

Abstract
A virtual screening approach based on a five-feature pharmacophoric model for negative modulators of GLI1 was applied to databases of commercially available compounds. The resulting quinoline derivatives showed significant ability to reduce the GLI1 protein level and were characterized by submicromolar antiproliferative activity toward human melanoma A375 and medulloblastoma DAOY cell lines. Decoration of the quinoline ring and chemical rigidification to an oxazino-quinoline scaffold allowed us to deduce SAR considerations for future ligand optimization.
Keywords: GLI1 negative modulators, quinoline, Hedgehog pathway, anticancer agents, pharmacophoric model, virtual screening
The canonical Hedgehog-GLI (Hh) pathway is initiated by binding of Hh ligands to the receptor Patched (PTCH1), which releases the inhibition of the transmembrane G protein coupled receptor Smoothened (SMO). The latter, in turn, triggers an intracellular cascade, which leads to the activation of the GLI transcription factors (TFs). There is also a noncanonical activation of the GLI TFs, which can be PTCH-SMO-independent and is triggered by several oncogenic signaling pathways.1 The Hh signaling can be inhibited at the level of SMO or the TFs GLI1/2. SMO is the main target of Hh inhibition. So far, five inhibitors of SMO (namely, NVP-LDE225, referred to as Sonidegib or Erismodegib, and NVP-LEQ506, referred to as LEQ506, both by Novartis; PF-04449913, referred to as Glasdegib by Pfizer; BMS-833923, referred to as XL139 by Exelixis and out-licensed to Bristol-Myers Squibb; GDC-0449, referred to as Vismodegib by Genentec) accessed clinical trials and instilled hope for the treatment of human cancers such as medulloblastoma and basal cell carcinoma, which depend on the canonical Hh pathway activation. On the contrary, the same compounds were unsuccessful in treating many solid tumors that are dependent on the noncanonical activation of GLI1.1 Indeed, negative modulators of GLI1 are currently represented by small molecules that can interfere with both canonical and noncanonical Hh signaling. Based on these results and considering that SMO inhibitors often select resistant mutations (primary or secondary resistance),2,3 many efforts were focused on the identification of small molecules able to directly target GLI1 and negatively modulate its activity.4−10 Few GLI1/2 inhibitors were identified thus far. The first GLI1/2 inhibitor described was GANT61, which blocks translocation of GLI1 to the nucleus and its binding to DNA.11,12 Arsenic trioxide, which is not a specific GLI inhibitor, was shown to abrogate ciliary accumulation of GLI213 and to reduce GLI1 mRNA and protein levels.14 Moreover, polyunsaturated fatty acids showed the ability to inhibit GLI.15 Additional small molecules identified by virtual screening approaches were also described in recent years. Unfortunately, toxicity and poor drug likeness are two major limitations of currently available GLI1 inhibitors.4,6,16
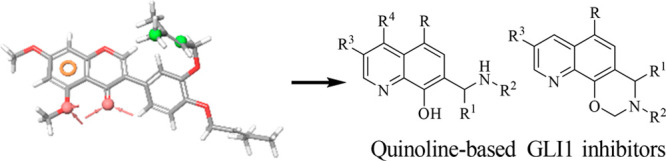
Recently, glabrescione B (GlaB, Table 1)17 was used by us as the initial scaffold for the generation of a five-feature pharmacophoric model that led to the identification of negative modulators of GLI1 belonging to the classes of thiophene and pyrazolo-pyrimidine small molecules that populated databases of commercially available compounds.18 In addition, the same pharmacophore allowed the prioritization of several 8-hydroxyquinoline derivatives as putative GLI1 binders that are described here. Such compounds showed a scaffold similar to that of an 8-hydroxyquinoline derivative identified by a structure-based virtual screening protocol based on biased molecular docking simulations that was focused on the binding site of GANT61 within the structural region of GLI1 comprised between zinc-fingers 2 and 3.19
Table 1. Chemical Structure, Antiproliferative Activity, and Effect on GLI1 Properties of the Quinoline and Oxazino-Quinoline Derivatives.

