

Abstract
Purpose:
We report final antitumor efficacy results from a phase II study of trilaciclib, an intravenous cyclin-dependent kinase 4/6 (CDK4/6) inhibitor, administered prior to gemcitabine plus carboplatin (GCb) in patients with metastatic triple-negative breast cancer (NCT02978716).
Patients and Methods:
Patients were randomized (1:1:1) to group 1 [GCb (days 1, 8); n = 34], group 2 [trilaciclib prior to GCb (days 1, 8); n = 33], or group 3 [trilaciclib (days 1, 8) and trilaciclib prior to GCb (days 2, 9); n = 35]. Subgroup analyses were performed according to CDK4/6 dependence, level of programmed death-ligand 1 (PD-L1) expression, and RNA-based immune signatures using proportional hazards regression. T-cell receptor (TCR) β CDR3 regions were amplified and sequenced to identify, quantify, and compare the abundance of each unique TCRβ CDR3 at baseline and on treatment.
Results:
Median overall survival (OS) was 12.6 months in group 1, not reached in group 2 (HR = 0.31; P = 0.0016), 17.8 months in group 3 (HR = 0.40; P = 0.0004), and 19.8 months in groups 2 and 3 combined (HR = 0.37; P < 0.0001). Efficacy outcomes were comparable regardless of cancer CDK4/6 dependence status and immune signatures. Administering trilaciclib prior to GCb prolonged OS irrespective of PD-L1 status but had greater benefit in the PD-L1–positive population. T-cell activation was enhanced in patients receiving trilaciclib.
Conclusions:
Administering trilaciclib prior to GCb enhanced antitumor efficacy, with significant improvements in OS. Efficacy outcomes in immunologic subgroups and enhancements in T-cell activation suggest these improvements may be mediated via immunologic mechanisms.
Translational Relevance.
Novel therapies are needed for patients with metastatic triple-negative breast cancer that have similar or better antitumor efficacy than immune checkpoint inhibitors (ICI) and chemotherapy but without the associated high-grade toxicities. Trilaciclib is a first-in-class cyclin-dependent kinase 4/6 (CDK4/6) inhibitor that transiently arrests hematopoietic stem and progenitor and immune cells during chemotherapy exposure to protect them from chemotherapy-induced damage. Mature data from this phase II study in patients with metastatic triple-negative breast cancer confirm that administering trilaciclib prior to gemcitabine plus carboplatin results in statistically significant improvements in overall survival versus chemotherapy alone; efficacy benefits were observed regardless of CDK4/6 dependence status and programmed death-ligand 1 expression, but with more pronounced effects in patients with more immunogenic tumors. Overall, the findings support further investigation into whether the addition of trilaciclib can improve the antitumor effects of chemotherapy or chemotherapy/ICI combinations.
Introduction
Triple-negative breast cancer (TNBC) has an aggressive clinical course and is associated with poorer outcomes than other breast cancer subtypes (1). Although progress has been made in the management of metastatic TNBC (mTNBC), chemotherapy remains a major component of treatment (2, 3).
Compared with other breast cancer subtypes, TNBC is characterized by higher genomic instability, rendering the tumor immunogenic and amenable to immunotherapeutic intervention (4). For patients with programmed death-ligand 1 (PD-L1)–positive mTNBC, the combination of chemotherapy plus immune checkpoint inhibitors (ICI) is the preferred first-line therapy. Accordingly, pembrolizumab has been approved in combination with chemotherapy by the FDA for the treatment of patients with unresectable locally advanced TNBC/mTNBC whose tumors express PD-L1 (5). Although the combination of ICIs with chemotherapy has provided meaningful advances in the treatment of patients with PD-L1–positive disease, the TNBC patient population still represents an area of high medical need. Owing to potential treatment toxicities associated with ICIs, not all patients with PD-L1–positive mTNBC are appropriate candidates for ICI therapies. Moreover, patients with PD-L1–negative mTNBC do not derive clinical benefit from treatment with ICIs (6). Novel therapeutic options that can offer similar or better antitumor efficacy without the associated high-grade toxicities are therefore needed.
Chemotherapy treatment often results in dose-limiting, cumulative myelosuppression and weakened immune systems, with chemotherapy-induced immunosuppression potentially compromising antitumor efficacy owing to an inability of the host immune system to effectively mount a response against the cancer. Trilaciclib is an intravenous cyclin-dependent kinase 4/6 (CDK4/6) inhibitor that transiently arrests hematopoietic stem and progenitor and immune cells in the G1 phase of the cell cycle during chemotherapy exposure, thereby protecting them from chemotherapy-induced damage, and potentially enhancing immune activity (7–9). To assess the safety and efficacy of administering trilaciclib prior to gemcitabine plus carboplatin (GCb) in patients with mTNBC, a randomized phase II trial was conducted. Patients who received trilaciclib had improvements in overall survival (OS) compared with those who received GCb alone. Preliminary data showed that the addition of trilaciclib did not impair the antitumor efficacy of GCb in patients with CDK4/6-dependent, -independent, or -indeterminate cancers (10).
In February 2021, trilaciclib was approved by the FDA to decrease the incidence of chemotherapy-induced myelosuppression in adult patients receiving etoposide/platinum (E/P)- or topotecan-containing chemotherapy regimens for the treatment of extensive-stage small cell lung cancer (SCLC), on the basis of the results from three phase II trials (11–13). Clinical evidence of enhanced immune activity with trilaciclib was demonstrated in two of these trials (9, 12). Administering trilaciclib prior to E/P in patients with extensive-stage SCLC protected and increased lymphocyte counts and enhanced T-cell activation, as evidenced by peripheral blood immunophenotyping and T-cell clonal expansion (9). In addition, administering trilaciclib prior to E/P plus atezolizumab (E/P/A) increased the number of circulating activated T cells and the ratio of effector T cells to regulatory T cells (Treg), with patients receiving trilaciclib plus E/P/A also having significantly higher numbers of expanded T-cell clones (12).
