

Abstract
The massive amount of experimental DNA and RNA sequence information provides an encyclopedia for cell biology that requires computational tools for efficient interpretation. The ability to write and apply simple computing scripts propels the investigator beyond the boundaries of online analysis tools to more broadly interrogate laboratory experimental data and to integrate them with all available datasets to test and challenge hypotheses. Here we describe robust prototypic bash and C++ scripts with metrics and methods for validation that we have made publicly available to address the roles of non-B DNA-forming motifs in eliciting genetic instability and to query The Cancer Genome Atlas. Importantly, the methods presented provide practical data interpretation tools to examine fundamental relationships and to enable insights and correlations between alterations in gene expression patterns and patient outcome. The exemplary source codes described are simple and can be efficiently modified, elaborated, and applied to other relationships and areas of investigation.
Keywords: non-B DNA, cancer genome, TCGA analyses, parallel computing, Bash, Kaplan-Meier survival, tumor normal pair, gene expression correlation analysis, custom scripts
1. Introduction
From searching for reagents to analyzing data, computers have become an integral part of a molecular biology laboratory, and the integration of “wet” laboratory data with bioinformatics and metadata analyses has become a powerful mean for casting experimental information into a broader spectrum of knowledge to test, challenge, and validate novel paradigms (1–4). The massive amount of experimental data generated by The Cancer Genome Atlas effort may in practical terms only be harnessed though computational means (5,6). Likewise, both the processing and biological interpretation of next-generation sequencing and RNA-Seq data are achieved through computational programming (7–10). The widespread use of computing resources is also generating an increasing need for the molecular biology laboratory to not only acquire the skills necessary for installing and running off-the-shelf software or use online analytical tools, but also to devise ad-hoc computational scripts to interrogate database information in a manner suited to specific needs that emerge during laboratory projects. Exemplary evidence for this need was captured by a recent seminar, where the speaker commented that “an investigator who knows even just the basics of a computer language owns the data”: this insight makes a qualitative distinction between the “button pusher” and the active interrogator and interpreter of the data that are being queried. Indeed, it is often of clear advantage to be able to write even simple codes to address specific questions or to process large data files in batch mode, so as to avoid manual mistakes and ensure reproducibility.
Here we present few enabling robust scripts in bash and C++ tailored to support the molecular biologist to address two types of questions: 1) the roles of non-B DNA in genomic instability and 2) RNA-Seq analyses of TCGA data, which we have used to help elucidate the interface between DNA repair (and its deficiencies) and DNA replication and transcription. Although distinct, these two areas of investigation are intimately interconnected, since DNA secondary structures are arising as powerful partners with the DNA repair arsenal to thrive a normal cell towards a malignant state (11–14). There are many helpful tools available to search for non-B DNA-forming sequences (15–19) and to explore TCGA data online, including among others cBioPortal (https://www.cbioportal.org), the Xena browser (https://xena.ucsc.edu), Mexpress (https://mexpress.be), TCGA Wanderer (http://maplab.imppc.org/wanderer/). The source codes presented here are available at https://github.com/abacolla. They are not a substitute for other available resources; rather, their goal is to stimulate and enable investigator interest in using and writing ad-hoc codes to adapt the output of a search to specific needs; in other words, to “own”, interrogate and integrate experimental laboratory data with public datasets.
2. Materials
We assume a basic knowledge of command line syntax and, preferably, access to a High Performance Computing (HPC) cluster. The intent of this chapter is to provide access to scripts and selected sample files on which to run the scripts. A flow-through of the steps described here may also be found in the README.md files at the GitHub site. The scripts used in this article are available at https://github.com/abacolla. Files containing the TCGA RNA-Seq normalized Rsem and patient clinical data may be downloaded from https://www.researchgate.net/profile/Albino_Bacolla, under the TCGA Analyses project. These files have been obtained using the TCGA Assembler-2 (https://github.com/compgenome365/TCGA-Assembler-2). Other utilities include twoBitToFa from http://hgdownload.cse.ucsc.edu/admin/exe and the human reference genome sequence contained in the hg38.2bit file, from http://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/.
3. Methods
3.1. Compile C++ programs
1 Inspect the Makefile associated with each cpp file, edit if needed and compile running “make”. This will generate a “build” directory containing the executable (see Note 1).
3.2. Search for non-B DNA-forming sequences in fasta files
This method aids at determining whether non-B DNA-forming repeats may be enriched at particular genomic loci. It searches for non-B DNA-forming motifs in fasta files and reports their total number. It also returns the distance between the center of each non-B DNA-forming motif and the middle position of the DNA fasta sequence, which is meant to assess whether non-B DNA-forming motifs occur more often near junctions (breakpoints) of genomic rearrangements, such as deletions, duplications, inversions and translocations than expected by chance (14,20–22). Scripts are contained in directories nonB-DB and submitMpi at https://github.com/abacolla.
1. Generate file fastaList.fa containing a list of 31 fasta records on which to perform a search of non-B DNA-forming motifs by executing “twoBitToFa -noMask -seqList=fastaList hg38.2bit fastaList.fa” (see Note 2).
2. Run each non-B DNA search script on fastaList.fa, i.e. “./dr_get.sh fastaList.fa”, to search for direct repeats (dr), “./h_get.sh fastaList.fa” to search for triplex-forming repeats (h), “./g4_get.sh fastaList.fa” to retrieve G4-forming repeats (g4), “./z_get.sh fastaList.fa” to obtain Z DNA-forming repeats (z) and “./ir_get.sh fastaList.fa” for inverted repeats, which can form cruciforms (ir). Running the searches on a single processor is inefficient; to speed up the process use parallel computing, as follows.
