

Abstract
FAM3A is a recently identified mitochondrial protein that stimulates pancreatic-duodenal homeobox 1 (PDX1) and insulin expressions by promoting ATP release in islet β cells. In this study, the role of intracellular ATP in FAM3A-induced PDX1 expression in pancreatic β cells was further examined. Acute FAM3A inhibition using siRNA transfection in mouse pancreatic islets significantly reduced PDX1 expression, impaired insulin secretion, and caused glucose intolerance in normal mice. In vitro , FAM3A overexpression elevated both intracellular and extracellular ATP contents and promoted PDX1 expression and insulin secretion. FAM3A-induced increase in cellular calcium (Ca 2+ ) levels, PDX1 expression, and insulin secretion, while these were significantly repressed by inhibitors of P2 receptors or the L-type Ca 2+ channels. FAM3A-induced PDX1 expression was abolished by a calmodulin inhibitor. Likewise, FAM3A-induced β-cell proliferation was also inhibited by a P2 receptor inhibitor and an L-type Ca 2+ channels inhibitor. Both intracellular and extracellular ATP contributed to FAM3A-induced PDX1 expression, insulin secretion, and proliferation of pancreatic β cells.
Key words: FAM3A, pancreatic β cells, intracellular ATP, PDX1, insulin secretion
Introduction
Diabetes mellitus (DM) is mainly characterized by chronic hyperglycemia. The number of DM patients is expected to reach 642 million by 2040 1 . Generally, DM is caused by a relative or absolute deficiency of insulin secretion. Pancreatic β-cell dysregulation has been identified as a major factor contributing to insufficient insulin secretion. In the pathogenesis of DM, chronic exposure to high concentrations of glucose and free fatty acids can lead to oxidative stress 2 , inflammation 3 , autophagy 4 , and senescence 5 , all of which contribute to pancreatic β-cell dysfunction and death.
Reduction of ATP synthesis is the key feature of mitochondrial dysfunction, which is the core event causing β-cell dysfunction and diabetes. Both intracellular and extracellular ATP regulates β-cell functions. The increase in intracellular ATP/ADP ratio leads to the closure of ATP-sensitive potassium (K + ) channels and opening of L-type calcium (Ca 2+ ) channels, resulting in an increase in intracellular free Ca 2+ concentration and triggering of exocytosis of insulin secretory granules 6 . In diabetic islets, the ATP content was noted to be significantly lower than that in normal islets. Likewise, lipid stress was found to activate uncoupling protein 2 (UCP2) expression in pancreatic β cells and impair ATP synthesis, which could eventually lead to insulin secretion disorders 7 . Insulin secretory dysfunction caused by glucose and lipid overloads is negatively correlated with ATP synthesis 8 9 . The expression of ATP synthase β subunit (ATPSβ) and increase in ATP synthesis is activated by leucine, which then improves insulin secretion disorder in human diabetic islets 10 . In one study, Genipin could increase ATP production in islets of diabetic mice by inhibiting UCP2, which improved insulin secretory dysfunction 11 . ATP is co-secreted with insulin from pancreatic β cells; released ATP, in turn, acts as a signaling molecule that regulates β-cell function by activating the purine 2 (P2) receptor located on the plasma membranes of β cells 8 . The Ca 2+ signaling pathway has been identified as an important downstream pathway of the P2 receptor signaling pathway. Extracellular ATP has often been shown to increase th e intracellular free Ca 2+ concentration 12 13 14 , which, in turn, not only promotes insulin secretion but also activates the calmodulin (CaM)-Protein kinase B (Akt) signaling pathway in various cell types including pancreatic β cells 15 16 .
Family with sequence similarity 3, member A (FAM3A) is the first member of the FAM3 gene family. It is ubiquitously expressed in the tissues of humans and rodents 17 . We had previously demonstrated that FAM3A is a new mitochondrial protein that enhances the production and release of ATP. FAM3A overexpression inhibits hepatic gluconeogenesis and lipogenesis by activating the ATP-Ca 2+ -Akt pathway 18] , which protects against hepatic ischemia/reperfusion injury 19 and neuronal oxidative stress 20 . FAM3A is also known to facilitate vascular smooth muscle cell proliferation 21 and adipocyte differentiation 22 by promoting ATP production and release. FAM3A-induced ATP release in vascular smooth muscle cells plays a crucial role in regulating vasoconstriction and blood pressure 23 . In our previous study, we also showed that insulin secretion is significantly impaired in pancreatic β cell-specific knockout FAM3A mice. Mechanistically, FAM3A-induced ATP release activates the CaM-forkhead box protein A2 (FOXA2) pathway to directly induce the expression of pancreatic-duodenal homeobox 1 (PDX1), the key regulator of insulin gene expression, and β-cell growth and proliferation 24 . However, the role of intracellular ATP in FAM3A-induced PDX1 expression and insulin secretion remains unclear. Although we demonstrated earlier that FAM3A is vital in controlling PDX1 expression and insulin secretion in genetically modulated FAM3A-deficient mice, the effect of acute inhibition of FAM3A in pancreatic islets on PDX1 expression, insulin secretion, and glucose metabolism remain to be examined.
In the current study, we aimed to determine the effects of acute inhibition of FAM3A on islet functions in vivo and also probed the contribution of intracellular ATP to FAM3A-induced PDX1 expression and insulin secretion in pancreatic β cells.
Materials and Methods
Animals
C57BL/6, FAM3A flox/flox control mice (Con), and β cell-specific FAM3A gene knockout (BKO) mice were used in the study. All mice were male and 8–10 weeks old. Mice were housed with unrestricted access to food and water. All animal protocols complied with all relevant ethical regulations and were approved by the Institutional Animal Care and Use Committee, the Experimental Animal Center, Fuwai Hospital, National Center for Cardiovascular Diseases, China.
Knockdown of FAM3A in the pancreas
To knock down pancreatic FAM3A expression in mice, we mixed three sets of siFAM3A (synthesized by Invitrogen). Using hydrodynamic injections, 400 μg of siFAM3A dissolved in 0.8 mL PBS was rapidly injected in the tail vein as described previously 25 . The sequences of the three sets of siFAM3A are provided in Supplemental Table 1 .
Oral glucose tolerance test (OGTT)
After 12 h of fasting, mice were subjected to an OGTT. Blood samples were collected, and glucose levels were measured using a glucometer at indicated time points (0, 15, 30, 60, 90, and 120 min) after glucose ingestion. The dose of D-glucose administration was 2 g/kg of body weight. For determination of insulin levels, blood samples were collected at 0, 15, 30, and 60 min after glucose administration, and insulin levels were measured using a rat/mouse insulin ELISA kit.
