

ABSTRACT
Despite their common use in research, monoclonal antibodies are currently not systematically sequenced. This can lead to issues with reproducibility and the occasional loss of antibodies with loss of cell lines. Hybridoma cell lines have been the primary means of generating monoclonal antibodies from immunized animals, including mice, rats, rabbits, and alpacas. Excluding therapeutic antibodies, few hybridoma-derived antibody sequences are known. Sanger sequencing has been “the gold standard” for antibody gene sequencing, but this method relies on the availability of species-specific degenerate primer sets for amplification of light and heavy antibody genes and it requires lengthy and expensive cDNA preparation. Here, we leveraged recent improvements in long-read Oxford Nanopore Technologies (ONT) sequencing to develop Nanopore Antibody sequencing (NAb-seq): a three-day, species-independent, and cost-effective workflow to characterize paired full-length immunoglobulin light- and heavy-chain genes from hybridoma cell lines. When compared to Sanger sequencing of two hybridoma cell lines, long-read ONT sequencing was highly accurate, reliable, and amenable to high throughput. We further show that the method is applicable to single cells, allowing efficient antibody discovery in rare populations such as memory B cells. In summary, NAb-seq promises to accelerate identification and validation of hybridoma antibodies as well as antibodies from single B cells used in research, diagnostics, and therapeutics.
KEYWORDS: Antibody sequencing, B cell, hybridoma, long-read, nanopore sequencing, rat B cell cloning, single cell, workflow
Introduction
Antibodies are essential tools for research as well as for diagnostic and therapeutic applications because they can bind specific targets. However, many research antibodies are poorly characterized, and around half in fact lack specificity for their reported targets.1,2 Poor quality antibodies waste resources and confound scientific research when experiments using such reagents cannot be reproduced by others.3–5 There are very few instances where hybridoma antibody genes have been sequenced, due in part to the added costs and complexity of antibody sequencing.
To improve the reliability of antibodies The Antibody Society6 and leading scientists4,7 have recommended collaboration and funding to define antibodies by their DNA sequence. Sequencing provides a basis on which to validate antibody specificity and sensitivity across all relevant applications. It also allows the generation of well-defined recombinant antibodies by incorporating the genes into plasmid DNA followed by expression in mammalian or bacterial cells.8
Conventional sequencing of antibody genes from hybridoma cell lines involves PCR amplification of antibody variable regions (VH and VL) followed by low-throughput and partial-length Sanger sequencing. Primer sets for antibody gene amplification must account for variability at the 5’ (leader sequence of framework region 1 (FR1)) and 3’ end (FR4, hinge, or constant region) on the transcripts. This requires the intricate design of many primer pairs and/or degenerate primers. Although they do not guarantee unbiased amplification, validated primer sets are available for mouse and human loci. In other species, however, the lack of such primer sets limits conventional antibody sequencing. The use of 5’ RACE (Rapid Amplification of cDNA ends), or the more recent but conceptually equivalent template-switching, halves the complexity, as it only requires primers specific to the 3’ ends of the transcripts (FR4, hinge, or constant region). This technique has been successfully applied to sequence rat, mouse, and human antibody transcripts,9–11 including from single B cells.12 In addition to these complexities of variable region PCR, Sanger sequencing by commercial providers is costly and slow: around US$800 (2 weeks) for the variable regions (VH and VL) and US$2,000 (4 weeks) for the full-length antibody (variable and constant regions). Even when performing Sanger sequencing in-house, there is still an estimated turnaround time of 5 days and cost of US$70–120 per antibody.10 Sanger sequencing is further complicated by the potential presence of multiple heavy- and light-chain transcripts in the same cell. An estimated 30% of all hybridomas express more than one productive heavy or light chain.13
Illumina sequencing has also been incorporated into antibody discovery platforms for high-throughput short-read sequencing of antibody heavy and light chains.14–17 However, these protocols also rely on species-specific primers and generate partial-length reads (up to 600 bp) which require assembly, while the high throughput is not well-suited to monoclonal antibody sequencing for a limited number of cell lines.
Long-read sequencing allows for the full-length sequencing of antibodies, but a relatively high error rate compared with the above short-read sequencing approaches has limited its application in antibody sequencing thus far.18 PacBio long-read sequencing has been applied to the sequencing of single B cells,19 phage display libraries,20,21 and repertoire sequencing,22 but its high cost makes it difficult to implement routinely. By contrast, Oxford Nanopore Technologies (ONT) sequencing has a much lower capital cost and flexible throughput, with raw-read accuracy having improved in recent years to >95%, enabling its use for the sequencing of phage display outputs.23 However, the remaining errors in ONT, notably systematic homopolymer errors (insertions/deletions in tracts of >5 identical bases) can lead to frameshifts, complicating translation of nanopore-derived sequences into accurate antibody protein sequences.24 Improvements in both sequencing accuracy and error correction techniques25 have enabled the study of antibody fragments26 and B cells27–29 but these studies use complex and costly error correction methods,28 are specific to cancerous cells,27 or are too high throughput to be realistically used for hybridoma sequencing.29
To bring about the transition to sequence-defined recombinant antibodies, we developed Nanopore Antibody sequencing (NAb-seq), a simplified experimental and computational workflow based on ONT sequencing. We used NAb-seq to obtain full-length antibody sequences from two rat hybridoma cell lines and compared the results to outsourced Sanger sequencing results. One million full-length cDNA reads were generated from multiplexed hybridomas on an ONT Flongle flow cell and assembled into 100% accurate antibody chains. With feasibility to upscale to 24 hybridomas per flow cell, consumables are estimated at approximately US$30 per antibody within a three-day turnaround (1 d library preparation, 1 d sequencing, 1 d analysis). We further show that NAb-seq can be applied to the antibody sequencing of single B cells. NAb-seq is thus a valuable tool for recombinant antibody production, humanization of therapeutic antibodies, and protection of intellectual property.
Results
Long-read sequencing of antibody genes from Rattus norvegicus hybridoma cells
Antibody sequencing requires high accuracy due to the crucial role of somatic mutation of variable regions in antibody specificity and affinity for the target. Recent improvements in the accuracy of ONT long-read data prompted us to test its efficacy in rapid sequencing of hybridoma antibody genes. We tested two hybridoma cell lines that had been developed in-house and whose antibody genes had been Sanger sequenced by commercial sources. The hybridoma cell lines express 7D10 and 3C10 antibodies that specifically bind and trigger conformation change in BAK and mitochondrial BAX, respectively, as shown by a range of biochemical and structural assays.30–32
The workflow for NAb-seq is outlined in Figure 1, with further details described in Materials and Methods and online (see below). Full-length cDNA libraries A and B were prepared from total RNA of two hybridoma cell lines via oligo-dT primed reverse transcription and template-switching, using the ONT PCR-cDNA barcoding kit (see Supplementary Table 1 for primer sequences). Analysis of the libraries in a DNA TapeStation (Figure 2a) showed bands at 1600 and 900 kb, the expected size for full-length antibody heavy and light chain. The two cDNA libraries were then pooled for parallel long-read sequencing using the ONT Flongle flow cell, which generated ~1 million raw reads in 24 hours. The sequence data were basecalled in super-high accuracy mode and aligned to the reference Rattus norvegicus antibody gene sequences obtained from the international ImMunoGeneTics information system® (IMGT, http://www.imgt.org/ligmdb/).33 The number of pass (Q score >10) reads were ~300,000 and 200,000 for library A and B (Figure 2b), with read length distribution shown in Figure 2c. Thus, even without enrichment, 2.04% of pass reads in library A and 3.51% of pass reads in library B corresponded to antibody transcripts (Table 1), providing more than enough antibody reads (~460-3700) for consensus sequence generation. Empirically, as few as 5 reads could generate a 100% accurate consensus for a heavy chain. This means that there is scope to multiplex up to 24 hybridomas per Flongle flow cell (using the recent SQK-PCB111.24 nanopore library preparation kit, capacity 1 million reads) and still generate ~42,000 reads per hybridoma with 2–3% (~800-1200) of those being antibody transcripts.
Figure 1.
NAb-seq workflow for parallel sequencing of full-length antibody heavy and light chain sequences from hybridoma cell lines and single B cells.