Here, we report final antitumor efficacy results from the phase II mTNBC trial for the whole study population, as well as analyses of antitumor efficacy outcomes by subgroups according to CDK4/6 dependence and immune subtyping, including levels of PD-L1 expression. Data illustrating the T-cell–mediated effects of trilaciclib in patients with TNBC are also presented.
Patients and Methods
Study design and participants
This was a multicenter, randomized, open-label, phase II trial (NCT02978716) of trilaciclib administered prior to GCb in patients aged ≥18 years with mTNBC. Patients must have received ≤2 prior chemotherapy regimens for locally recurrent/mTNBC (noncytotoxic therapies were not considered). For a regimen to be a line of therapy, the patient must have had disease progression after that therapy prior to the start of the next therapy or enrollment in this study. Therapy given in the neoadjuvant/adjuvant setting where the patient had recurrent disease more than 12 months after the last dose of therapy was not considered a line of therapy in the locally recurrent/metastatic setting. Patients were randomized between February 2017 and May 2018. Full details of the study design have been published previously (10).
Patients were randomized (1:1:1) to one of three treatments, given in 21-day cycles: group 1 received GCb alone on days 1 and 8; group 2 received trilaciclib prior to GCb on days 1 and 8; and group 3 received trilaciclib alone on days 1 and 8, and trilaciclib before GCb on days 2 and 9. Gemcitabine was administered at 1,000 mg/m2 and carboplatin at AUC 2 (both intravenous administration). Intravenous trilaciclib 240 mg/m2 was administered within 4 hours prior to GCb. Treatment was continued until disease progression, unacceptable toxicity, withdrawal of consent, or discontinuation by the investigator.
The study was designed and conducted in accordance with the Declaration of Helsinki and the Good Clinical Practice guidelines of the International Council for Harmonisation. The protocol and all study-related materials were approved by the institutional review board or independent ethics committee of each investigational site, and all patients provided written informed consent.
Antitumor efficacy endpoints and assessments
Prespecified secondary endpoints included objective response rate (ORR; confirmed complete or partial response), progression-free survival (PFS), and OS. Tumor response was assessed by the investigator according to RECIST version 1.1. CT or MRI was required at screening and at protocol-specified intervals until progression, withdrawal of consent, or subsequent anticancer therapy.
Assessments of immunologic markers, and genetic and/or expression (RNA/protein) biomarkers in blood and tumors were included in the protocol as exploratory objectives. These analyses were not prespecified but were performed posthoc to further interrogate the observed efficacy outcomes from the primary analysis.
Analysis of CDK dependence/independence
Archival breast cancer tissue was collected at screening, and DNA and RNA were isolated from formalin-fixed, paraffin-embedded (FFPE) tissue sections using the Allprep DNA/RNA FFPE kit (QIAGEN), which is designed to simultaneously purify genomic DNA and total RNA. DNA and RNA were released sequentially by differential solubilization of the same FFPE sample. RNA quality was assessed by RNA integrity number, proportion of fragments greater than 200 nucleotides, and concentration. Libraries were prepared using the TruSeq RNA Exome kit (Illumina), and cluster generation and sequencing performed on the Illumina HiSeq system, as described previously (10). Specifically, libraries were sequenced using the sequencing-by-synthesis platform, with a sequencing protocol of 50-bp paired-end sequencing and total read depth of 25 million reads per sample. Expression values were estimated using RNA sequencing (RNA-seq) by Expectation Maximization software.
Patient samples were retrospectively characterized as CDK4/6-independent, -indeterminate, or -dependent (Supplementary Table S1). According to the Prediction Analysis of Microarray 50 (PAM50) signature (14, 15), CDK4/6 independence correlates with basal-like tumors, which generally manifest functional retinoblastoma tumor suppressor deficiency (16–19). Because their reliance on the CDK4/6 pathway for proliferation is either unknown or heterogeneous, the remaining PAM50 signature groups (including human EGFR2, normal-like, luminal A, and luminal B) were categorized as CDK4/6-indeterminate (14). Classification of the PAM50 subtype for each sample was determined using the Genefu package (20), which was applied to log-transformed, Fragments Per Kilobase Million (FPKM)-normalized RNA-seq–derived expression data. Using the Lehmann TNBC subtyping signatures (21), the luminal-androgen receptor (LAR) subtype of TNBC is highly sensitive to CDK4/6 inhibition both in vitro and in vivo, indicating CDK4/6 dependence (22, 23). The remaining Lehmann TNBC signature groups (including basal-like and mesenchymal) were therefore categorized as CDK4/6-indeterminate for the same reason as outlined above. TNBC type-4 classifications were assigned to samples using a random forest classifier trained on log-transformed, upper-quartile–normalized data from The Cancer Genome Atlas (TCGA; refs. 22, 23).
PD-L1 IHC
PD-L1 expression was assessed in archival tumor tissue samples from each patient using the Ventana SP142 PD-L1 assay (Ventana Medical Systems, Inc.; ref. 24). Consistent with the standard approach for evaluating PD-L1 in TNBC, expression was scored as negative or positive if <1% or ≥1% of the total tumor area contained PD-L1–labelled immune cells, respectively (24).
Immune subtyping analysis
RNA was isolated from FFPE tissue sections and sequenced as described above. Three RNA-based immune signatures for evaluating immunogenicity were assessed: (i) Ayers IFNγ signature (25)—a six-gene signature, used to classify patients as having high or low gene expression; (ii) Ayers expanded IFNγ signature (25)—an 18-gene signature, used to classify patients as having high or low gene expression; and (iii) Thorsson six-gene signature (26)—an immune signature based on six identified immune response subtypes, used to classify patients as being IFNγ-dominant (class 2; Thorsson C2) or not.