Isolation and culture of islets
As described previously 24 , mice were anesthetized, and the upper part of the common bile duct near the hepatic portal vein was isolated and ligated. The duodenum was pulled with forceps and perfused with 3–5 mL of collagenase V [Sigma, 0.5 mg/mL in Hank’s Balanced Salt Solution (HBSS)] in the lower part of the common bile duct using an injection syringe. The pancreas was then isolated, collected in a 15 mL centrifuge tube containing 5 mL 0.5 mg/mL collagenase V, and digested for 10 min at 38 °C. Subsequently, about 5 mL 10% fetal bovine serum (FBS in HBSS) was added to the tube containing the digested pancreas, and the tube was then placed on ice to stop digestion. The mixture was then filtered and centrifuged (1500 rpm, 2 min at 4 °C); the process was repeated thrice. Then, the precipitate was resuspended in HBSS, and the islets were viewed under a stereoscopic microscope. The islets were cultured in 10% FBS Roswell Park Memorial Institute Medium (RPMI) 1640 for 24 h and later transferred to a low attachment 24-well plate for glucose-stimulated insulin secretion experiments or insulin and protein analysis.
Immunohistochemical and immunofluorescent staining
Pancreas tissues were fixed in 4% paraformaldehyde and paraffin sections (5 μm) were prepared. For immunohistochemistry, sections were stained with anti-FAM3A (Sigma-Aldrich, SAB1102488), anti-insulin (Abcam, ab181547), and anti-PCNA (Cell Signaling Technology, 2586) primary antibodies. After antibody incubation overnight, diaminobenzidine staining was performed. For immunofluorescent staining, HIT-T15 cells were fixed, permeabilized, blocked, and incubated with anti-FOXO1 antibodies (Cell Signaling Technology, 2880) at 4 °C overnight. On the next day, cells were stained with goat anti-rabbit Alexa Fluor 594. After nuclear staining with DAPI, images were observed under a confocal laser scanning microscope, and images were acquired.
Real-time PCR
Total RNA was extracted from 30 mg of liver tissues in TRIzol reagent (Invitrogen, USA) according to the manufacturer’s instructions. RNA (3–5 μg) was converted to cDNA using a cDNA synthesis kit (Thermo Scientific, USA). The target genes were quantified using SYBR Green PCR Master Mix (TOYOBO, Japan). β-actin was used as an endogenous control. The primer sequences are listed in Supplemental Table 2 .
Western blotting
Cells or tissues were lysed in RIPA lysis buffer in the presence of proteinase inhibitors. Protein samples were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane. The membranes were then blocked in 5% milk (in tris-buffered saline with Tween 20) for 1 h at room temperature and then incubated with primary antibodies [anti-FAM3A (Sigma-Aldrich, SAB1102488), anti-pAkt (Cell Signaling Technology, 9271), anti-Akt (Cell Signaling Technology, 9272), anti-pFOXO1 (Cell Signaling Technology, 9461), anti-FOXO1 (Cell Signaling Technology, 2880), anti-PDX1 (ABclonal, A3070), anti-β-actin (ZSGB Biotechnology, TA-09), and anti-GAPDH (Cell Signaling Technology, 5174)] overnight at 4 °C. On the next day, the membranes were washed and incubated with secondary antibodies for 2 h. Blots were visualized using enhanced chemiluminescence.
Cell culture and treatment
HIT-T15 cells and INS-1 cells were maintained in RPMI 1640 medium supplemented with 10% FBS. MIN6 cells were cultured in Dulbecco’s Modified Eagle Medium (with high glucose) supplemented with 15% FBS. To inhibit the expression of the P2 receptors or Ca 2+ signaling, the cells were incubated with 50 μM pyridoxalphosphate-6-azophenyl-2',4'-disulfonic acid (PPADS), 50 μM suramin, and 50 μM Chlorpromazine (CPZ) or 10 μM nifedipine for another 2 h after Ad-FAM3A infection for 24 h. To detect insulin secretion, HIT-T15 cells were incubated in Krebs-Ringer bicarbonate (KRB; 115 mM NaCl, 24 mM NaHCO 3 , 5 mM KCl, 1 mM MgCl 2 , 2.5 mM CaCl 2 , 25 mM HEPES, 0.1% BSA, pH 7.4) in the absence of glucose for 1 h after infection with Ad-FAM3A for 24 h. They were then incubated in KRB with different doses of glucose (0, 5 mM, 20 mM) and KCl (30 mM). After 1 h of incubation, the supernatant was removed for insulin detection, and the insulin values were normalized by protein levels.
Determination of ATP content
To determine the ATP content, an ATP-Lite Assay Kit (Vigorous Biotechnology Beijing Co., Ltd) was used 18 . Cell culture media and cellular components were all collected. The ATP standard curve was prepared with standard ATP samples and a luminometer. The ATP content of the samples was determined using the ATP standard curve and normalized to their protein level.
Determination of free cellular calcium levels
HIT-T15 cells were infected with Ad-GFP or Ad-FAM3A for 24 h. This was followed by incubation in KRB in the absence of glucose for 1 hour. HIT-T15 cells were incubated in KRB with different doses of glucose (0, 5 mM, 20 mM) and KCl (30 mM). Then, 1 μM of Fura-2 acetoxymethyl (AM) was added and was further incubated for 30 min. Ca 2+ levels were measured using an Olympus ix71 fluorescence microscope. For inhibition of the P2 receptors, L-type Ca 2+ channels, or Ca 2+ signaling, the cells were treated with 50 μM PPADS, 50 μM suramin, 50 μM CPZ, or 10 μM nifedipine for 1 h before Fura-2 AM treatment.
Cell cycle analysis
Cells were infected with different adenoviruses for 24 h. For inhibition of the P2 receptors, cells were treated with 50 μM suramin for another 2 h, and then stained with propidium iodide using a CycleTEST PLUS DNA Reagent Kit (Becton Dickinson, USA). Cell cycles were measured with flow cytometry.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay
Cells were seeded into 96-well plates and infected with different adenoviruses for 24 h. For inhibition of the P2 receptors, cells were treated with 50 μM suramin for another 2 h. Then, 0.5 mg/mL of MTT was added to each well. After 4 h of incubation at 37 °C, the supernatant was discarded, and the precipitate was dissolved with dimethyl sulfoxide for 15 min. Absorbance was measured at 490 nm and normalized to the control values.
Statistical analysis
Data are presented as the mean ± standard error of the mean. The normal distribution of data was determined with a Shapiro-Wilk test. Statistical significance of differences between the groups was analyzed with a t -test or Mann–Whitney test (two groups) or with a one-way ANOVA/Kruskal-Wallis test, followed by Bonferroni’s or Dunn’s post-hoc analysis, respectively (multiple groups). P-values<0.05 were considered statistically significant.