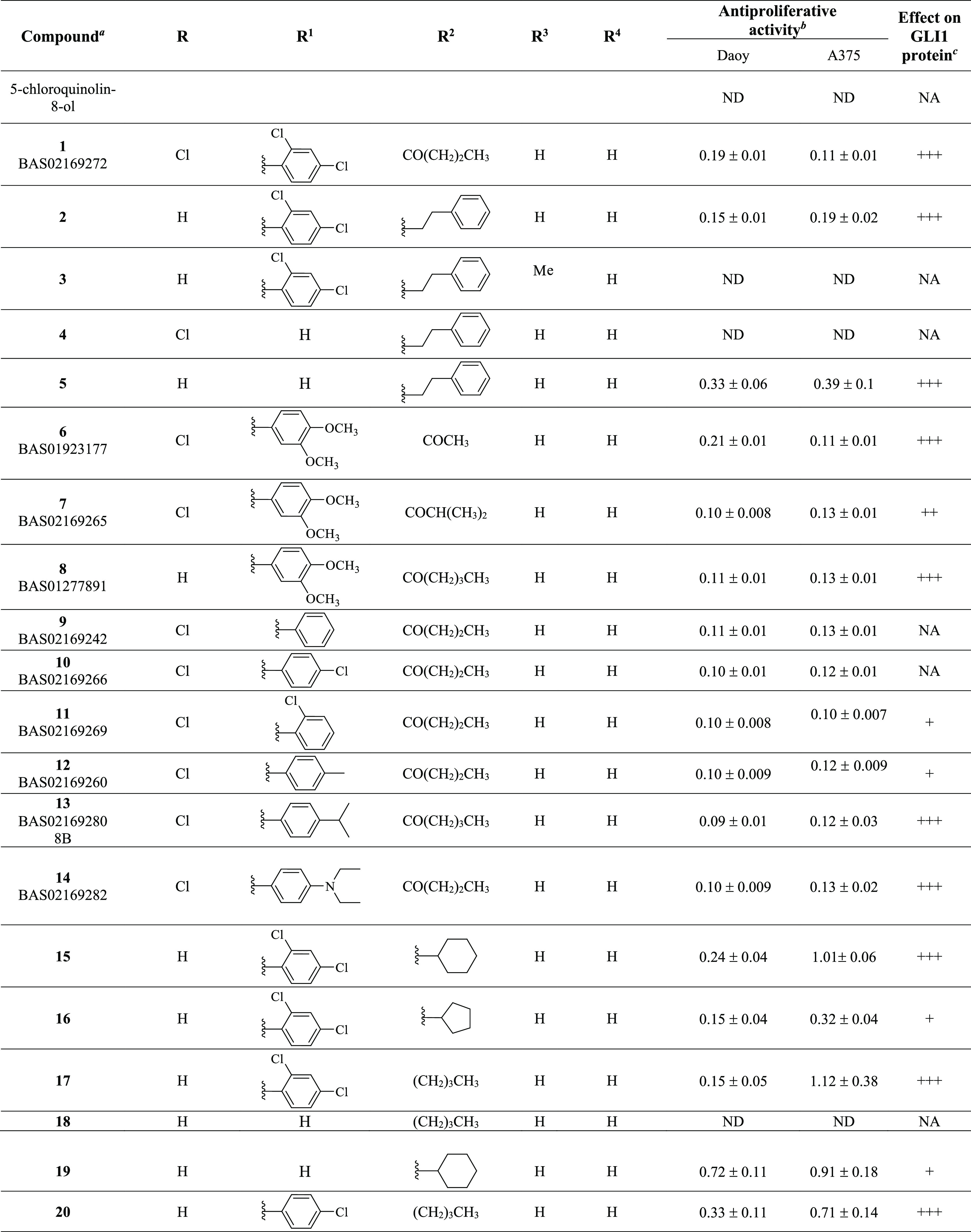
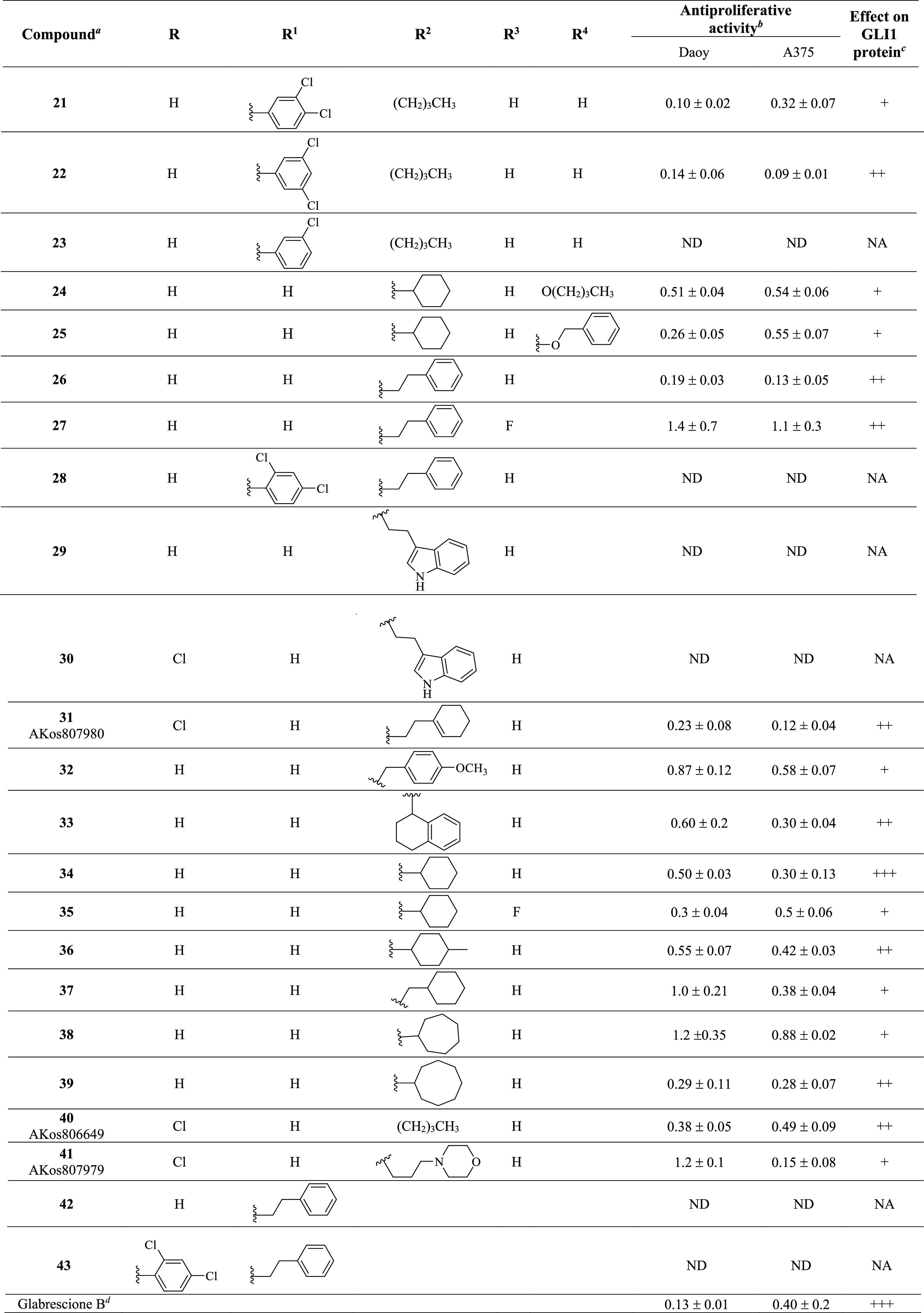
Compounds also labeled with BAS and AKos notations were purchased from Asinex and AKos vendors, respectively.
