

Supplemental Digital Content is Available in the Text.
Key Words: islatravir, doravirine, HIV-1, treatment naive, randomized controlled trial
Background:
Islatravir (MK-8591) is a nucleoside reverse transcriptase translocation inhibitor in development for treatment and prevention of HIV-1. We present efficacy and safety data for islatravir and doravirine (DOR) through 96 weeks of the phase 2b trial (NCT03272347).
Methods:
In this randomized, double-blind, dose-ranging trial, participants initially received islatravir (0.25, 0.75, or 2.25 mg) with doravirine (100 mg) and lamivudine (3TC, 300 mg) or a fixed-dose combination of doravirine, 3TC, and tenofovir disoproxil fumarate (DOR/3TC/TDF) daily. Beginning at week 24, participants receiving islatravir stopped 3TC if HIV-1 RNA from the prior visit was <50 copies per milliliter and continued taking the assigned islatravir dose (still blinded) with doravirine. All islatravir groups transitioned to open-label use of 0.75 mg between weeks 60 and 84. Efficacy end points at week 96 included the proportion of participants maintaining HIV-1 RNA of <50 copies per milliliter (FDA Snapshot). Safety was assessed by adverse event (AE) reporting.
Results:
One hundred twenty-one treatment-naive participants received the study drugs and were included in the analyses. Through week 96, HIV-1 RNA<50 copies per milliliter was maintained in 86.2% (25/29), 90.0% (27/30), and 67.7% (21/31) of participants in the 0.25-, 0.75-, and 2.25-mg islatravir groups, respectively, 81.1% (73/90) of the combined islatravir group, and 80.6% (25/31) of the DOR/3TC/TDF group. One participant in the 2.25-mg islatravir group had Protocol-Defined Virologic Failure after week 48. Drug-related AE rates were higher for DOR/3TC/TDF participants (22.6%) compared with islatravir (combined 7.8%). Two participants (2.2%) receiving islatravir with doravirine and one (3.2%) receiving DOR/3TC/TDF discontinued because of an AE.
Conclusions:
Treatment regimens containing islatravir and doravirine maintained viral suppression through week 96 and were well tolerated regardless of dose.
INTRODUCTION
Islatravir (ISL, MK-8591) is the first nucleoside reverse transcriptase translocation inhibitor in development for the treatment and prevention of HIV-1 infection.1,2 Islatravir inhibits reverse transcriptase (RT) through multiple mechanisms of action, which include its mechanism as an RT translocation inhibitor.1,3–5 Doravirine (DOR) is a nonnucleoside reverse transcriptase inhibitor approved for the treatment of HIV-1, with a favorable safety profile in phase 2 and phase 3 studies.6–9 The potent antiviral activity and complimentary in vitro resistance profile of doravirine and islatravir create the potential for a highly efficacious 2-drug regimen that would have a high barrier to resistance and a favorable safety profile. Previously, we showed the first clinical data from a phase 2b dose ranging study evaluating the safety and efficacy of islatravir and doravirine through 48 weeks. Here, we show efficacy and safety of doravirine- and islatravir-based antiretroviral regimens through 96 weeks.
METHODS
Trial Design and Participants
Protocol MK-8591-011 (NCT03272347) trial design, eligibility criteria, and participant randomization have been previously described10 and are briefly summarized in Supplementary Methods and Supplementary Figure 1, Supplemental Digital Content, http://links.lww.com/QAI/B770.
Assessments, End Points, and Statistical Analysis
Full methodology was previously described10 and is briefly summarized in Supplementary Methods, Supplemental Digital Content, http://links.lww.com/QAI/B770. The primary efficacy end points of this trial were the proportions of participants achieving HIV-1 RNA of <50 copies per milliliter at week 24 and at week 48 (using the FDA Snapshot approach).11 Here, we present the efficacy end point of proportions of participants achieving HIV-1 RNA of <50 copies per milliliter at week 96 (using the FDA Snapshot approach). As an additional efficacy analysis, a post hoc viral blip analysis was performed for participants who entered part 2 of the trial through week 96. For this analysis, a viral blip was defined as an HIV-1 RNA ≥50 copies per milliliter value observed between 2 values of <50 copies per milliliter after achieving initial response (see Supplementary Methods, Supplemental Digital Content, http://links.lww.com/QAI/B770 for details).