Results
Acute FAM3A knockdown significantly impairs insulin secretion
To evaluate the effect of acute FAM3A knockdown in pancreatic islets on insulin secretion, FAM3A expression in mouse pancreatic islets was transiently inhibited by hydrodynamic siRNA transfection in vivo . Before injecting siRNA, mice were randomly categorized into two groups. No significant difference was noted in terms of glucose tolerance between the two groups ( Fig. 1a ). On day 3 post siFAM3A transfection, although the fasting glucose level in the two groups of mice did not exhibit significant difference, siFAM3A-treated mice exhibited significant glucose intolerance when compared to the scramble-treated mice ( Fig. 1b ). Acute pancreatic FAM3A knockdown significantly decreased fasting serum insulin levels and weakened insulin secretion after a glucose load ( Fig. 1c ). We then examined the islets of acute FAM3A inhibited mice and observed decreased insulin content and impaired glucose-stimulated insulin secretion compared to those in the scramble group ( Figs. 1d–e ). These findings suggest that acute inhibition of pancreatic FAM3A significantly impaired insulin secretion.
Fig. 1.
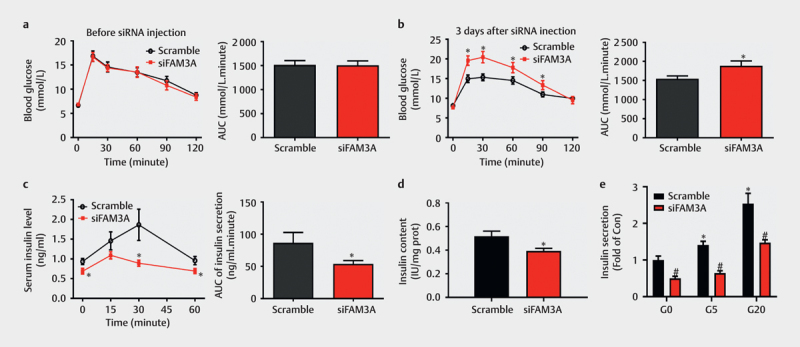
Acute FAM3A knockdown significantly impaired insulin secretion. Male C57BL/6 mice were injected with scramble or siFAM3A via tail vein hydrodynamic injection. ( a ) An oral glucose tolerance test (OGTT) was performed before siRNA injection. The area under the curve (AUC) of OGTT (day 0) is presented in the right panel. ( b ) OGTT on day 3 after tail injection of scramble or siFAM3A is shown. AUC of OGTT (day 3) is presented in the right panel. ( c ) Three days after siRNA injection, blood samples were collected from the tail vein at the first four time points (0, 15, 30, and 60 min) of OGTT, and serum insulin levels were measured. The AUC of serum insulin is presented in the right panel. ( d ) Insulin content in islets of mice with acute FAM3A inhibition and the scramble group. ( e ) Glucose-stimulated insulin secretion in the scramble and siFAM3A groups. For (a-c), N=14–18. For (d-e), N=6. For (a-d), *P<0.05 vs. scramble group. For ( e ), *P<0.05 vs. scramble islets (G0), #P<0.05 between siFAM3A islets vs. corresponding scramble group under G0, G5, and G20 stimulation separately. G0: 0 mmol/L, G5: 5 mmol/L, and G20: 20 mmol/L glucose.
FAM3A knockdown reduced PDX1 expression in the pancreas
Immunohistochemical staining, real-time PCR, and immunoblotting assays revealed significantly lower expression of FAM3A in pancreatic islets of siFAM3A-treated mice than that in the control mice ( Figs. 2a–c ). This repression of FAM3A expression led to significantly reduced insulin expression in the pancreases of siFAM3A-treated mice compared to that in the control mice ( Figs. 2a–b ). Importantly, acute FAM3A repression caused a decrease in PDX1 mRNA and protein expressions in the pancreases of the mice ( Figs. 2b–c ). The Akt-FOXO1 pathway plays a vital role in regulating β-cell functions 26 27 28 , therefore, we further evaluated whether acute FAM3A repression also influenced the Akt-FOXO1 pathway. A western blot assay indicated acute FAM3A knockdown in the pancreas significantly repressed Akt phosphorylation and FOXO1 phosphorylation and increased non-phosphorylated FOXO1 levels ( Fig. 2c ). Thus, acute FAM3A inhibition reduced PDX1 and insulin gene expressions in the pancreas of the mice.
Fig. 2.
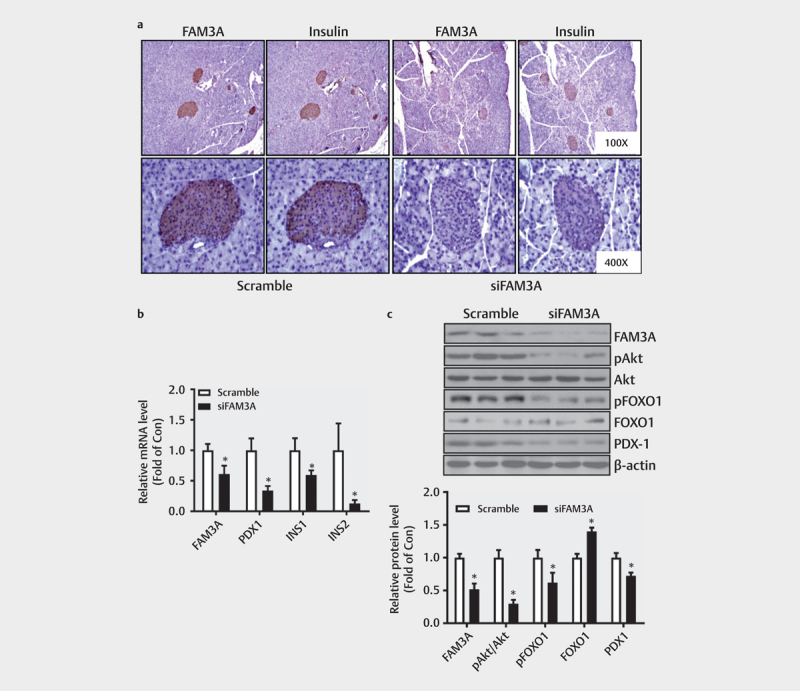
FAM3A knockdown reduced the expression of PDX1 in the pancreas. ( a ) FAM3A and insulin expressions in mouse pancreatic islets were measured by immunohistochemical analysis. ( b ) Changes in FAM3A, PDX1, INS1, and INS2 mRNA levels in the pancreas after siFAM3A injection. ( c ) FAM3A knockdown decreased pAkt and PDX1 protein expressions in the pancreas. The upper panel shows representative images and the lower panel shows statistics graphs. N=7–8. *P<0.05 vs. scramble.