Long Description: A series of 12 steps are numbered 1 to 12. Step 1 depicts hybridomas or FACS sorted B cells (with images of hybridoma cells in culture and of B cells being sorted). The remaining steps are 2) RNA extraction (test tube plus RNA), 3) cDNA synthesis (test tube plus DNA), 4) barcoding (five DNA strands with colored ends), 5) adapter ligation (five DNA strands with blue added to each end), 6) Nanopore sequencing (images of a matchbox-shaped Flongle and of DNA strand passing through a protein nanopore), 7) sequencing quality control using nanoQC (graphs), 8) identification of antibody reads using minimap2 (colored DNA strands), 9) adapter and polyA tail trimming using porechop and cutadapt (scissors cutting a DNA strand), 10) grouping by germline genes using minimap2 and IgBLAST (parallel lines representing VH and VL sequences), 11) consensus calling using racon and medaka (VH and VL sequences with red crosses representing errors that are resolved to single sequence). Step 12 is final annotation and analysis using minimap2 and IgBLAST (green and blue line labeled with descriptors of an antibody gene sequence).
Figure 1 A series of images numbered 1 to 12 with word descriptions for each, and with arrows between images to indicate the workflow involved in Nanopore antibody sequencing.
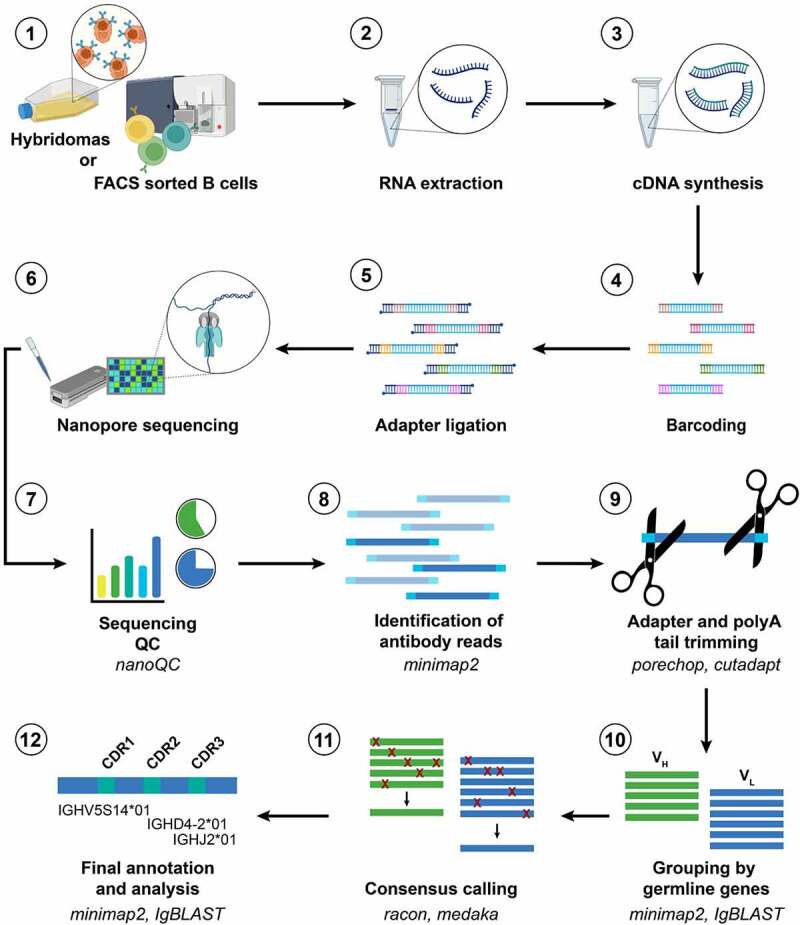
Figure 2.
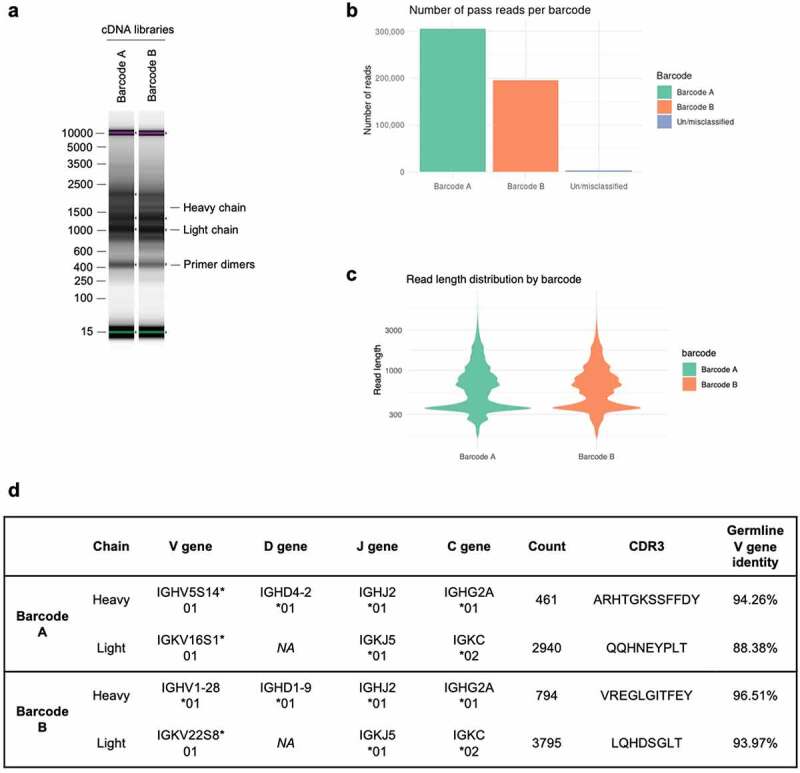
NAb-seq of two hybridoma cell lines revealed antibody sequences. (a) cDNA library size and amplification. Total RNA from two hybridoma cell lines was extracted and converted to cDNA followed by amplification and barcoding, generating whole transcriptome cDNA libraries A and B. (b) Basecalling in Guppy’s super-high accuracy mode yielded approximately 0.5 million total pass reads. (c) Read length of pass reads varied from ~400 bp to ~3000 bp. (d) Sequence analysis of cDNA libraries A and B reveals V(D)J recombination, C gene usage and complementarity-determining region 3 (CDR3) amino acid sequence. Consensus calling of antibody transcripts revealed IgG isotype.
Table 1.
Summary of antibody gene sequencing using NAb-seq.
| Cell type | Hybridoma |
Single B cell |
||||
|---|---|---|---|---|---|---|
| cDNA library | A | B | I | II | III | IV |
| Cells for cDNA library construction | 4 million | 4 million | 1 | 1 | 1 | 1 |
| cDNA amplification and barcoding | Oxford Nanopore PCR-cDNA Barcoding kit (SQK-PCB109) | NEBNext® single cell/low input cDNA synthesis & amplification module, NEBNext Ultra II end repair/dA-tailing module, Oxford Nanopore Native Barcoding Expansion (PCR-free) kit (EXP-NBD114), Oxford Nanopore blunt/TA ligase master mix | ||||
| Adapter ligation | Oxford Nanopore Rapid Adapters (RAP; SQK-PCB109) | Oxford Nanopore Adapter Mix II (AMII; EXP-NBD114), NEBNext® quick ligation reaction mix, NEB Quick T4 DNA ligase enzyme | ||||
| Nanopore flow cell | Flongle R9.4.1 | MinION R9.4.1 | ||||
| Sequencer | MinION Mk1C | |||||
| Number of raw reads generated | 1,000,000 | 5,000,000 | ||||
| Total number of pass reads | 305,833 | 195,324 | 678,218 | 1,204,561 | 450,238 | 441,738 |
| Number of antibody transcripts (heavy; light) | 1111; 5122 | 1385; 5463 | 2319; 6281 | 6851; 15133 | 27; 161 | 10; 140 |
| Number of top clone reads (heavy; light) | 461; 2940 | 794; 3795 | 923; 5129 | 3436; 7440 | 5; 114 | 3; 45 |
To generate accurate full-length antibody sequences, reads trimmed of their polyA tails were aligned against germline antibody sequences using three tools: IgBLAST,34 IMGT/V-QUEST33 and minimap2.35 Only antibody transcripts with identical V(D)J and C genes were grouped together for consensus calling to avoid generating a chimeric consensus. The consensus sequences with the most abundant V(D)J and C gene combinations accounted for at least 97% of the productive antibody sequences (Table 2) and were taken as the heavy and light chain sequences for each cell. The heavy chain isotype of both hybridomas was IgG2A (Figure 2d).