Expressed as IC50 values (micromolar concentrations) calculated using GraphPad Prism V6 from triplicate experiments in melanoma (A375) and medulloblastoma (DAOY) cells. ND: not determined.
The GLI1 protein level was determined by Western blotting (see Figure 4 for details) in murine NIH3T3 cells treated with SAG (100 nM), each compound (1 μM), and GANT61 (5 μM) for 48 h. NA: not affected; weakly reduced: +; reduced: ++; strongly reduced: +++.
Activity data taken from ref (18).
As a result of our pharmacophore-based virtual screening on databases of commercially available compounds, 1 was identified as a small molecule able to fully match the five pharmacophoric features (Figure 1): the quinoline nitrogen and the oxygen atom of the phenol substituent represented the hydrogen bond acceptors (HBAs), while the terminal propyl chain together with the 2-Cl substituent at the pendant phenyl ring were superposed to the hydrophobic features (HYs). Finally, the ring aromatic (RA) feature was fitted by the pyridine ring. Such a compound strongly reduced the GLI1 protein level in murine NIH3T3 cells treated with 100 nM SAG and 1 μM test compound for 48 h. Moreover, a submicromolar antiproliferative activity was found toward the medulloblastoma DAOY and the melanoma A375 cell lines (IC50 = 0.19 and 0.11 μM, respectively, Table 1). Overall activity found for 1 was comparable to that of GlaB (IC50 = 0.13 and 0.40 μM, respectively, and a strong reduction of GLI1 level, Table 1).
Figure 1.
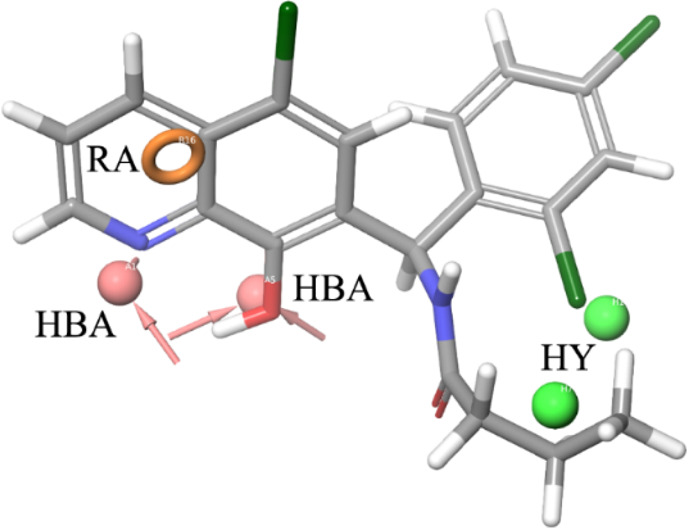
Graphical representation of 1 superposed to the five-feature pharmacophoric model for GLI1 negative modulators. The hydrogen bond acceptor (HBA) groups are represented by the quinoline nitrogen atom and the oxygen atom at C8 of the quinoline ring. The terminal propyl substituent and the 2-Cl at the pendant phenyl ring fit the hydrophobic regions (HYs). Finally, the pyridine ring corresponds to the ring aromatic (RA) feature.
A computational protocol previously described by us20−22 was used to identify putative binding sites on GLI1 and to prioritize new ligands for these pockets. In details, the SiteMap routine of the Glide software was applied to the crystallographic five-finger structure of GLI23 with the aim of finding putative binding sites for small molecule ligands. Molecular docking simulations of 1 were then focused into the two putative binding sites identified by SiteMap. As a result, a best scored binding pose was characterized by an extended network of hydrogen bonds (Figure 2, left, and Figure 3). In particular, the pyridine ring made a π–cation interaction with Arg223 of the zinc-finger 4 (numbers of amino acids refer to the 2gli structure reported in the Protein Data Bank; the corresponding UNIPROT numbers can be obtained by adding 131 units: as an example, the Arg223 reported in the Protein Data Bank corresponds to Arg354 of the UNIPROT P08151 sequence), and the quinoline nitrogen atom gave a hydrogen bond with the NH backbone moiety of His220 (one of the residues involved in coordination of a zinc ion). Moreover, the OH group of the ligand interacted with both the imidazole ring NH of His220 and with the hydroxyl group of Thr224, which in turn gave a hydrogen bond with the carbonyl oxygen of the ligand amide group. This binding pose, however, did not account for the hydrophobic features predicted by the pharmacophoric model. Among the best scored binding modes of 1, an alternative pose was found (Figure 2, right, and Figure 3), where the ligand interacted with amino acids of the zinc-fingers 1 and 3, within the same binding pocket already found for another 8-hydroxyquinoline derivative19 and for the known GLI1 inhibitor GANT61.12 The major anchor points were represented by hydrogen bonds between both the quinoline nitrogen and the 8-OH substituent with the terminal edge of Lys171, between the 8-OH group and the carboxyl moiety of Glu119, and between the amide oxygen of the ligand and the terminal NH2 group of Gln118. Hydrophobic contacts were also found between the propyl terminal of 1 and the central portion of the Gln118 side chain as well as between the pendant phenyl ring and the central part of the Lys168 side chain.
