightly biased toward GCGR albeit with reduced activity. Further point mutations by incorporating Aib (U) at positions 2 and 16 were reported to enhance peptide stability against the DPP-IV enzyme,20 helicity, and potency.19 This led to 2 with increased GLP1 and GCG activities. However, an isoelectric point (pI) of 7.0 gave this compound poor solubility in PBS (0.08 mg/mL),19 discouraging its progression as a development candidate. Based on homology model analysis, we also explored C-terminal modifications including fatty acid acylation for half-life extension but failed to tune in GLP1 activity (data no shown). However, in an effort to modulate its physicochemical property, we identified the KγE extension that was able to maintain the activities on both receptors and more importantly lowered pI by almost 2 units (peptide 3, pI = 5.2), setting the stage for further SAR optimization.
As part of a comprehensive effort to discover a dual agonist as a potential drug candidate, we decided next to address the underexplored chemical and metabolic stability issues that are inherent to the peptide sequence. A qualitative bioanalytical study of an analogue (not shown) of peptide 3 revealed multiple hotspots susceptible for potential degradation. That includes (a) Met27 oxidative metabolism, (b) Asn28 deamidation (especially at higher pH), and (c) Arg17-Arg18 dipeptide proteolytic cleavage. Rather than performing an extensive screening at those locations, we leveraged judicious SAR information that was already available in the literature to quickly test the compatibility of the reported mutation options. Replacement of Met27 with Leu in peptide 4 showed remarkable potency improvement at both GCGR and GLP1R with an almost equally balanced tone (1.2) in human. Next, Ala, originated from the GLP1 sequence, was identified as a suitable replacement for Arg18 in peptide 5. Such a mutation effectively lowered the pI by 0.5 units and ameliorated the metabolic stability of the Arg17-Arg18 dipeptide.8,21 In an attempt to minimize deamidation of Asn28, we reasoned that an Asp replacement would be tolerated based on receptor–peptide interaction in the homology model. Indeed, the Asn28Asp mutation in peptide 6 was found to slightly promote receptor activities. Moreover, it afforded a significant reduction of pI (4.1), a value within the desired range to provide high solution at physiological pH conditions. However, the receptor selectivity ratio in rhesus monkey (rhTone 9.1) was no longer balanced. We envisioned that the superior solubility benefit from the C-terminal KγE could be compensated by the newly introduced negative charge from Asp28. Therefore, peptide 7 was made without C-terminal KγE extension at no cost of pI shift and it showed improved receptor selectivity ratios in human but not in rhesus monkey. Although Aib2 was widely used to effectively circumvent DPP-IV mediated degradation and to increase GLP1R potency in the context of glucagon-based scaffolds,22 with the goal of developing a more balanced dual agonist in both human and rhesus monkey for lower translational risk, we switched to d-Ser at position 2, which was reported to be tolerated and showed resistance to cleavage by DPP-IV.23−25 Indeed, peptide 8 with d-Ser2 showed comparable receptor activity to that of peptide 7 but with reversed receptor selectivity ratio in rhesus monkey (rhTone 0.5). Nevertheless, considering the potential synthetic challenge associated with sterically hindered Aib, we decided to advance our SAR with d-Ser2. In addition, we also performed a quick scan at Aib16 to look for a potential replacement that could lower the synthetic barrier and provide ideal receptor selectivity ratios across species. As summarized in Table 1, peptides 9–12 having various mutations at position 16, including Ser from native GCG sequence, were able to adjust tones in human and rhesus monkey without significantly impact receptor potency. In particular, peptide 12 with Ala16 provided potent and most balanced dual agonism for both human and rhesus monkey (hTone of 0.9 and rhTone of 0.8, respectively). Peptide 12 has expected poor solubility at pH 3–5 which is consistent with a pI of 4.1, but its solubility increases significantly at pH 6 or higher (>20 mg/mL at pH 7 and >80 mg/mL at pH 8). Peptide 12 also demonstrated superior stability as there was no decrease in bioactivity following overnight incubation at 37 °C with DPP-IV enzyme.
Table 1. Biological Activities of Selected Dual Agonistsa.