RESULTS
Participants
A total of 121 participants were randomly assigned, treated, and included in the week 96 analyses (see Supplementary Figure. 2, Supplemental Digital Content, http://links.lww.com/QAI/B770); the last participant visit for the week 96 database lock occurred on April 16, 2020. Key demographic and baseline clinical characteristics were well balanced between all treatment groups, as previously reported10 (see Supplementary Table 1, Supplemental Digital Content, http://links.lww.com/QAI/B770). Most participants in the islatravir groups (87.8%) achieved HIV-1 RNA of <50 copies per milliliter and stopped taking lamivudine (3TC) and transitioned to part 2 of the trial by week 24.10 During the first 48 weeks of the trial,10 14 participants discontinued from the trial (see Supplementary Figure 2, Supplemental Digital Content, http://links.lww.com/QAI/B770). In part 3, the islatravir groups transitioned (from weeks 60-84) to open-label use of 0.75 mg islatravir with 100 mg doravirine through week 144. During weeks 48 through 96, an additional 9 participants discontinued from the trial: 2 (6.9%) in the 0.25-mg islatravir group, 2 (6.7%) in the 0.75-mg islatravir group, 4 (12.9%) in the 2.25-mg islatravir group, and 1 (3.2%) in the group receiving the fixed combination of doravirine, lamivudine, and tenofovir disoproxil fumarate (DOR/3TC/TDF). Overall, the most common reasons for discontinuation were lack of efficacy (7 participants) and lost to follow-up (5 participants) (see Supplementary Figure 2, Supplemental Digital Content, http://links.lww.com/QAI/B770).
Efficacy
At week 96, HIV-1 RNA of <50 copies per milliliter was maintained in 86.2% (25/29), 90.0% (27/30), 67.7% (21/31), and 81.1% (73/90) of participants in the 0.25-mg, 0.75-mg, 2.25-mg, and combined islatravir groups, respectively, compared with 80.6% (25/31) of participants who received DOR/3TC/TDF [difference (95% CI), 6.0 (−13.3 to 25.2), 9.5 (−9.3 to 28.3), −12.3 (−34.7 to 10.1), 0.9 (−15.8 to 17.6)] (FDA Snapshot approach) (Fig. 1A and see Supplementary Table 2, Supplemental Digital Content, http://links.lww.com/QAI/B770). Among participants who entered part 2 of the trial (and switched to the 2-drug regimen in the islatravir groups), the proportion with HIV-1 RNA of <50 copies per milliliter at 96 weeks was similar across islatravir doses [86.2% (25/29), 90.0% (27/30), and 77.8% (21/27), respectively], the combined islatravir group [84.9% (73/86)], and the DOR/3TC/TDF group [89.3% (25/28)] (FDA Snapshot approach) (Fig. 1B and see Supplementary Table 2, Supplemental Digital Content, http://links.lww.com/QAI/B770). Among participants with baseline HIV-1 RNA of >100,000 copies per milliliter, the proportion with HIV-1 RNA of <50 copies per milliliter at week 96 was 83.3% (5/6), 80.0% (4/5), and 57.1% (4/7) in the 0.25-mg, 0.75-mg, and 2.25-mg islatravir groups, 72.2% (13/18) in the combined islatravir group, and 75.0% (3/4) in the DOR/3TC/TDF group, respectively (Observed Failure Approach). The immunologic response as measured by mean change in CD4+ T-cell count from baseline to week 96 was similar for all groups with a mean increase of 251, 173, and 171 cells per cubic millimeter in the 0.25, 0.75, and 2.25 mg islatravir dose groups, respectively, and an increase of 290 cells per cubic millimeter in the DOR/3TC/TDF group.
FIGURE 1.
