

Abstract
Biosynthesis of proteinogenic amino acids in the extremely halophilic archaeon Haloarcula hispanica was explored by using biosynthetically directed fractional 13C labeling with a mixture of 90% unlabeled and 10% uniformly 13C-labeled glycerol. The resulting 13C-labeling patterns in the amino acids were analyzed by two-dimensional 13C,1H correlation spectroscopy. The experimental data provided evidence for a split pathway for isoleucine biosynthesis, with 56% of the total Ile originating from threonine and pyruvate via the threonine pathway and 44% originating from pyruvate and acetyl coenzyme A via the pyruvate pathway. In addition, the diaminopimelate pathway involving diaminopimelate dehydrogenase was shown to lead to lysine biosynthesis and an analysis of the 13C-labeling pattern in tyrosine indicated novel biosynthetic pathways that have so far not been further characterized. For the 17 other proteinogenic amino acids, the data were consistent with data for commonly found biosynthetic pathways. A comparison of our data with the amino acid metabolisms of eucarya and bacteria supports the theory that pathways for synthesis of proteinogenic amino acids were established before ancient cells diverged into archaea, bacteria, and eucarya.
Halophilic archaea are aerobic chemo-organotrophs that grow on a variety of carbon sources. The central carbon metabolisms of some species are relatively well explored (15, 50), while comprehensive investigations of amino acid metabolism have so far been pursued only for organisms belonging to other phylogenetic groups (40) within the domain of the archaea (76), i.e., various methanogens (22, 24, 25, 61) and the anaerobic, extremely thermophilic Thermoproteus neutrophilus (54). These studies indicated that most amino acids in thermophilic and methanogenic archaea are synthesized via pathways that had previously been described for bacteria and eucarya (27, 46, 68, 69). An extension of such studies to halophilic archaea is thus of interest for obtaining new insights into the evolution of carbon metabolism in general. Furthermore, organisms living under extreme environmental conditions (extremophiles) are gaining increasing importance for biotechnological applications (17) and the analysis of their metabolism constitutes a prerequisite for possible future metabolic engineering (2).
In this paper we investigated amino acid biosynthesis in the halophilic archaeon Haloarcula hispanica. H. hispanica was selected for its potential biotechnology interest, since it can efficiently use glycerol for amino acid synthesis (31). We primarily employed biosynthetically directed fractional 13C labeling (53, 58, 63–66, 77) with glycerol as the sole carbon source, combined with two-dimensional (2D) 13C,1H correlation nuclear magnetic resonance (NMR) spectroscopy for the analysis of the resulting nonrandom 13C-labeling patterns. In this approach, contiguous carbon fragments arising from a single carbon source molecule are traced through a cellular bioreaction network. Since the patterns of intact carbon fragments observed for a given metabolite are very often different depending on which pathway is employed for its synthesis, we are able to analyze both the topological structure of the bioreaction network, i.e., the locations of nodes at which one substance is either a substrate for two branching reactions or a product of two converging reactions, and the relative contributions of alternative pathways to the generation of amino acids (63, 64, 66).
MATERIALS AND METHODS
Labeling strategy.
The biosynthetic pathways were explored by biosynthetically directed fractional 13C labeling of the proteinogenic amino acids (53, 58, 63–66, 77), with glycerol as the sole carbon source. Fractional 13C labeling was achieved by growing H. hispanica in a minimal medium containing approximately 10% uniformly 13C-labeled glycerol and 90% glycerol containing 13C at natural abundance. Incorporation of intact two- or three-carbon fragments from the uniformly 13C-labeled carbon source leads to nonrandom 13C-labeling patterns in the amino acids. These are identified from 13C—13C scalar-coupling fine structures in 2D 13C,1H correlation spectroscopy (COSY) (11). With a set of probabilistic equations (63), the observed 13C fine structures then yield the relative abundances of intact glycerol carbon fragments in the carbon skeletons of the amino acids. This approach allows for a comprehensive analysis of the bioreaction network (63, 64, 66). Cells were grown in a batch culture and harvested in the mid-exponential and the early stationary phases in order to assess possible changes of the metabolic state during growth. Since all relevant peaks of the individual amino acids are well resolved in the 2D NMR spectrum, a separation of the amino acids prior to NMR analysis is not required (63–66, 77). To verify the threonine pathway for isoleucine biosynthesis, the labeling experiment was repeated with a growth medium containing [13C4]threonine instead of [13C3]glycerol, which allowed us to directly trace the incorporation of 13C—13C units from threonine into isoleucine.