FAM3A overexpression promoted ATP synthesis and insulin secretion in pancreatic β cells
Considering that FAM3A is a mitochondrial protein that increases ATP production in various cell lines, we evaluated the effects of FAM3A on ATP synthesis and insulin secretion in pancreatic β cells. First, we examined the efficacy of FAM3A adenovirus overexpression in HIT-T15 cells 24 h post-infection through a western blot assay ( Fig. 3a ); for this experiment, we chose viruses with an MOI of 25. FAM3A overexpression upregulated mRNA levels of PDX1 , INS1 , and INS2 genes ( Fig. 3b ) in HIT-T15 cells and also led to increased cellular ATP content with the presence of different concentrations of glucose (0 mmol/L glucose, G0; 20 mmol/L, G20), and KCl compared to those in control cells ( Fig. 3c ). Overexpression of FAM3A also augmented ATP release with or without a glucose challenge in HIT-T15 cells. ( Fig. 3d ) and significantly promoted insulin secretion with or without glucose stimulation, but had little effect on insulin content in HIT-T15 cells ( Figs. 3e–f ). The unchanged insulin content after FAM3A overexpression is likely due to a higher secretion rate than that in control cells. These findings suggest that FAM3A overexpression promoted ATP synthesis and insulin secretion with or without glucose stimulation in HIT-T15 cells.
Fig. 3.
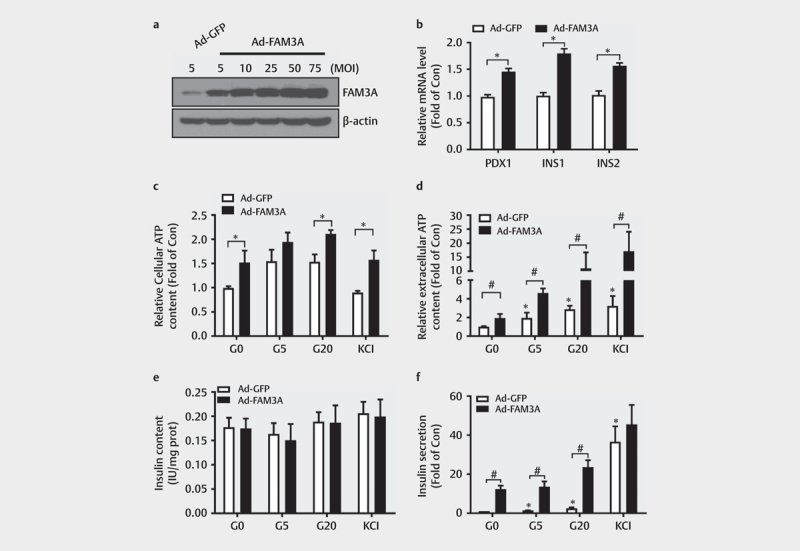
Overexpression of FAM3A promoted ATP synthesis and insulin secretion in pancreatic β cells. ( a ) A western blot assay showing Ad-FAM3A infection efficacy in HIT-T15 cells. ( b ) mRNA levels of PDX1, INS-1, and INS-2 in Ad-GFP, and Ad-FAM3A in HIT-T15 cells. *P<0.05 vs. indicated control. ( c ) FAM3A overexpression increased intracellular ATP levels in HIT-T15 cells. *P<0.05 vs. indicated control. ( d ) The effect of FAM3A overexpression on extracellular ATP content in HIT-T15 cells. *P<0.05 vs. Ad-GFP (G0); #P<0.05 vs. indicated controls. Effect of FAM3A overexpression on ( e ) insulin content and ( f ) insulin secretion in HIT-T15 cells. *P<0.05 vs. Ad-GFP (G0); #P<0.05 vs. indicated control. G0: 0 mmol/L, G5: 5 mmol/L, and G20: 20 mmol/L glucose. N=5–7.
Intracellular ATP contributed to FAM3A-induced increase in Ca 2+ levels and insulin secretion in HIT-T15 cells
To evaluate the roles of intracellular and extracellular ATP on FAM3A-induced Ca 2+ levels and insulin secretion, the roles of nifedipine and P2 receptor inhibitors were evaluated in HIT-T15 cells. We found elevated Ca 2+ levels in HIT-T15 cells treated with glucose at 5 mmol/L and 20 mmol/L when compared to 0 mmol/L ( Fig. 4a ). FAM3A overexpression caused an increase in intracellular free Ca 2+ levels with or without glucose stimulation ( Fig. 4a ). Importantly, inhibition of P2 receptors using PPADS or suramin, and L-type Ca 2+ channels using nifedipine, weakened FAM3A-induced increase in intracellular Ca 2+ levels in HIT-T15 cells at both 0 mmol/L and 20 mmol/L glucose concentration ( Figs. 4b–c ). In contrast, inhibition of CaM using CPZ failed to affect FAM3A-promoted elevation of cellular Ca 2+ levels in the presence of 0 mmol/L and 20 mmol/L glucose in HIT-T15 cells ( Figs. 4b–c ). However, inhibition of P2 receptors, CaM, and L-type Ca 2+ channels all impaired FAM3A-promoted insulin secretion in HIT-T15 cells in the presence of 20 mmol/L glucose ( Fig. 4d ). Overall, these findings suggest that both intracellular and extracellular ATP contribute to FAM3A-induced increase in Ca 2+ levels and insulin secretion.
Fig. 4.
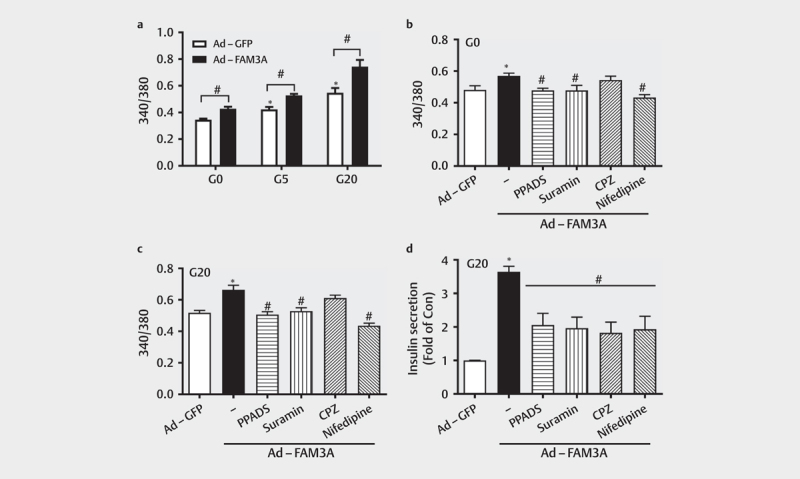
Intracellular ATP contributed to FAM3A-induced increase in Ca 2+ level and insulin secretion in HIT-T15 cells. ( a ) Effect of FAM3A overexpression of cellular calcium levels under different stimulants (G0, G5, and G20). *P<0.05 vs. Ad-GFP (G0); #P<0.05 vs. indicated control. ( b-c ) The effect of P2 receptor inhibitors (PPADS and suramin), CaM inhibitor (CPZ), and L-type calcium channels inhibitor (nifedipine) on FAM3A-induced cellular calcium elevation in different concentrations of glucose (G0 and G20) in HIT-T15 cells. *P<0.05 vs. Ad-GFP; #P<0.05 vs. Ad-FAM3A. ( d ) The effect of PPADS, suramin, CPZ, and nifedipine on FAM3A-promoted insulin secretion in HIT-T15 cells. *P<0.05 vs. Ad-GFP; #P<0.05 vs. Ad-FAM3A. G0: 0 mmol/L, G5: 5 mmol/L, and G20: 20 mmol/L glucose. N=5.