3. Split fastaList.fa into individual fasta records by executing the “csplit” command reported in Step 2 of the README.md file at https://github.com/abacolla/nonB-DNA; delete file bin_000 (“rm bin_000”). This generates 31 fasta files named bin_001 to bin_031.
4. Make bash files to process bin_001 to bin_031 for all non-B DNA search scripts by executing “./makeFile.sh dr”; “./makeFile.sh z”; “./makeFile.sh g4”; “./makeFile.sh h”; and “./makeFile.sh ir”. This generates 5 sets of 31 bash files each (drdna_1.sh to drdna_31.sh; zdna_1.sh to zdna_31.sh; g4dna_1.sh to g4dna_31.sh; hdna_1.sh to hdna_31.sh; and irdna_1.sh to irdna_31.sh).
5. Launch the job using the directive “ibrun (or mpirun depending on HPC instructions) vga_submitMpiJob drdna_”, where drdna_ is the prefix of the 31 bash files generated in step 4 (see launchJob.sh for an example template) (see Note 3). The time required to complete a job will depend on the type of script used, the total number of tasks (fasta files to process) and the number of processors that have been requested per node (Figure 1).
Figure 1.

HPC performance for processing nonB DNA-related batch jobs. A total of 125 individual fasta files, 10 kb of DNA sequence each, were used to search for nonB DNA-forming repeats using vga_submitMpi on variable number of nodes. Except where indicated, jobs were run on Bridges2 (RM nodes, 64 cores/node) at the Pittsburgh Supercomputing Center, Pittsburgh, PA. Stampede2 (Knights Landing compute nodes, 68 cores/node) and Lonestar5 (24 cores/node) were from the Texas Advanced Computing Center, Austin, TX.
6. Process the output. The output consists of a number of files containing: chromosome number, hg38 coordinate, length, distance from start and end of the sequence to the center of the fasta sequence, DNA sequence (for dr, g4, z); and chromosome number, hg38 coordinate, length of stem, length of loop, distance from the center loop to the center of the fasta sequence, and the sequence of both stems (for h, ir). Use nonB_getRes.sh to extract the number of tracts. Its usage is: nonB_getRes.sh file_suffix(dr | ir | q1k | z1k | triplex); i.e. “nonB_getRes.sh triplex”. A comparison of all results should show that the number of inverted repeats is significantly higher in bin_22 than in the other bins. For studies aimed at assessing whether non-B DNA-forming repeats are enriched at rearrangement junctions, such as in cancer genomes, plot the distance of the tracts (or all the non-B DNA-forming bases) from the center positions of the fasta files (Figure 2).
Figure 2.
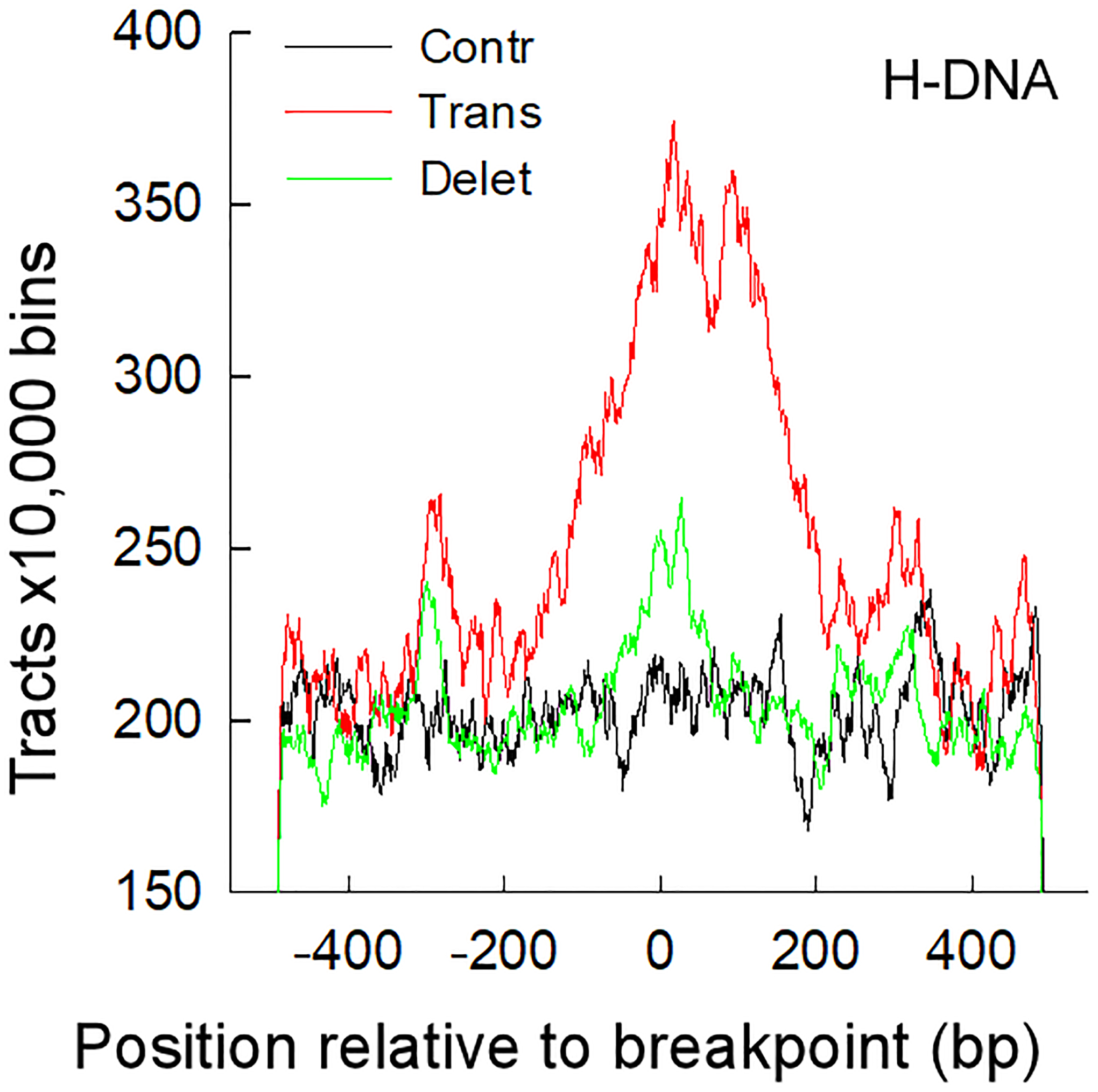
Triplex DNA-forming repeats are enriched at translocation junctions in cancer genomes. Line plots of number of triplex-forming repeats flanking the junction breakpoints of translocations, deletions and control sets from the Catalogue Of Somatic Mutations In Cancer (COSMIC, https://cancer.sanger.ac.uk/cosmic). Reproduced from (14) by permission of Oxford University Press.