Figure 2.

Graphical representation of the best-docked binding poses of 1 (ball & stick and element + gray carbon notations) within the structure of GLI1. A complex network of hydrogen bonds (light green dashed lines) is formed by GLI1 and the ligand. Left: The quinoline nitrogen, the 8-OH group, and the oxygen of the ester moiety form hydrogen bonds with the backbone and the side chain NH of His220 as well as with the side chain OH of Thr224. Moreover, the pyridine ring and the guanidino group of Arg223 give a π–cation interaction (green dashed line). Right: In an alternative binding pose, the terminal ammonium group of Lys171 makes hydrogen bonds with both the quinoline nitrogen and the 8-OH group of 1. The latter substituent gives an additional hydrogen bond with the terminal carboxyl group of Glu119. The amide oxygen of the ligand and the terminal NH2 of Gln118 provide another hydrogen bond. The propyl chain and the pendant phenyl ring make hydrophobic interactions with the target.
Figure 3.
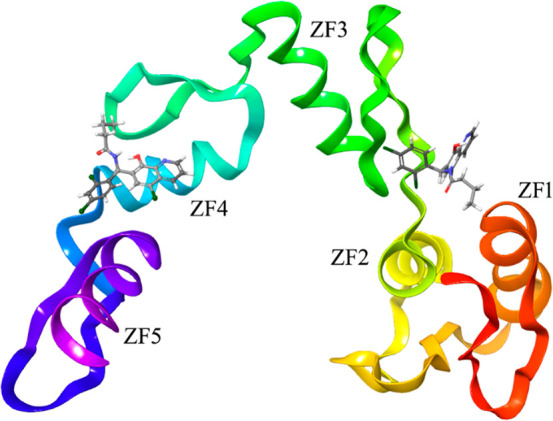
Graphical representation of the zinc-finger portion of the GLI1 structure, as stored in the entry 2gli of the Protein Data Bank.23 Two alternative best-docked binding poses of the pyridine derivatives are also represented to show the locations of the different binding sites identified by the SiteMap routine of the Schrodinger suite. One binding site is located within the zinc-finger 4, while an alternative binding region is between the zinc-fingers 1 and 3, and it was hypothesized as the binding region of GANT6112 and another 8-hydroxyquinoline derivative.19
Starting from the commercially available parent compound 1, which was prioritized by the virtual screening approach, a focused library was built around the quinoline scaffold in the attempt to improve the biological profile and to deduce SAR considerations for further optimization. Table 1 summarizes the structure, antiproliferative activity, and ability to affect the GLI1 protein level of the new compounds. To assess the effects of compounds on GLI1 protein level, we used murine NIH3T3 cells, a well-established model to study the modulation of Hh signaling. Treatment of these cells with the SMO agonist SAG activates the endogenous Hh pathway. In Figure 4, we reported representative examples of compounds with different effects on the GLI1 protein level. For instance, 6, 15, and 17 strongly reduced GLI1 protein levels compared to SAG-treated NIH3T3 cells and were classified as “+++” (see Table 1). Compounds 7, 26, 36, 39, and 22 led to a moderate reduction of GLI1 protein expression and were classified as “++”. Compound 19 weakly reduced GLI1 protein expression and was classified as “+”. On the other hand, compound 18 did not affect GLI1 protein levels and was classified as “NA” (not affected).
Figure 4.

Representative Western blot of endogenous GLI1 in NIH3T3 cells stimulated with the Smoothened agonist SAG (100 nM) and treated with vehicle (DMSO), our putative GLI1 inhibitors (1 μM), and GANT61 (5 μM) for 48 h. HSP90 was used as loading control. See Table 1 for details.
Increasing the size and bulkiness of the propyl ester moiety into a phenylethylamino appendage and removing the chloride substituent at the C5 of 1 led to 2, which retained a similar antiproliferative profile in DAOY and A375 cancer cells (IC50 = 0.15 and 0.19 μM) and a similar degree of reduction of GLI1. Insertion of a methyl group at the quinoline C3 as in 3 abrogated the activity on the GLI1 level. In a similar way, removal of the pendant phenyl ring as in 4 completely abrogated reduction of GLI1 protein level that was fully restored by further removal of the C5 chlorine substituent as in 5, together with a submicromolar antiproliferative activity (IC50 = 0.33 and 0.39 μM, respectively in DAOY and A375 cells).
Replacement of the dichloro substituents with a 3,4-dimethoxy substitution, independently from the length of the ester chain (methyl, isopropyl, and butyl as in 6, 7, and 8, respectively) and the presence of a C5 chlorine substitution (R group) led to an antiproliferative activity in the submicromolar range (between 0.10 and 0.21 μM) and a significant reduction of the GLI1 protein level. Worth noting, 6 strongly inhibited GLI1 expression (Figure 4). Further changing either the substituents or the substitution pattern on the pendant phenyl ring (at R1) resulted in propyl or butyl esters that maintained the submicromolar antiproliferative activity, while the effect on GLI1 protein level was variable. As examples, the unsubstituted derivative 9 and the corresponding 4-Cl analogue 10 did not affect the GLI1 protein level, while the 2-Cl 11 and 4-Me 12 analogues showed weak reduction. A submicromolar antiproliferative activity and a strong reduction of the GLI1 protein level were also found for both the 4-iPr and 4-diethylamino derivatives 13 and 14, respectively.