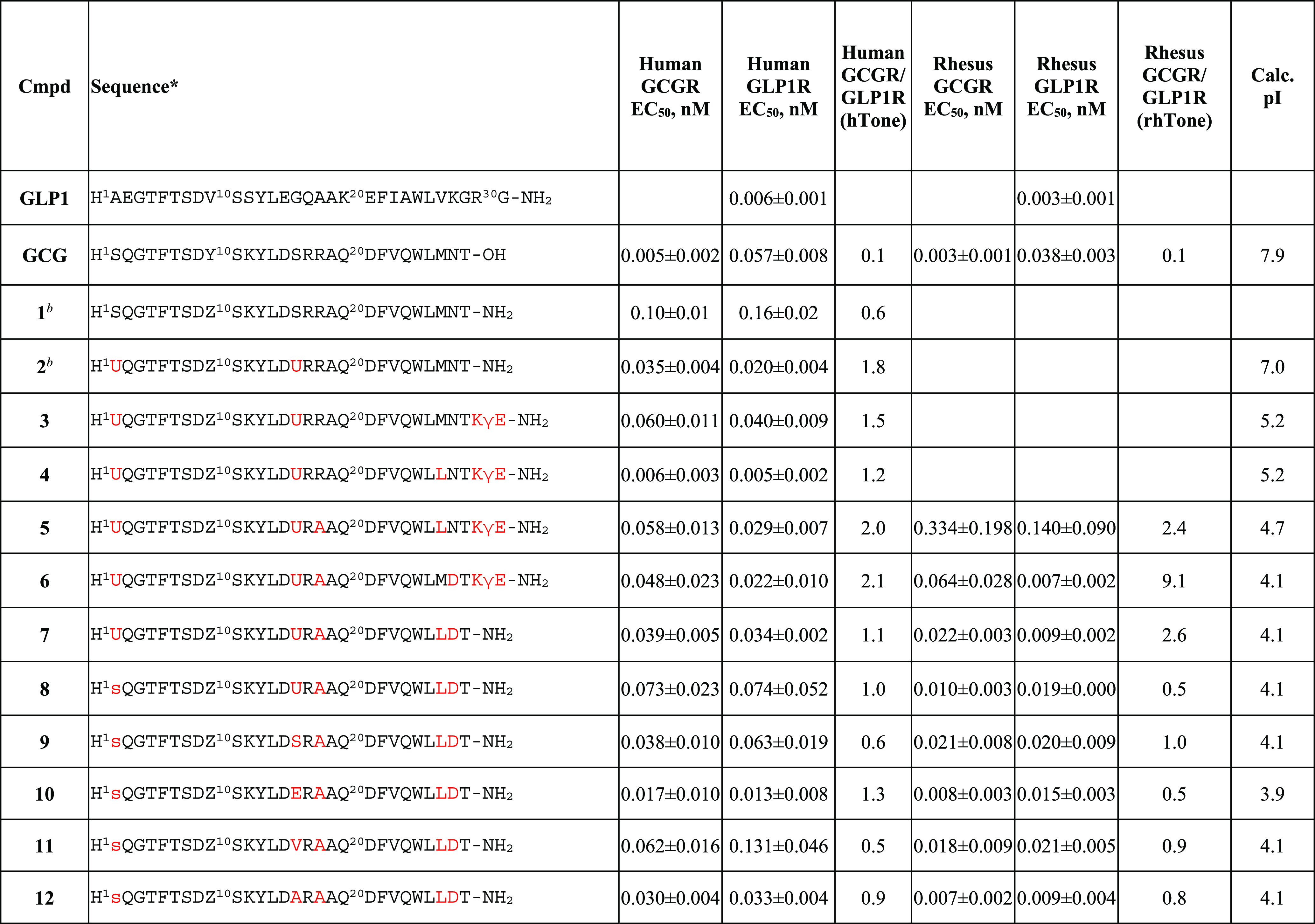
Lower case letters represent d-amino acids. Z = K(γE-γE-C16), U = Aib. Calc. pI, calculated isoelectric point. In all cases (except 12, n = 6), three replicates were measured.
Potency values were taken from literature.19
To demonstrate superior weight loss with balanced dual agonism relative to GLP1 agonist, we selected the T2DM nonhuman primate (NHP) rhesus monkey model for preclinical efficacy studies. In this study, we tested our novel GLP1R/GCGR dual agonist 12, in a cohort of diabetic rhesus monkeys. This peptide was compared to vehicle or the benchmark liraglutide (in-house rhesus receptor potency: GCGR EC50 > 2 nM, GLP1R EC50 0.06 nM) at a dose of 30 μg/kg after daily dosing for 3 weeks. The liraglutide dose was selected at a dose comparable to the 3 mg/day dose given for the treatment of obesity in humans.26
We performed this study in T2DM (noninsulin dependent) rhesus monkey because type 2 diabetic humans are known to be more refractory to weight loss than their nondiabetic, obese counterparts. Our studies demonstrated that the balanced GLP1R/GCGR dual agonist 12 dosed at 3 μg/kg/day provided superior weight loss relative to liraglutide (7.6% vs 3.3%, respectively, Figure 3A). Liraglutide reduced body weight by 1.7%, 1%, and 0.6% per week in the first, second, and third week of treatment, respectively, suggesting a deacceleration of the weight loss induced by the GLP1R selective agonist. This weight loss profile was distinct from that of 12 which reduced the NHP body weight at a constant rate throughout the entire study.