Table 2.
Summary of productive heavy and light chains identified for the hybridoma cell lines 7D10 and 3C10.
| Cell line | Chain | V gene | D gene | J gene | C gene | CDR3 | Count | Percentage abundance |
|---|---|---|---|---|---|---|---|---|
| 7D10 | H | IGHV5S14*01 | IGHD4-2*01 | IGHJ2*01 | IGHG2A*01 | ARHTGKSSFFDY | 604 | 100% |
| L | IGKV16S1*01 | NA | IGKJ5*01 | IGKC*02 | QQHNEYPLT | 3007 | 99.27% | |
| IGKV3S19*01 | NA | IGKJ1*01 | IGKC*01 | QHIRELT | 22 | 0.73% | ||
| 3C10 | H | IGHV1-28*01 | IGHD1-9*01 | IGHJ2*01 | IGHG2A*01 | VREGLGITFEY | 972 | 97.00% |
| IGHV1-28*01 | IGHD1-10*01 | IGHJ2*01 | IGHG2A*01 | VREGLDITFEY | 20 | 2.00% | ||
| IGHV1-28*01 | IGHD1-5*01 | IGHJ2*01 | IGHG2A*01 | VREGLGTFEY | 10 | 1.00% | ||
| L | IGKV22S8*01 | NA | IGKJ5*01 | IGKC*02 | LQHDSGLT | 3811 | 99.71% | |
| IGKV3S19*01 | NA | IGKJ1*01 | IGKC*02 | QHIRELT | 11 | 0.29% |
The heavy and light chain sequences from libraries A and B aligned with 100% accuracy to the Sanger-generated sequences of 7D10 and 3C10, respectively (Figure 3 a,b and Supplementary Figures S1-3). Further analysis using BLAST36 showed that additional sequences at the 5’ and 3’ ends of the heavy and light chains matched with the 5’ and 3’ UTR sequences of a Rattus norvegicus antibody (Figure 3 a,b and Supplementary Figures S1-3).
Figure 3.
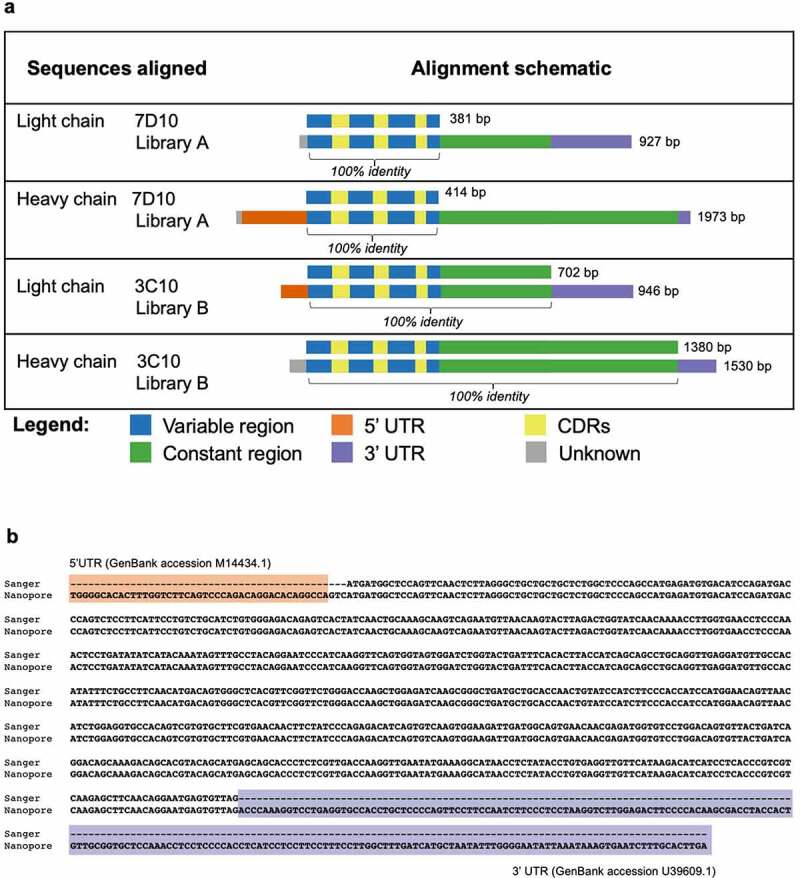
Antibody sequences from libraries A and B align with 100% accuracy to the 7D10 and 3C10 sequences. (a) Schematic of alignment of 7D10 and 3C10 antibody chains, as derived from Sanger (top row) and Nanopore (bottom row) sequencing methods. Additional bases present in the Nanopore sequence have been annotated with BLAST as indicated. Short sequences at the beginning of reads (gray) were sometimes unable to be annotated with BLAST, nor did they match the primer sequences used during library preparation. (b) Sequence alignment of the light chain sequence of 3C10 as derived from Sanger (top row) and Nanopore (bottom row) sequencing methods. Additional bases present in the Nanopore sequence have been annotated with BLAST. Regions highlighted as in Figure 3a. See Supplementary Figures 1–3 for other chains of 7D10 and 3C10.
In addition to the heavy- and light-chain sequences of 7D10 and 3C10 that were 100% identical to that obtained from Sanger sequencing, NAb-seq was able to identify additional productive sequences (Table 2). For the 3C10 cell line, the two additional productive heavy chains were much less abundant than the dominant heavy chain (representing 2.00% and 1.00% of all productive heavy chain reads, respectively), but their complementarity-determining region 3 (CDR3) sequences were highly similar to that of the dominant heavy chain, with only a single amino acid difference in both (Table 2). It is therefore possible that these sequences represent an error during reverse transcription, PCR amplification or sequencing rather than being true heavy-chain transcripts present in the 3C10 cells. Nevertheless, this demonstrates NAb-seq’s ability to distinguish between highly similar antibody transcripts in the same cell. It may be beneficial to test the binding affinity of such variants in order to validate that the most abundant heavy chain is indeed the correct one. For the 3C10 antibody, the most abundant heavy chain (that was also detected by Sanger sequencing) is confirmed by electron density in the crystal structure (pdb 5W5Z31).
Both the 7D10 and 3C10 hybridoma cell lines contained the same additional light-chain transcript (CDR3 sequence QHIRELT) at low abundance (0.73% and 0.29%, respectively; Table 2). A BLAST search revealed that these sequences were highly similar (99% identical) to the endogenous mouse kappa chain present in the SP2/0 myeloma cell line used to generate hybridomas, but with an indel that corrects the frameshift mutation (and has been previously reported37). Such sequences are thus unlikely to generate the antigen-specific antibody of interest.
Long-read sequencing of antibody genes from Rattus norvegicus single B cells
Following the successful application of NAb-seq to bulk hybridoma cell-line samples, we assessed if this approach could also recover antibody genes from single primary cells that produce antibodies, such as purified rat B cells. Splenocytes from rats immunized with BAX peptide were harvested, enriched, and sorted to isolate B cells with antibodies specific for the corresponding region in BAX (for further details see Methods). As cell sorting provided only four cells that may have been mature B cells expressing the desired antibodies, long-read sequencing of single cells was pursued.
Full-length cDNA nanopore libraries (libraries I–IV) were prepared from the four individual B cells. Cells were lysed, total RNA extracted, and full-length cDNA amplified via oligo-dT primed reverse transcription and template-switching using the NEBNext® Single cell/Low Input kit followed by native barcoding. Bands at the expected size for full-length antibody heavy- and light-chain genes were not evident after analysis on a DNA TapeStation (Figure 4a), suggesting low expression of antibody genes. The cDNA libraries were sequenced using a MinION flow cell to provide increased sequencing depth compared to the Flongle, as we could not predict the expression levels of antibody genes nor the efficiency of the cDNA preparation. Accordingly, ~5 million pass reads (Q score >10) were generated during a 72-hour run (Figure 4b-c). Basecalling was again performed in super-high accuracy mode.