Intracellular ATP contributed to FAM3A-induced Akt activation and PDX1 upregulation
Activation of Akt and PDX1 is critical for maintaining pancreatic β-cell mass and function 29 30 31 . In vivo , FAM3A knockdown markedly repressed the Akt-FOXO1 pathway and PDX1 expression, therefore, we further evaluated the effects of both intracellular and extracellular ATP on FAM3A-induced Akt activation in cultured cells. In HIT-T15 cells, FAM3A overexpression significantly promoted Akt and FOXO1 phosphorylation, increased FOXO1 levels, and upregulated PDX1 protein levels, but this effect was reversed by the CaM inhibitor CPZ ( Fig. 5a ). Moreover, inhibitors of P2 receptors (PPADS and suramin) and the L-type Ca 2+ channels (nifedipine) could repress FAM3A-induced elevations of pAkt, pFOXO1, and PDX1 protein levels ( Figs. 5a–b ). Collectively, intracellular ATP also contributed to FAM3A-induced Akt activation and PDX1 upregulation, likely by closing the ATP-sensitive potassium channels to open the L-type Ca 2+ channels.
Fig. 5.
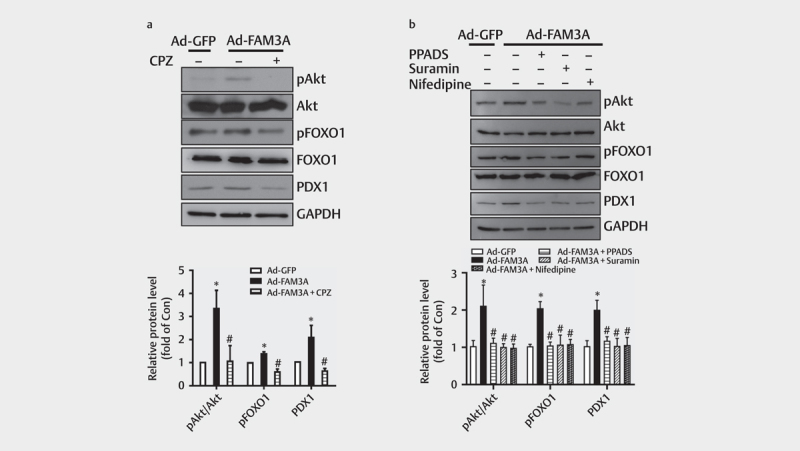
Intracellular ATP contributed to FAM3A-induced Akt activation and PDX1 upregulation. ( a ) The effect of CaM inhibitor (CPZ) on FAM3A-induced Akt-FOXO1 activation and PDX1 upregulation in HIT-T15 cells. The upper panel shows a representative image, and the lower panel shows a statistical graph. ( b ) Extended exposure to PPADS, suramin, and nifedipine for 24 h weakened FAM3A-induced Akt-FOXO1 phosphorylation and PDX1 upregulation. The upper panel shows a representative image and the lower panel shows a statistical graph. N=4–5, *P<0.05 vs. Ad-GFP, #P<0.05 vs. Ad-FAM3A.
FOXO1 can inhibit PDX1 expression by interfering with the binding of FOXA2 to the promoter region of the PDX1 gene 32 , therefore, the role of FOXO1 inactivation in FAM3A-induced PDX1 upregulation was evaluated. Consistent with phosphorylation, FAM3A overexpression reduced the nuclear distribution of FOXO1, which was inhibited by an inhibitor of P2 receptors (suramin) and CaM inhibitor (CPZ) in HIT-T15 cells ( Fig. 6a ). However, FOXO1 overexpression was determined to have no significant effect on PDX1 protein expression in three independent pancreatic β-cell lines (HIT-T15 cells, INS-1 cells, and MIN6 cells; Figs. 6b–d ). Thus, as per these findings, Akt-mediated FOXO1 inactivation is not likely involved in FAM3A-induced PDX1 activation in pancreatic β cells.
Fig. 6.
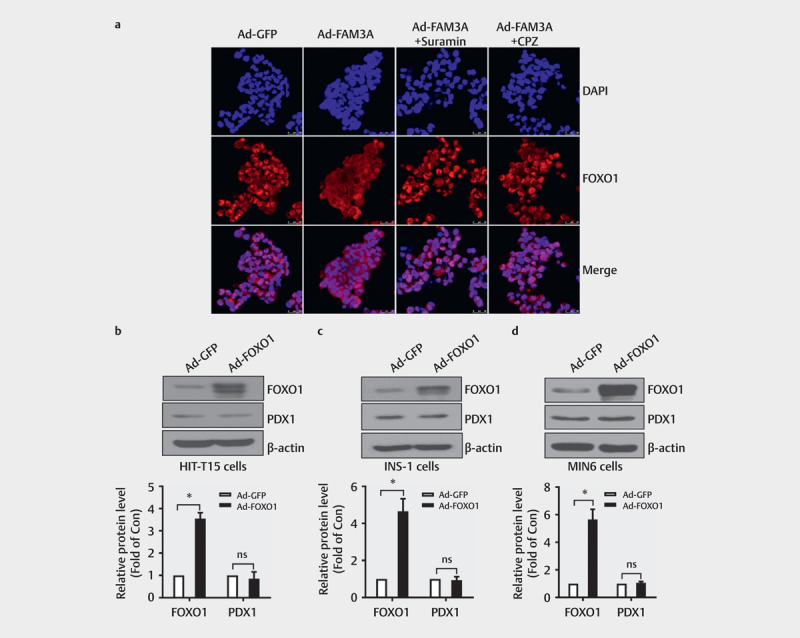
Akt-mediated FOXO1 inactivation is not involved in FAM3A-induced PDX1 upregulation. ( a ) Representative immunofluorescent staining for FOXO1 with different treatments. Nuclei stained with DAPI are in blue. ( b–d ) Western blot assay to analyze FOXO1 and PDX1 protein levels after Ad-FOXO1 infection in ( b ) HIT-T15 cells, ( c ) INS-1 cells, and ( d ) MIN6 cells. The upper panel shows representative images and the lower panel shows statistical graphs. N=3–4, *P<0.05 vs. Ad-GFP. ns: no significant difference.