3.3. Assess TCGA gene expression levels between tumor and normal controls
RNA-Seq gene expression data for the TCGA repository are widely available and several online tools exist to visualize the data for a specific gene of interest. However, it is often desirable to create custom-made graphs where one can control aesthetic features for publication-quality figures. In addition, it may be necessary to compare the data for large sets of genes, and this can be accomplished most easily from custom-generated flat text files. Here we present vga_makeBoxPlotRsem.sh, a bash script that accomplishes these tasks easily. Scripts are contained in the directory tcgaAnalyses at https://github.com/abacolla.
1 vga_makeBoxPlotRsem.sh is a script that generates a high-quality png box plot using mRNA expression of a given gene for 15 TCGA tumor and normal matched controls suitable for publication with minimal editing. The number of tumor/normal pairs is limited to those cancer sets with at least 10 normal controls. Its usage is: vga_makeBoxPlotRsem.sh <geneName>, where <geneName> is an official gene name in capital letters, i.e. vga_makeBoxPlotRsem.sh BLM. Before running the script edit lines 8–11 to load any module required for the R language and edit DIR0 on line 13 to point to the RNA gene expression files. A copy of these files may be found in directory TCGA Analyses at https://www.researchgate.net/profile/Albino_Bacolla, which were obtained using the TCGA-Assembler v.2.0 (https://github.com/compgenome365/TCGA-Assembler-2). Box plots are drawn according to the list on lines 63–77; to change the ranking, such as plotting according to p-values, change the order of tumor/normal pairs on lines 63–77.
2 vga_makeBoxPlotRsem.sh calls automatically the R program vga_pngBoxPlotRsem.R. Options in vga_pngBoxPlotRsem.R that control main aesthetic features include the y-axis range on line 52 (ylim), p-values (on, off) on line 53 (stats_compare_means), colors for the plots on line 73 (scale_fill_manual), the x-axis line (axis.line.x) on line 71, and notch (true, false) on line 46.
3 vga_makeBoxPlotRsem.sh generates two files: a box plot and a text file named after the input file, i.e. “boxPlotGeneExprBLM.png” and “pngBoxPlotBLM.out”, containing the statistical data (see Note 4). For BLM, which encodes the BLM helicase with functions in DNA replication and repair, and whose deficiency is associated with the autosomal recessive Bloom syndrome, the data show that the gene is significantly upregulated in all types of cancer (Figure 3) (see Note 5).
Figure 3.