T-cell receptor analysis
To assess the effect of trilaciclib on the peripheral T-cell compartment and clonal expansion, complementary determining regions 3 of T-cell receptor (TCR)β chains (TCRβ CDR3) were amplified and sequenced from purified genomic DNA in peripheral blood mononuclear cells isolated from whole blood samples on day 1 of cycles 1, 3, and 5 using the immunoSEQ Assay (Adaptive Biotechnologies). Newly detected expanded clones were defined as clones that were not detected at baseline but were measurable after treatment. A Simpson clonality score quantified the average proportional abundance of TCR clones, whereby high values indicated an even distribution of TCR clones, and low values indicated an enrichment of clones (27, 28).
Statistical methods
OS was analyzed following the final database lock on July 17, 2020; other endpoints (ORR, PFS) were based on a data cut-off of May 15, 2020. PFS and OS were assessed in the intention-to-treat population, and ORR in response-evaluable patients (patients in the intention-to-treat population who received at least one dose of study drug, had measurable disease at baseline, and either had at least one postbaseline tumor assessment, investigator-determined clinical progression before the first postbaseline scan, or died due to disease progression before the first postbaseline scan).
Kaplan–Meier methodology was used to estimate median PFS and OS. Treatment group differences in PFS and OS were evaluated using a stratified log-rank test, with HRs and their 95% confidence intervals (CI; trilaciclib prior to GCb vs. GCb alone) generated using a Cox proportional hazard model that included number of lines of prior therapy (0 vs. 1 or 2) and liver involvement (yes vs. no) as stratification factors. Stratification factors were not included in any of the models for the subgroup analyses. Association of CDK4/6 dependence, PD-L1 expression, and immune signatures with antitumor efficacy was assessed using proportional hazards regression, with data restricted to only those patients in the relevant strata. Due to the small sample sizes, comparisons for subgroup analysis are presented between the combined trilaciclib groups (groups 2 and 3) and group 1. Individual group comparisons are included in the Supplementary Material.
Newly expanded T-cell clones (defined as increased frequency in posttreatment vs. pretreatment samples in a given patient) were computationally identified as described previously (29). A binomial model with Benjamini–Hochberg correction for multiple comparisons at the amino acid level was used to identify clones with significantly different frequencies. Survival was assessed with Cox proportional hazard regression analysis and the Wald test to determine statistical significance.
Data availability statement
The datasets generated in this study are available from the corresponding author upon reasonable request.
Results
Participants and treatment
In total, 102 eligible patients were randomized: 34 to group 1 (GCb on days 1 and 8), 33 to group 2 (trilaciclib and GCb on days 1 and 8), and 35 to group 3 (trilaciclib days 1 and 8, and trilaciclib and GCb on days 2 and 9). The proportion of tumor samples from primary (breast, chest wall, or regional nodes) versus distant metastatic sites was 27:30 (90.0%) versus 3:30 (10.0%) samples for group 1, 27:32 (84.4%) versus 5:32 (15.6%) samples for group 2, and 28:34 (82.4%) versus 6:34 (17.6%) samples for group 3. As of July 17, 2020, median (range) duration of follow-up was 8.4 (0.1–25.7) months for group 1, 14.0 (1.3–33.6) months for group 2, and 15.3 (3.5–33.7) months for group 3.
Subsequent anticancer treatments received after discontinuation of on-study treatment are summarized in Supplementary Table S2.
Antitumor efficacy in the overall study population
Patients in the trilaciclib groups continued to have numerically higher ORRs and longer PFS than those receiving GCb alone (Table 1). Compared with group 1, OS was statistically significantly improved in both trilaciclib groups, both individually and combined; median OS was 12.6 months for group 1, not reached for group 2, 17.8 months for group 3, and 19.8 months for groups 2 and 3 combined (Table 1; Fig. 1).
Table 1.
Antitumor efficacy outcomes in the overall study population: tumor response, PFS, and OS.
| Group 1 | Group 2 | Group 3 | Groups 2 and 3 | |
|---|---|---|---|---|
| Patients, n | 34 | 33 | 35 | 68 |
| ORR,an/N (%) | 7/24 (29.2) | 15/30 (50.0) | 12/31 (38.7) | 27/61 (44.3) |
| Median PFS,a months (95% CI) | 5.7 (3.3–9.9) | 9.4 (6.1–11.9) | 7.3 (6.2–13.9) | 9.0 (6.4–11.3) |
| HR (95% CI) | — | 0.62 (0.3–1.2) | 0.63 (0.3–1.2) | 0.62 (0.4–1.1) |
| P value | — | 0.2099 | 0.1816 | 0.1291 |
| Median OS,b months (95% CI) | 12.6 (6.3–15.6) | NR (10.2–NR) | 17.8 (12.9–32.7) | 19.8 (14.0–NR) |
| HR (95% CI) | — | 0.31 (0.2–0.6) | 0.40 (0.2–0.7) | 0.37 (0.2–0.6) |
| P value | — | 0.0016 | 0.0004 | <0.0001 |
Note: Group 1, chemotherapy on days 1 and 8; group 2, trilaciclib prior to chemotherapy on days 1 and 8; group 3, trilaciclib alone on days 1 and 8 and prior to chemotherapy on days 2 and 9. HR and P values are for comparisons between group 2 and group 1, between group 3 and group 1, and between groups 2 and 3 combined and group 1.
Abbreviation: NR, not reached.
aORR/PFS data are from the May 15, 2020 data cut-off.
bOS data are from the final database lock, with a data cut-off of July 17, 2020.