Moreover, replacement of the ester or aryl side chain at R2 with an alkyl or cycloalkyl substituent gave interesting results. As an example, 15 and its cyclopentyl and butyl analogues (16 and 17, respectively) significantly affected the GLI1 protein level (Figure 4) and showed micromolar antiproliferative activity, while the butyl analogue 18, which was deprived of the pendant phenyl ring, was inactive. This result seemed to suggest the benzyl moiety at the quinoline C7 as an important substituent for activity, although the simplified analogue of 15 (namely, 19) maintained micromolar antiproliferative activity and weak GLI1 protein level reduction. Moreover, the phenylethylamino derivative 4 was also inactive, while a good biological profile was restored when its C5 chlorine substituent was removed as in 5.
Among butylamino analogues, compounds where the 2,4-diCl substituents and substitution pattern at R1 were changed (as in 20, 21, and 22), were characterized by a submicromolar antiproliferative activity and variable reduction of the GLI1 protein level. The sole exception was represented by the 3-Cl analogue 23 that was inactive.
Worth noting, insertion of an alkoxy substituent at the quinoline C4 of the cyclohexylamino derivative 19 (R4) led to 24 and 25, which showed a slightly better antiproliferative activity (IC50 between 0.26 and 0.55 μM), with only a modest reduction of the GLI1 protein level.
Biological data on 8-hydroxyquinoline derivatives suggested several SAR considerations. The antiproliferative activity seemed to be slightly dependent on the substituents and substitution pattern on the phenyl ring of the C7 benzyl moiety. In fact, IC50 values were in the single-digit micromolar or submicromolar range. On the contrary, the effect on the GLI1 protein level was affected by these molecular features. As an example, unsubstituted compounds (as 9) or those bearing a small substituent (such as a Cl or Me as found in 10, 11, 23, and 12) showed low or no ability to affect the GLI1 protein level. An exception was represented by 20 that almost abrogated GLI1 protein expression. However, the C7 benzyl moiety was not a mandatory structural feature for compound activity. In fact, although the butyl derivative 18 was inactive, increasing the size of the R2 substituent to a cyclohexyl ring (as in 19, 25, and 24) gradually restored activity. Finally, the simple 5-chloroquinolyl-8-ol was inactive.
Following a structural rigidification strategy, the methanamino moiety and the hydroxyl substituents present at the quinoline C7 and C8, respectively, were connected to give a condensed dihydro-1,3-oxazine ring. The N-phenylethyl analogue 26 showed submicromolar antiproliferative activity toward both cell lines (IC50 = 0.19 and 0.13 μM, respectively, Table 1) and a significant reduction of the GLI1 protein level (Figure 4). Docking simulations showed a binding pose where the heteroatoms of the condensed scaffold were involved in a network of hydrogen bonds (Figure 5). In particular, the nitrogen atom of the quinoline system interacted with the imidazole ring of His220, while the oxygen and nitrogen atoms of the oxazine ring gave hydrogen bonds with the hydroxyl groups of Thr224 and Thr243, respectively. Similarly to the 8-hydroxyquinoline derivatives, also the oxazino-quinoline showed an alternative best scored docking pose within the putative binding site of GANT61 (not shown).
Figure 5.
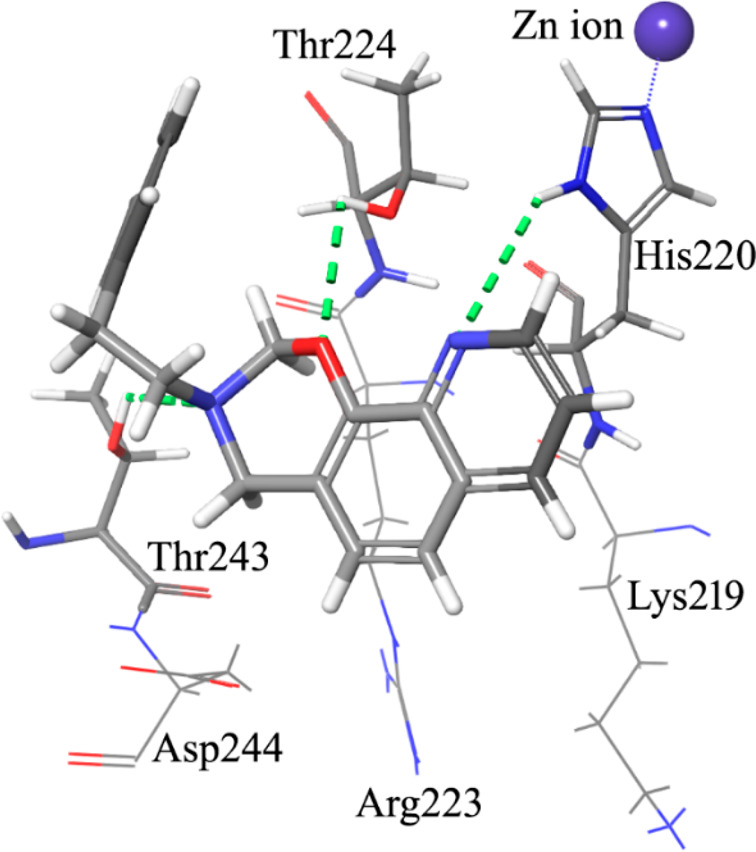
Graphical representation of the docked pose of the oxazino-quinoline derivative 26. The quinoline nitrogen gives a hydrogen bond with the imidazole ring of His220. Two additional hydrogen bonds are made by the oxygen and the nitrogen atom of the oxazine ring with the side chain OH groups of Thr224 and Thr243.