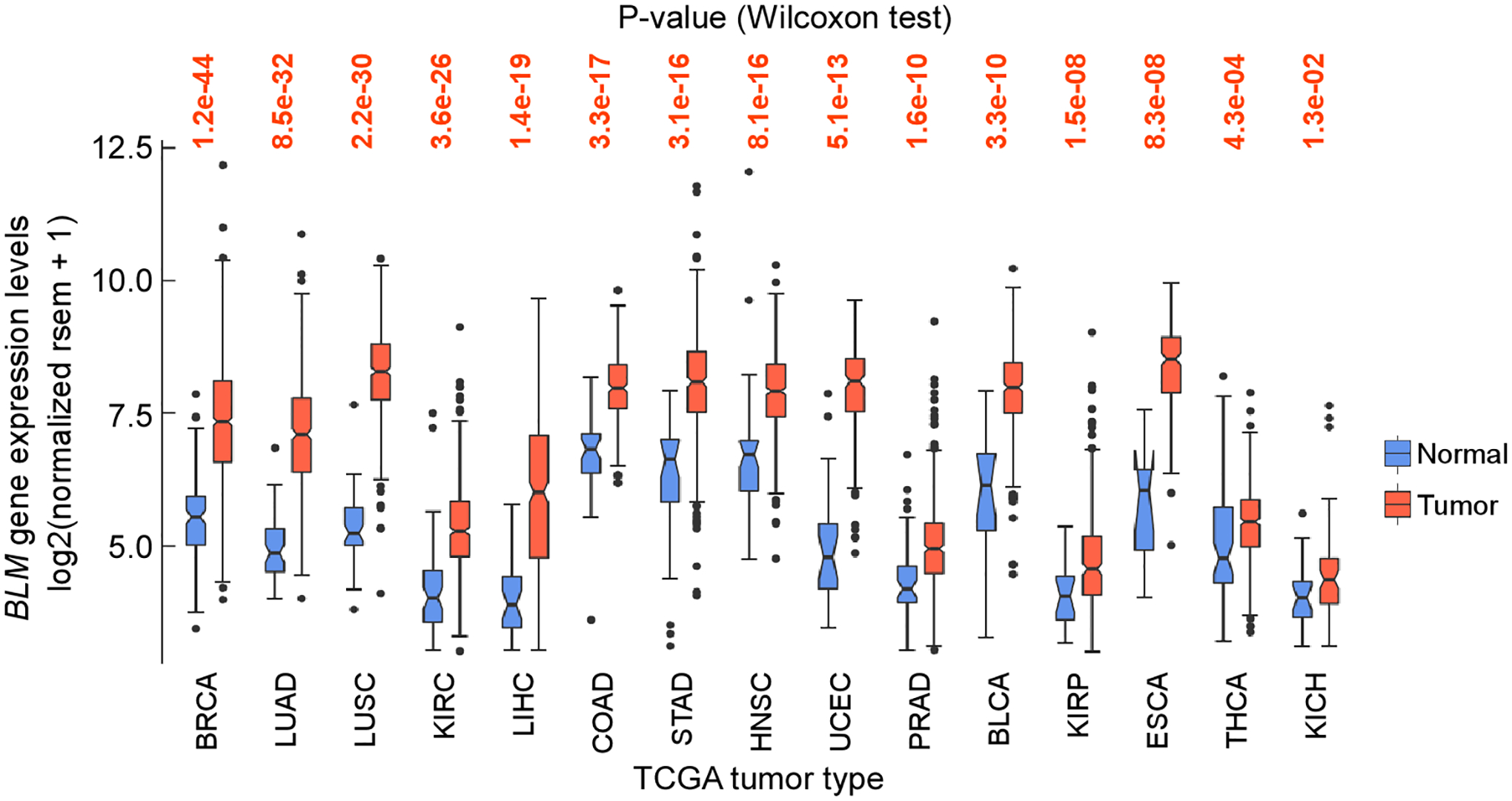
Box plot of TCGA BLM mRNA levels between tumor and matched controls. The core plot generated with vga_makeBoxPlotRsem.sh was edited with Canvas (https://www.canvasgfx.com)
4 vga_makeBoxPlotRsem.sh can be scaled-up using vga_submitMpiJob, which is detailed in directory submitMpi and in Section 3.1, and the associated .out files, which can be queried in batch mode to assess statistical trends.
3.4. Kaplan-Meier survival curves in TCGA patients with high and low mRNA levels
It can be critically enabling to create custom graphs for Kaplan-Meier survival curve analyses of gene expression and to use the associated flat text files for large-scale analyses. Script “vga_survivalCurve.sh” in the https://github.com/abacolla/tcgaAnalyses repository serves this purpose.
1 vga_survivalCurve.sh generates a Kaplan-Meier survival curve comparing patients with high (above mean) versus low (below mean) expression levels for a given gene. Its usage is: vga_survivalCurve.sh <TCGA_TUMOR> <GENE_NAME>, where TCGA_TUMOR is the TCGA tumor code and GENE_NAME an official gene name, both in capital letters; i.e. vga_survivalCurve.sh KIRC ERCC1. Edit lines 8–11 to load any module required in R. Line 19 launches the vga_spotLight binary, its path needs to be specified; the --optFdat option points to the TCGA gene expression files, edit the path. Edit line 21 to point to the TCGA clinical data files, which can be extracted from https://www.researchgate.net/profile/Albino_Bacolla. Line 32 calls vga_survival.R, verify its path. The example above will generate a graphic file named kirc_ercc1.png and a text file named survival_ercc1_kirc.out.
2 Use vga_submitMpiJob, which is detailed in directory https://github.com/abacolla/submitMpi, to scale-up vga_survivalCurve.sh to process more (or all) genes for the 33 TCGA tumor types and use the associated .out files for statistical comparisons (23) (Figure 4).
Figure 4.
