

ABSTRACT
The small GTPase RhoA controls many important cellular processes through its ability to activate multiple downstream effector pathways. Most RhoA effectors contain a Rho-binding domain (RBD), and interaction between active RhoA and the RBD typically induces a conformational change in effectors that stimulates their recruitment or activity. Isolated GTPase binding domains fused to GST have been widely used in so-called pulldown assays to measure the activation state of other GTPases in cell lysates. Similarly, GST fusions containing the RBD of the RhoA effector Rhotekin have been widely adopted as a standardized tool for the measurement of RhoA activation. RBDs have also been used to generate fluorescent reporter constructs to localize sites of GTPase activation in intact cells. In this report, we demonstrate that not all forms of active RhoA are capable of interacting with the Rhotekin RBD. A constitutively active RhoA-G14V mutant, which interacted with the RBDs of ROCK2 and mDIA1, was unable to bind the Rhotekin RBD as evidenced by both conventional GST pulldown assay and our newly established BRET assay. Furthermore, active RhoA induced by different stimuli in cells also displayed binding preference for its diverse effectors. Our data demonstrate that RhoA may undergo effector-specific activation for differential regulation of its downstream pathways, and that RhoA activation should not be defined solely by its interaction with Rhotekin.
KEYWORDS: RhoA, Rhotekin, mDia1, ROCK2, Rho-binding domain, RhoA activation assay, GST pulldown, BRET
Introduction
Ras homolog member A (RhoA) is one of the oldest and best characterized small GTPases. RhoA plays crucial roles in many biological processes ranging from actin cytoskeleton reorganization to regulation of cell polarity, migration, morphogenesis, transformation, cell cycle progression, and transcription [1,2]. Acting downstream of diverse cell surface receptors, RhoA can be activated by a variety of stimuli including cytokines, growth factors, serum, lysophosphatidic acid (LPA), mechanical force and many others [3–5]. Upon activation, RhoA becomes GTP-bound and generally translocates from the cytosol to cellular membranes, where it interacts with effector proteins to advance signal transduction [6,7]. Mutations in RhoA have been reported in many human cancers where they contribute to malignant processes [8].
Under physiological conditions, RhoA interconverts between two molecular states: a GTP-bound active state and a GDP-bound inactive state. Activation is mediated and fine-tuned by Rho-specific guanine nucleotide exchange factors (RhoGEFs), while inactivation is mediated by a large family of Rho GTPase activating proteins (RhoGAPs). Rho guanine nucleotide dissociation inhibitors (RhoGDIs) bind GDP-bound RhoA and maintain it in a soluble state by sequestering C-terminal prenyl groups [2,6,9–11]. Aside from these three classes of regulators, RhoA activity can be further modulated by post-translational modifications [9,12,13].
GTP-bound RhoA activates downstream signalling by interacting with Rho-binding domains (RBDs) of diverse effector proteins [3,6,7,14]. These effectors include Rhotekin (RTKN), mammalian diaphanous homolog 1 and 2 (mDia1/2), Rho-associated coiled-coil kinases 1 and 2 (ROCK1/2), serine/threonine-protein kinase N (PKN), and many others [7,15–19]. The interaction sites for the majority of these structurally diverse effectors are located in the RhoA Switch I (amino acids 29–42) and Switch II (amino acids 62–68) domains, which are also common contact sites for various RhoGEFs, RhoGAPs and RhoGDIs [6]. Interestingly, many RhoA mutations identified in human cancers are either within or adjacent to these domains [8]. While some of the mutations affect its GTPase activity, others alter interaction of RhoA with a subset of its regulators and/or effectors, thus contributing to pathological processes including cancer.
The notion that the Rhotekin RBD selectively binds to GTP-bound RhoA and inhibits RhoA GTPase activity has been adopted as the molecular basis for assays to detect RhoA activation [3,18]. The first and most widely used assay uses a Glutathione S-transferase (GST)-fusion of the Rhotekin RBD (GST-RBD) as a bait to capture active RhoA, thus allowing precipitation of active RhoA in response to various stimuli, and its quantitation by immunoblotting [3]. Fluorescence resonance energy transfer (FRET)-based assays have also been developed for the spatiotemporal detection of RhoA activation in living cells by microscopic imaging [11,20].
Here, we report that not all forms of active RhoA interact with Rhotekin. We demonstrate that two different constitutively active RhoA mutants, RhoA-G14V and RhoA-Q63L differentially interact with the Rhotekin RBD as determined by GST-pulldown assay using GST-RBD (hereafter as GST-RTKN-RBD) beads. This result was further confirmed by our newly established bioluminescence resonance energy transfer (BRET) assay. We also demonstrate that measurements of RhoA activation kinetics may differ depending upon the reagents used for their analysis. Specifically, we observed distinct activation kinetics in response to lysophosphatidic acid (LPA) using Rho-binding domains derived from Rhotekin vs mDia1.
Results and discussion
Like many other GTPases, activation of Rho family GTPases is primarily quantified using pulldown assays. Assays using GST-RTKN-RBD as the bait have long been used as a standard and popular methodology for detection of RhoA activation under various conditions, and almost all commercially available RhoA activation assay kits are based exclusively on the interaction between GTP-bound RhoA and the RBD of Rhotekin. However, such assays rely on an assumption that RhoA will always associate with Rhotekin upon activation, discounting the possibility that RhoA may undergo effector-specific activation under certain physiological circumstances.