The antiproliferative activity decreased by about 10-fold in the C8 (the R3 substituent) fluoro derivative 27 (IC50 between 1.4 and 1.1 μM, respectively), and the insertion of a substituted phenyl ring at the oxazine C4 was further detrimental for the activity of 28 that was inactive. Replacing the pendant phenyl ring of 26 with an indolyl moiety resulted in 29 and 30 that were both inactive too. On the other hand, partial reduction of the phenyl moiety to a cyclohexene ring, shortening to a benzyl chain, and rigidification to a tetrahydronaphthyl ring led to 31, 32, and 33, respectively, which showed submicromolar antiproliferative activity toward both cell lines (IC50 between 0.12 and 0.87 μM) and a significant reduction of the GLI1 protein level. Interestingly, when the condensed phenyl ring of 33 was simplified to have the corresponding cyclohexyl analogue 34, the antiproliferative activity was maintained, while the GLI1 protein level was strongly reduced. Insertion of a fluorine substituent at the quinoline C8 resulted in 35 with a reduced ability to affect the GLI1 protein level. Methylation at the cyclohexyl C4 of 34 resulted in 36 with a reduced activity. In a similar way, lengthening the R2 side chain by insertion of a methylene bridge as in 37 maintained a micromolar antiproliferative activity but weakly affected the GLI1 protein level. Moreover, homologation of the cyclohexyl moiety of 34 into a cycloheptyl ring as in 38 caused a reduction of the antiproliferative activity and ability to affect the GLI1 protein level. An improvement of the biological profile was obtained with further enlargement to the octyl ring of 39 that showed IC50 = 0.29 and 0.28 μM, respectively, and a significant reduction of GLI1 protein level. Simplification of the R2 substituent to a butyl chain (as in 40) retained interesting activity, while adding a morpholinyl appendage as in 41 negatively affected both the antiproliferative activity toward DAOY cells and the ability to reduce GLI1 protein level.
Among oxazino-quinoline condensed compounds, as a general rule, the presence of a C8 substituent was detrimental for activity (compare 34 with 35 and 26 with 27), as well as the presence of a substituent at the C4 of the oxazine ring (compare 26 with 28). On the other hand, the presence of a substituent at the C6 (the R substituent) did not affect the activity (compare 29 to 30).
Finally, transformation of the dihydro-1,3-oxazine ring into the corresponding N-phenylethyl cyclic carbamates yielded 42 and 43 that were inactive.
Overall, many of the quinoline derivatives showed an antiproliferative activity in the submicromolar range, significantly better (in general, at least 1 order of magnitude higher) in comparison to the thiophene and pyrazolo-pyrimidine compounds previously identified by means of the same pharmacophore model.18 Cyclization of the C7–C8 quinoline substituents to a condensed oxazine ring allowed a further enlargement of SAR considerations.
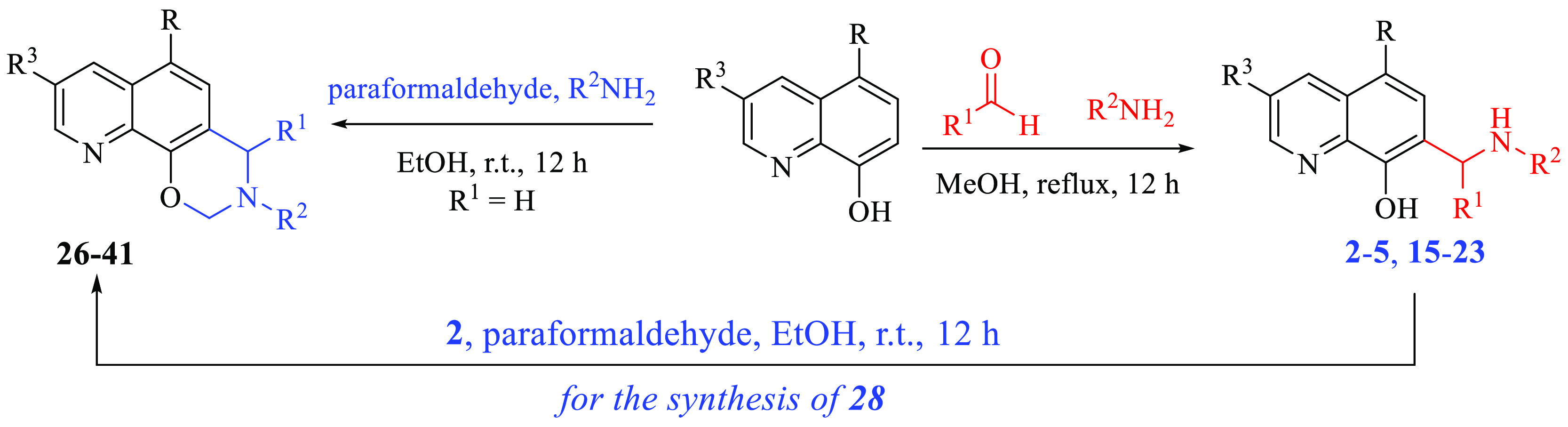
Expected derivatives 2–5 and 15–23 were efficiently synthesized starting from commercially available properly substituted 8-hydroxyquinolines by treatment with different aldehydes and amines in MeOH at reflux (Scheme 1). Tricyclic compounds 26–41 were obtained from the same starting materials by using paraformaldehyde and proper amines at room temperature in EtOH (Scheme 1).