Figure 1.

Kaplan–Meier curve for OS in the intention-to-treat population. HR and P values are for comparisons between group 2 and group 1, and between group 3 and group 1.
Subgroup analysis: CDK4/6 dependence
CDK4/6 dependence status was assessed in 22 of 34 (64.7%) patients in group 1 and 53 of 68 (77.9%) patients in groups 2 and 3 combined. Efficacy outcomes were similar across tumors categorized as CDK4/6-dependent, -independent, or -indeterminate. Trilaciclib did not impair the efficacy of GCb in patients with CDK4/6-dependent tumors (Lehmann LAR), and outcomes were similar in patients with CDK4/6-independent tumors (PAM50 basal-like) or tumors with CDK4/6-indeterminate dependency (Lehmann basal-like 1/2 or mesenchymal, PAM50 non–basal-like; Supplementary Tables S3–S5).
Subgroup analysis: PD-L1 expression
PD-L1 status was available for 27 of 34 (79.4%) patients in group 1 and 58 of 68 (85.3%) patients in groups 2 and 3 combined. Among these patients, expression of PD-L1 was positive in 49 of 85 (57.6%) tumor tissue samples across the three treatment groups, including 32 of 58 (55.2%) samples in the trilaciclib groups and 17 of 27 (63.0%) samples in the GCb group. Baseline characteristics were generally similar between the PD-L1–positive and –negative patient populations, except that, of those with PD-L1–negative tumors, fewer patients had received prior cytotoxic chemotherapy, more patients had acquired TNBC (history of prior estrogen/progesterone receptor–positive status), and slightly more patients had an Eastern Cooperative Oncology Group performance status of 1, compared with patients with PD-L1–positive tumors (Supplementary Table S6).
Administering trilaciclib prior to GCb prolonged OS irrespective of PD-L1 status but with a larger OS benefit in the PD-L1–positive population (Table 2; Supplementary Table S7; Fig. 2). Administering trilaciclib prior to GCb also increased ORRs and extended PFS in the PD-L1–positive population (Table 2; Supplementary Table S7).
Table 2.
Tumor response, PFS, and OS according to PD-L1 status.
| PD-L1–positive | PD-L1–negative | |||
|---|---|---|---|---|
| Group 1 | Groups 2 and 3 | Group 1 | Groups 2 and 3 | |
| Patients analyzed, n | 17 | 32 | 10 | 26 |
| ORR, n (%) | 4 (23.5) | 15 (46.9) | 3 (30.0) | 8 (30.7) |
| Median PFS, months (95% CI) | 5.4 (3.3–NR) | 9.7 (6.2–15.5) | 9.2 (8.3–NR) | 9.4 (6.5–14.6) |
| HR (95% CI) | — | 0.57 (0.3–1.2) | — | 0.97 (0.4–2.5) |
| Median OS, months (95% CI) | 10.5 (6.3–18.8) | 32.7 (17.7–NR) | 13.9 (12.6–NR) | 17.8 (13.1–NR) |
| HR (95% CI) | — | 0.34 (0.2–0.7) | — | 0.48 (0.2–1.2) |
Note: Group 1, chemotherapy on days 1 and 8; group 2, trilaciclib prior to chemotherapy on days 1 and 8; group 3, trilaciclib alone on days 1 and 8 and prior to chemotherapy on days 2 and 9. HRs are for comparisons between groups 2 and 3 combined and group 1.
Figure 2.

Kaplan–Meier curves for OS in group 1 and groups 2 and 3 combined for patients with PD-L1–positive (A) and PD-L1–negative (B) tumors.
Subgroup analysis: immune subtypes
Immune subtyping analysis was performed on samples from 22 of 34 (64.7%) patients in group 1 and 53 of 68 (77.9%) patients in groups 2 and 3 combined. The addition of trilaciclib prior to GCb enhanced OS irrespective of immune signature (Table 3; Supplementary Table S8). There was a larger PFS benefit among patients in the trilaciclib groups with high Ayers IFNγ gene expression signatures, compared with in those receiving GCb alone.
Table 3.
Tumor response, PFS, and OS according to immune subtypes.
| Ayers IFNγ signature | Ayers expanded IFNγ signature | Thorsson six-class immune signature | ||||
|---|---|---|---|---|---|---|
| Outcome (groups 2 and 3 vs. group 1) | High | Low | High | Low | Class 2 | Not class 2 |
| ORR, % | 56.5 vs. 38.5 | 36.7 vs. 22.2 | 50.0 vs. 38.5 | 41.4 vs. 22.2 | 48.6 vs. 30.0 | 38.9 vs. 33.3 |
| Median PFS, months | 11.3 vs. 5.7 | 8.8 vs. 8.3 | 9.7 vs. 5.7 | 9.4 vs. 8.3 | 10.9 vs. 9.2 | 9.4 vs. 5.4 |
| HR (95% CI) | 0.49 (0.2–1.11) | 0.87 (0.3–2.2) | 0.47 (0.2–1.1) | 1.1 (0.4–2.7) | 0.69 (0.3–1.7) | 0.76 (0.3–1.8) |
| Median OS, months | 22.3 vs. 12.8 | 15.6 vs. 8.3 | 20.1 vs. 12.8 | 15.6 vs. 9.1 | 32.7 vs. 12.8 | 13.1 vs. 10.2 |
| HR (95% CI) | 0.40 (0.2–0.9) | 0.37 (0.2–0.9) | 0.41 (0.2–0.9) | 0.40 (0.2–0.9) | 0.46 (0.2–1.0) | 0.49 (0.2–1.0) |
Note: Group 1, chemotherapy on days 1 and 8; group 2, trilaciclib prior to chemotherapy on days 1 and 8; group 3, trilaciclib alone on days 1 and 8 and prior to chemotherapy on days 2 and 9. HRs are for comparisons between groups 2 and 3 combined and group 1. Class 2 was defined as IFNγ-dominant. Not adjusted for multiplicity.