When using GST pulldown assays for the detection of RhoA activation in our experiments, we found, to our surprise, that a constitutively active mutant widely used as a positive control, RhoA-G14V, was not pulled down by GST-RTKN-RBD. To determine whether this was specific for the Rhotekin RBD, we generated GST-fusions of RBDs from two other well-known effectors of RhoA, mDia1 and ROCK2 (GST-mDia1-RBD and GST-ROCK2-RBD), and purified them in parallel with the GST-RTKN-RBD (Figure 1(a and b)). We then tested four different forms of RhoA, including wild-type (RhoA-WT), two constitutively active mutants caused by different mutations (RhoA-G14V and RhoA-Q63L), and a dominant negative mutant RhoA-T19N. All forms of RhoA contained the influenza haemagglutinin (HA) epitope tag at their N-terminus. These RhoA variants were ectopically expressed in HEK293 cells and subjected to parallel pulldown assays using equivalent amounts of each of the three RBD fusions. As shown in Figure 1(c and d), RhoA-WT interacted with all three RBDs but with different binding affinities: strongly with RTKN-RBD and mDia1-RBD but weakly with ROCK2-RBD. RhoA-Q63L interacted strongly with all three RBDs (Figure 1(c and d)). As expected, no interaction was observed of any RBD with the dominant negative RhoA-T19N (Figure 1(c and d)). Surprisingly, we found that the active mutant RhoA-G14V interacted strongly with mDia1-RBD and weakly with ROCK2-RBD, but failed to interact with Rhotekin RBD (Figure 1(c)). Interestingly, RhoA-G14V/F25N, which carries an additional point mutation in the Switch I domain of RhoA [6], exhibited robust binding to the mDia1-RBD but no interaction with ROCK2-RBD (Figure 1(d)). This is in agreement with previous observations that point mutations in effector-binding domains of GTPases can differentially affect interactions with downstream effectors [7].
Figure 1.
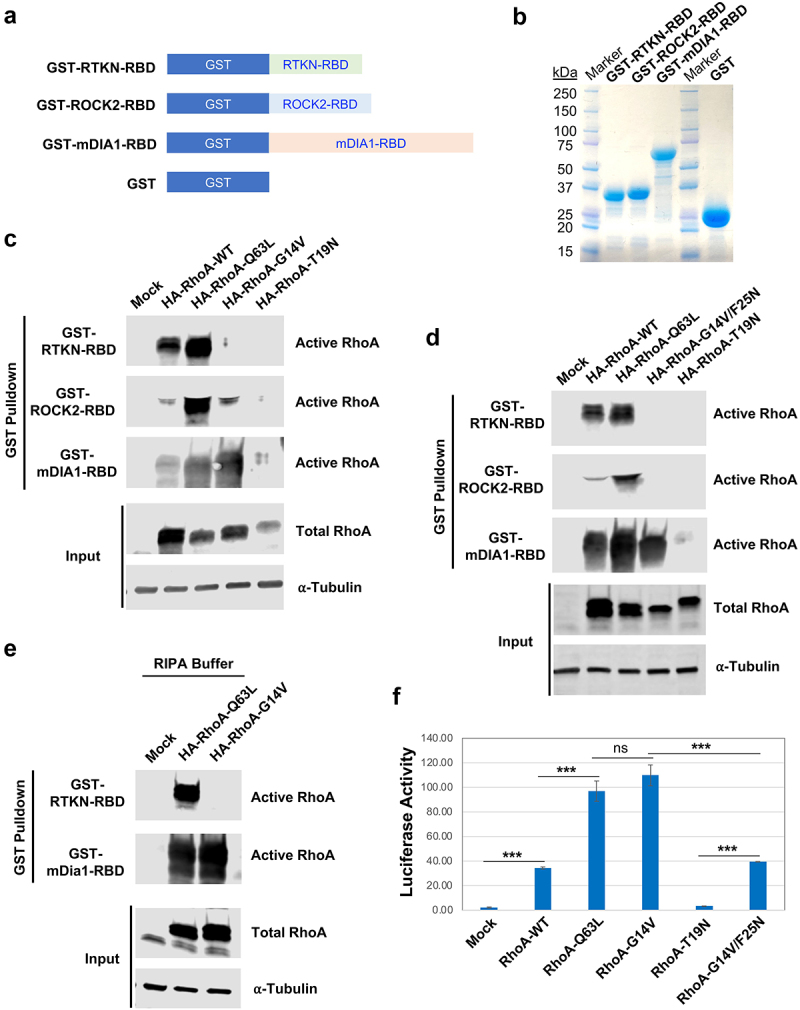
RhoA-G14V activates mDIA1 and ROCK2 but not Rhotekin. A) Schematic illustration of the constructs for producing GST-fusion effector RBDs. RTKN-RBD: RBD of mouse Rhotekin (amino acids 7–89); ROCK2-RBD: RBD of human ROCK2 (amino acids 979–1068); mDIA1-RBD: RBD of human mDIA1 (amino acids 69–451). B) Production of GST-fusion RBDs. The indicated GST fusion proteins were produced in BL21 E.coli and captured by Glutathione Sepharose beads. The proteins eluted from the beads were stained with GelCode Blue after electrophoresis on an SDS-PAGE gel. C-D) Detection of active RhoA by GST pulldown assay. HEK293 cells were transfected with HA-tagged wild-type (WT) and mutant RhoA expression vectors as indicated. Lysates from the transfected cells were extracted and subjected to GST pulldown assay using the GST-RTKN-RBD, GST-ROCK2-RBD, or GST-mDIA1-RBD beads. Both active (GTP-bound) and total RhoA were detected by anti-RhoA antibody, and ⍺-Tubulin was probed for loading controls. E) Detection of RhoA activation by GST pulldown assay using RIPA buffer [3]. HEK293 cells transfected with Mock, HA-RhoA-Q63L, or HA-RhoA-G14V were lysed in the RIPA buffer, and subjected to GST pulldown assay using GST-RTKN-RBD or GST-mDia1-RBD beads. F) Assessment of RhoA-induced SRF-driven luciferase reporter activity in HEK293 cells co-transfected with SRF-Luc, Renilla, and the indicated RhoA expression vector. The assay was performed in triplicate, and the bars represent average luciferase activity. Error bars represent standard deviation (***: p < 0.0005; ns: not significant).