Figure 3.

(A) Chronic weight loss and (B) glucose lowering results after oral administration of 3 μg/kg of 12 relative to Liraglutide dosed at 30 μg/kg in TDM NHPs.
The body weight loss was due to a combined loss of fat and lean mass as is expected during a dynamic period of significant weight loss (data not shown). Cumulative food intake was reduced by 32% in the Lira group relative to baseline over 21 days reflective of the described anorexic effects of GLP1 agonists. Interestingly, food intake was reduced to a similar degree with 12 (−30% relative to baseline) but showed significantly greater weight loss than Lira.
Compound 12 reduced fasting glucose relative to vehicle but glucose lowering was inferior to that with liraglutide treatment in T2DM NHPs (Figure 3B). As expected, liraglutide corrected fasting plasma glucose by about 30 mg/dL after 3 weeks of treatment relative to baseline. The relative amount of glucose lowering is in good agreement with clinical observations suggesting that the effects of GLP1R agonism translates well between diabetic rhesus and humans.
Next, we sought to characterize the biomarkers that may link to the superior weight loss effect of GLP1R/GCGR dual agonists. In T2DM NHPs, 12 selectively and gradually increased FGF21 by more than 3-fold after 21 days of dosing without evidence of reaching a plateau (Figure 4A); however, the change was not statistically different due to high baseline variability. FGF21 was not changed with liraglutide relative to baseline. FGF21 has been implicated in mediating specific actions of GCG including elevation of fatty acid oxidation in the liver, lipolysis, lipid metabolism, and insulin independent glucose uptake in muscle, all together mimicking prolonged fasting conditions and associated with a caloric deficit.8,21
Figure 4.

Biomarker results. (A) FGF21, (B) β-hydroxybutyrate, (C) FFA, (D) insulin, and (E) adiponectin changes after 21 day treatment with peptide 12 dosed at 3 μg/kg in T2DM NHPs.
Consistent with GCG and FGF21 biology, 12 treatment increased markers for hepatic fatty acid oxidation and lipolysis, respectively, in T2DM animals. Fasting levels of the ketone β-hydroxybutyrate were increased ∼4-fold relative to baseline after 21 days of treatment reflective of GCGR activation in the liver (Figure 4B). Conversely, a selective elevation of free fatty acids by treatment of 12 (+63%) indicates an increase in lipolysis from adipose tissue (Figure 4C).
Coinciding with a state of energy conservation, fasting insulin was also suppressed after chronic dosing by ∼50% with 12 relative to vehicle treatment (Figure 4D). Analogously, HOMA-IR was also reduced in T2DM NHP after 21 days of dosing with 12 (vehicle 17.2 ± 4 vs 12 9.6 ± 1.9), indicating a sensitization to insulin. Liraglutide did not affect the HOMA-IR. Furthermore, total adiponectin levels gradually increased significantly with 12 but not with liraglutide relative to vehicle treatment in T2DM NHPs, concurrently with weight loss (Figure 4E). Adiponectin levels correlate inversely with adiposity and insulin sensitivity.
Given the favorable overall in vitro and in vivo profiles of 12, additional characterization was conducted (Figure 5). Detailed pharmacokinetics (PK) studies were evaluated in male Wistar Han naïve rats (n = 3 per group) and rhesus monkeys (n = 3 per group) following intravenous (IV) or subcutaneous (SC) administration. Notably, 12 exhibited low systemic clearance in both rat (0.42 mL/min/kg) and rhesus monkey (0.12 mL/min/kg) with an elimination half-life of 3 and 16 h, respectively. The volume of distribution was 0.10 L/kg in rat and 0.16 L/kg in rhesus monkey. The subcutaneous bioavailability of 12 was 18% in rat and 63% in rhesus monkey. The observed good PK in both rat and rhesus monkey supported a robust human PK predication. A target human AUC0–24h of 68 nM·h was estimated based on modeling of corresponding exposures associated with the efficacy established for glucose reduction in a rhesus monkey model. Allometric scaling of rhesus monkey CLp was used to project the PK of 12 in humans. With the assumption of a similar bioavailability of 63% and unbound fraction as observed in rhesus monkey, a dose of 180 μg QD (once daily) is projected to provide the required target AUC after SC injection in humans for glucose reduction (Figure 6).