Intracellular ATP contributed to FAM3A-induced proliferation of pancreatic β cells
The proliferation of various cell types is stimulated by Akt 19 21 22 ; likewise, the contributions of both intracellular and extracellular ATP in FAM3A-induced proliferation of pancreatic β cells were further evaluated. In HIT-T15 cells, FAM3A overexpression elevated the proportion of S phase (DNA synthesis phase) cells and decreased G1/G2 phase cells as determined by cell cycle analysis ( Fig. 7a ). Besides, the MTT assay also indicated stimulation of the proliferation of β cells by FAM3A overexpression ( Fig. 7b ). In MIN6 cells, FAM3A overexpression also increased the percentage of S phase cells but decreased that of G1/G2 phase cells, which was inhibited by suramin ( Figs. 7c–d ). The results of MTT assay also revealed that FAM3A overexpression stimulated MIN6 cell proliferation, but was inhibited by a P2 receptor inhibitor (suramin) and L-type Ca 2+ channels inhibitor (nifedipine; Fig. 7e ). Immunohistochemical staining showed inhibition of β-cell proliferation in both acute FAM3A knockdown and BKO mice ( Figs. 7f–g ). Overall, both intracellular and extracellular ATP contributed to the FAM3A-induced proliferation of pancreatic β cells.
Fig. 7.
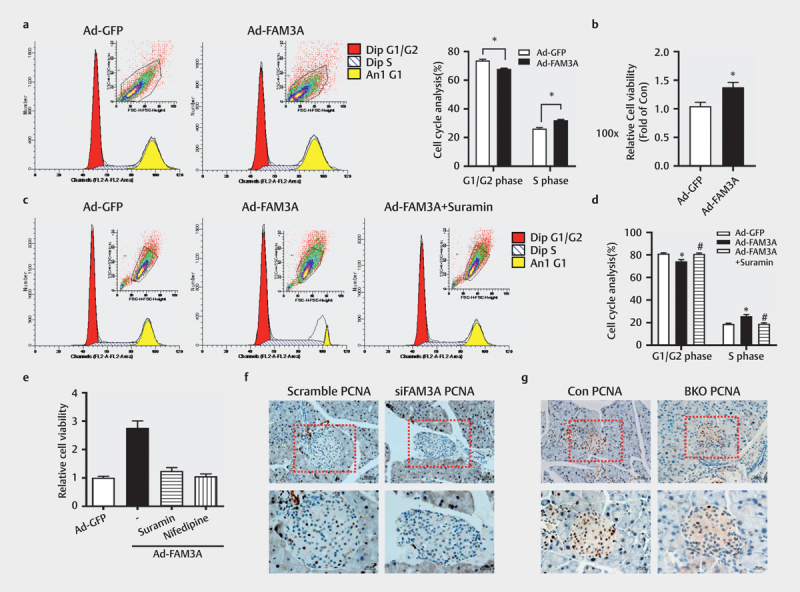
Intracellular ATP contributed to FAM3A-induced proliferation of pancreatic β cells. ( a ) The cell cycle analysis was performed after Ad-FAM3A infection in HIT-T15 cells. The left panel shows a representative image and the right panel shows a statistical graph. ( b ) MTT assay to analyze the effect of FAM3A overexpression on cell viability in HIT-T15 cells. In a MIN6 cell line, analysis of cell cycle showed the effect of suramin on FAM3A-induced cell proliferation. ( c ) Representative images of the cell cycle, ( d ) statistical graphs of panel ( c ). ( e ) Cells exposed to suramin and nifedipine for 24 h weakened the effect of FAM3A on cell viability. Immunohistochemical staining of PCNA in ( f ) acute FAM3A knockdown mice and ( g ) β cell-specific FAM3A gene knockout (BKO) mice. N=4–5, *P<0.05 vs. Ad-GFP, #P<0.05 vs. Ad-FAM3A.
Discussion
In this current study, we have provided new evidence that acute FAM3A inhibition in the pancreas impaired insulin synthesis and secretion. Although gene knockout animals are powerful models for exploring gene functions, they always lead to genetic compensation 33 34 35 . Moreover, inhibition, but not knockout, can mimic the effects of target gene repression in the progression of diseases. RNAi has been widely used in gene function analysis and gene therapy 36 . Acute FAM3A knockdown in the pancreas significantly repressed insulin expression and secretion and caused glucose intolerance in mice, further supporting our previous findings using β cell-specific FAM3A gene knockout mice 24 . We found that the responsiveness and ability to secrete insulin of isolated islets toward different doses of glucose in the siFAM3A group were damaged. Overall, both genetic deficiency and pathophysiological repression of FAM3A in pancreatic islets were found to affect cellular insulin content and secretion.
We showed that intracellular ATP also contributed to FAM3A-induced PDX1 expression and insulin secretion. FAM3A-promoted elevation of cellular ATP also increased cellular Ca 2+ levels likely by closing ATP-sensitive potassium channels and opening L-type Ca 2+ channels. Intracellular ATP elevation can trigger the opening of L-type Ca 2+ channels to increase cellular Ca 2+ concentrations, while extracellular ATP can be secreted as a signaling molecule to produce a variety of biological effects 37 38 . The P2 receptor is the receptor of exogenous ATP and is of two types, P2X and P2Y receptors. P2X receptor subtypes are ligand-gated ion channel type receptors that allow Ca 2+ to pass through. A P2Y receptor isoform is a G protein-coupled receptor, which can increase the level of IP3 and cause the release of Ca 2+ from the endoplasmic reticulum 38 39 . Glucagon-like peptide-1 has been shown to promote insulin secretion through augmentation of ATP production and increase in Ca 2+ levels 40 . In this study, we found that inhibitors of P2 receptors (PPADS and suramin), and the L-type Ca 2+ channels (nifedipine) weakened FAM3A-induced elevation of cellular Ca 2+ levels and insulin secretion. These results, along with the previous findings, suggest that both intracellular and extracellular ATP contribute to FAM3A-induced increase in cellular Ca 2+ levels, PDX1 upregulation, and insulin secretion.
Overexpression of FAM3A not only elevated the mRNA levels of insulin genes but also increased intracellular ATP and Ca 2+ levels. These results indicate that FAM3A could promote both insulin synthesis and secretion. Consistent with our previous findings in MIN6 cells 24 , we did not observe any increase in the intracellular insulin content. This might be because most of the additional synthetic insulin being released from the cells is promoted by ATP and Ca 2+ elevation. A study in rat islets also showed that one week of leucine treatment also promoted glucose-induced insulin secretion, but reduced the cellular insulin content 41 .