As noted above, RhoA-G14V has previously been reported to interact with the Rhotekin RBD [3], prompting us to examine potential causes of this discrepancy. Because our assay buffer differed slightly from that used by Ren et al., we repeated pulldowns using identical buffer conditions (RIPA buffer). Again, we found that even in RIPA buffer RhoA-G14V failed to interact with GST-RTKN-RBD, but still interacted strongly with GST-mDia1-RBD (Figure 1(e)), demonstrating that the discrepancy was not caused by the use of different buffers. All constructs were sequenced and correspond to sequences in publicly available databases. These observations indicate that care must be taken when interpreting phenotypes of cells expressing activating Rho mutants – while both G12V and Q63L are constitutively active, they apparently bind to different arrays of downstream effectors. An alignment of the three RBD sequences is provided in Supplementary Figure. S1. This alignment shows virtually no homology among the three sequences, suggesting that each RBD binds RhoA through distinct mechanisms.
To determine whether there is a meaningful correlation between these pulldown assays and functional activities of RhoA in intact cells, we assayed stress fibre formation, one of the best-known activities of RhoA, after ectopic expression of the RhoA wild type and different mutants in Hela cells (Supplementary Figure. S2). Robust stress fibre formation can be observed in the cells transfected with the WT, Q63L, and G14V RhoA, as compared to mock transfected controls or cells expressing dominant negative RhoA-T19N. However, we observed that the stress fibres induced by WT and G14V were much more intense than those induced by RhoA-Q63L (Supplementary Figure. S2), which showed the strongest binding to the ROCK2-RBD (Figure 1(c and d)). Of note, most of the cells transfected with RhoA-Q63L appeared to be rounded up, suggesting that its overexpression is toxic to the cells. Taken together, these data indicate that stress fibre formation does not directly correlate with the pulldown assay.
In addition to its effects on the actin cytoskeleton, RhoA can also modulate the transcription of downstream target genes through its indirect activation of serum response factor (SRF) [21]. To determine if SRF activation coincided with RhoA binding to a specific effector, we used a luciferase-based SRF reporter assay to measure the transcriptional activity of wild-type and mutant RhoA constructs. We found that ectopic expression of WT RhoA induced detectable SRF activation, which was enhanced significantly and equivalently by both RhoA-G14V and RhoA-Q63L (figure 1(f)). As expected, the dominant negative mutant RhoA-T19N had virtually no activity (figure 1(f)). Interestingly, RhoA-G14V/F25N displayed a significantly reduced transcriptional activity compared to RhoA-G14V, but equivalent to WT RhoA (figure 1(f)). These results indicate that the RhoA-induced SRF-driven transcriptional activity is not primarily mediated by Rhotekin, but does coincide with the ability to bind mDia.
We next compared the efficiency of the three GST-RBD constructs in pulldown assays for endogenous RhoA, in two different cells lines, HEK293 and Hela. For these assays, we used serum-starved cells that were treated with two different stimuli, lysophosphatidic acid (LPA, 1 µM), which activates RhoA via a G-protein coupled receptor, and serum (FBS, 5%) that activates RhoA via multiple pathways. Cells were incubated with LPA or FBS for either 5 or 10 min, lysed and RhoA activity was measured using each of the three GST-RBD fusions. As shown in Figure 2(a and b), both GST-RTKN-RBD and GST-mDia1-RBD were able to precipitate endogenous active RhoA efficiently. However, no detectable recovery was observed for GST-ROCK2-RBD, even after 30–60 min of LPA or FBS stimulation (Figure 2(a-b); Supplementary Figure. S3). Interestingly, the kinetics of activation appeared to be different, depending on the RBD used in the assay. Most prominently, RhoA activity increased in Hela cells between 5 min and 10 min after LPA treatment when GST-RTKN-RBD was used. In contrast, RhoA activity was stimulated at 5 min, but consistently declined 10 min after LPA stimulation in Hela cells when GST- mDia-RBD was used (Figure 2(a-b)). We also tested a very different stimulus, colchicine, a microtubule depolymerizing drug that has been shown to activate RhoA by activating the RhoGEF GEF-H1 [3,22]. In both cell lines, we found that colchicine-induced RhoA binding to Rhotekin and mDia1 but not that of ROCK2 (Figure 2(c-d)). In this instance, RhoA activation occurred with similar kinetics whether GST-RTKN-RBD or GST-mDia-RBD was used, and was overall similar in both cell lines. These results suggest that different stimuli may activate RhoA with different effector-binding preferences, thus leading to effector-specific pathway activation and eliciting biological activity that is unique to the particular stimulus.
Figure 2.
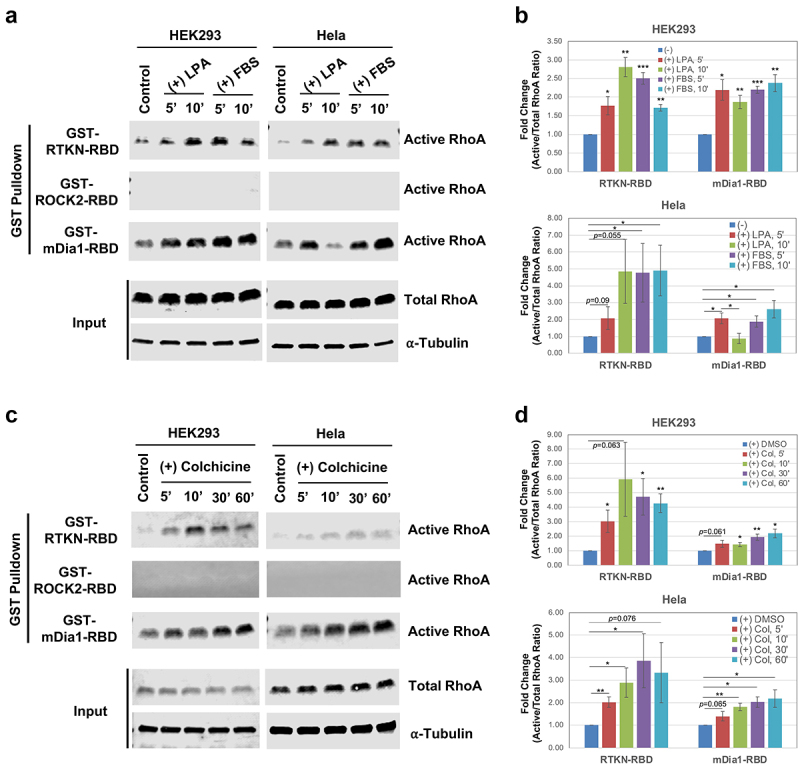
RhoA downstream effectors are differentially activated by different stimuli. A) Representative Western blots showing detection of LPA- and FBS-induced RhoA activation by three different effector-based GST pulldown assays. HEK293 or Hela cells were stimulated with LPA (1 μM) or FBS (5%) for 5 min and 10 min after serum-starvation, and the cell lysates were subjected to GST pulldown assay described above. B) Quantification of RhoA activation derived from three independent data sets. Box bars represent fold changes of the ratio of active RhoA/total RhoA by comparing stimulated vs control. Error bars represent standard error. Statistical significance was determined by student t-test (*: p < 0.05; **: p < 0.005; ***: p < 0.0005). C) Representative Western blots showing detection of RhoA activation upon colchicine treatment. GST pulldown assays were performed with the lysates of HEK293 and Hela cells treated with colchicine (10 μg/ml) for 5’, 10’, 30’, and 60’. As controls, cells were treated with DMSO (the solvent for colchicine). D) Quantification of three independent RhoA activation assays upon colchicine treatment.