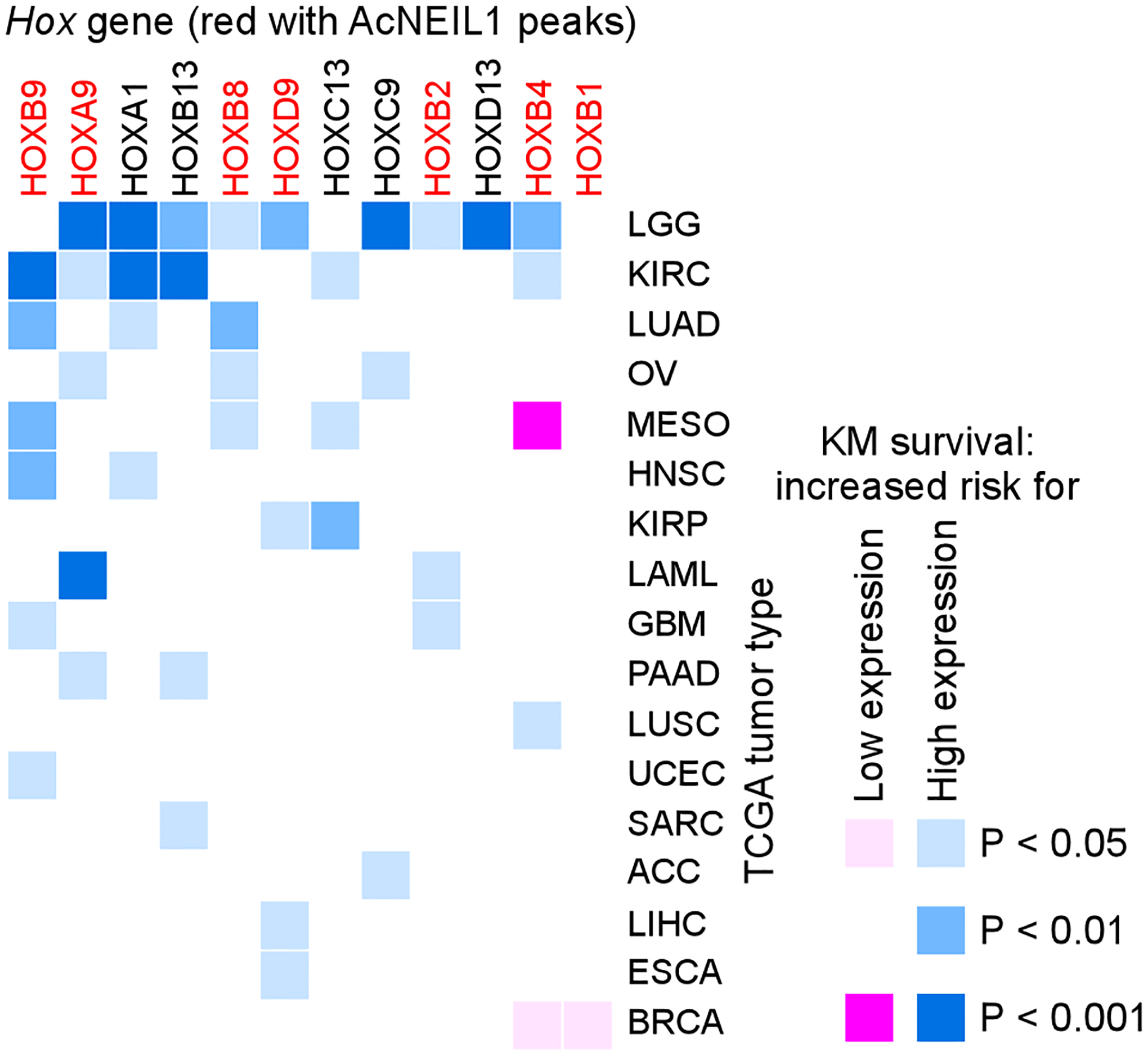
Heat map of the effect of HOX gene expression on survival in TCGA patients. In adult tissues HOX genes display tissue-restricted expression, and hyper- or re-activation of HOX gene expression in tumors is generally associated with poor survival in most cases. Red, HOX genes actively repaired from oxidative DNA damage by acetylated base excision repair DNA glycosylase NEIL1. Reproduced from (23) by permission of Oxford University Press.
3.5. Gene correlation expression analyses (GCEA)
The program vga_geneExprMain.cpp at https://github.com/abacolla/tcgaAnalyses generates the vga_spotLight binary, a utility for performing gene expression correlation analyses (GCEA) and other analyses using TCGA RNA-Seq gene expression data.
1 Edit Makefile to point to the BOOST library and preload any module required for MPI. Edit lines 96 and 97 of vga_geneExprUsage.hpp to point to the directories containing the gene expression and mutation data (files for the mutation data are not included and are not required for gene expression analyses). File testStart.sh may be used as a guide to test the vga_spotLight compiled binary.
2 Use option A is to find a correlation between the expression of 2 genes. The usage is: vga_spotLight --optAdat <dataset> --optAgene1 <GENE1> --optAgene2 <GENE2>, where <dataset> is the TCGA gene expression file and <GENE1> and <GENE2> are official gene names. For example, launching “ibrun -n 1 vga_spotLight --optAdat ACC__geneExprT.txt --optAgene1 GRB2 --optAgene2 FGFR2” will generate an output file named “ACC_GRB2_FGFR2_expr.txt” containing the gene symbols, patient codes, log2 of normalized Rsem values for the genes, i.e. log2(norm rsem + 1), the regression coefficient and p-value (see Note 6).
3 Use option B to compute the correlation between one gene and all other genes (~20500) of a given dataset. The usage is: vga_spotLight --optBdat <dataset> --optBgene <GENE>, where <dataset> is a TCGA gene expression file and <GENE> an official gene name. For example, “ibrun -n 1 vga_spotLight --optBdat ACC__geneExprT.txt --optBgene GRB2” will generate file “GRB2_toAll_ACC_T.txt”, returning input gene name, test gene name, entrez gene record, number of observations, linear regression coefficient and p-value. The program returns 2 and 1 in place of the linear regression coefficient and p-value when there is an insufficient number of observations (see Note 7). Use this option to explore co-expression patterns between a test gene and potential members of its pathways, as exemplified for KAT8 encoding MOF, a member of the MYST histone acetyltransferase protein family, in which the co-expression data support a function in chromosome segregation (24).
4 Option F returns the gene expression results for one gene in a given dataset and its use is: vga_spotLight --optFdat <dataset> --optFgene <GENE>. Using “ibrun -n 1 vga_spotLight --optFdat ACC__geneExprT.txt --optFgene GRB2” will generate file “ACC_GRB2_exprOne.txt” with patient code and log2(norm rsem + 1) values.