Figure 5.

Plasma concentrations vs time profiles of 12 in rat (A: IV and B: SC) and rhesus monkey (C: IV and D: SC). 12 was formulated in mannitol buffer (5% mannitol, 5 mM sodium phosphate, pH 8), n = 3 animals/group. Concentrations of 12 were determined by LCMS assay.
Figure 6.

Rat and rhesus monkey PK of 12 and human dose prediction. *The value was assumed similar to that in rhesus following SC administration.
In summary, a literature inspired medicinal chemistry effort enabled the expedited identification of peptide 12 with potent and well-balanced receptor activities at both GLP1R and GCGG. Importantly, it provided superior solubility and stability to support the progression of this molecule in advanced studies. Efficacy studies in healthy and diseased NHPs have indicated that 12 has the potential to provide significant improvement over GLP1R-selective agonists in several markers associated with metabolic syndrome. Compound 12 afforded a 2-fold greater reduction in body weight, a greater than 3-fold increase in FGF21, compared with liraglutide. The overall preclinical safety profile supported continued development of this compound. Compound 12 was evaluated in cardiovascular studies with no significant effect on hERG current, and an overall reasonable clean profile was observed in dose limiting toxicology studies in rat and monkey. As such, based on the overall preclinical characteristics including potency, selectivity, PK, in vivo efficacy, and safety, peptide 12 was granted as development candidate MK-1462 and was advanced into the clinic.
Acknowledgments
We would like to thank Tomi Sawyer, Tom Tucker, Ryan Vargo, Sandra Souza, Keefe Chng, Qi Jiang, Lori Mixson, Lingkang Huang, Peggy Wong, Al Gilotti, Rachel Liu, Daphne Szeto, Franklin Li, Olga Price, Carlos G. Rodríguez, Jiyan Xu, Stephen Previs, Gail Forrest, Michael Crutchlow, and Eunkyung Kauh for their significant scientific contribution and discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00217.
Author Present Address
† Janssen Pharmaceuticals Inc., 1400 McKean Road, Spring House, PA 19477, USA
This work was funded entirely by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
The authors declare no competing financial interest.
Supplementary Material
References
- Müller T. D.; Blüher M.; Tschöp M. H.; DiMarchi R. D. Anti-obesity drug discovery: advances and challenges. Nat. Rev. Drug Discovery 2022, 21 (3), 201–223. 10.1038/s41573-021-00337-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauck M. A.; Wefers J.; Meier J. J. Treatment of type 2 diabetes: challenges, hopes, and anticipated successes. Lancet Diabetes & Endocrinology 2021, 9 (8), 525–544. 10.1016/S2213-8587(21)00113-3. [DOI] [PubMed] [Google Scholar]
- Nauck M. A.; Quast D. R.; Wefers J.; Meier J. J. GLP-1 receptor agonists in the treatment of type 2 diabetes - state-of-the-art. Molecular Metabolism 2021, 46, 101102. 10.1016/j.molmet.2020.101102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consoli A.; Formoso G. Potential side effects to GLP-1 agonists: understanding their safety and tolerability. Expert Opin. Drug Saf. 2015, 14 (2), 207–218. 10.1517/14740338.2015.987122. [DOI] [PubMed] [Google Scholar]
- Pirro V.; Roth K. D.; Lin Y.; Willency J. A.; Milligan P. L.; Wilson J. M.; Ruotolo G.; Haupt A.; Newgard C. B.; Duffin K. L. Effects of Tirzepatide, a Dual GIP and GLP-1 RA, on Lipid and Metabolite Profiles in Subjects With Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2022, 107 (2), 363–378. 10.1210/clinem/dgab722. [DOI] [PubMed] [Google Scholar]
- https://www.fda.gov/news-events/press-announcements/fda-approves-novel-dual-targeted-treatment-type-2-diabetes.