Figure 4.
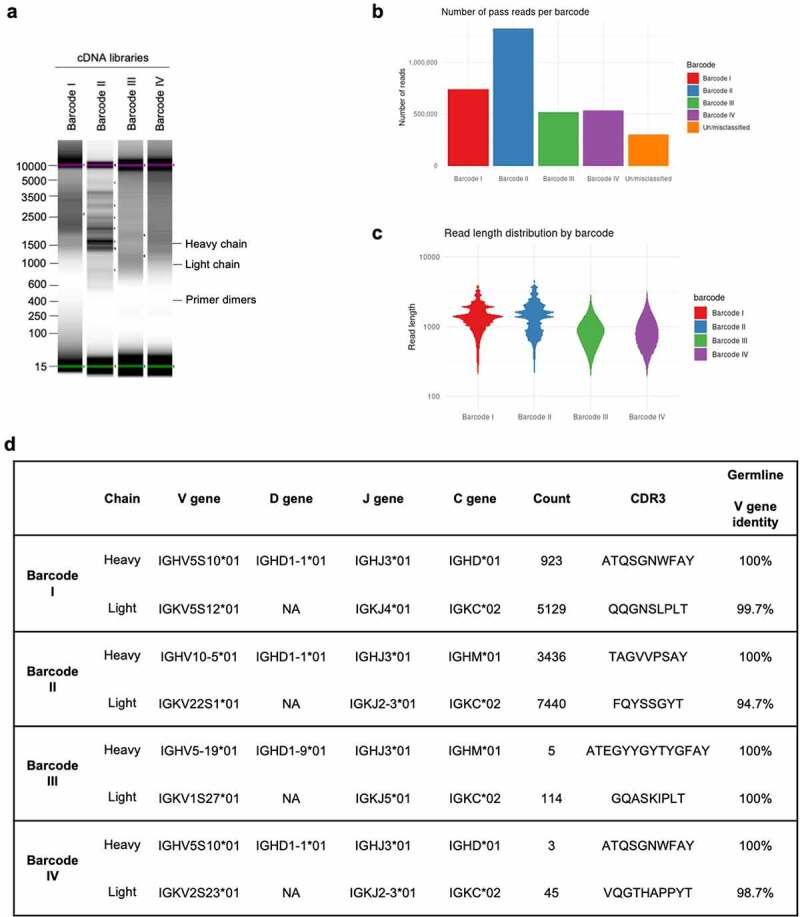
NAb-seq of four single B cells revealed antibody sequences. (a) cDNA library size and amplification. Total RNA from four sorted single B cells were extracted and converted to whole transcriptome cDNA libraries I–IV. (b) Basecalling in Guppy’s super-high accuracy mode yielded approximately 5 million total pass reads. (c) Read length of pass reads varied from ~400 bp to ~5000 bp. (d) Sequence analysis of cDNA libraries I to IV reveals V(D)J recombination, C gene usage and complementarity-determining region 3 (CDR3) amino acid sequence. Consensus calling of antibody transcripts revealed IgM and IgD isotypes.
Alignment of the pass reads to the reference Rattus norvegicus antibody gene sequences deposited in IMGT33 revealed that 1.27% of pass reads in library I and 1.82% of pass reads in library II corresponded to antibody transcripts, similar to that found for hybridoma transcripts (Table 1). However, a low percentage of pass reads in libraries III and IV corresponded to antibody transcripts (0.04% and 0.03%, respectively). The low number of antibody transcripts in cDNA libraries III and IV is a consequence of less efficient PCR amplification resulting in a high proportion of artifacts, with few reads overall aligning to the rat transcriptome (7.28% for library III and 3.26% for library IV vs ~80% for libraries I and II). Nonetheless, more than 100 antibody reads were obtained for each library, and consensus sequences could be generated for all four libraries. Upon alignment to germline sequences using IMGT/V-QUEST,33 the isotype of the heavy chains was identified to be either IgM or IgD, rather than IgG (Figure 4d). This indicated that all four cells were likely to be naïve, rather than mature switched memory B cells. In addition, consensus calling revealed near 100% identity with germline sequences (Figure 4d), indicative of antibodies expressed on naïve B cells. The small degree of sequence mismatch observed in the light chain V regions (Figure 4d) is likely due to genetic variation between the rat strain used for this study (Wistar) and the rat reference genome generated in the BN/SsNHsd strain. Regardless, these data show that, in addition to the characterization of hybridoma cell lines, the NAb-seq workflow could identify antibody sequence and isotype with near 100% accuracy from individual B cells and quickly determine that the sorting procedure for antibodies specific to BAX had failed.
Discussion
The widespread adoption of high-quality recombinant antibodies is essential for improving biological research quality and reproducibility and requires an accurate and cost-effective means of sequencing antibody genes from pre-existing and future hybridomas. Here, we have developed a new long-read sequencing workflow, NAb-seq, to provide full-length sequences of antibody heavy and light chains from hybridoma cell lines, as well the sequence of antibody genes from single naïve rat B cells. High-quality, in-frame antibody consensus sequences were generated by basecalling raw reads using guppy’s super high accuracy model, followed by a two-step consensus-calling process using Racon38 and Medaka (https://github.com/nanoporetech/medaka) for the removal of sequencing errors, including those associated with homopolymers. This was achieved with as few as five copies, an improvement on previous studies which indicated up to 25 may be required.25 Thus, NAb-seq generated antibody gene sequences with perfect accuracy (100%).
By sequencing the whole transcriptome, NAb-seq offers a simple protocol that obviates the need for species-specific customized primers. NAb-seq could thus be used for the determination of antibody sequences from any species. This simple and universal protocol is a major advantage over all targeted sequencing techniques (Sanger, Illumina, PacBio/ONT10,14,21,26). It is also much simpler, cheaper and less prone to chimerism than whole-transcriptome concatemer nanopore sequencing.28 In addition, the sequencing of the whole transcriptome using NAb-seq allows for the identification of additional productive heavy and light chains (Table 2), as hybridomas frequently produce more than one VH and VL combination.13
The trade-off for the whole-transcriptome sequencing strategy of NAb-seq is that most of the reads are not antibody transcripts and are therefore discarded. Still, we found that about 2% of the reads sequenced from hybridoma cell lines or single B cells correspond to antibody transcripts. Combined with the capacity of a nanopore Flongle flow cell to generate in excess of a million reads, this allows multiplexing dozens of samples in a single run. The use of sample barcodes in NAb-seq prior to pooling samples for sequencing also allows the pairing of heavy- and light-chain sequences in each hybridoma or single B cell. The achieved throughput is far superior to Sanger sequencing, while the ability to tune sequencing throughput (using different size nanopore flow cells, washing and reusing flow cells) makes it more flexible than Illumina or PacBio sequencing.
NAb-seq also benefits from a streamlined bioinformatics workflow. By contrast to short read Sanger or Illumina sequencing, there is no need for assembly, as long reads span the entire antibody transcript.39 With improvements in both basecalling accuracy and error correction,25 NAb-seq improves on previous efforts to sequence antibodies with nanopore. Without error correction steps, Lowden and Henry26 found that CDR3s could not be identified in 75–80% of antibody fragment reads. Error correction methods that rely on the sequencing of concatemers from rolling circle amplification can successfully reconstruct B cell receptor sequences from single B cells,28 but they require more time (both in wet and dry lab) and computational power than NAb-seq. Similarly, the assembly-based approach used to characterize cancerous B cells,27 though effective, is slower and more computationally intensive than the simple consensus strategy implemented by NAb-seq. To facilitate uptake of NAb-seq without extensive bioinformatics experience, we provide a detailed step-by-step tutorial with example data (https://kzeglinski.github.io/nab-seq/NAb_seq_vignette.html).