To further confirm the GST pulldown assay results, we developed a BRET-based assay to measure the interaction between active RhoA and its downstream effectors (Figure 3(a)). BRET assays have been extensively utilized in the study of GPCRs and their downstream signalling including G proteins and arrestins and allow quantitative measurement of signalling output as well as real-time monitoring of temporospatial signalling dynamics [23–25]. In principle, BRET assays rely on energy transfer from a luminescent donor (such as Luciferase) to a fluorescent acceptor (for instance, GFP or its derivatives), which occurs when the two molecules physically interact or are in very close proximity (<10 nm), thus leading to emission of fluorescent signal in the absence of photo excitation. In this study, the donor vectors were constructed by fusing RhoA with NanoLuc luciferase (Nluc), and the acceptor vectors were constructed by fusion of effector RBDs with mVenus (a derivative of GFP), either N-terminally or C-terminally linked by a flexible peptide (Figure 3(b)). Expression of all constructs was confirmed by Western blot analysis (Figure 3(c and d)). We performed BRET assays following co-transfection of the donor and acceptor expression plasmids (1:100 ratio) in HEK293 cells. Venus.L-RTKN-RBD (where L connotes the linker peptide, Figure 3(b)) co-transfected with the N-terminally tagged Nluc.L-RhoA-WT or -RhoA-Q63L resulted in 4.3-fold and 3.8-fold net BRET signal induction, respectively, compared to that of the Venus controls (Figure 3(e); Supplementary Figure. S4); however, no such induction was observed for the Nluc.L-RhoA-G14V or, as expected, RhoA-T19N (Figure 3(e)). This result confirms the lack of interaction between the Rhotekin RBD and RhoA-G14V or RhoA-T19N. In contrast, Venus.L-mDia1-RBD co-transfected with Nluc.L-RhoA-G14V resulted in a near 2.9-fold induction of net BRET, versus 1.4-fold with -RhoA-WT and 1.3-fold with -RhoA-Q63L (Figure 3(e); Supplementary Figure. S4). These BRET data are largely in agreement with the results obtained from the GST pulldown assay (Figure 1), confirming that RhoA-G14V interacts with mDia1 but not with Rhotekin.
Figure 3.
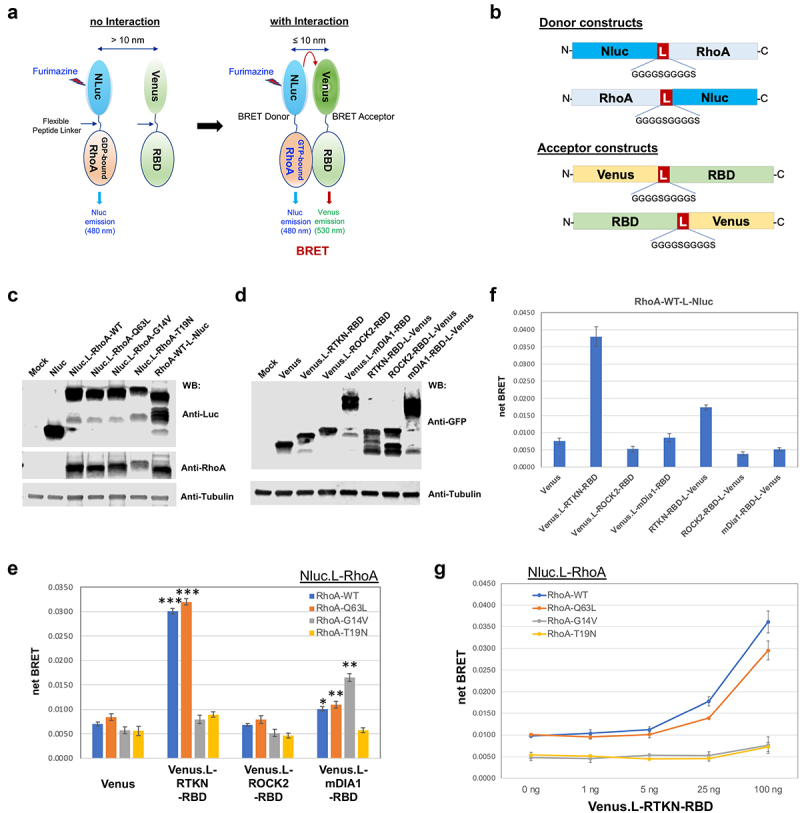
BRET assay development. A) Diagram depicting the principle of the BRET assay. B) Construct design for BRET assay donors and acceptors. Donors were constructed by fusion of RhoA to the N-terminus or C-terminus of Nluc, and the acceptors were constructed by fusion of effector RBD to the N-terminus or C-terminus of mVenus. All fusion constructs were linked by an in-frame flexible peptide linker (GGGGSGGGGS). C-D) Western blot detection of the indicated donor and acceptor proteins. Nluc and its fusion proteins were detected by anti-Luc antibody, and the mVenus and its fusion products were detected by anti-GFP antibody. Loading controls were probed with anti-⍺ Tubulin. E) Detection of RhoA activation by BRET assay in HEK293 cells after co-transfection of the indicated N-terminally linked Nluc-fused RhoA (Nluc.L-RhoA, WT and mutants) with the N-terminally linked Venus-fused effector RBDs (Venus.L-RBDs). Statistical significance was determined by student t-test, comparing the corresponding RhoA co-transfected with Venus-fused RTKN-RBD or mDIA1-RBD to those co-transfected with the backbone Venus (*: p < 0.05; **: p < 0.005; ***: p < 0.0005). F) BRET assay for the C-terminally linked Nluc-fused wild type RhoA (RhoA-WT-L-Nluc) co-transfected with the indicated Venus-fused RBD vectors (both N-terminally and C-terminally linked). G) Titration of Venus.L-RTKN-RBD (0, 1 ng, 5 ng, 25 ng, and 100 ng) for detection of BRET activity after co-transfection with the Nluc.L-RhoA (WT and mutants) (1 ng) in HEK293 cells. The amount of Venus vector used for each transfection was same (total 100 ng) after adjusted with backbone Venus accordingly.