Immunomodulatory effects
TCR analysis was performed for 17 of 34 (50.0%) patients in group 1 and 36 of 68 (52.9%) patients in groups 2 and 3 combined. There was a significant decrease in Simpson clonality among patients who received trilaciclib prior to GCb compared with those who received GCb alone across both postbaseline timepoints (Pinteraction = 0.012; Fig. 3A). When patients were stratified above or below median Simpson clonality, there was a trend for improved OS among patients with decreased peripheral clonality, with a statistically significant improvement among patients receiving trilaciclib (P = 0.02; Fig. 3B).
Figure 3.
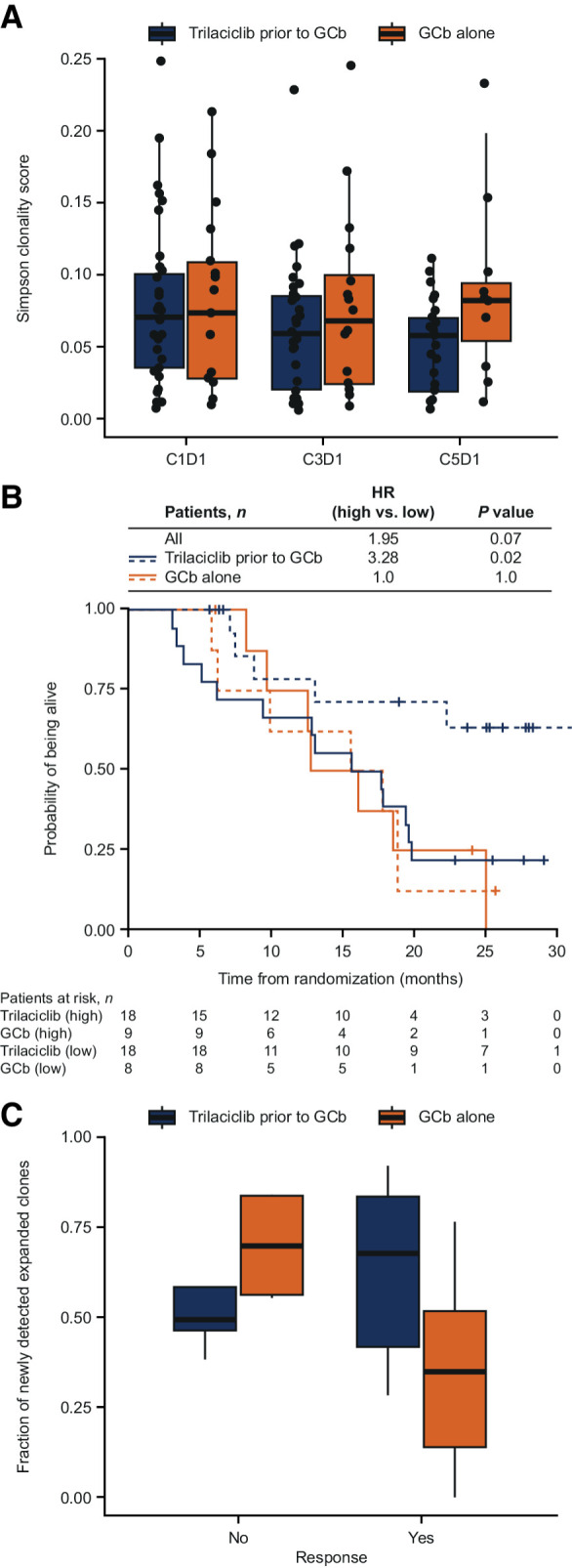
TCR clonality and expansion. A and C, Median values with 25% and 75% quartiles. B, Patients stratified by high (equal to or above median; solid lines) and low (below median; dashed lines) Simpson clonality scores. HR indicates ratio of high relative to low score. C, Data are from cycle 3, day 1. C, cycle; D, day.
At cycle 3 day 1, both responders (complete or partial response) and nonresponders (stable or progressive disease) receiving trilaciclib in groups 2 and 3 maintained a high fraction of newly detected expanded clones (P = 0.79). Patients who responded to GCb plus trilaciclib had a higher fraction of newly expanded clones than patients who responded to GCb alone (P = 0.09; Fig. 3C).
Discussion
Mature results for secondary antitumor efficacy outcomes in the overall population were consistent with the primary analysis (10), confirming that administering trilaciclib prior to GCb enhanced antitumor efficacy, with statistically significant improvements in OS.
Prespecified assessment of efficacy outcomes according to CDK dependence was conducted on the basis of the theoretical risk that CDK4/6 inhibition with trilaciclib could antagonize the intended effects of cytotoxic chemotherapy in CDK4/6-dependent tumors (30). Previously, we have shown that administering trilaciclib prior to chemotherapy did not decrease the antitumor activity of chemotherapy in CDK4/6-dependent breast cancer patient–derived xenografts (31), and clinical evidence to date has shown either no detriment to, or enhancement of, chemotherapy efficacy with the addition of trilaciclib to chemotherapy (10–13). The current analysis demonstrated that administering trilaciclib prior to GCb improved antitumor efficacy regardless of cancer CDK4/6 dependence status classified using PAM50 and Lehmann signatures (14, 22, 23).