We conclude that NAb-seq is ideally suited for quickly and cheaply generating high-accuracy antibody sequences from between 1 and 24 hybridoma cell lines or single B cells. The low up-front cost of the minION nanopore sequencing instrument (US$1000), low cost per run (~US$30 per sample), universal protocol, flexible throughput and quick turnaround enable NAb-seq to be performed in-house and easily integrated into existing workflows. For much larger sample numbers (e.g., hundreds or thousands of hybridomas or single B cells using 10X single-cell approaches), targeted methods like RAGE-Seq which sequence only heavy/light chain amplicons become more efficient.29 Still, the computational error correction (consensus) approach implemented in NAb-seq could also be applied to targeted methods to generate high accuracy consensus sequences.
In summary, NAb-seq could help to deliver well-defined antibody products for research, diagnostic and therapeutic applications. In addition, it could facilitate the supply of all antibodies as recombinant proteins to improve reproducibility, an approach some manufacturers have started to implement.7
Materials and methods
Hybridoma cell culture and total RNA extraction
Hybridomas expressing the rat monoclonal antibody clones 7D10 and 3C10 (WEHI Antibody Facility) were cultured in Hybridoma serum-free medium (12045076; Gibco) supplemented with 10% fetal bovine serum (FBS; F9423; Sigma) at 37°C in 8% humidified CO2. When expanded for purification, the FBS was reduced to 1% FBS, and 100 U/ml interleukin-6 added to increase antibody expression.40 Hybridomas were harvested and spun down at 300 × g for 5 min. Pelleted cells were resuspended in sterile 1x Dulbecco’s phosphate-buffered saline (DPBS; 14190144; Gibco). Total RNA was extracted using the Qiagen RNeasy mini kit (74104; Qiagen) as per the manufacturer’s protocol. Briefly, 4 × 106 hybridoma cells were pelleted and resuspended in 350 µl lysis buffer (provided with the kit), loaded onto QIAshredder spin columns and centrifuged at 16000 × g for 2 min. The flow through was collected as homogenized lysates and transferred to gDNA eliminator spin columns to remove genomic DNA by centrifugation at 16000 × g for 30 s. Ethanol (350 µl 70%) was added to the flow through and mixed well. Samples (700 µl) were transferred to RNeasy spin columns and centrifuged at 16000 × g for 15 s. The flow through was discarded and wash buffer (700 µl buffer RW1 provided with the kit) added and columns centrifuged at 16000 × g for 15 s, followed by two further washes (with buffer RPE provided with the kit). RNA was eluted by adding 30 µl DNase-free water directly to the spin column membranes and centrifuging at 16000 × g for 1 min. Total RNA extracted from 7D10 and 3C10 expressing hybridoma cells was 37 and 60 µg, respectively, as measured by nanodrop. 50 ng total RNA from each hybridoma was taken directly for cDNA synthesis.
cDNA synthesis and library construction from hybridomas
To sequence antibody genes from the two hybridomas, whole-transcriptome cDNA libraries were first constructed from extracted mRNA using a PCR-cDNA barcoding kit (SQK-PCB109; Oxford Nanopore Technologies). Briefly, 50 ng total RNA (~1 ng (polyA)+ mRNA) from each hybridoma was taken in RNase-free PCR tubes containing Maxima H Minus reverse transcriptase enzyme (EP0751; ThermoFisher Scientific), VN primer (variant of oligo (dT) with complementary nucleotides for annealing of barcode primers) and strand-switching primer (see Supplementary Table 1 for sequences). Samples were then incubated at 42°C for 90 min (one cycle) followed by enzyme inactivation at 85°C for 1 min (one cycle) in a thermocycler for the generation of full-length cDNA from poly(A)+ messenger RNA.
cDNA from each of the two hybridomas then underwent full-length amplification and sample barcoding using 12 cycles of PCR. Each PCR reaction mix consisted of 25 µl 2x LongAmp Taq master mix (M0287; New England Biolabs), 1.5 µl barcoded primers A or B (for ease, barcodes BP01 and BP02 from the kit are referred to here as A and B), 18.5 µl nuclease-free water, and 5 µl (~0.25 ng) cDNA from 7D10 or 3C10-expressing hybridoma cells. The following PCR cycling conditions were used: initial denaturation at 95°C for 30 s (1 cycle), denaturation at 95°C for 15 s (12 cycles), annealing at 62°C for 15 s (12 cycles), extension at 65°C for 4 min 10 s (12 cycles), and final extension at 65°C for 6 min (1 cycle). (The long extension time of 4 min 10 s was to selectively amplify cDNAs up to ~5 kb in length.)
To clean up the cDNA libraries, PCR reactions with the same barcode (A or B) were pooled in two 1.5 ml Eppendorf® DNA LoBind tubes (EP0030108051; Merck) and primer dimers removed using 0.8x volume equivalent Agencourt® AMPure® XP beads (A63880; Beckman Coulter). Briefly, beads (80 µl) were added to each pooled sample, incubated on a hula mixer for 5 min at room temperature, and spun and pelleted on a magnet. Supernatants were pipetted off and the resulting beads washed with 70% ethanol (200 µl freshly prepared using nuclease-free water) without disturbing the pellet. The ethanol was removed using a pipette and the beads washed again with ethanol, and the pelleted beads spun down and placed back on the magnet. Residual ethanol was pipetted off and the beads briefly allowed to dry. While the beads still appeared glossy (with no cracking) they were resuspended in 12 µl elution buffer (provided with the PCR-cDNA barcoding kit) to recover the cDNA libraries. Roughly 100 ng of each cleaned up cDNA libraries A and B, as quantified using Qubit dsDNA HS kit (Q32851; ThermoFisher Scientific), was taken forward for sequencing. Size distribution of the amplified cDNAs was analyzed using High Sensitivity D5000 ScreenTape (50675592; Agilent Technologies) in a 4200 TapeStation (Agilent). DNA length for cDNA libraries A and B ranged from 400 bp to 2500 bp (see Figure 2a). The concentration (11–13 ng/µl) was consistent with the Qubit and translated to a molarity of 11.5–13.6 nM.
Long-read sequencing of hybridoma cDNA libraries
Prior to loading the barcoded cDNA libraries A and B onto the Flongle flow cell for long-read sequencing, they were ligated with adapters. The two barcoded libraries (25 fmol of each) were pooled in a 1.5 ml Eppendorf® DNA LoBind tube, combined with Rapid adapters (SQK-PCB109 kit; Oxford Nanopore Technologies), and incubated at room temperature for 5 min. The adapter-ligated pooled cDNA library was transferred to a fresh tube and combined with loading beads and sequencing buffer (SQK-PCB109 kit) to form the sequencing mix. The Flongle flow cell R9.4.1 (FLO-FLG001; Oxford Nanopore Technologies) was primed with pre-mixed flush buffer and flush tether (EXP-FLP002; Oxford Nanopore Technologies), then loaded with the sequencing mix and run for 24 hours on a MinION Mk1C sequencing device. Upon completion, guppy (v5.0.11) was used to perform super high accuracy basecalling and demultiplexing of the data.
Single rat B cell sort
Single rat B cells were sourced from a separate project designed to generate antibodies to a specific region in the pro-apoptotic protein BAX. Two Wistar rats had been immunized with keyhole limpet hemocyanin-conjugated BAX peptide and splenocytes used to generate hybridomas, with excess splenocytes frozen. As the hybridomas had not generated the desired antibodies to a specific region in BAX, we pursued the possibility that memory B cells that recognized this region in BAX were rare, but could be isolated from stored splenocytes using rat B cell cloning, especially if B cells were selected for binding to the BAX peptide during flow cytometry. As rat B cell cloning had not been reported, the mouse B cell cloning procedure41 was modified by sourcing several antibodies specific for rat B cell surface markers. In addition, long-read sequencing was pursued to avoid the need for rat-specific primers.