5 Options C-E were implemented to explore correlations between gene expression and mutation loads; we found correlations among common sets of genes in various types of tumor, including cell cycle and DNA repair in 4 tumors (KICH, LUAD, PRAD and LGG), mitochondrial respiration in 3 tumors (STAD, THCA and CHOL), antigen processing and presentation in CESC, reactivation of olfactory receptor genes in SKCM, and the unfolded protein response in BRCA (20) (see Note 8).
3.6. Basic utilities
When working with custom scripts on repetitive tasks, it is helpful to create utilities that perform routine operations, such as using the information from file 1 to extract matching information in file 2 or running t-tests or linear regressions, without the need for user intervention. Here we present few scripts that may help automating these tasks.
1 Intersect two files. Let’s assume we have RNA-Seq data and we wish to assess whether there is a correlation between differentially up and down regulated transcripts and gene length. Because gene length information is not contained in the analyses of RNA-Seq output files, we need to obtain it from additional sources, such as in file knownGenes.txt from http://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/. We can simplify our gene length file by executing: “awk ‘{ print $1 “\t” $5-$4 }’ knownGene.txt | sed ‘s/\.[0–9]*//’ > knownGenes_ens_length.txt” (see Note 9) to only contain ENS number and gene length:
| ENST00000619216 | 68 | |
| ENST00000473358 | 1544 | |
| ENST00000499289 | 843 | (“knownGenes_ens_length.txt”, first 3 lines) |
Given our files “rnaSeq_up.txt” and “rnaSeq_down.txt” containing upregulated and downregulated transcripts, respectively, each with the list of significantly differentially expressed transcripts containing 3 fields, ESN number, geneID and gene name:
| ENST00000225964 | 1277 | COL1A1–201 | |
| ENST00000358171 | 871 | SERPINH1–201 | |
| ENST00000250383 | 10202 | DHRS2–201 | (“rnaSeq_up.txt”, first 3 lines) |
we use the first field of “rnaSeq_up.txt” to extract its matching ENS number from “knownGenes_ens_length.txt”. We could use Unix “grep”; however, “grep” is slow, particularly if the -w option is used and files are large. Script vga_intersect2×3.cpp at https://github.com/abacolla/intersect is a convenient alternative; its usage is: vga_intersect2×3 file1 file2 file3, where file1 is in our case “knownGenes_ens_length.txt”, file2 is for example “rnaSeq_up.txt” and file3 is the output file (up_output, for example). The vga_intersect2×3 script will be run separately on both up ad downregulated RNA-Seq files. The output file returned by vga_intersect2×3 contains all 5 fields from the 2 input files so as to verify that the match is correct. The 2 output files may be used to run a t-test (or a Wilcoxon test) to assess for statistical significance in gene (transcript) length as follows.
2 Run a t-test on the 2 output files obtained from vga_intersect2×3 above. Take the log10 of the second field from up_output and down_output (awk ‘{ print log($2)/log(10) }’ up_output > logf2_up; awk ‘{ print log($2)/log(10) }’ down_output > logf2_down) and run script vga_tTest as “vga_tTest logf2_up logf2_down”. The t-test script may be found at https://github.com/abacolla/tTest.
3 Use vga_linearRegression to embed a linear regression analysis from an input file containing paired x and y coordinates and vga_fisherTest to compute Fisher exact tests from an input file containing one or more sets of data. The scripts may be found at https://github.com/abacolla/linearRegression and https://github.com/abacolla/fisherTest, respectively. All the commands above can be inserted directly into a single bash or other script and run without user intervention, thereby fulfilling our goal of simplifying the workflow and maintaining a record for reproducibility purposes.
4. Notes
1. We suggest moving the binaries to ~/bin and other scripts such as bash and R to ~/sbin, and to modify PATH in .bash_profile, i.e. “export PATH=$HOME/bin:$PATH”; “export PATH=$HOME/sbin:$PATH”. To compile use the appropriate compiler type and its associated commands: Intel and icpc, AMD (AOCC) and clang, Gnu and g++, PGI and pgc++.
2. The -noMask flag returns the DNA sequences in capital letters.
3. When scaling up jobs it us useful to use a utility, such as remora, to track CPU usage and optimize the number of nodes requested for a job.
4 It is generally most convenient to crop the box frame of the original .png file, import it into a graphic program and fill-in with any additional statistical data using the associated .out file.
5 In the R ggplot2 package notches for the box plots extend 1.58 * IQR / sqrt(n), where IQR is the interquartile range. Since these are asymmetrical with respect to the median, they can recurve when the data are asymmetrical, as seen in Figure 2 for the ESCA data in normal tissues. In these cases, the notch option on line 46 may be turned off.
6 Consult the user manual for the HPC system in use to compile and run MPI applications in C++.
7 It is common to find strong co-expression for genes located near each other on the same chromosome in tumors.
8 It is essential to be mindful that whereas all scripts reported here return results, the results obtained ought to be cast onto broader contexts, and extensive controls need to be used, to avoid incorrect biological interpretation. For example, we found that in cancer genomes several HOX genes display significant coexpression with two transcription factors, FOXM1 and MYBL2. However, when compared with the coexpression levels of all genes, which revealed -log10 P-values for the linear regressions down to ~10−250 and r-values up to 0.94, the results for the HOX genes were comparatively weak, and indeed the comprehensive comparison suggested indirect transactivation or perhaps non-casual relationships (23). Likewise, the range of Kaplan-Meier p-values is strongly dependent upon tumor types, and therefore a ”significant” p-value of 0.01, for example, may need to be interpreted with extreme caution. It is for this reasons that we think it is important to conduct broad-based investigations using in-house scripts rather than relying on limited analyses using exclusively on-line tools.