Growth of the organism and sample preparation.
By following the protocol developed in reference 49, H. hispanica (31) was grown in a medium containing, per liter, 200 g of NaCl, 36 g of MgSO4 · 7H2O, 6 g of Tris base, 4 g of KCl, 1 g of CaCl2 · 2H2O, 2 ml of FeSO4 · 7H2O (0.4% in 1 mM HCl), 2 ml of K2HPO4 (5% in distilled water), and 5 ml of NH4Cl (20% in distilled water). Glycerol (20 ml, 25% in water) was added, and the pH was adjusted to 7.5 with HCl prior to sterilization (filter pore size cutoff, 0.45 μm). In the standard experiments, 10% of the glycerol was uniformly 13C labeled. In the experiment with 13C-labeled threonine, no labeled glycerol was used and 307 mg of [13C4]threonine per liter was added under otherwise identical conditions. Cells were grown in six 35-ml cultures that were shaken at 100 rpm in 100-ml Erlenmeyer flasks for 7 days at 40°C until the early stationary phase was reached. Cells from cultures in the mid-exponential growth phase were harvested after 3 days. After centrifugation at 3,000 × g, the cells were taken up in 10 ml of water and frozen in liquid nitrogen. No DNase was added in order to prevent contamination of the halobacterial proteins with unlabeled amino acids. After being warmed to 0°C, the slurry was centrifuged at 20,000 × g and the protein in the clear supernatant was precipitated with 65% ethanol at a temperature of −20°C overnight. After centrifugation, the pellet was lyophilized and hydrolyzed after addition of 3 ml of 6 M HCl at 80°C for 2 days in a sealed Pyrex tube, yielding about 70 mg of dried biomass.
NMR spectroscopy.
NMR experiments were performed at 40°C. Proton-detected 2D 13C,1H-heteronuclear single-quantum correlation spectra were recorded with the pulse sequence devised by Bodenhausen and Ruben (11), which ensures that 1H—13C scalar couplings do not affect the 13C—13C scalar-coupling fine structure along ω1(13C) (Fig. 1 in reference 65). Pulsed-field gradients were employed for coherence pathway rejection (6, 75), and a 2-ms spin-lock pulse (47) was used to purge the magnetization arising from 12C-bound protons and the residual 2HOH signal. 13C decoupling during t2 was achieved with the composite-pulse decoupling scheme GARP (59), and quadrature detection in ω1 was accomplished with States-TPPI (41). For the samples obtained from the cultures grown with [13C3]glycerol, two spectra were recorded, i.e., one focused on the aliphatic carbons, with the 13C carrier set to 42.5 ppm relative to the chemical shift of 2,2-dimethyl-2-silapentane-5-sulfonate sodium salt (DSS), and one for the aromatic rings, with the 13C carrier set to 125.9 ppm. For the sample obtained from the culture grown with [13C4]threonine, only the spectrum focusing on the aliphatic carbons was recorded. The spectra for the aliphatic resonances were folded along ω1(13C), with a sweep width of 33.8 ppm.
Somewhat different experimental conditions were chosen for the individual measurements. For the sample harvested during the mid-exponential growth phase and the sample generated with [13C4]threonine, the aliphatic spectra were recorded at a 13C resonance frequency of 125.8 MHz, with a Bruker DRX500 spectrometer. The measurement time was 9 h per spectrum (1,706 × 256 complex points; maximal evolution times, t1max = 402 ms, t2max = 102 ms; relaxation delay between scans, 2 s). For the sample harvested in the early stationary growth phase, the aliphatic spectrum was recorded in 8 h at 188.6 MHz with a Bruker DRX750 spectrometer (2,559 × 512 complex points; t1max = 392 ms; t2max = 87 ms; relaxation delay between scans, 2.4 s). The aromatic spectra were recorded in 3 h with a Bruker DRX500 spectrometer (920 × 512 complex points; t1max = 392 ms; t2max = 87 ms; relaxation delay between scans, 1 s). The data were processed with the program PROSA (28). Before Fourier transformation, the time domain data were multiplied in t1 and t2 with sine-bell windows shifted by π/2 (19). The digital resolution after zero-filling was 1.0 Hz/point along ω1 and 2.4 Hz/point along ω2 for the aliphatic spectra at 125.8 MHz, and 0.6 Hz/point along ω1 and 5.8 Hz/point along ω2 for the aromatics. For the 188.6-MHz spectrum the digital resolution was 0.8 Hz/point along ω1 and 3.7 Hz/point along ω2.