Preclinical and clinical data suggest that the positive effects of trilaciclib on antitumor efficacy could be immune mediated and are hypothesized to be more likely to occur with more immunogenic chemotherapy regimens, tumors that are sensitive to immune modulation, and a favorable host immune system. In this regard, tumor-infiltrating lymphocytes (TIL), certain gene signatures, and expression of PD-L1 may serve as surrogates for immunogenicity in breast cancer and may predict immune-mediated responses to immunotherapies and other treatment modalities (32, 33). In our study, the larger OS benefit observed with trilaciclib in the PD-L1–positive mTNBC population reinforces the concept that the immune-mediated effects of trilaciclib may be more pronounced in more immunogenic tumors, providing further evidence for the immunomodulatory effect of trilaciclib on the tumor microenvironment. However, the findings must be considered hypothesis generating given the small number of patients across each subgroup. Most important, is the finding that the addition of trilaciclib to GCb enhanced OS irrespective of PD-L1 expression, suggesting that trilaciclib can improve GCb-associated antitumor efficacy in both PD-L1–positive and –negative patient populations. Similarly, survival benefits with trilaciclib were more pronounced in, but not exclusive to, patients with higher immune-related gene expression, suggesting that immune-mediated mechanisms may have contributed to the observed survival benefit.
Mechanistically, preclinical evidence suggests that trilaciclib enhances immune activation and promotes antitumor immunity by differentially arresting cytotoxic T-cell and Treg subsets and accelerating the recovery of cytotoxic T cells compared with Tregs (9). Additionally, within the tumor microenvironment, trilaciclib-induced transient cell-cycle arrest of immune cells also results in a more robust clonal expansion of T cells and an enrichment of proinflammatory gene signatures in preclinical models, ultimately resulting in enhanced T-cell effector function (9). Other CDK4/6 inhibitors have been shown to also activate antitumor immunity independently of their cell cycle effects (34, 35). For example, CDK4/6 inhibition increased PD-L1 expression and reduced CD3+ TILs in an in vivo breast cancer model (34, 36). Additionally, CDK4/6 inhibition modulates T-cell activation by derepressing nuclear factor of activated T-cell family proteins and their target genes, thereby increasing the production of cytokines that enhance immune system function (37). As shown here, the TCR analysis revealed that administering trilaciclib resulted in an enrichment of new T-cell clones and decreased Simpson clonality in peripheral blood, suggesting enhanced T-cell activation. Previous results from this trial suggested that administering trilaciclib prior to GCb did not preserve lymphocyte counts or enhance T-cell activation; however, there was a higher frequency of IFNγ-producing CD8+ T cells after ex-vivo stimulation in the trilaciclib groups, suggesting that trilaciclib had a positive impact on T-cell function (10). In the current analyses, there were significant increases in newly detected expanded clones among patients in groups 2 and 3 who responded to GCb, suggesting that trilaciclib may enhance antigen presentation, a phenomenon observed in preclinical studies with other CDK4/6 inhibitors (38). Moreover, patients with an enrichment of T-cell clones appeared to have greater improvement in survival with trilaciclib. Overall, these findings suggest that trilaciclib has the potential to enhance the efficacy of chemotherapy and chemotherapy/ICI combinations through a variety of mechanisms. Additional analyses of immune cell subsets and activation markers are underway using tumor samples from trilaciclib-treated patients to further elucidate the effects of trilaciclib on the antitumor immune response.
Limitations of this study include the small sample size, which meant that only large differences in OS and PFS would be detected. Moreover, antitumor efficacy outcomes were not the primary study endpoints. The sample size was powered to show superiority of group 3 over group 1 for at least one primary endpoint (duration of severe neutropenia in cycle 1 or occurrence of severe neutropenia during the treatment period). As such, comparisons of secondary endpoints (ORR, PFS, and OS) should be considered exploratory and interpreted with caution. Subgroup analyses according to CDK4/6 dependence were also exploratory. In addition, use of the doublet GCb backbone may restrict extrapolation to patients with mTNBC receiving single-agent chemotherapy. The observed immune effects of trilaciclib are also not yet fully understood and require further study in clinical trials. Nonetheless, these findings are hypothesis generating, prompting further study into the association between enhanced antitumor immunity and improved OS among patients with mTNBC receiving trilaciclib and chemotherapy. A pivotal phase III trial of trilaciclib or placebo in combination with first- or second-line GCb in patients with locally recurrent unresectable TNBC, or mTNBC is underway, with OS as the primary endpoint (NCT04799249). Trilaciclib will be evaluated in the first-line setting regardless of PD-L1 status, and in the second-line setting following progression on a PD-1/PD-L1 inhibitor in two independent cohorts. Exploratory endpoints will assess pharmacodynamic parameters, including those related to immune-based mechanisms.
Overall, our findings suggest that administering trilaciclib prior to GCb improves OS among patients with mTNBC, with a more pronounced effect in patients with more immunogenic tumors. We hypothesize that the effects of trilaciclib on antitumor immunity are two-fold and involve (i) the protection of lymphocyte populations from chemotherapy-induced damage, and (ii) the enhancement of T-cell immunity via multiple mechanisms, including increased antigen presentation, enhanced T-cell activation, and a more robust clonal expansion of T cells.