9 The alterative “sed” command: “awk ‘{ print $1 “\t” $5-$4 }’ knownGene.txt | sed ‘s/\.[0–9]\+//’ > knownGenes_ens_length.txt” may also work. Verify that fields are separated by a tab without additional white spaces; in “vim” you can use “:set list” to verify.
Figure 5.
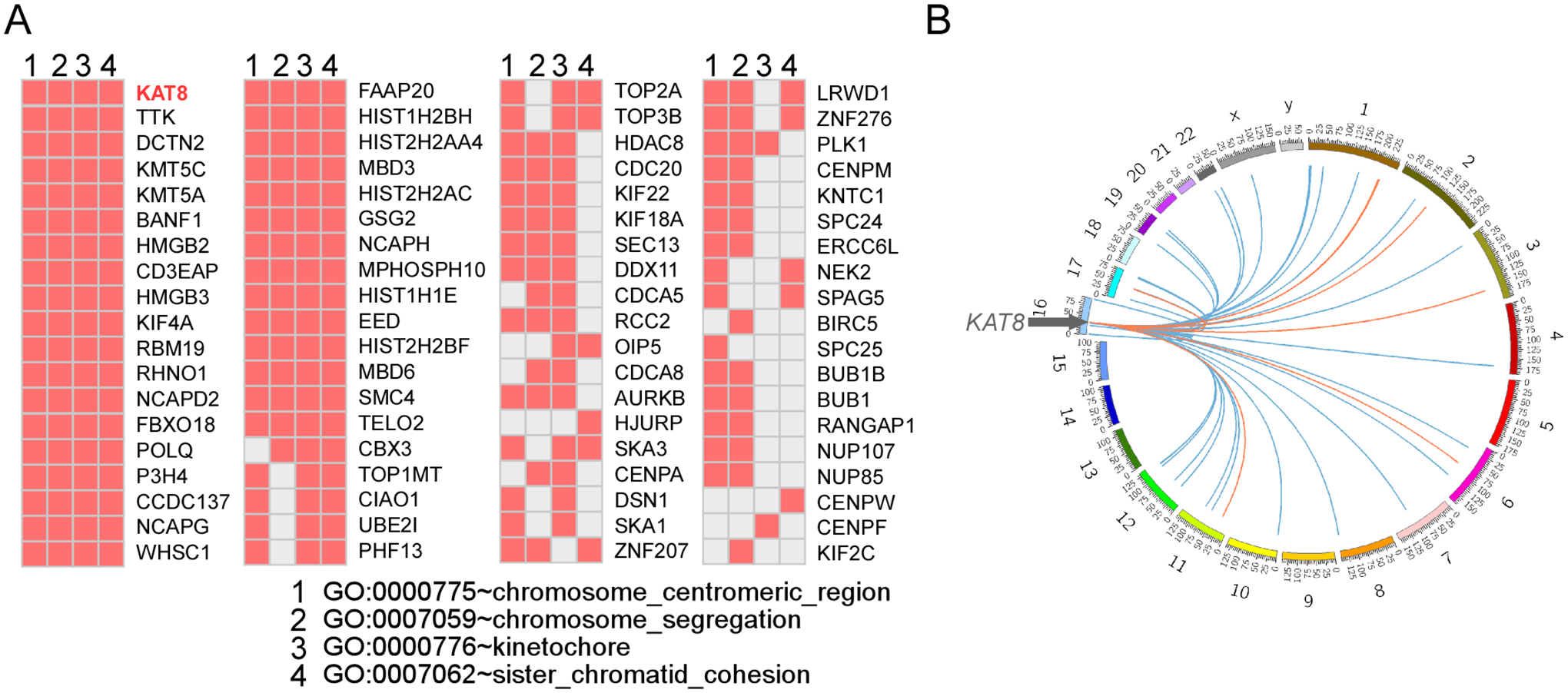
Using GCEA to explore gene pathways. A) List of genes highly coexpresssed with KAT8/MOF in TCGA comprising Gene Ontology terms related to “chromosome segregation”. B) Circos plot of chromosome location of genes significantly (P<0.01) coexpressed (blue links) or anticorrelated (orange links) with KAT8/MOF from panel A in KIRC patients. Adapted from (24) by permission of the American Society for Microbiology.
Acknowledgements
This work was supported by grants from the National Institutes of Health P01 CA092584, R35 CA220430, by the Cancer Prevention and Research Institute of Texas RP180813 and by a Robert A. Welch Chemistry Chair to J.A.T. The research used the Bridges/Bridges2 Pittsburgh Supercomputing Center through the Extreme Science and Engineering Discovery Environment (XSEDE), which are supported by the National Science Foundation grants ACI-1445606 and ACI-1548562, and the Texas Advanced Computing Center, supported by National Science Foundation grant ACI-1134872.