Scheme 1. Preparation of 2–5, 15–23, and 26–41.
For the preparation of 4-OR derivatives 24 and 25, a decarboxylation of xanthurenic acid under microwave irradiation at 255 °C followed by a selective protection of the phenol moiety in the C-8 position were required (Scheme 2). The intermediates obtained were alkylated by treatment with the proper alkyl halide, deprotected, and functionalized at their C7 in the same reaction conditions reported for 2–5 and 15-23.
Scheme 2. Preparation of 4-OR Derivatives 24 and 25.

Tricyclic carbamates 42 and 43 were prepared from 5 and 2, respectively, by treatment with N,N′-carbonyldiimidazole (CDI) in the presence of Et3N at reflux (Scheme 3).
Scheme 3. Preparation of Tricyclic Carbamates 42 and 43.

In conclusion, a virtual screening procedure based on a five-feature pharmacophoric model led to prioritizing 8-hydroxyquinoline derivatives as negative modulators of GLI1 with submicromolar antiproliferative activity toward both human melanoma and medulloblastoma cell lines. Many of these compounds also showed ability to strongly reduce the GLI1 protein level in NIH3T3 cells. Decoration of the quinoline ring and its rigidification to an oxazino-quinoline moiety allowed to deduce SAR considerations that could be usefully applied for future compound optimization.
Acknowledgments
The authors would like to thank Dr. G. Battistuzzi (R&D Alfasigma S.p.A., formerly Sigma-Tau IFR S.p.A) for assistance with a few aspects of this project, Dr. A. Noseda (Leadiant Biosciences SA) for facilitating this study, as well as Dr. E. Monciatti and F. Migliorini for some NMR analyses. Glabrescione B was a gift from B. Botta and L. Di Marcotullio, University of Rome La Sapienza.
Glossary
Abbreviations
- Hh
Hedgehog
- PTCH1
Patched
- SMO
Smoothened
- TF
transcription factor
- GlaB
glabrescione B
- CDI
N,N′-carbonyldiimidazole
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00249.
Details of the computational protocols, synthetic procedures, analytical data, and biological assays (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported in part by the research grant from the AIRC foundation (grant IG 2017 n. 20758), by the project “Development and application of QM/MM technologies for the design of light responsive proteins or protein-mimics based on rhodopsin architecture” within the program “Dipartimenti di Eccellenza -2018–2022 financed by MIUR (Roma, Italy) and by Bando Ricerca Salute 2018 financed by Tuscany Region (project GLI SELTHER). We are grateful for funding obtained from the Institute for Cancer Research, Prevention and Clinical Network (ISPRO). L. Maresca is supported by a postdoctoral fellowship from the Italian Association for Cancer Research (AIRC, project n. 22644). Additional financial support was provided by Leadiant Biosciences S.A. (Mendrisio, CH) (formerly Sigma-Tau Research Switzerland, S.A.). The MIUR research project 2015LZE994 (Insights into the functions of DNA damage processing and repair factors to design novel selective anticancer drugs) is also acknowledged.
The authors declare the following competing financial interest(s): G. Giannini participated in the project as Project Leader, thanks to the agreement signed between Leadiant Bioscience and Sigma-Tau IFR SpA (now Alfasigma SpA). Part of the results described herein has been the subject of a patent application: "GLI inhibitors and uses thereof" Application No EP 17166194.5 April 12, 2017. EP3388419A1. Giannini, G.; Taddei, M.; Manetti, F.; Petricci, E.; Stecca, B.
Supplementary Material
References
- Pietrobono S.; Gagliardi S.; Stecca B. Non-canonical Hedgehog signaling pathway in cancer: activation of GLI transcription factors beyond Smoothened. Front. Genet. 2019, 10, 556. 10.3389/fgene.2019.00556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danhof R.; Lewis K.; Brown M. Small molecule inhibitors of the Hedgehog pathway in the treatment of basal cell carcinoma of the skin. Am. J. Clin. Dermatol. 2018, 19, 195–207. 10.1007/s40257-017-0319-4. [DOI] [PubMed] [Google Scholar]
- Nguyen N. M.; Cho J. Hedgehog pathway inhibitors as targeted cancer therapy and strategies to overcome drug resistance. Int. J. Mol. Sci. 2022, 23, 1733. 10.3390/ijms23031733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery J. T.; Zhang R.; Boohaker R. J. GLI1: a therapeutic target for cancer. Front. Oncol. 2021, 11, 673154. 10.3389/fonc.2021.673154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peer E.; Tesanovic S.; Aberger F. Next-generation Hedgehog/GLI pathway inhibitors for cancer therapy. Cancers 2019, 11, 538. 10.3390/cancers11040538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.; Ma J.; Avery J. T.; Sambandam V.; Nguyen T. H.; Xu B.; Suto M. J.; Boohaker R. J. GLI1 inhibitor SRI-38832 attenuates chemotherapeutic resistance by downregulating NBS1 transcription in BRAFV600E colorectal cancer. Front. Oncol. 2020, 10, 241. 10.3389/fonc.2020.00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lospinoso Severini L.; Ghirga F.; Bufalieri F.; Quaglio D.; Infante P.; Di Marcotullio L. The SHH/GLI signaling pathway: a therapeutic target for medulloblastoma. Expert Opin. Ther. Targets 2020, 24, 1159–1181. 10.1080/14728222.2020.1823967. [DOI] [PubMed] [Google Scholar]