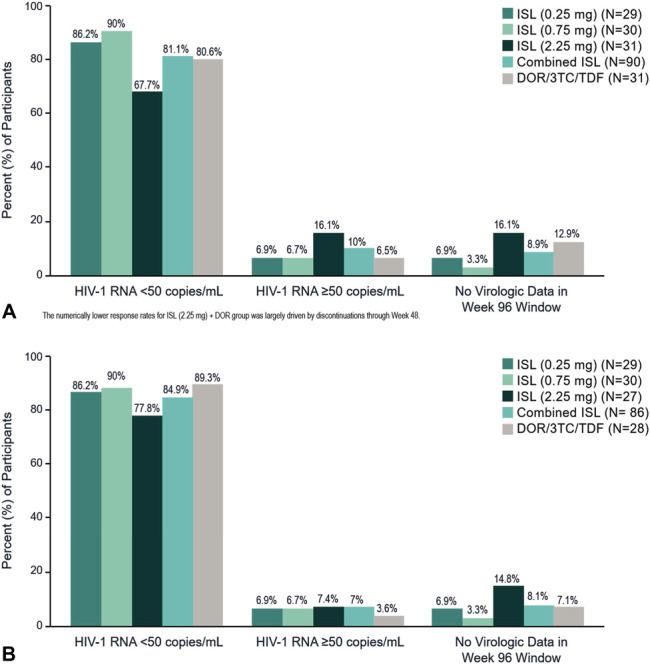
Virologic outcomes at week 96 (FDA Snapshot approach). A, All participants; B, Participants entering part 2 (ISL groups switch to the 2-drug regimen).
Through week 96, a total of 7 participants met the criteria for protocol-defined virologic failure (PDVF). Six of these participants met the criteria for PDVF during the first 48 weeks of the trial and were previously described10 (see Supplementary Table 3 and Supplementary Figure 3A, Supplemental Digital Content, http://links.lww.com/QAI/B770). One participant met the criteria for PDVF between weeks 48 and 96. This participant was randomized to the 2.25-mg group and met PDVF criteria as a rebounder with an initial HIV-1 RNA level of 79 copies per milliliter and a confirmation level of 70 copies per milliliter (see Supplementary Figure 3B, Supplemental Digital Content, http://links.lww.com/QAI/B770). This participant had an additional HIV-1 RNA value of <50 copies per milliliter before switching to a new treatment regimen of DOR/3TC/TDF and remained suppressed after switching to the new regimen. Similar to this participant with PDVF, we previously reported that 4 of the 6 participants with PDVF during the first 48 weeks of the trial had an additional HIV-1 RNA level of <50 copies per milliliter before switching to a new regimen.10 We previously reported that through week 48, all HIV-1 RNA levels at the confirmatory visit were less than 80 copies per milliliter for all participants with PDVF; similarly, the only participant with PDVF between weeks 48 and 96 also had a confirmation level below 80 copies per milliliter. None of the participants who discontinued with PDVF met the threshold for resistance testing (HIV-1 RNA levels of 400 copies/mL or higher). Of the 7 participants discontinuing because of PDVF, 5 participants had baseline HIV-1 RNA of ≥100,000 copies per milliliter, with no distinction among islatravir groups or the DOR/3TC/TDF group (see Supplementary Figure 3A, Supplemental Digital Content, http://links.lww.com/QAI/B770). Four participants who did not meet the criteria for PDVF (3 in the 2.25-mg islatravir group and 1 in the DOR/3TC/TDF group) had HIV-1 RNA ≥50 copies per milliliter at the time of discontinuation; all discontinued before week 48 (see Supplementary Table 3, Supplemental Digital Content, http://links.lww.com/QAI/B770).10 For participants who entered part 2 of the trial, viral blips were relatively rare for all treatment groups (see Supplementary Table 4, Supplemental Digital Content, http://links.lww.com/QAI/B770). In the islatravir groups, HIV-1 RNA was <200 copies per milliliter during all blip episodes. In the DOR/3TC/TDF group, 1 of 4 blip episodes was HIV-1 RNA of ≥ 200 copies per milliliter and in the other 3 episodes HIV-1 RNA was <200 copies per milliliter. No participant who experienced a viral blip subsequently experienced protocol-defined virologic failure.