References
- 1.Pucker B, Schilbert HM and Schumacher SF (2019) Integrating molecular biology and bioinformatics education. J Integr Bioinform 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Houl JH, Ye Z, Brosey CA, Balapiti-Modarage LPF, Namjoshi S, Bacolla A, Laverty D, Walker BL, Pourfarjam Y, Warden LS et al. (2019) Selective small molecule PARG inhibitor causes replication fork stalling and cancer cell death. Nat Commun 10: 5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eckelmann BJ, Bacolla A, Wang H, Ye Z, Guerrero EN, Jiang W, El-Zein R, Hegde ML, Tomkinson AE, Tainer JA et al. (2020) XRCC1 promotes replication restart, nascent fork degradation and mutagenic DNA repair in BRCA2-deficient cells. NAR Cancer 2: zcaa013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lees-Miller JP, Cobban A, Katsonis P, Bacolla A, Tsutakawa SE, Hammel M, Meek K, Anderson DW, Lichtarge O, Tainer JA et al. (2020) Uncovering DNA-PKcs ancient phylogeny, unique sequence motifs and insights for human disease. Prog Biophys Mol Biol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Consortium ITP-CAoWG (2020) Pan-cancer analysis of whole genomes. Nature 578: 82–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerstung M, Jolly C, Leshchiner I, Dentro SC, Gonzalez S, Rosebrock D, Mitchell TJ, Rubanova Y, Anur P, Yu K et al. (2020) The evolutionary history of 2,658 cancers. Nature 578: 122–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J et al. (2013) From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43: 11 10 11–11 10 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Franke KR and Crowgey EL (2020) Accelerating next generation sequencing data analysis: an evaluation of optimized best practices for Genome Analysis Toolkit algorithms. Genomics Inform 18: e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hesketh AR (2019) RNA sequencing best practices: Experimental protocol and data analysis. Methods Mol Biol 2049: 113–129 [DOI] [PubMed] [Google Scholar]
- 10.Vieth B, Parekh S, Ziegenhain C, Enard W and Hellmann I (2019) A systematic evaluation of single cell RNA-seq analysis pipelines. Nat Commun 10: 4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Wietmarschen N, Sridharan S, Nathan WJ, Tubbs A, Chan EM, Callen E, Wu W, Belinky F, Tripathi V, Wong N et al. (2020) Repeat expansions confer WRN dependence in microsatellite-unstable cancers. Nature 586: 292–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKinney JA, Wang G and Vasquez KM (2020) Distinct mechanisms of mutagenic processing of alternative DNA structures by repair proteins. Mol Cell Oncol 7: 1743807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berroyer A and Kim N (2020) The functional consequences of eukaryotic topoisomerase 1 interaction with G-quadruplex DNA. Genes 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bacolla A, Tainer JA, Vasquez KM and Cooper DN (2016) Translocation and deletion breakpoints in cancer genomes are associated with potential non-B DNA-forming sequences. Nucleic Acids Res 44: 5673–5688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Puig Lombardi E and Londono-Vallejo A (2020) A guide to computational methods for G-quadruplex prediction. Nucleic Acids Res 48: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cer RZ, Donohue DE, Mudunuri US, Temiz NA, Loss MA, Starner NJ, Halusa GN, Volfovsky N, Yi M, Luke BT et al. (2013) Non-B DB v2.0: a database of predicted non-B DNA-forming motifs and its associated tools. Nucleic Acids Res 41: D94–D100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brazda V, Kolomaznik J, Lysek J, Haronikova L, Coufal J and St’astny J (2016) Palindrome analyser - A new web-based server for predicting and evaluating inverted repeats in nucleotide sequences. Biochem Biophys Res Commun 478: 1739–1745 [DOI] [PubMed] [Google Scholar]
- 18.Buske FA, Bauer DC, Mattick JS and Bailey TL (2012) Triplexator: detecting nucleic acid triple helices in genomic and transcriptomic data. Genome Res 22: 1372–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hon J, Martinek T, Rajdl K and Lexa M (2013) Triplex: an R/Bioconductor package for identification and visualization of potential intramolecular triplex patterns in DNA sequences. Bioinformatics 29: 1900–1901 [DOI] [PubMed] [Google Scholar]
- 20.Bacolla A, Ye Z, Ahmed Z and Tainer JA (2019) Cancer mutational burden is shaped by G4 DNA, replication stress and mitochondrial dysfunction. Prog Biophys Mol Biol 147: 47–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao J, Wang G, Del Mundo IM, McKinney JA, Lu X, Bacolla A, Boulware SB, Zhang C, Zhang H, Ren P et al. (2018) Distinct Mechanisms of Nuclease-Directed DNA-Structure-Induced Genetic Instability in Cancer Genomes. Cell Rep 22: 1200–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seo SH, Bacolla A, Yoo D, Koo YJ, Cho SI, Kim MJ, Seong MW, Kim HJ, Kim JM, Tainer JA et al. (2020) Replication-based rearrangements are a common mechanism for SNCA duplication in Parkinson’s disease. Mov Disord 35: 868–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bacolla A, Sengupta S, Ye Z, Yang C, Mitra J, De-Paula RB, Hegde ML, Ahmed Z, Mort M, Cooper DN et al. (2021) Heritable pattern of oxidized DNA base repair coincides with pre-targeting of repair complexes to open chromatin. Nucleic Acids Res 49: 221–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh M, Bacolla A, Chaudhary S, Hunt CR, Pandita S, Chauhan R, Gupta A, Tainer JA and Pandita TK (2020) Histone Acetyltransferase MOF Orchestrates Outcomes at the Crossroad of Oncogenesis, DNA Damage Response, Proliferation, and Stem Cell Development. Mol Cell Biol 40 [DOI] [PMC free article] [PubMed] [Google Scholar]