Unlike RTKN-RBD and mDia1-RBD, however, no net BRET signal was detected from the co-transfection of Venus.L-ROCK2-RBD with any of the RhoA donor vectors including the RhoA-Q63L (Figure 3(e)), which displayed a strong interaction with ROCK2-RBD in the GST pulldown assay (Figure 1(c and d)).
To determine whether fusion orientation affects the assay, we also tested co-transfection of Nluc.L-RhoAs with C-terminally fused RBDs (RTKN-RBD-L-Venus, ROCK2-RBD-L-Venus, and mDia1-RBD-L-Venus). However, in this configuration, we observed only weak net BRET (figure 3(f)). The C-terminally fused RhoA-WT-L-Nluc showed a similar activity as that of N-terminally fused Nluc.L-RhoA-WT when tested in combination with both N-terminally and C-terminally fused RBDs (Figure 3(e-g)). Even though prenylation at the RhoA C-terminus is required for its association with cellular membranes, these results indicate that the orientation of the RhoA/Nluc fusion does not affect the assay.
To find an optimal donor/acceptor ratio for the assay, we then performed co-transfection of a fixed amount of Nluc.L-RhoAs (1 ng) with an increasing amount of Venus.L-RBDs (0, 1, 5, 20, or 100 ng). For Venus.L-RTKN-RBD, a dose-dependent increase of net BRET signal was detected from its co-transfection with Nluc.L-RhoA-WT or -Q63L but not with -G14V or T19N, and the optimal ratio among all tested was 1:100 (Figure 3(g)). It is worth noting that the dynamic range of the BRET signal resulting from Venus.L-mDia1-RBD was significantly smaller than that from Venus.L-RTKN-RBD (Figure 3(e)). One explanation for this difference is that the mDia1-RBD is much larger than the RTKN-RBD (Figure 1(a and b)) and may thus undergo less efficient energy transfer from Nluc to Venus.
Finally, we also assessed whether the newly established BRET assay is suitable for the detection of stimulus-induced RhoA activation. After donor-acceptor co-transfection, HEK293 cells were stimulated with or without LPA (1 μM), FBS (5%), or colchicine (10 μg/ml) for 5 or 10 min, followed by BRET measurement. Somewhat surprisingly, no further increase of net BRET signal was detected, upon any stimulation (Figure 4); this is likely because the dynamic range between ectopically expressed RhoA-WT and its fully activated form (as active as RhoA-Q63L) is not sufficiently large (Figure 3(e and g)).
Figure 4.
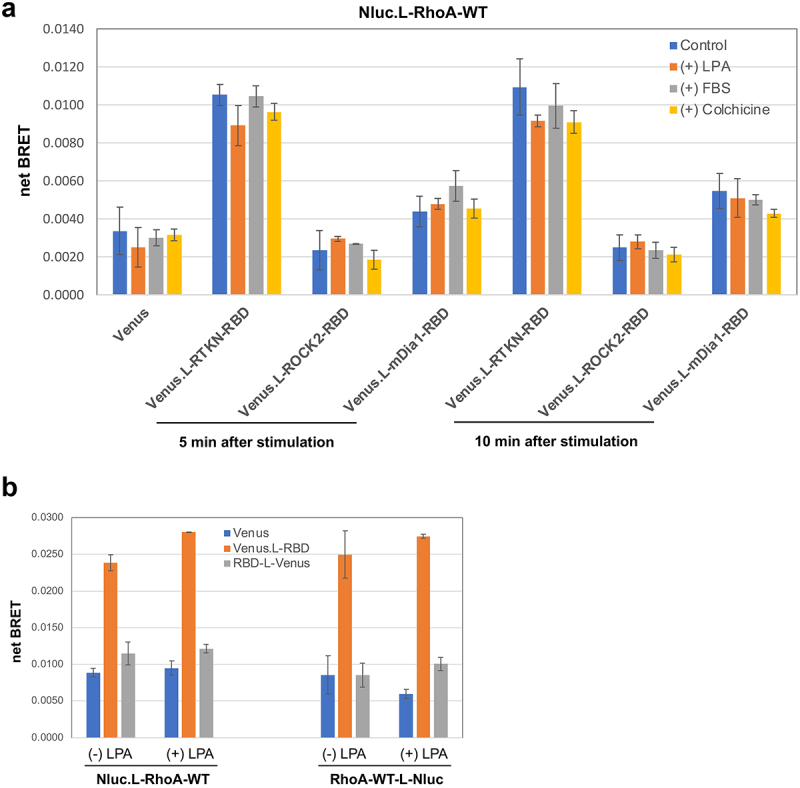
Assessment of stimulus-induced RhoA activation by BRET assay. A) HEK293 cells were co-transfected with Nluc.L-RhoA-WT and the indicated Venus fusion vectors. BRET activity was measured at 5 min and 10 min after cells were stimulated with LPA (1 μM), FBS (5%), or colchicine (10 μg/ml). B) BRET assay for HEK293 cells with the indicated four different combination of transfection with/without LPA (1 μM) stimulation for 5 min.