Safety
Through week 96, no deaths were reported. Overall rates of any adverse event (AE), drug-related AEs, and discontinuation because of AEs through week 96 were low and consistent across treatment groups and consistent with the previously reported week-48 data (Table 1). A higher rate of drug-related AEs was reported for DOR/3TC/TDF (22.6%) participants compared with any of the doses of islatravir (combined 7.8%) (Table 1). No additional drug-related serious AEs were reported in any group between weeks 48 and 96. Only 1 additional participant discontinued because of an AE after week 48 (nondrug-related Burkitt lymphoma at approximately study week 96). The most common AEs with incidence of >10% in 1 or more treatment groups are listed in Supplementary Table 5, Supplemental Digital Content, http://links.lww.com/QAI/B770. Consistent with week 48, diarrhea was more frequently reported in the DOR/3TC/TDF group (19.4%) compared with the combined islatravir group (combined 7.8%), whereas headache was more common in islatravir groups (combined 11.1%) compared with the DOR/3TC/TDF group (6.5%). Of the most common AEs, only headache, nausea, diarrhea, vomiting, and rash had a subset of occurrences that were classified as drug related by the investigator. No islatravir dose-dependent difference was observed in reported AEs. The frequency of grade 3 or 4 laboratory abnormalities (according to Division of AIDS criteria) was low and similar between the treatment groups (see Supplementary Table 6, Supplemental Digital Content, http://links.lww.com/QAI/B770). All grade 3 and 4 creatine kinase abnormalities/elevations resolved while on study drug; one case was attributed to a newly diagnosed hepatitis C infection, and the others were confirmed as exercise related. No islatravir dose-dependent difference was observed in reported grade 3 or 4 laboratory abnormalities.
TABLE 1.
AE Summary (Week 0–96)
| ISL (0.25 mg) + DOR* QD | ISL (0.75 mg) + DOR* QD | ISL (2.25 mg) + DOR* QD | Combined ISL + DOR* QD | DOR/3TC/TDF QD | |
| Number of participants, N | 29 | 30 | 31 | 90 | 31 |
| ≥1 AE, n (%) | 25 (86.2) | 27 (90.0) | 22 (71.0) | 74 (82.2) | 27 (87.1) |
| Drug-related AE, n (%) | 0 (0.0) | 3 (10.0) | 4 (12.9) | 7 (7.8) | 7 (22.6) |
| Serious AE, n (%) | 1 (3.4) | 3 (10.0) | 1 (3.2) | 5 (5.6) | 3 (9.7) |
| Discontinued because of AE, n (%) | 1 (3.4) | 0 (0.0) | 2 (6.5) | 3 (3.3) | 1 (3.2) |
Participants initially received ISL + DOR + 3TC and switched to ISL + DOR during the week 24–48 period of the study.
DISCUSSION
This phase 2b trial of islatravir in combination with doravirine is the first, double-blind, randomized, clinical trial to show that an islatravir-based antiviral regimen maintained viral suppression up to 96 weeks. Previously, we reported that a high proportion of participants achieved HIV-1 RNA of <50 copies per milliliter within the first 24 weeks of initiating antiviral therapy on islatravir and doravirine with 3TC, and that response rates were similar regardless of islatravir dose and were similar to the proportion observed with DOR/3TC/TDF; we also showed that high efficacy was observed for all treatment groups through week 48, after participants in the islatravir arms switched to the 2-drug regimen of islatravir plus doravirine.10 Here, we show that islatravir in combination with doravirine maintained high efficacy as measured by the proportion of participants with HIV-1 RNA of <50 copies per milliliter through 96 weeks of treatment.
The 2.25-mg group had a disproportionate number of discontinuations when compared with the other groups; however, these were not because of dose-related toxicity. This imbalance resulted in the 2.25-mg group having a numerically lower proportion of participants with HIV-1 RNA of <50 copies per milliliter at week 96 (67.7%) based on the FDA Snapshot approach for missing data, and because this study has a small sample size, a few discontinuations have a large effect on the efficacy.