To select for mature B cells, splenocytes underwent a negative selection for non-B cells, followed by B cell cloning41 using rat-specific antibodies. Splenocytes from two immunized rats were rapidly thawed in a 37°C water bath, combined, pelleted, and resuspended in fluorescence-activated cell sorting (FACS) buffer (1x DPBS supplemented with 2% FBS and 1 mM EDTA) and stored on ice for cell counting. 5 × 107 splenocytes were centrifuged (300 × g, 4 min) and resuspended in 1 ml FACS buffer. Non-B cells were then negatively selected using EasySep® rat B cell isolation kit (19644; STEMCELL Technologies) according to manufacturer’s instructions. In brief, the splenocytes were taken in a 5 ml polystyrene round-bottom tube (352058; Corning) and incubated with an antibody cocktail (50 µl) to label non-B cell surface markers for 10 min at room temperature. Labeled splenocytes were then mixed with 25 µl of RapidSpheresTM (magnetic particles) and topped up to 2.5 ml with culture medium. The tube was placed in an EasySepTM magnet (18000; STEMCELL Technologies), incubated for 3 min at room temperature, and the negatively selected splenocytes transferred into a new tube. The magnetic selection step was performed two additional times to obtain splenocytes depleted of non-B cells.
To then FACS-sort for memory cells, the partially enriched B cells were resuspended at 1–2 × 107 cells per 100 µl and incubated at 4°C with a mixture of antibodies to rat B and non-B cells. The mixture included antibodies to CD3 to stain T cells (PE; 201411; BioLegend; 0.5 µg per 100 µl cells); CD11b/c to stain monocytes, granulocytes, macrophages, dendritic cells, and natural killer cells (PE; 201807; BioLegend; 0.25 µg per 100 µl cells), CD32 to stain cells with FcγII receptor (Fc block; 556970; BD Biosciences; 1 µg per 100 µl cells), IgM to stain immature B cells (FITC; 408905; BioLegend; 1 µg per 100 µl cells), and CD45RA to stain mature B cells (APC; 202314; BioLegend; 0.25 µg per 100 µl cells). To also select for B cells that bind to a specific region in BAX, the mixture contained a biotinylated BAX peptide conjugated to fluorescently tagged streptavidin, either brilliant violet (BV) 510 streptavidin (SA/BV510; 405234; BioLegend) or BV 785 streptavidin (SA/BV785; 405249; BioLegend), as for mouse B cell cloning.41
Prior to FACS sorting, stained cells were washed twice with FACS buffer and resuspended at 8 × 106 cells/ml. Cells that were CD3− CD11b/c− IgM−, as well as CD45RA+ BAX peptide+, were sorted using FACSAria III (Becton Dickinson) into 1x DPBS in a 96-well semi skirt PCR plate (342000; Interpath Services). This preliminary rat B cell cloning approach sorted only four B cells, each of which was shown by long-read sequencing to be naïve. Future development of rat B cell cloning may benefit from additional antibodies to differentiate between naïve and mature B cells and also between the different immunoglobulin isotypes.
cDNA synthesis and library preparation from single rat B cells
Whole-transcriptome cDNA libraries from each of the four sorted single B cells were prepared using the NEBNext® single cell/low input cDNA synthesis and amplification module (E6421S; New England BioLabs Inc.). As a single mammalian cell contains only 0.1 pg mRNA,42 this module is required to amplify cDNA to levels sufficient for long-read sequencing (100–200 fmol, assuming a 1 kb average size). To lyse the single cells, NEBNext® cell lysis buffer (10X), murine RNase inhibitor and nuclease-free water were added to wells and incubated at room temperature for 5 min. NEBNext® single cell RT primer mix consisting of oligo (dT) primers (see Supplementary Table 1 for sequences) was added and the plate incubated in a thermocycler at 70°C for 5 min for primer annealing. For reverse transcription and template switching, single cell RT buffer, template switching oligo, single cell RT enzyme mix, and nuclease-free water (all from the NEBNext® kit) were added and the plate incubated in a thermocycler at 42°C for 90 min followed by enzyme inactivation at 70°C for 10 min. The resulting cDNAs were amplified via 21 PCR cycles using NEBNext® single cell cDNA PCR master mix and NEBNext® single cell cDNA PCR primer mix (E6421S; New England BioLabs Inc.). The following PCR cycling conditions were used: initial denaturation at 98°C for 45 s (1 cycle), denaturation at 98°C for 10 s (21 cycles), annealing at 62°C for 15 s (21 cycles), extension at 72°C for 6 min (21 cycles) and final extension at 72°C for 5 min (1 cycle). The long extension time should allow amplification of long transcripts of cDNA (for instance, whole antibody gene transcripts). The libraries were cleaned up using 0.8x volume equivalent Agencourt® AMPure® XP beads, as for the hybridoma libraries. The amount of cDNAs in the amplified libraries ranged from 2 to 12 ng (Qubit, ~10 fmol).
As nanopore barcoding by ligation requires ~100–200 fmol starting material, a second round of PCR amplification was performed with 15 cycles and extension time of 7 min, yielding 550–990 ng cDNAs from a starting material of 1 ng cDNAs from the first PCR. The lengths of the whole transcriptome cDNAs in the amplified libraries ranged from 600 to 3500 bp (see Figure 4a) as analyzed using High Sensitivity D5000 ScreenTape (50675592; Agilent Technologies) in a 4200 TapeStation (Agilent). The cDNA libraries were taken in fresh 1.5 ml Eppendorf® DNA LoBind tubes and end-prepped for barcoding using NEBNext® Ultra II end repair/dA-tailing module (E7546S; New England BioLabs Inc.). The end-prep reactions were incubated at 20°C for 15 min followed by enzyme inactivation at 65°C for 15 min. The end-prepped cDNA libraries were cleaned up using 1x volume equivalent Agencourt® AMPure® XP beads, taken to fresh 1.5 ml Eppendorf® DNA LoBind tubes and barcoded using native barcode primers (EXP-NBD114; Oxford Nanopore Technologies) and blunt/TA ligase master mix (M0367S; Oxford Nanopore Technologies). Barcoding reactions were incubated at room temperature for 30 min and cleaned up using 1x volume equivalent Agencourt® AMPure® XP beads. The barcoded primers BP15, BP16, BP18 and BP19 from the native barcoding expansion kit (EXP-NBD114; Oxford Nanopore Technologies) are referred to here as I, II, III and IV.
Barcoded cDNA libraries I–IV were pooled to obtain ~100-200 fmol final. Adapters were ligated to the pooled library using adapter mix II (AMII; EXP-NBD114; Oxford Nanopore Technologies), NEBNext® quick ligation reaction mix (E6056S; New England BioLabs Inc.) and Quick T4 DNA ligase (E6056S; New England BioLabs Inc.) by incubation at room temperature for 10 min. The adapter-ligated libraries were cleaned up using 1x volume equivalent Agencourt® AMPure® XP beads and the total sample taken to long-read sequencing.
Long-read sequencing of single B cell cDNA libraries
Long-read sequencing of single B cells used the Ligation sequencing kit (SQK-LSK109; Oxford Nanopore Technologies). Before loading of the cDNA libraries onto a MinION R9.4.1 flow cell (FLO-MIN106D; Oxford Nanopore Technologies) in a MinION Mk1C sequencing device, the flow cell was primed, as for the Flongle flow cell above. The sequencing mix was prepared in a fresh 1.5 ml Eppendorf® DNA LoBind with sequencing buffer, loading beads (provided in the SQK-LSK109 kit) and the pooled libraries. After quality checking the flow cell, the sequencing mix was added and the MinION flow cell run for 72 hours. Upon completion, guppy (v5.0.11) was used to perform super high accuracy basecalling and demultiplexing of the data.
Bioinformatic analysis
Quality control checks of read counts and length were performed using NanoComp (v1.12.043). Sequencing adapters and polyA tails were trimmed from the reads using cutadapt (v3.444). All pass reads (Q score > 10, representing an accuracy of >90%) were aligned to reference Rattus norvegicus antibody gene sequences retrieved from IMGT33 using minimap2 (v2.17-r94135) with the map-ont preset (settings designed for error-prone long reads). Any read with an alignment to a germline antibody gene (variable or constant) was considered an antibody transcript and taken forward for annotation. Antibody transcripts were annotated to identify their germline V(D)J and C genes, as well as CDR sequences using IgBLAST (v1.17.134) with default parameters, and the results written out in AIRR format45 for processing in R (v4.0.546).