Nonetheless, we performed GST pulldown assays to confirm the activity of the RhoA fusions used in the BRET assay. The Nluc.L-RhoA-WT, -Q63L, and -G14V displayed very similar activities as their HA-tagged counterparts for interaction with both RTKN-RBD and mDia1-RBD (Figure 1(c) and Supplementary Figure. S5A). We also performed GST pulldown assays to examine the activity of Nluc.L-RhoA-WT upon stimulation. Even though LPA and FBS stimulation were able to induce activation of endogenous RhoA (Supplementary Figure. S5D; also, as seen in Figure 2(a-b)), they did not appear to significantly enhance the activation of ectopically expressed Nluc.L-RhoA-WT (Supplementary Figure. S5C). It therefore appears that spontaneous activation of ectopically expressed RhoA renders the newly established BRET assay insufficiently useful for measurement of stimulus-induced RhoA activation, unless this obstacle can be resolved.
Taken together, this report unveils an effector-specific activation of RhoA, evidenced by the lack of interaction between active RhoA-G14V and Rhotekin as well as the differential activation of RhoA and its downstream effectors by different stimuli. Although the mechanism underlying such differential activation is unclear, it is possible that different stimuli would lead to the activation of RhoA in different cellular contexts, thus allowing preferential interaction with certain downstream effectors. Such differences likely arise through coordinated action with RhoA regulators including RhoGEFs, RhoGAPs, RhoGDIs, membrane lipids and other modulators [2,9,10]. The specificity of RhoA activation of its downstream effectors is likely dictated by when/where/how and to what extent these regulators are activated or inactivated upon stimulation with a particular stimulus. Given that the majority of the effectors and regulators interact with RhoA through narrowly defined sequences (e.g. Switch I and II in RhoA) [6], competition among effectors might also play a role in differential signalling.
These data emphasize that there is a necessity to be cautious when interpreting RhoA activation results solely based on its interaction with the Rhotekin RBD. Thus, additional confirmation might be necessary, such as performing a parallel assay using GST-mDia1-RBD, which displays a broader and stronger binding capacity for active RhoA than GST-RTKN-RBD.
Materials and methods
Cell line
Human HEK293 and Hela cells were originally obtained from the American Type Culture Collection (ATCC) and maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% foetal bovine serum (FBS) and 1% penicillin/streptomycin/Fungizone (Gibco Antibiotic-Antimycotic) at 37°C in 5% CO2 incubator.
Plasmids, construction and cloning
The Rho-binding domain (RBD) of ROCK2 (amino acids 979–1068) [26] was amplified from total RNA extracted from HEK293 cells by RT-PCR using the following primer pair: 5’-CGCGGATCCACTAGTGATGTTGCCAATCTTGC-3’ (forward) and 5’-CCGGAATTCTACATATGTAGCTTTCTATTCTCCTTC-3’ (reverse). The RBD of mDia1 (amino acids 69–451) [16] was amplified from the mEmerald-mDia1-N14 (Addgene plasmid #54157, a gift from Michael Davidson) using the following primers: 5’-GAAGATCTAGAAATTCTTCTGCATCATATGGG-3’ (forward) and 5’-CCGGAATTCTAGTCAGGATCAGCCCCGTTCTTG-3’ (reverse). For construction of GST-ROCK2-RBD and GST-mDia1-RBD expression vectors, the fragments of ROCK2-RBD and mDia1-RBD were inserted between the BamHI and EcoRI sites of the pGEX-2 T vector, respectively. The expression plasmid for GST-RTKN-RBD, which contains RBD of Rhotekin (RTKN) in pGEX-2 T vector, was kindly provided by Dr Martin A Schwartz (Yale University) [3].
The HA-tagged RhoA expression plasmids HA-RhoA-WT, HA-RhoA-G14V/F25N, and HA-RhoA-T19N were also provided by Dr Martin A Schwartz (Yale University). Using HA-RhoA-WT as the template, HA-RhoA-Q63L was created using the primer pair of 5’-GGGACACAGCTGGGCTGGAAGATTATGATCG-3’ (sense)/5’- CGATCATAATCTTCCAGCCCAGCTGTGTCCC-3’ (antisense), and HA-RhoA-G14V was created using the primer pair of 5’-TGATTGTTGGTGATGTAGCCTGTGGAAAGAC-3’ (sense)/5’-GTCTTTCCACAGGCTACATCACCAACAATCA-3’ (antisense) by site-directed mutagenesis.
Nluc.L-RhoA-WT was constructed by inserting wild-type RhoA coding regions into pCDH-CMV (Addgene plasmid #72265, a gift from Kazuhiro Oka), followed by the insertion of a flexible peptide linker (GGGGSGGGGS) between NanoLuc (Nluc) and RhoA by site-directed mutagenesis using the following primer pair: 5’-GTGCGAACGCATTCTGGCGGGAGGTGGAGGTTCTGGAGGTGGAGGTTCTATGGCTGCCATCCGGAAGAA-3’ (sense) and 5’-TTCTTCCGGATGGCAGCCATAGAACCTCCACCTCCAGAACCTCCACCTCCCGCCAGAATGCGTTCGCAC-3’ (antisense). Using Nluc.L-RhoA-WT as the template, the Nluc.L-RhoA-Q63L and Nluc.L-RhoA-G14V were created by site-directed mutagenesis using the primer pairs described above, whereas the Nluc.L-RhoA-T19N was created using the following primers: 5’-GAGCCTGTGGAAAGAACTGCTTGCTCATAGTC-3’ (sense) and 5’-GACTATGAGCAAGCAGTTCTTTCCACAGGCTC-3’ (antisense). For construction of RhoA-WT-L-Nluc with a fusion of RhoA to the N-terminus of Nluc, RhoA-WT and the flexible peptide linker (L) were introduced into the pCDH-CMV vector in front of the Nluc coding sequences.