Through trial week 96, 7 participants discontinued because of PDVF, one of which occurred between weeks 48 and 96. Consistent with the first 6 participants with PDVF, this new participant with PDVF had an HIV-1 RNA level at the viral confirmation visit of <80 copies per milliliter, which is below the level of 200 copies per milliliter often considered clinically significant. Furthermore, this participant with PDVF had an additional HIV-1 RNA level of <50 copies per milliliter before switching to a new regimen in the follow-up period. PDVF was conservatively defined using HIV-1 RNA of ≥50 copies per milliliter as the threshold for failure. This threshold was recommended when designing the trial because both islatravir and doravirine were classified as investigational compounds with limited clinical data at that time.10 For the remainder of this trial and for additional clinical trials evaluating DOR/ISL, an HIV-1 RNA threshold of 200 copies per milliliter for PDVF has been instituted so that participants with lower-level viremia can remain in the trial and be followed instead of being mandated to discontinue from the trial. None of the participants who discontinued with or without PDVF met the threshold for resistance testing, and therefore, no viral drug resistance was identified.
The week 96 safety data support the safety and tolerability profile of islatravir plus doravirine established at week 48.10 Overall safety and tolerability profiles through week 96 were similar for both the islatravir-based regimens and the DOR/3TC/TDF regimen. Islatravir in combination with doravirine was generally well tolerated by trial participants throughout the trial. No additional islatravir participants reported drug-related AEs between weeks 48 and 96. No additional drug-related serious AEs were reported in any group during weeks 48 through 96. Only 1 additional discontinuation because of an AE occurred but was not considered to be related to study drug. Drug-related AEs occurred predominately in the first 48 weeks for all groups. Among the 90 participants taking islatravir, no specific drug-related AE (at either the system-organ-class or preferred term level) occurred in more than 5% of combined islatravir participants.
The 96-week results of this phase 2b trial further illustrate that islatravir plus doravirine has high antiviral efficacy and a favorable safety profile. Additionally, this combination may offer a potential new option for people living with HIV.
Note added in proof: In light of lymphocyte reductions observed in other clinical studies of islatravir, a post hoc analysis of total lymphocyte counts was conducted while this article was in production. At week 96, the mean change from baseline in total lymphocytes was 0.24, –0.13, and –0.13 ×109 cells/L for the islatravir 0.25-, 0.75-, and 2.25-mg groups, respectively, and 0.32 ×109 cells/L for the DOR/3TC/TDF group.
DATA AVAILABILITY STATEMENT
The data sharing policy, including restrictions, of Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA, is available at http://engagezone.msd.com/ds_documentation.php. Requests for access to the clinical study data can be submitted through the Engage Zone site or via email to dataaccess@merck.com.
ACKNOWLEDGMENTS
Medical writing support was provided by Dean Campbell, PhD, and editorial assistance was provided by Carol Zecca, BS, from Merck & Co., Inc., Rahway, NJ.
Footnotes
Merck, Sharp, & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, provided financial support for the trial. Clinical Trials Registration: NCT03272347.
J.M.M.: reports grants from Gilead, Merck, ViiV, Sanofi; Y.Y., A.A.S., C.B., C.C.A.: nothing to report; S.O.K., A.G., K.E., D.H., M.N.R., C.H., G.J.H., T.C.: employees of Merck, Sharp, & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.jaids.com).
Contributor Information
Jean-Michel Molina, Email: jean-michel.molina@aphp.fr.
Yazdan Yazdanpanah, Email: yazdan.yazdanpanah@aphp.fr.
Alejandro Afani Saud, Email: aafani@vtr.net.
Christopher Bettacchi, Email: chris.bettacchi@ntidc.org.
Carolina Chahin Anania, Email: carolina.chahin@gmail.com.
Stephanie O. Klopfer, Email: stephanie_klopfer@merck.com.