- Pocai A.; Carrington P. E.; Adams J. R.; Wright M.; Eiermann G.; Zhu L.; Du X.; Petrov A.; Lassman M. E.; Jiang G.; Liu F.; Miller C.; Tota L. M.; Zhou G.; Zhang X.; Sountis M. M.; Santoprete A.; Capito E.; Chicchi G. G.; Thornberry N.; Bianchi E.; Pessi A.; Marsh D. J.; SinhaRoy R. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes 2009, 58 (10), 2258–66. 10.2337/db09-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day J. W.; Ottaway N.; Patterson J. T.; Gelfanov V.; Smiley D.; Gidda J.; Findeisen H.; Bruemmer D.; Drucker D. J.; Chaudhary N.; Holland J.; Hembree J.; Abplanalp W.; Grant E.; Ruehl J.; Wilson H.; Kirchner H.; Lockie S. H.; Hofmann S.; Woods S. C.; Nogueiras R.; Pfluger P. T.; Perez-Tilve D.; DiMarchi R.; Tschöp M. H. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat. Chem. Biol. 2009, 5 (10), 749–757. 10.1038/nchembio.209. [DOI] [PubMed] [Google Scholar]
- Spezani R.; Mandarim-de-Lacerda C. A. The current significance and prospects for the use of dual receptor agonism GLP-1/Glucagon. Life Sci. 2022, 288, 120188. 10.1016/j.lfs.2021.120188. [DOI] [PubMed] [Google Scholar]
- Yang P.-Y.; Zou H.; Amso Z.; Lee C.; Huang D.; Woods A. K.; Nguyen-Tran V. T. B.; Schultz P. G.; Shen W. New Generation Oxyntomodulin Peptides with Improved Pharmacokinetic Profiles Exhibit Weight Reducing and Anti-Steatotic Properties in Mice. Bioconjugate Chem. 2020, 31, 1167–1176. 10.1021/acs.bioconjchem.0c00093. [DOI] [PubMed] [Google Scholar]
- More V. R.; Lao J.; McLaren D. G.; Cumiskey A.-M.; Murphy B. A.; Chen Y.; Previs S.; Stout S.; Patel R.; Satapati S.; Li W.; Kowalik E.; Szeto D.; Nawrocki A.; Pocai A.; Wang L.; Carrington P. Glucagon like receptor 1/ glucagon dual agonist acutely enhanced hepatic lipid clearance and suppressed de novo lipogenesis in mice. PLoS One 2017, 12 (10), e0186586 10.1371/journal.pone.0186586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson S. J.; Konkar A.; Hornigold D. C.; Trevaskis J. L.; Jackson R.; Fritsch Fredin M.; Jansson-Löfmark R.; Naylor J.; Rossi A.; Bednarek M. A.; Bhagroo N.; Salari H.; Will S.; Oldham S.; Hansen G.; Feigh M.; Klein T.; Grimsby J.; Maguire S.; Jermutus L.; Rondinone C. M.; Coghlan M. P. Robust anti-obesity and metabolic effects of a dual GLP-1/glucagon receptor peptide agonist in rodents and non-human primates. Diabetes Obes. Metab. 2016, 18 (12), 1176–1190. 10.1111/dom.12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynne K.; Park A. J.; Small C. J.; Meeran K.; Ghatei M. A.; Frost G. S.; Bloom S. R. Oxyntomodulin increases energy expenditure in addition to decreasing energy intake in overweight and obese humans: a randomised controlled trial. Int. J. Obes. (London) 2006, 30 (12), 1729–36. 10.1038/sj.ijo.0803344. [DOI] [PubMed] [Google Scholar]
- Kosinski J. R.; Hubert J.; Carrington P. E.; Chicchi G. G.; Mu J.; Miller C.; Cao J.; Bianchi E.; Pessi A.; SinhaRoy R.; Marsh D. J.; Pocai A. The Glucagon Receptor Is Involved in Mediating the Body Weight-Lowering Effects of Oxyntomodulin. Obesity 2012, 20, 1566–1571. 10.1038/oby.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Qiao A.; Yang L.; Van Eps N.; Frederiksen K. S.; Yang D.; Dai A.; Cai X.; Zhang H.; Yi C.; Cao C.; He L.; Yang H.; Lau J.; Ernst O. P.; Hanson M. A.; Stevens R. C.; Wang M.-W.; Reedtz-Runge S.; Jiang H.; Zhao Q.; Wu B. Structure of the glucagon receptor in complex with a glucagon analogue. Nature 2018, 553 (7686), 106–110. 10.1038/nature25153. [DOI] [PubMed] [Google Scholar]