The mVenus-C1 and mVenus-N1 (Addgene plasmids #54651 and #54640, gifts from Michael Davidson & Atsushi Miyawaki) [27] were used as the backbones for creating C-terminal and N-terminal fusions of Venus constructs, respectively. Venus.L-RTKN-RBD, Venus.L-ROCK2-RBD, and Venus.L-mDia1-RBD were generated by insertion of the RBD fragment amplified from each corresponding gene into the mVenus-C1 vector between XhoI and EcoRI sites, followed by an in-frame insertion of a flexible peptide linker (L) between Venus and RBD into each construct. For RTKN-RBD-L-Venus, ROCK2-RBD-Venus, and mDia1-RBD-Venus, a flexible peptide linker (L) was first inserted into the mVenus-N1 in front of Venus, followed by in-frame insertion of each RBD fragment (containing also Kozak sequences and start codon) in front of the linker to complete the constructions. All constructs were confirmed by Sanger Sequencing (Genewiz).
Production and preparation of GST fusion proteins
GST fusion proteins were produced and prepared from BL21 E. coli transformed with each GST expression vector described above. Briefly, bacteria cultures with OD600 between 0.6 and 0.8 were induced with 1 mM of IPTG for 3 hours at 30°C for GST-RTKN-RBD and GST-mDia1-RBD or at 37°C for GST-ROCK2-RBD. The bacteria were lysed and sonicated in ice-cold bacteria lysis buffer consisting of 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 5 mM MgCl2, 1% Triton X-100, supplemented with 1 mM DTT, 1 mM PMSF, 10 µg/ml aprotinin, and 10 µg/ml Leupeptin. After centrifugation, the supernatant of bacteria lysates was incubated with Glutathione Sepharose 4B beads (GE Healthcare) for 2 hours at 4°C with gentle rotation. The GST-bound beads were washed extensively with ice-cold Washing Buffer consisting of 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 5 mM MgCl2, 0.5% Triton X-100, supplemented with 1 mM DTT, 1 mM PMSF, 10 µg/ml aprotinin, and 10 µg/ml Leupeptin. The beads were then resuspended in the Washing Buffer plus 10% glycerol, aliquoted, and stored at −80°C. Protein concentration in the purified GST beads was estimated by electrophoresis in a 4–20% gradient gel (Bio-Rad), followed by staining with GelCode Blue Stain Reagent (Thermo Fisher Scientific).
Western blot analysis
Cell lysates were extracted in ice-cold Lysis Buffer consisting of 1% Triton X-100, 25 mM HEPES, 150 mM NaCl, 10 mM MgCl2, 1 mM EDTA, and Protease inhibitor cocktails (Roche). The lysates were mixed with equal volume of 2xLaemmli Buffer (Sigma), denatured at 95°C for 10 min, separated in 4–20% gradient gel by electrophoresis, and transferred onto a PVDF membrane. Western blot analysis was performed by incubating the membrane with primary antibodies overnight at 4°C, followed by incubation with IRDye-conjugated secondary antibodies for 1 hour at room temperature. The proteins were detected using an Odyssey Imaging System (LI-COR Biosciences). The primary antibodies used for Western blot analysis include: mouse anti-RhoA (26C4; Santa Cruz), rabbit anti-GFP (Invitrogen), mouse anti-Nluc (R&D System), and mouse anti-α-Tubulin (Invitrogen).
RhoA activation analysis by GST pulldown assay
HEK293 cells (5x106 cells) were seeded in 10 cm dishes and transiently transfected with 2.5 µg of backbone or RhoA expression plasmids using PolyJet In Vitro DNA Transfection Reagent (SignaGen Laboratories) the next day. Forty-eight hours after transfection, cell lysates were extracted from 1 ml of RBD Lysis/Binding Buffer consisting of 50 mM Tris-HCl pH7.2, 150 mM NaCl, 10 mM MgCl2, 1% Triton X-100, supplemented with 1 mM PMSF, 10 µg/ml aprotinin, and 10 µg/ml Leupeptin. The beads with purified GST-RTKN-RBD, GST-ROCK2-RBD, and GST-mDia1-RBD were incubated with 300 µl of lysates for each reaction at 4°C for 1 hour, and then washed in 0.5 ml of RBD Lysis/Binding Buffer for 5 min for a total of 3 times. Bound protein complexes were eluted in 2xLaemmli Buffer and subjected to Western blot analysis for the detection of active RhoA (GTP-bound) using anti-RhoA antibody.
Luciferase reporter assay
HEK293 cells (30,000 cells/well) were seeded in CellStar 96-well white Cell Culture Microplate (Greiner Bio-one) after coating with Fibronectin (Sigma-Aldrich). Cells were co-transfected the next day with 50 ng of pGL4.34[luc2P/SRF-RE/Hygro] firefly luciferase vector (Promega) and 10 ng of Renilla luciferase vector (pRL-TK) with/without a RhoA expression vector (10 ng) using the PolyJet transfection reagent (SignaGen Laboratories). Five hours after transfection, medium was replaced with DMEM supplemented with 0.5% FBS. Luciferase reporter activity was quantitated the next day with Dual-Glo Luciferase Assay System (Promega) using a Cytation-1 Cell Imaging Multi-Mode Reader (BioTek).
Bioluminescence resonance energy transfer (BRET) assay
BRET assays were performed in CellStar 96-well white cell culture plates (Greiner Bio-One). Briefly, Fibronectin-treated plates were seeded with HEK293 cells (30,000 cells/well). The next day, cells were transfected with 1 ng of Nluc expression plasmid together with 100 ng of Venus expression plasmid using PolyJet, and refreshed with DMEM plus 0.5% FBS 24 hours after transfection. Nluc (460/40 nm) and Venus (540/25 nm) emissions were measured without excitation on Day 4 with Nano-Glo Luciferase assay System (Promega) using a Cytation-1 Cell Imaging Multi-Mode Reader (BioTek). Net BRET signal was calculated as a ratio of Venus emission/Nluc emission after background subtraction.