Authors' Disclosures
A.R. Tan reports personal fees from G1 Therapeutics, Inc. during the conduct of the study; A.R. Tan also reports grants from Merck and Pfizer, as well as personal fees from Athenex, AstraZeneca, Eisai, Novartis, Immunomedics, Pfizer, Merck, Puma, and Genentech outside the submitted work. G.S. Wright reports non-financial support and other support from G1 Therapeutics, Inc. during the conduct of the study, as well as other support from AbbVie, Astellas, AstraZeneca, F. Hoffmann-La Roche, Innocrin, GlaxoSmithKline, H3 BioMedicine, G1 Therapeutics, Inc., Daiichi Sankyo, Sermonix, Taiho Oncology, Seattle Genetics, ImmunoGen, Incyte, Genentech, Novartis, Eli Lilly and Company, Janssen, Celgene, Bristol-Myers Squibb, Boehringer Ingelheim, Medivation, MacroGenics, Merrimack, Tesaro, Pfizer, Dana-Farber Cancer Institute, Celldex Therapeutics, NanoString Technologies, Cascadian Therapeutics, Odonate Therapeutics, and Nucana outside the submitted work. A.R. Thummala reports other support from G1 Therapeutics, Inc. during the conduct of the study. L. Popovic reports personal fees from F. Hoffmann-La Roche, Pfizer, Novartis, MSD, Merck-Serono, Astellas, Janssen, Sanofi Genzyme, Gilead, Bristol-Myers Squibb, and Takeda outside the submitted work. T.J. Pluard reports other support from G1 Therapeutics, Inc. during the conduct of the study. T.J. Pluard also reports personal fees and other support from Novartis, SeaGen, Pfizer, H3B, Gilead, AstraZeneca, and Sermonix; personal fees from Tempus; and other support from Zymeworks outside the submitted work. H.S. Han reports personal fees from Eli Lilly and Company; grants from Department of Defense; and other support from Arvinas, AbbVie, BMS, Daiichi Sankyo, GSK, Horizon, G1 Therapeutics, Inc., Marker Therapeutics, Pfizer, Seagen, and Zymeworks outside the submitted work. J. Sorrentino reports other support from G1 Therapeutics, Inc. during the conduct of the study. J.O'Shaughnessy reports personal fees from AbbVie, Amgen, Aptitude Health, Bayer, Bristol-Myers Squibb, Carrick Therapeutics, Celgene Corporation, Daiichi Sankyo, Eisai, G1 Therapeutics, Inc., Gilead, GRAIL, Halozyme Therapeutics, Immunomedics, Ipsen Biopharmaceuticals, Eli Lilly and Company, Merck, Myriad, Novartis, Ontada, Pfizer, Pharmacyclics, Pierre Fabre Pharmaceuticals, Puma, Prime Oncology, F. Hoffmann-La Roche, Samsung Bioepis, Sanofi-Aventis, SeaGen, Takeda, and Synthon outside the submitted work. No disclosures were reported by the other authors.
Supplementary Material
Acknowledgments
We thank and acknowledge all the patients, their families, and study personnel for participating in the study. Medical writing assistance was provided by Fiona Bolland, Alligent Europe (Envision Pharma Group), funded by G1 Therapeutics, Inc.
This work was supported by G1 Therapeutics, Inc. Medical writing support was funded by G1 Therapeutics, Inc.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
A.R. Tan: Conceptualization, investigation, methodology, writing–review and editing. G.S. Wright: Investigation, writing–review and editing. A.R. Thummala: Investigation, writing–review and editing. M.A. Danso: Investigation, writing–review and editing. L. Popovic: Investigation, writing–review and editing. T.J. Pluard: Investigation, writing–review and editing. H.S. Han: Investigation, writing–review and editing. Z. Vojnović: Investigation, writing–review and editing. N. Vasev: Investigation, writing–review and editing. L. Ma: Investigation, writing–review and editing. D.A. Richards: Investigation, writing–review and editing. S.T. Wilks: Investigation, writing–review and editing. D. Milenković: Investigation, writing–review and editing. J. Xiao: Formal analysis, methodology, writing–review and editing. J. Sorrentino: Conceptualization, formal analysis, investigation, methodology, writing–review and editing. J. Horton: Conceptualization, formal analysis, methodology, writing–review and editing. J. O'Shaughnessy: Conceptualization, investigation, writing–review and editing.
References
- 1. Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 2007;13:4429–34. [DOI] [PubMed] [Google Scholar]
- 2. Twelves C, Jove M, Gombos A, Awada A. Cytotoxic chemotherapy: still the mainstay of clinical practice for all subtypes metastatic breast cancer. Crit Rev Oncol Hematol 2016;100:74–87. [DOI] [PubMed] [Google Scholar]
- 3. Garrido-Castro AC, Lin NU, Polyak K. Insights into molecular classifications of triple-negative breast cancer: improving patient selection for treatment. Cancer Discov 2019;9:176–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Park JH, Ahn JH, Kim SB. How shall we treat early triple-negative breast cancer (TNBC): from the current standard to upcoming immuno-molecular strategies. ESMO Open 2018;3:e000357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cortes J, Cescon DW, Rugo HS, Nowecki Z, Im SA, Yusof MM, et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020;396:1817–28. [DOI] [PubMed] [Google Scholar]
- 6. Emens LA, Adams S, Barrios CH, Diéras V, Iwata H, Loi S, et al. First-line atezolizumab plus nab-paclitaxel for unresectable, locally advanced, or metastatic triple-negative breast cancer: IMpassion130 final overall survival analysis. Ann Oncol 2021;32:983–93. [DOI] [PubMed] [Google Scholar]
- 7. Bisi JE, Sorrentino JA, Roberts PJ, Tavares FX, Strum JC. Preclinical characterization of G1T28: a novel CDK4/6 inhibitor for reduction of chemotherapy-induced myelosuppression. Mol Cancer Ther 2016;15:783–93. [DOI] [PubMed] [Google Scholar]
- 8. He S, Roberts PJ, Sorrentino JA, Bisi JE, Storrie-White H, Tiessen RG, et al. Transient CDK4/6 inhibition protects hematopoietic stem cells from chemotherapy-induced exhaustion. Sci Transl Med 2017;9:eaal3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lai AY, Sorrentino JA, Dragnev KH, Weiss JM, Owonikoko TK, Rytlewski JA, et al. CDK4/6 inhibition enhances antitumor efficacy of chemotherapy and immune checkpoint inhibitor combinations in preclinical models and enhances T-cell activation in patients with SCLC receiving chemotherapy. J Immunother Cancer 2020;8:e000847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tan AR, Wright GS, Thummala AR, Danso MA, Popovic L, Pluard TJ, et al. Trilaciclib plus chemotherapy versus chemotherapy alone in patients with metastatic triple-negative breast cancer: a multicentre, randomised, open-label, phase 2 trial. Lancet Oncol 2019;20:1587–601. [DOI] [PubMed] [Google Scholar]