In order to correct sequencing errors, antibody transcripts with identical V(D)J and C genes were grouped, and a separate consensus was called for each group with at least 5 reads (as this was empirically determined to be the number of reads required for a high-accuracy consensus sequence). Grouping the reads by their germline genes prevents a chimeric consensus (that merges together multiple antibody sequences) from being generated, as there are often multiple different antibody transcripts in one cell (e.g., resulting from leaky transcription from the second allele, PCR chimeras or hybridomas that express multiple antibody sequences).13 Error-corrected consensus sequences were generated using a two-step process: one round of Racon (v1.4.3, using the parameters -w 5000 -t 4 -u -g − 8 -x − 6 -m 8 – no-trimming38) followed by one round of Medaka (v1.4.2, using default parameters (https://github.com/nanoporetech/medaka)). These software tools use multiple reads of the same transcript to correct errors by taking the consensus of the reads. Finally, the error corrected heavy and light chain sequences for each cell were re-analyzed using IgBLAST (v1.17.134), minimap2 (v2.17-r941, to identify constant genes35) and the IMGT/V-QUEST33 webserver in order to determine their properties (e.g. percentage identity to germline genes, CDR sequences). All code is available on GitHub (https://github.com/kzeglinski/nab-seq) as is an in-depth bioinformatic tutorial (https://kzeglinski.github.io/nab-seq/NAb_seq_vignette.html).
Supplementary Material
Acknowledgments
We thank Sarah MacRaild, Stephen Wilcox and the WEHI Genomics Platform for help with ONT sequencing, Paul Masendycz, Kaye Wycherley and the WEHI Antibody Facility for providing hybridoma cell lines and stored rat splenocytes, and Michelle Miller for advice on the structure of 3C10 antibody.
Funding Statement
This work was supported by a Program Grant (GNT1113133) to RMK, a Fellowship (1155342) to SLN, and Investigator Grant (2007996) to QG from the Australian National Health and Medical Research Council. Work in the laboratories of the authors was made possible through the Victorian State Government Operational Infrastructure Support and the Australian Government NHMRC IRISS (9000587).
Abbreviations.
| APC | allophycocyanin |
| BAX | BCL-2-associated X protein |
| BLAST | basic local alignment search tool |
| BV | brilliant violet |
| DPBS | Dulbecco’s phosphate-buffered saline |
| FACS | fluorescence-activated cell sorting |
| FBS | fetal bovine serum |
| Fc | fragment crystallizable region |
| FR1 | framework region 1 |
| FR4 | framework region 4 |
| Ig | immunoglobulin |
| IMGT® | international ImMunoGeneTics information system® |
| NAb-seq | Nanopore Antibody sequencing |
| ONT | Oxford Nanopore Technologies |
| PCR | polymerase chain reaction |
| PE | phycoerythrin |
| RACE | rapid amplification of cDNA ends |
| RAGE-seq | repertoire and gene expression by sequencing |
| VH | variable heavy chain |
| VL | variable light chain |
| V-QUEST | V-QUEry and Standardization |
Author contributions
HPSS, RU, KZ, QG, and RK conceived and designed the study. HPSS and RU assembled the team. HPSS performed the experiments and KZ the analysis. SN consulted the possible applications, and RU, SN, and MR advised throughout the study. HPSS, KZ, QG, and RK wrote the manuscript, RU, SN, and MR edited and contributed to the final draft. All authors reviewed and approved the final draft.
Disclosure statement
The authors report there are no competing interests to declare.
Data availability statement
All data generated in this study (FASTQ and FAST5) is available from the European Nucleotide Archive (ENA) under accession number PRJEB51442.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2022.2106621
References
- 1.Berglund L, Bjorling E, Oksvold P, Fagerberg L, Asplund A, Szigyarto CA, Persson A, Ottosson J, Wernerus H, Nilsson P, et al. A genecentric human protein atlas for expression profiles based on antibodies. Mol Cell Proteomics. 2008;7(10):2019–12. doi: 10.1074/mcp.R800013-MCP200. [DOI] [PubMed] [Google Scholar]
- 2.Slaastad H, Wu W, Goullart L, Kanderova V, Tjonnfjord G, Stuchly J, Kalina T, Holm A, Lund-Johansen F.. Multiplexed immuno-precipitation with 1725 commercially available antibodies to cellular proteins. Proteomics. 2011;11(23):4578–82. doi: 10.1002/pmic.201000744. [DOI] [PubMed] [Google Scholar]
- 3.Begley CG. Six red flags for suspect work. Nature. 2013;497(7450):433–34. doi: 10.1038/497433a. [DOI] [PubMed] [Google Scholar]
- 4.Bradbury A, Pluckthun A. Reproducibility: standardize antibodies used in research. Nature. 2015;518(7537):27–29. doi: 10.1038/518027a. [DOI] [PubMed] [Google Scholar]
- 5.Bradbury AR, Pluckthun A. Getting to reproducible antibodies: the rationale for sequenced recombinant characterized reagents. Protein Eng Des Sel. 2015;28(10):303–05. doi: 10.1093/protein/gzv051. [DOI] [PubMed] [Google Scholar]
- 6.Voskuil JLA, Bandrowski A, Begley CG, Bradbury ARM, Chalmers AD, Gomes AV, Hardcastle T, Lund-Johansen F, Pluckthun A, Roncador G, et al. The antibody society’s antibody validation webinar series. MAbs. 2020;12:1794421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bradbury ARM, Dubel S, Knappik A, Pluckthun A. Animal- versus in vitro -derived antibodies: avoiding the extremes. MAbs. 2021;13(1):1950265. doi: 10.1080/19420862.2021.1950265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Polakiewicz RD. Antibodies: the solution is validation. Nature. 2015;518(7540):483. doi: 10.1038/518483b. [DOI] [PubMed] [Google Scholar]
- 9.Doenecke A, Winnacker EL, Hallek M. Rapid amplification of cDNA ends (RACE) improves the PCR-based isolation of immunoglobulin variable region genes from murine and human lymphoma cells and cell lines. Leukemia. 1997;11(10):1787–92. doi: 10.1038/sj.leu.2400781. [DOI] [PubMed] [Google Scholar]
- 10.Meyer L, Lopez T, Espinosa R, Arias CF, Vollmers C, DuBois RM, Gill AC. A simplified workflow for monoclonal antibody sequencing. PLoS One. 2019;14(6):e0218717. doi: 10.1371/journal.pone.0218717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruberti F, Cattaneo A, Bradbury A. The use of the RACE method to clone hybridoma cDNA when V region primers fail. J Immunol Methods. 1994;173(1):33–39. doi: 10.1016/0022-1759(94)90280-1. [DOI] [PubMed] [Google Scholar]
- 12.Ozawa T, Kishi H, Muraguchi A. Amplification and analysis of cDNA generated from a single cell by 5’-RACE: application to isolation of antibody heavy and light chain variable gene sequences from single B cells. Biotechniques. 2006;40(4):469–70, 72, 74 passim. doi: 10.2144/000112123. [DOI] [PubMed] [Google Scholar]
- 13.Bradbury ARM, Trinklein ND, Thie H, Wilkinson IC, Tandon AK, Anderson S, Bladen CL, Jones B, Aldred SF, Bestagno M, et al. When monoclonal antibodies are not monospecific: hybridomas frequently express additional functional variable regions. MAbs. 2018;10(4):539–46. doi: 10.1080/19420862.2018.1445456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y, Kim SH, Shang Y, Guillory J, Stinson J, Zhang Q, Hotzel I, Hoi KH. Barcoded sequencing workflow for high throughput digitization of hybridoma antibody variable domain sequences. J Immunol Methods. 2018;455:88–94. doi: 10.1016/j.jim.2018.01.004. [DOI] [PubMed] [Google Scholar]