Supplementary Material
Acknowledgments
We thank Dr Martin A Schwartz for providing HA-tagged RhoA expression plasmids and GST-RTKN-RBD vector. We also thank current and former Casanova lab members for general support and assistance. The work was funded by an NIH RO1 grant to JEC (R01AI136073).
Funding Statement
This work was supported by the NIH, NIAID [R01AI136073].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
No data were deposited in a public database. Request for reagents used in this study should be addressed to J.E. Casanova (jec9e@virginia.edu)
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/21541248.2022.2111945
References
- [1].Etienne-Manneville S, Hall A.. Rho GTPases in cell biology. Nature. 2002;420(6916):629–635. [DOI] [PubMed] [Google Scholar]
- [2].Mosaddeghzadeh N, Ahmadian MR. The RHO family GTPases: mechanisms of regulation and signaling. Cells. 2021;10(7):1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 1999;18(3):578–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bros M, et al. RhoA as a key regulator of innate and adaptive immunity. Cells. 2019;8(7):733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lessey EC, Guilluy C, Burridge K. From mechanical force to RhoA activation. Biochemistry. 2012;51(38):7420–7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dvorsky R, Ahmadian MR. Always look on the bright site of Rho: structural implications for a conserved intermolecular interface. EMBO Rep. 2004;5(12):1130–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sahai E, Alberts AS, Treisman R. RhoA effector mutants reveal distinct effector pathways for cytoskeletal reorganization, SRF activation and transformation. EMBO J. 1998;17(5):1350–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kataoka K, Ogawa S. Variegated RHOA mutations in human cancers. Exp Hematol. 2016;44(12):1123–1129. [DOI] [PubMed] [Google Scholar]
- [9].Hodge RG, Ridley AJ. Regulating Rho GTPases and their regulators. Nat Rev Mol Cell Biol. 2016;17(8):496–510. [DOI] [PubMed] [Google Scholar]
- [10].Bagci H, Sriskandarajah N, Robert A, et al. Mapping the proximity interaction network of the Rho-family GTPases reveals signalling pathways and regulatory mechanisms. Nat Cell Biol. 2020;22(1):120–134. [DOI] [PubMed] [Google Scholar]
- [11].Pertz O, Hodgson L, Klemke RL, et al. Spatiotemporal dynamics of RhoA activity in migrating cells. Nature. 2006;440(7087):1069–1072. [DOI] [PubMed] [Google Scholar]
- [12].Tong J, et al. Phosphorylation and activation of RhoA by ERK in response to epidermal growth factor stimulation. PLoS One. 2016;11(1):e0147103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Rolli-Derkinderen M, Toumaniantz G, Pacaud P, et al. RhoA phosphorylation induces Rac1 release from guanine dissociation inhibitor alpha and stimulation of vascular smooth muscle cell migration. Mol Cell Biol. 2010;30(20):4786–4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fujisawa K, Madaule P, Ishizaki T, et al. Different regions of Rho determine Rho-selective binding of different classes of Rho target molecules. J Biol Chem. 1998;273(30):18943–18949. [DOI] [PubMed] [Google Scholar]
- [15].Lammers M, et al. Specificity of interactions between mDia isoforms and Rho proteins. J Biol Chem. 2008;283(50):35236–35246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rose R, Weyand M, Lammers M, et al. Structural and mechanistic insights into the interaction between Rho and mammalian Dia. Nature. 2005;435(7041):513–518. [DOI] [PubMed] [Google Scholar]
- [17].Maesaki R, Ihara K, Shimizu T, et al. The structural basis of Rho effector recognition revealed by the crystal structure of human RhoA complexed with the effector domain of PKN/PRK1. Mol Cell. 1999;4(5):793–803. [DOI] [PubMed] [Google Scholar]
- [18].Reid T, Furuyashiki T, Ishizaki T, et al. Rhotekin, a new putative target for Rho bearing homology to a serine/threonine kinase, PKN, and rhophilin in the rho-binding domain. J Biol Chem. 1996;271(23):13556–13560. [DOI] [PubMed] [Google Scholar]
- [19].Watanabe G, et al. Protein kinase N (PKN) and PKN-related protein rhophilin as targets of small GTPase Rho. Science. 1996;271(5249):645–648. [DOI] [PubMed] [Google Scholar]
- [20].Fritz RD, Letzelter M, Reimann A, et al. A versatile toolkit to produce sensitive FRET biosensors to visualize signaling in time and space. Sci Signal. 2013;6(285):rs12. [DOI] [PubMed] [Google Scholar]
- [21].Kim JG, Islam R, Cho JY, et al. Regulation of RhoA GTPase and various transcription factors in the RhoA pathway. J Cell Physiol. 2018;233(9):6381–6392. [DOI] [PubMed] [Google Scholar]
- [22].Fine N, Gracey E, Dimitriou I, et al. GEF-H1 is required for colchicine inhibition of neutrophil rolling and recruitment in mouse models of gout. J Immunol. 2020;205(12):3300–3310. [DOI] [PubMed] [Google Scholar]
- [23].Kobayashi H, Picard L-P, Schönegge A-M, et al. Bioluminescence resonance energy transfer-based imaging of protein-protein interactions in living cells. Nat Protoc. 2019;14(4):1084–1107. [DOI] [PubMed] [Google Scholar]
- [24].Dale NC, Johnstone EKM, White CW, et al. NanoBRET: the bright future of proximity-based assays. Front Bioeng Biotechnol. 2019;7:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Laschet C, Dupuis N, Hanson J. A dynamic and screening-compatible nanoluciferase-based complementation assay enables profiling of individual GPCR-G protein interactions. J Biol Chem. 2019;294(11):4079–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Leung T, et al. A novel serine/threonine kinase binding the Ras-related RhoA GTPase which translocates the kinase to peripheral membranes. J Biol Chem. 1995;270(49):29051–29054. [DOI] [PubMed] [Google Scholar]
- [27].Nagai T, et al. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol. 2002;20(1):87–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No data were deposited in a public database. Request for reagents used in this study should be addressed to J.E. Casanova (jec9e@virginia.edu)