FAM3A induces Akt activation in various cell types to alleviate diabetes and fatty liver, protect against neuronal oxidative stress, and promote vascular smooth muscle cell proliferation 18 20 21 . FOXO1 is a direct downstream target gene of Akt. The Akt-FOXO1 pathway mediates the protection of FAM3A in hepatic ischemia/reperfusion injury 19 and promotes osteoprotegerin-induced β-cell proliferation by 42 . Liraglutide protects against apoptosis and facilitates the survival of pancreatic β cells in an Akt-dependent manner 26 . Akt-FOXO1 was also found to mediate quercetin’s effect on restoring β-cell mass and functions 28 . PDX1 is also a key regulator of β-cell proliferation and differentiation. Loss of PDX1 led to mouse pancreatic agenesis 43 , while PDX1 deficiency in acinar was found to cause cell senescence 44 . Our results also showed that acute FAM3A repression and β cell-specific FAM3A gene knockout could impair pancreatic β-cell proliferation.
FOXO1 can interfere with the binding of FOXA2 to the promoter region of the PDX1 gene 32 . Although we found that FAM3A inactivated FOXO1, overexpression of FOXO1 failed to inhibit PDX1 in various pancreatic β-cell lines. This suggests that the inactivation of FOXO1 does not possibly have a role in FAM3A-induced PDX1 and insulin expressions in pancreatic β cells.
In summary, our current and previous findings reveal that both intracellular and extracellular ATP contributed to FAM3A-induced increase in cellular Ca 2+ levels, PDX1 expression, and insulin secretion. Moreover, FAM3A potentially influences the proliferation of pancreatic β cells. Thus, FAM3A is a potential target for the treatment of islet dysfunction and diabetes.
Author Contributions
H.Y. performed experiments and contributed to the data collection and discussion. Z.C. drafted the original manuscript and provided technical assistance. H.Z., W.Y., J.Y., and X.L. contributed to the data analysis. Y.M. and R.X. were involved in solving methodology issues and animal preparation. Z.W. revised the manuscript. Y.C. and J.Y. designed the experiments and assisted in funding acquisition and checking the data authenticity. All authors have read and approved the manuscript.
Funding Statement
Funding This research was funded by Beijing Natural Science Foundation (7212123/7171006/7204320), the National Natural Science Foundation of China (82070844/81670787/81670748/81471035/818 70551/81800367/81900492), the National Key Research Program of China (2016YFC0903000/2017YFC0909600) and the State Key Laboratory of Cardiovascular Disease Grant (2018fk-02).
Footnotes
Conflict of Interest The authors declare that they have no conflict of interest.
Supplementary Material
References
- 1.Guariguata L, Whiting D R, Hambleton I. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract. 2014;103:137–149. doi: 10.1016/j.diabres.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Gerber P A, Rutter G A. The role of oxidative stress and hypoxia in pancreatic beta-cell dysfunction in diabetes mellitus. Antioxid Redox Signal. 2017;26:501–518. doi: 10.1089/ars.2016.6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berchtold L A, Prause M, Størling J. Cytokines and pancreatic β-cell apoptosis. Adv Clin Chem. 2016;75:99–158. doi: 10.1016/bs.acc.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Marasco M R, Linnemann A K. β-cell autophagy in diabetes pathogenesis. Endocrinology. 2018;159:2127–2141. doi: 10.1210/en.2017-03273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Helman A, Avrahami D, Klochendler A. Effects of ageing and senescence on pancreatic β-cell function. Diabetes Obes Metab. 2016;18 Suppl 1:58–62. doi: 10.1111/dom.12719. [DOI] [PubMed] [Google Scholar]
- 6.Gerencser A A. Metabolic activation-driven mitochondrial hyperpolarization predicts insulin secretion in human pancreatic beta-cells. Biochim Biophys Acta Bioenerg. 2018;1859:817–828. doi: 10.1016/j.bbabio.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang C Y, Baffy G, Perret P. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, β cell dysfunction, and type 2 diabetes. Cell. 2001;105:745–755. doi: 10.1016/s0092-8674(01)00378-6. [DOI] [PubMed] [Google Scholar]
- 8.Wang C, Geng B, Cui Q. Intracellular and extracellular adenosine triphosphate in regulation of insulin secretion from pancreatic β cells (β) J Diabetes. 2014;6:113–119. doi: 10.1111/1753-0407.12098. [DOI] [PubMed] [Google Scholar]
- 9.Köhnke R, Mei J, Park M. Fatty acids and glucose in high concentration down-regulates ATP synthase beta-subunit protein expression in INS-1 cells. Nutr Neurosci. 2007;10:273–278. doi: 10.1080/10284150701745910. [DOI] [PubMed] [Google Scholar]
- 10.Yang J, Chi Y, Burkhardt B R. Leucine metabolism in regulation of insulin secretion from pancreatic beta cells. Nutr Rev. 2010;68:270–279. doi: 10.1111/j.1753-4887.2010.00282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang C Y, Parton L E, Ye C P. Genipin inhibits UCP2-mediated proton leak and acutely reverses obesity- and high glucose-induced beta cell dysfunction in isolated pancreatic islets. Cell Metab. 2006;3:417–427. doi: 10.1016/j.cmet.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 12.Geschwind J F, Hiriart M, Glennon M C. Selective activation of Ca2+ influx by extracellular ATP in a pancreatic beta-cell line (HIT) Biochim Biophys Acta. 1989;1012:107–115. doi: 10.1016/0167-4889(89)90018-9. [DOI] [PubMed] [Google Scholar]
- 13.Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1β release by the ATP-gated P2X7 receptor. EMBO J. 2006;25:5071–5082. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orellana J A, Montero T D, von Bernhardi R. Astrocytes inhibit nitric oxide-dependent Ca(2+) dynamics in activated microglia: involvement of ATP released via pannexin 1 channels. Glia. 2013;61:2023–2037. doi: 10.1002/glia.22573. [DOI] [PubMed] [Google Scholar]
- 15.Zhu L, Almaça J, Dadi P K. β-arrestin-2 is an essential regulator of pancreatic β-cell function under physiological and pathophysiological conditions. Nat Commun. 2017;8:14295. doi: 10.1038/ncomms14295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alejandro E U, Bozadjieva N, Blandino-Rosano M. Overexpression of kinase-dead mTOR impairs glucose homeostasis by regulating insulin secretion and not β-cell mass. Diabetes. 2017;66:2150–2162. doi: 10.2337/db16-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu Y, Xu G, Patel A. Cloning, expression, and initial characterization of a novel cytokine-like gene family. Genomics. 2002;80:144–150. doi: 10.1006/geno.2002.6816. [DOI] [PubMed] [Google Scholar]