Anjana Grandhi, Email: anjana.grandhi@merck.com.
Karen Eves, Email: karen_eves@merck.com.
Deborah Hepler, Email: dahepler@comcast.net.
Michael N. Robertson, Email: michael.robertson@merck.com.
Carey Hwang, Email: carey.hwang@gmail.com.
George J. Hanna, Email: georgejhannamd@yahoo.com.
REFERENCES
- 1.Markowitz M, Sarafianos SG. 4'-Ethynyl-2-fluoro-2'-deoxyadenosine, MK-8591: a novel HIV-1 reverse transcriptase translocation inhibitor. Curr Opin HIV AIDS. 2018;13:294–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grobler JHQ, Hazuda D, Lai M, et al. Efficacy of MK-8591 against diverse HIV-1 subtypes and NRTI-resistant clinical isolates. Presented at: HIV Glasgow 2018; October 28–31, 2018; Glasgow, UK.
- 3.Salie ZL, Kirby KA, Michailidis E, et al. Structural basis of HIV inhibition by translocation-defective RT inhibitor 4'-ethynyl-2-fluoro-2'-deoxyadenosine (EFdA). Proc Natl Acad Sci U S A. 2016;113:9274–9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kawamoto A, Kodama E, Sarafianos SG, et al. 2'-deoxy-4'-C-ethynyl-2-halo-adenosines active against drug-resistant human immunodeficiency virus type 1 variants. Int J Biochem Cel Biol. 2008;40:2410–2420. [DOI] [PubMed] [Google Scholar]
- 5.Michailidis E, Huber AD, Ryan EM, et al. 4'-Ethynyl-2-fluoro-2'-deoxyadenosine (EFdA) inhibits HIV-1 reverse transcriptase with multiple mechanisms. J Biol Chem. 2014;289:24533–24548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Molina JM, Squires K, Sax PE, et al. Doravirine versus ritonavir-boosted darunavir in antiretroviral-naive adults with HIV-1 (DRIVE-FORWARD): 48-week results of a randomised, double-blind, phase 3, non-inferiority trial. Lancet HIV. 2018;5:e211–e220. [DOI] [PubMed] [Google Scholar]
- 7.Wong A, Goldstein D, Mallolas J, et al. Efficacy and safety of doravirine/lamivudine/tenofovir disoproxil fumarate (DOR/3TC/TDF) in treatment-naive adults with HIV-1 and transmitted nonnucleoside reverse transcriptase inhibitor resistance mutations. J Acquir Immune Defic Syndr. 2019;82:e47–e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orkin C, Squires KE, Molina JM, et al. Doravirine/lamivudine/tenofovir disoproxil fumarate is non-inferior to efavirenz/emtricitabine/tenofovir disoproxil fumarate in treatment-naive adults with human immunodeficiency virus-1 infection: week 48 results of the DRIVE-AHEAD trial. Clin Infect Dis. 2019;68:535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson M, Kumar P, Molina JM, et al. Switching to doravirine/lamivudine/tenofovir disoproxil fumarate (DOR/3TC/TDF) maintains HIV-1 virologic suppression through 48 Weeks: results of the DRIVE-SHIFT trial. J Acquir Immune Defic Syndr. 2019;81:463–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Molina JM, Yazdanpanah Y, Afani Saud A, et al. Islatravir in combination with doravirine for treatment-naive adults with HIV-1 infection receiving initial treatment with islatravir, doravirine, and lamivudine: a phase 2b, randomised, double-blind, dose-ranging trial. Lancet HIV. 2021;8:e324–e333. [DOI] [PubMed] [Google Scholar]
- 11.US Food and Drug Administration. Human immunodeficiency virus-1 infection: developing anti-retroviral drugs for treatment, guidance for industry. Available at: https://www.fda.gov/media/86284/download. Accessed May 18, 2022.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data sharing policy, including restrictions, of Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA, is available at http://engagezone.msd.com/ds_documentation.php. Requests for access to the clinical study data can be submitted through the Engage Zone site or via email to dataaccess@merck.com.