- 11. Weiss JM, Csoszi T, Maglakelidze M, Hoyer RJ, Beck JT, Domine Gomez M, et al. Myelopreservation with the CDK4/6 inhibitor trilaciclib in patients with small-cell lung cancer receiving first-line chemotherapy: a phase Ib/randomized phase II trial. Ann Oncol 2019;30:1613–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Daniel D, Kuchava V, Bondarenko I, Ivashchuk O, Reddy S, Jaal J, et al. Trilaciclib prior to chemotherapy and atezolizumab in patients with newly diagnosed extensive-stage small cell lung cancer: a multicentre, randomised, double-blind, placebo-controlled phase II trial. Int J Cancer 2020;148:2557–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hart LL, Ferrarotto R, Andric ZG, Beck JT, Subramanian J, Radosavljevic DZ, et al. Myelopreservation with trilaciclib in patients receiving topotecan for small cell lung cancer: results from a randomized, double-blind, placebo-controlled phase II study. Adv Ther 2021;38:350–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Prat A, Bianchini G, Thomas M, Belousov A, Cheang MC, Koehler A, et al. Research-based PAM50 subtype predictor identifies higher responses and improved survival outcomes in HER2-positive breast cancer in the NOAH study. Clin Cancer Res 2014;20:511–21. [DOI] [PubMed] [Google Scholar]
- 15. Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009;27:1160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012;490:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ertel A, Dean JL, Rui H, Liu C, Witkiewicz AK, Knudsen KE, et al. RB-pathway disruption in breast cancer: differential association with disease subtypes, disease-specific prognosis and therapeutic response. Cell Cycle 2010;9:4153–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pernas S, Tolaney SM, Winer EP, Goel S. CDK4/6 inhibition in breast cancer: current practice and future directions. Ther Adv Med Oncol 2018;10:1758835918786451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sobhani N, D'Angelo A, Pittacolo M, Roviello G, Miccoli A, Corona SP, et al. Updates on the CDK4/6 inhibitory strategy and combinations in breast cancer. Cells 2019;8:321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gendoo DM, Ratanasirigulchai N, Schröder MS, Paré L, Parker JS, Prat A, et al. Genefu: an R/Bioconductor package for computation of gene expression-based signatures in breast cancer. Bioinformatics 2016;32:1097–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 2011;121:2750–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lehmann BD, Jovanović B, Chen X, Estrada MV, Johnson KN, Shyr Y, et al. Refinement of triple-negative breast cancer molecular subtypes: implications for neoadjuvant chemotherapy selection. PLoS One 2016;11:e0157368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Asghar US, Barr AR, Cutts R, Beaney M, Babina I, Sampath D, et al. Single-cell dynamics determines response to CDK4/6 inhibition in triple-negative breast cancer. Clin Cancer Res 2017;23:5561–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. U.S. Food and Drug Administration. VENTANA PD-L1 (SP142) Assay. FDA; 2019. Available from: https://www.accessdata.fda.gov/cdrh_docs/pdf16/p160002s009c.pdf.
- 25. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 2017;127:2930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The immune landscape of cancer. Immunity 2018;48:812–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Simpson E. Measurement of diversity. Nature 1949;163:688. [Google Scholar]
- 28. Aversa I, Malanga D, Fiume G, Palmieri C. Molecular T-cell repertoire analysis as source of prognostic and predictive biomarkers for checkpoint blockade immunotherapy. Int J Mol Sci 2020;21:2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. DeWitt WS, Emerson RO, Lindau P, Vignali M, Snyder TM, Desmarais C, et al. Dynamics of the cytotoxic T cell response to a model of acute viral infection. J Virol 2015;89:4517–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roberts PJ, Kumarasamy V, Witkiewicz AK, Knudsen ES. Chemotherapy and CDK4/6 inhibitors: unexpected bedfellows. Mol Cancer Ther 2020;19:1575–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sorrentino JA, Bisi JE, Thompson D. Trilaciclib, a CDK4/6 inhibitor, does not impair the efficacy of chemotherapy in CDK4/6-dependent tumor models [abstract]. In: Proceedings of the 30th European Organisation for Research and Treatment of Cancer, National Cancer Institute, and American Association for Cancer Research Symposium; 2018 Nov 13–16; Dublin, Ireland. 2018. Abstract nr. 377. [Google Scholar]
- 32. Marra A, Viale G, Curigliano G. Recent advances in triple negative breast cancer: the immunotherapy era. BMC Med 2019;17:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. de Melo Gagliato D, Buzaid AC, Perez-Garcia J, Cortes J. Immunotherapy in breast cancer: current practice and clinical challenges. BioDrugs 2020;34:611–23. [DOI] [PubMed] [Google Scholar]
- 34. Chaikovsky AC, Sage J. Beyond the cell cycle: enhancing the immune surveillance of tumors via CDK4/6 inhibition. Mol Cancer Res 2018;16:1454–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Laphanuwat P, Jirawatnotai S. Immunomodulatory roles of cell cycle regulators. Front Cell Dev Biol 2019;7:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 2018;553:91–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Deng J, Wang ES, Jenkins RW, Li S, Dries R, Yates K, et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov 2018;8:216–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017;548:471–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated in this study are available from the corresponding author upon reasonable request.