- 15.DeKosky BJ, Ippolito GC, Deschner RP, Lavinder JJ, Wine Y, Rawlings BM, Varadarajan N, Giesecke C, Dorner T, Andrews SF, et al. High-throughput sequencing of the paired human immunoglobulin heavy and light chain repertoire. Nat Biotechnol. 2013;31(2):166–69. doi: 10.1038/nbt.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldstein LD, Chen YJ, Wu J, Chaudhuri S, Hsiao YC, Schneider K, Hoi KH, Lin Z, Guerrero S, Jaiswal BS, et al. Massively parallel single-cell B-cell receptor sequencing enables rapid discovery of diverse antigen-reactive antibodies. Commun Biol. 2019;2(1):304. doi: 10.1038/s42003-019-0551-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rouet R, Jackson KJL, Langley DB, Christ D. Next-generation sequencing of antibody display repertoires. Front Immunol. 2018;9:118. doi: 10.3389/fimmu.2018.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Philpott M, Watson J, Thakurta A, Brown T Jr., Brown T Sr., Oppermann U, Cribbs AP. Nanopore sequencing of single-cell transcriptomes with scCOLOR-seq. Nat Biotechnol. 2021;39(12):1517–20. doi: 10.1038/s41587-021-00965-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vollmers C, Penland L, Kanbar JN, Quake SR, Fugmann SD. Novel exons and splice variants in the human antibody heavy chain identified by single cell and single molecule sequencing. PLoS One. 2015;10(1):e0117050. doi: 10.1371/journal.pone.0117050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han SY, Antoine A, Howard D, Chang B, Chang WS, Slein M, Deikus G, Kossida S, Duroux P, Lefranc MP, et al. Coupling of single molecule, long read sequencing with IMGT/HighV-QUEST analysis expedites identification of SIV gp140-specific antibodies from scFv phage display libraries. Front Immunol. 2018;9:329. doi: 10.3389/fimmu.2018.00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hemadou A, Giudicelli V, Smith ML, Lefranc MP, Duroux P, Kossida S, Heiner C, Hepler NL, Kuijpers J, Groppi A, et al. Pacific biosciences sequencing and IMGT/HighV-QUEST analysis of full-length single chain fragment variable from an in vivo selected phage-display combinatorial library. Front Immunol. 2017;8:1796. doi: 10.3389/fimmu.2017.01796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brochu HN, Tseng E, Smith E, Thomas MJ, Jones AM, Diveley KR, Law L, Hansen SG, Picker LJ, Gale M Jr., et al. Systematic profiling of full-length Ig and TCR repertoire diversity in rhesus macaque through long read transcriptome sequencing. J Immunol. 2020;204(12):3434–44. doi: 10.4049/jimmunol.1901256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raftery LJ, Howard CB, Grewal YS, Vaidyanathan R, Jones ML, Anderson W, Korbie D, Duarte T, Cao MD, Nguyen SH, et al. Retooling phage display with electrohydrodynamic nanomixing and nanopore sequencing. Lab Chip. 2019;19(24):4083–92. doi: 10.1039/C9LC00978G. [DOI] [PubMed] [Google Scholar]
- 24.Hackl T, Trigodet F, Eren AM, Biller SJ, Eppley JM, Luo E, Burger A, DeLong EF, FM G. proovframe: frameshift-correction for long-read (meta)genomics. bioRxiv. 2021. doi: 10.1101/2021.08.23.457338. [DOI] [Google Scholar]
- 25.Karst SM, Ziels RM, Kirkegaard RH, Sorensen EA, McDonald D, Zhu Q, Knight R, Albertsen M. High-accuracy long-read amplicon sequences using unique molecular identifiers with Nanopore or PacBio sequencing. Nat Methods. 2021;18(2):165–69. doi: 10.1038/s41592-020-01041-y. [DOI] [PubMed] [Google Scholar]
- 26.Lowden MJ, Henry KA. Oxford nanopore sequencing enables rapid discovery of single-domain antibodies from phage display libraries. Biotechniques. 2018;65(6):351–56. doi: 10.2144/btn-2018-0123. [DOI] [PubMed] [Google Scholar]
- 27.Minervini CF, Cumbo C, Redavid I, Conserva MR, Orsini P, Zagaria A, Anelli L, Coccaro N, Tota G, Impera L, et al. Nanopore sequencing approach for immunoglobulin gene analysis in chronic lymphocytic leukemia. Sci Rep. 2021;11(1):17668. doi: 10.1038/s41598-021-97198-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Volden R, Vollmers C. Single-cell isoform analysis in human immune cells. Genome Biol. 2022;23(1):47. doi: 10.1186/s13059-022-02615-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh M, Al-Eryani G, Carswell S, Ferguson JM, Blackburn J, Barton K, Roden D, Luciani F, Giang Phan T, Junankar S, et al. High-throughput targeted long-read single cell sequencing reveals the clonal and transcriptional landscape of lymphocytes. Nat Commun. 2019;10(1):3120. doi: 10.1038/s41467-019-11049-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iyer S, Anwari K, Alsop AE, Yuen WS, Huang DC, Carroll J, Smith NA, Smith BJ, Dewson G, Kluck RM. Identification of an activation site in Bak and mitochondrial Bax triggered by antibodies. Nat Commun. 2016;7(1):11734. doi: 10.1038/ncomms11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robin AY, Iyer S, Birkinshaw RW, Sandow J, Wardak A, Luo CS, Shi M, Webb AI, Czabotar PE, Kluck RM, et al. Ensemble properties of Bax determine its function. Structure. 2018;26(10):1346–59.e5. doi: 10.1016/j.str.2018.07.006. [DOI] [PubMed] [Google Scholar]
- 32.Robin AY, Miller MS, Iyer S, Shi MX, Wardak AZ, Lio D, Smith NA, Smith BJ, Birkinshaw RW, Czabotar PE, et al. Structure of the BAK-activating antibody 7D10 bound to BAK reveals an unexpected role for the alpha1-alpha2 loop in BAK activation. Cell Death Differ. 2022. doi: 10.1038/s41418-022-00961-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lefranc MP, Giudicelli V, Duroux P, Jabado-Michaloud J, Folch G, Aouinti S, Carillon E, Duvergey H, Houles A, Paysan-Lafosse T, et al. IMGT®, the international ImMunoGeneTics information system® 25 years on. Nucleic Acids Res. 2015;43(D1):D413–22. doi: 10.1093/nar/gku1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ye J, Ma N, Madden TL, Ostell JM. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 2013;41(W1):W34–40. doi: 10.1093/nar/gkt382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Birol I. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34(18):3094–100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 37.Pornnoppadol G, Zhang B, Desai AA, Berardi A, Remmer HA, Tessier PM, Greineder CF, Murugan AK. A hybridoma-derived monoclonal antibody with high homology to the aberrant myeloma light chain. PLoS One. 2021;16(10):e0252558. doi: 10.1371/journal.pone.0252558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vaser R, Sovic I, Nagarajan N, Sikic M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017;27(5):737–46. doi: 10.1101/gr.214270.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amarasinghe SL, Su S, Dong X, Zappia L, Ritchie ME, Gouil Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020;21(1):30. doi: 10.1186/s13059-020-1935-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bazin R, Lemieux R. Increased proportion of B cell hybridomas secreting monoclonal antibodies of desired specificity in cultures containing macrophage-derived hybridoma growth factor (IL-6). J Immunol Methods. 1989;116(2):245–49. doi: 10.1016/0022-1759(89)90210-X. [DOI] [PubMed] [Google Scholar]
- 41.von Boehmer L, Liu C, Ackerman S, Gitlin AD, Wang Q, Gazumyan A, Nussenzweig MC. Sequencing and cloning of antigen-specific antibodies from mouse memory B cells. Nat Protoc. 2016;11(10):1908–23. doi: 10.1038/nprot.2016.102. [DOI] [PubMed] [Google Scholar]
- 42.Huang XT, Li X, Qin PZ, Zhu Y, Xu SN, Chen JP. Technical advances in single-cell RNA sequencing and applications in normal and malignant hematopoiesis. Front Oncol. 2018;8:582. doi: 10.3389/fonc.2018.00582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Coster W, D’Hert S, Schultz DT, Cruts M, Van Broeckhoven C, Berger B. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 2018;34(15):2666–69. doi: 10.1093/bioinformatics/bty149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011;17(1):10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 45.Vander Heiden JA, Marquez S, Marthandan N, Bukhari SAC, Busse CE, Corrie B, Hershberg U, Kleinstein SH, Iv FA M, Ralph DK, et al. AIRR Community Standardized Representations for Annotated Immune Repertoires. Front Immunol. 2018;9:2206. doi: 10.3389/fimmu.2018.02206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.R Development Core Team : a language and environment for statistical computing. 2021.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated in this study (FASTQ and FAST5) is available from the European Nucleotide Archive (ENA) under accession number PRJEB51442.