- 18.Wang C, Chi Y, Li J. FAM3A activates PI3K p110α/Akt signaling to ameliorate hepatic gluconeogenesis and lipogenesis. Hepatology. 2014;59:1779–1790. doi: 10.1002/hep.26945. [DOI] [PubMed] [Google Scholar]
- 19.Chen Z, Wang J, Yang W. FAM3A mediates PPARγ’s protection in liver ischemia-reperfusion injury by activating Akt survival pathway and repressing inflammation and oxidative stress. Oncotarget. 2017;8:49882–49896. doi: 10.18632/oncotarget.17805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song Q, Gou W L, Zhang R. FAM3A protects HT22 cells against hydrogen peroxide-induced oxidative stress through activation of PI3K/Akt but not MEK/ERK pathway. Cell Physiol Biochem. 2015;37:1431–1441. doi: 10.1159/000438512. [DOI] [PubMed] [Google Scholar]
- 21.Jia S, Chen Z, Li J. FAM3A promotes vascular smooth muscle cell proliferation and migration and exacerbates neointima formation in rat artery after balloon injury. J Mol Cell Cardiol. 2014;74:173–182. doi: 10.1016/j.yjmcc.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 22.Chi Y, Li J, Li N. FAM3A enhances adipogenesis of 3T3-L1 preadipocytes via activation of ATP-P2 receptor-Akt signaling pathway. Oncotarget. 2017;8:45862–45873. doi: 10.18632/oncotarget.17578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ishihara H, Asano T, Tsukuda K. Pancreatic beta cell line MIN6 exhibits characteristics of glucose metabolism and glucose-stimulated insulin secretion similar to those of normal islets. Diabetologia. 1993;36:1139–1145. doi: 10.1007/BF00401058. [DOI] [PubMed] [Google Scholar]
- 24.Yang W, Chi Y, Meng Y. FAM3A plays crucial roles in controlling PDX1 and insulin expressions in pancreatic beta cells. FASEB J. 2020;34:3915–3931. doi: 10.1096/fj.201902368RR. [DOI] [PubMed] [Google Scholar]
- 25.Bradley S P, Rastellini C, da Costa M A. Gene silencing in the endocrine pancreas mediated by short-interfering RNA. Pancreas. 2005;31:373–379. doi: 10.1097/01.mpa.0000179730.69081.64. [DOI] [PubMed] [Google Scholar]
- 26.Kapodistria K, Tsilibary E P, Kotsopoulou E. Liraglutide, a human glucagon-like peptide-1 analogue, stimulates AKT-dependent survival signalling and inhibits pancreatic beta-cell apoptosis. J Cell Mol Med. 2018;22:2970–2980. doi: 10.1111/jcmm.13259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong J C, Vo V, Gorjala P, Fiscus R R. Pancreatic-β-cell survival and proliferation are promoted by protein kinase G type Iα and downstream regulation of AKT/FOXO1. Diab Vasc Dis Res. 2017;14:434–449. doi: 10.1177/1479164117713947. [DOI] [PubMed] [Google Scholar]
- 28.Li J M, Wang W, Fan C Y. Quercetin preserves β-cell mass and function in fructose-induced hyperinsulinemia through modulating pancreatic Akt/FoxO1 activation. Evid Based Complement Alternat Med. 2013;2013:303902. doi: 10.1155/2013/303902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Wang H, Liu Y. Antihyperglycemic effect of ginsenoside Rh2 by inducing islet β-cell regeneration in mice. Horm Metab Res. 2012;44:33–40. doi: 10.1055/s-0031-1295416. [DOI] [PubMed] [Google Scholar]
- 30.Li Z, Shangguan Z, Liu Y. Puerarin protects pancreatic β-cell survival via PI3K/Akt signaling pathway. J Mol Endocrinol. 2014;53:71–79. doi: 10.1530/JME-13-0302. [DOI] [PubMed] [Google Scholar]
- 31.Bastidas-Ponce A, Roscioni S S, Burtscher I. Foxa2 and Pdx1 cooperatively regulate postnatal maturation of pancreatic β-cells. Mol Metab. 2017;6:524–534. doi: 10.1016/j.molmet.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kitamura T, Nakae J, Kitamura Y. The forkhead transcription factor FoxO1 links insulin signaling to Pdx1 regulation of pancreatic βcell growth. J Clin Invest. 2002;110:1839–1847. doi: 10.1172/JCI16857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu P, Ma Z, Guo L. Short body length phenotype is compensated by the upregulation of nidogen family members in a deleterious nid1a mutation of zebrafish. J Genet Genomics. 2017;44:553–556. doi: 10.1016/j.jgg.2017.09.011. [DOI] [PubMed] [Google Scholar]
- 34.Ma Z, Zhu P, Shi H. PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components. Nature. 2019;568:259–263. doi: 10.1038/s41586-019-1057-y. [DOI] [PubMed] [Google Scholar]
- 35.El-Brolosy M A, Kontarakis Z, Rossi A. Genetic compensation triggered by mutant mRNA degradation. Nature. 2019;568:193–197. doi: 10.1038/s41586-019-1064-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crooke S T, Witztum J L, Bennett C F. RNA-Targeted Therapeutics. Cell Metab. 2018;27:714–739. doi: 10.1016/j.cmet.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 37.Faas M M, Sáez T, de Vos P. Extracellular ATP and adenosine: The Yin and Yang in immune responses? Mol Aspects Med. 2017;55:9–19. doi: 10.1016/j.mam.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 38.Di Virgilio F, Sarti A C, Falzoni S. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat Rev Cancer. 2018;18:601–618. doi: 10.1038/s41568-018-0037-0. [DOI] [PubMed] [Google Scholar]
- 39.Emmett D S, Feranchak A, Kilic G. Characterization of ionotrophic purinergic receptors in hepatocytes. Hepatology. 2008;47:698–705. doi: 10.1002/hep.22035. [DOI] [PubMed] [Google Scholar]
- 40.MacDonald P E, El-Kholy W, Riedel M J. The multiple actions of GLP-1 on the process of glucose-stimulated insulin secretion. Diabetes. 2002;51:S434–S442. doi: 10.2337/diabetes.51.2007.s434. [DOI] [PubMed] [Google Scholar]
- 41.Yang J, Wong R K, Wang X. Leucine culture reveals that ATP synthase functions as a fuel sensor in pancreatic β-cells. J Biol Chem. 2004;279:53915–53923. doi: 10.1074/jbc.M405309200. [DOI] [PubMed] [Google Scholar]
- 42.Tang S, Xin Y, Yang M. Osteoprotegerin promotes islet β cell proliferation in intrauterine growth retardation rats through the PI3K/AKT/FoxO1 pathway. Int J Clin Exp Pathol. 2019;12:2324–2338. [PMC free article] [PubMed] [Google Scholar]
- 43.Ahlgren U, Jonsson J, Edlund H. The morphogenesis of the pancreatic mesenchyme is uncoupled from that of the pancreatic epithelium in IPF1/PDX1-deficient mice. Development. 1996;122:1409–1416. doi: 10.1242/dev.122.5.1409. [DOI] [PubMed] [Google Scholar]
- 44.Horiguchi M, Yoshida M, Hirata K. Senescence caused by inactivation of the homeodomain transcription factor Pdx1 in adult pancreatic acinar cells in mice. FEBS Lett. 2019;593:2226–2234. doi: 10.1002/1873-3468.13504. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.