- Bariwal J.; Kumar V.; Dong Y.; Mahato R. I. Design of Hedgehog pathway inhibitors for cancer treatment. Med. Res. Rev. 2019, 39, 1137–1204. 10.1002/med.21555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman J. M.; Firestone A. J.; Heine V. M.; Zhao Y.; Ocasio C. A.; Han K.; Sun M.; Rack P. G.; Sinha S.; Wu J. J.; Solow-Cordero D. E.; Jiang J.; Rowitch D. H.; Chen J. K. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 14132–14137. 10.1073/pnas.0907134106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrangelo E.; Milani M. Role and inhibition on GLI1 protein in cancer. Lung Cancer Targets Ther. 2018, 9, 35–43. 10.2147/LCTT.S124483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauth M.; Bergström A.; Shimokawa T.; Toftgård R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 8455–8460. 10.1073/pnas.0609699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agyeman A.; Jha B. K.; Mazumdar T.; Houghton J. A. Mode and specificity of binding of the small molecule GANT61 to GLI determines inhibition of GLI-DNA binding. Oncotarget 2014, 5, 4492–4503. 10.18632/oncotarget.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Lee J. J.; Kim J.; Gardner D.; Beachy P. A. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 13432–13437. 10.1073/pnas.1006822107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X.; Yu K.; Zhang L.; Li Y.; Li Q.; Yang Z.; Shen T.; Duan L.; Xiong W.; Wang W. Synergistic inhibition of colon carcinoma cell growth by Hedgehog-Gli1 inhibitor arsenic trioxide and phosphoinositide 3-kinase inhibitor LY294002. OncoTargets Ther. 2015, 8, 877–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comba A.; Almada L. L.; Tolosa E. J.; Iguchi E.; Marks D. L.; Vara Messler M.; Silva R.; Fernandez-Barrena M. G.; Enriquez-Hesles E.; Vrabel A. L.; Botta B.; Di Marcotullio L.; Ellenrieder V.; Eynard A. R.; Pasqualini M. E.; Fernandez-Zapico M. F. Nuclear factor of activated T cells-dependent down-regulation of the transcription factor glioma-associated protein 1 (GLI1) underlies the growth inhibitory properties of arachidonic acid. J. Biol. Chem. 2016, 291, 1933–1947. 10.1074/jbc.M115.691972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai J. Y.; Sugumar V.; Alshawsh M. A.; Wong W. F.; Arya A.; Chong P. P.; Looi C. Y. The role of Smoothened-dependent and -independent Hedgehog signaling pathway in tumorigenesis. Biomedicines 2021, 9, 1188. 10.3390/biomedicines9091188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infante P.; Mori M.; Alfonsi R.; Ghirga F.; Aiello F.; Toscano S.; Ingallina C.; Siler M.; Cucchi D.; Po A.; Miele E.; D’Amico D.; Canettieri G.; De Smaele E.; Ferretti E.; Screpanti I.; Uccello Barretta G.; Botta M.; Botta B.; Gulino A.; Di Marcotullio L. Gli1/DNA interaction is a druggable target for Hedgehog-dependent tumors. EMBO J. 2015, 34, 200–217. 10.15252/embj.201489213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manetti F.; Stecca B.; Santini R.; Maresca L.; Giannini G.; Taddei M.; Petricci E. Pharmacophore-based virtual screening for identification of negative modulators of GLI1 as potential anticancer agents. ACS Med. Chem. Lett. 2020, 11, 832–838. 10.1021/acsmedchemlett.9b00639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash R. C.; Wen J.; Zaino A. M.; Morel S. R.; Chau L. Q.; Wechsler-Reya R. J.; Hadden K. M. Structure-based virtual screening identifies an 8-hydroxyquinoline as a small molecule GLI1 inhibitor. Mol. Ther. Oncolytics 2021, 20, 265–276. 10.1016/j.omto.2021.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manetti F.; Faure H.; Roudaut H.; Gorojankina T.; Traiffort E.; Schoenfelder A.; Mann A.; Solinas A.; Taddei M.; Ruat M. Virtual screening-based discovery and mechanistic characterization of the acylthiourea MRT-10 family as Smoothened antagonists. Mol. Pharmacol. 2010, 78, 658–665. 10.1124/mol.110.065102. [DOI] [PubMed] [Google Scholar]
- Badolato M.; Carullo G.; Perri M.; Cione E.; Manetti F.; Di Gioia M. L.; Brizzi A.; Caroleo M. C.; Aiello F. Quercetin/oleic acid-based G-protein-coupled receptor 40 ligands as new insulin secretion modulators. Fut. Med. Chem. 2017, 9, 1873–1885. 10.4155/fmc-2017-0113. [DOI] [PubMed] [Google Scholar]
- Cione E.; Caroleo M. C.; Kagechika H.; Manetti F. Pharmacophore-guided repurposing of fibrates and retinoids as GPR40 allosteric ligands with activity on insulin release. J. Enz. Inhib. Med. Chem. 2021, 36, 377–383. 10.1080/14756366.2020.1864629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavletich N. P.; Pabo C. O. Crystal structure of a five-finger GLI-DNA complex: new perspectives on zinc fingers. Science 1993, 261, 1701–1707. 10.1126/science.8378770. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.