- Siu F. Y.; He M.; de Graaf C.; Han G. W.; Yang D.; Zhang Z.; Zhou C.; Xu Q.; Wacker D.; Joseph J. S.; Liu W.; Lau J.; Cherezov V.; Katritch V.; Wang M. W.; Stevens R. C. Structure of the human glucagon class B G-protein-coupled receptor. Nature 2013, 499 (7459), 444–9. 10.1038/nature12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koth C. M.; Murray J. M.; Mukund S.; Madjidi A.; Minn A.; Clarke H. J.; Wong T.; Chiang V.; Luis E.; Estevez A.; Rondon J.; Zhang Y.; Hötzel I.; Allan B. B. Molecular basis for negative regulation of the glucagon receptor. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (36), 14393–14398. 10.1073/pnas.1206734109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K.; Dockerill S.; Adamiak D. A.; Tickle I. J.; Blundell T. X-ray analysis of glucagon and its relationship to receptor binding. Nature 1975, 257 (5529), 751–757. 10.1038/257751a0. [DOI] [PubMed] [Google Scholar]
- Ward B. P.; Ottaway N. L.; Perez-Tilve D.; Ma D.; Gelfanov V. M.; Tschöp M. H.; DiMarchi R. D. Peptide lipidation stabilizes structure to enhance biological function. Molecular Metabolism 2013, 2 (4), 468–479. 10.1016/j.molmet.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon C. F.; Knudsen L. B.; Madsen K.; Wiberg F. C.; Jacobsen O.; Holst J. J. Dipeptidyl peptidase IV resistant analogues of glucagon-like peptide-1 which have extended metabolic stability and improved biological activity. Diabetologia 1998, 41 (3), 271–278. 10.1007/s001250050903. [DOI] [PubMed] [Google Scholar]
- Day J. W.; Gelfanov V.; Smiley D.; Carrington P. E.; Eiermann G.; Chicchi G.; Erion M. D.; Gidda J.; Thornberry N. A.; Tschöp M. H.; Marsh D. J.; SinhaRoy R.; DiMarchi R.; Pocai A. Optimization of co-agonism at GLP-1 and glucagon receptors to safely maximize weight reduction in DIO-rodents. Biopolymers 2012, 98 (5), 443–50. 10.1002/bip.22072. [DOI] [PubMed] [Google Scholar]
- Patterson J. T.; Day J. W.; Gelfanov V. M.; DiMarchi R. D. Functional association of the N-terminal residues with the central region in glucagon-related peptides. J. Pept. Sci. 2011, 17 (10), 659–66. 10.1002/psc.1385. [DOI] [PubMed] [Google Scholar]
- Evers A.; Haack T.; Lorenz M.; Bossart M.; Elvert R.; Henkel B.; Stengelin S.; Kurz M.; Glien M.; Dudda A.; Lorenz K.; Kadereit D.; Wagner M. Design of Novel Exendin-Based Dual Glucagon-like Peptide 1 (GLP-1)/Glucagon Receptor Agonists. J. Med. Chem. 2017, 60 (10), 4293–4303. 10.1021/acs.jmedchem.7b00174. [DOI] [PubMed] [Google Scholar]
- Kerr B. D.; Flatt P. R.; Gault V. A. (d-Ser2)Oxm[mPEG-PAL]: A novel chemically modified analogue of oxyntomodulin with antihyperglycaemic, insulinotropic and anorexigenic actions. Biochem. Pharmacol. 2010, 80 (11), 1727–1735. 10.1016/j.bcp.2010.08.010. [DOI] [PubMed] [Google Scholar]
- Santoprete A.; Capitò E.; Carrington P. E.; Finotto M.; Langella A.; Ingallinella P.; Zytko K.; Bufali S.; Cianetti S.; Veneziano M.; Bonelli F.; Zhu L.; Monteagudo E.; Marsh D. J.; Sinharoy R.; Bianchi E.; Pessi A. DPP-IV-resistant, long-acting oxyntomodulin derivatives. J. Peptide Sci. 2011, 17 (4), 270–280. 10.1002/psc.1328. [DOI] [PubMed] [Google Scholar]
- Astrup A.; Rössner S.; Van Gaal L.; Rissanen A.; Niskanen L.; Al Hakim M.; Madsen J.; Rasmussen M. F.; Lean M. E. J. Effects of liraglutide in the treatment of obesity: a randomised, double-blind, placebo-controlled study. Lancet 2009, 374 (9701), 1606–1616. 10.1016/S0140-6736(09)61375-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.