For the experiments using [13C3]glycerol as the 13C source, the overall degree of 13C labeling in the amino acids, denoted as p1 in the probabilistic equations of reference 63, was determined from the satellites of selected well-separated peaks in 1D 1H NMR spectra (tmax = 1.022 s; relaxation delay between scans, 10 s). p1 was 0.127 for both preparations used here.
Data analysis.
The individual multiplet components of the 13C—13C scalar-coupling fine structures were integrated with the program XEASY (4), and the observed relative multiplet intensities were used to calculate the relative abundances of intact carbon fragments (63). By following the definitions of references 63 and 66, f(1) represents the fraction of molecules in which the observed carbon atom and its neighboring carbons originate from different source molecules of glycerol and f(2) represents the fraction of molecules in which the observed carbon atom and at least one neighboring carbon originate from the same source molecule. For a central carbon in a C3 fragment that exhibits different 13C—13C scalar-coupling constants with the two attached carbons, f(2) represents the fraction of molecules for which the central carbon and the carbon with the smaller coupling come from the same source molecule, while f(2*) is used if the carbon with the larger coupling comes from the same source molecule as the observed carbon. f(3) denotes the fraction of molecules in which the observed carbon atom and both neighbors in the C3 fragment originate from the same glycerol molecule.
The relative abundances of intact carbon fragments in the amino acids that are expected for specific biosynthetic pathways can be calculated from the relative abundances found in the respective precursors (see Fig. 1 and 3). Here, we present only those equations that are required to explore the biosynthesis of isoleucine, lysine, and tyrosine.
FIG. 1.

Synthesis of erythrose-4-phosphate from glycerol via reversal of glycolytic reactions and the action of transketolase (EC 2.2.1.1). Thick lines indicate carbon-carbon connectivities arising from a single source molecule of glycerol (see the text).
FIG. 3.

Incorporation of oxalacetate or threonine, 2-oxoglutarate, pyruvate, and acetyl-CoA, for which carbon skeletons are schematically shown (A), into the carbon skeletons of isoleucine (B) and lysine (C), according to the biosynthetic pathways shown in Fig. 2 and 6. Note that threonine is synthesized from oxalacetate without rearrangement of the carbon skeleton (see the text). The notation of the carbon atoms follows IUPAC-IUB recommendations (30), i.e., −O2C(4)—C(3)H3—C(2)O—C(1)O2− for oxalacetate, −O2C(5)—C(4)H2—C(3)H2—C(2)O—C(1)O2− for 2-oxoglutarate, C(3)H3—C(2)O—C(1)O2− for pyruvate, and C(2)H3—C(1)O—SCoA for acetyl-CoA.
(i) Biosynthesis of isoleucine.
Cγ2—Cβ of Ile derives from pyruvate in all pathways considered (see Fig. 2 and 3B), and Ile-γ2 is therefore not used in the calculations.
FIG. 2.

Three routes for isoleucine biosynthesis (see the text). Pyruvate and acetyl-CoA are the precursors for Ile synthesis via the pyruvate pathway (route 1) (14), threonine and pyruvate are the precursors for Ile synthesis via the threonine pathway (route 2) (1, 68, 69), and glutamate and pyruvate are the precursors for Ile synthesis via the glutamate pathway (route 3) (36, 48). α-Ketobutyrate is a common intermediate in these three pathways.
(a) Pyruvate pathway. Pyruvate and acetyl coenzyme A (acetyl-CoA) are assessed via Ala-β and Leu-α, respectively.
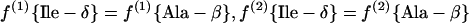 |
1 |
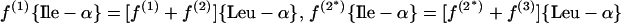 |
2 |
(b) Threonine pathway. Threonine is directly assessed.
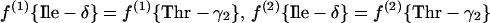 |
3 |
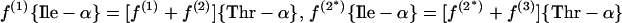 |
4 |
(c) Glutamate pathway. Glutamate is directly assessed.
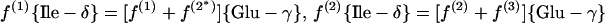 |
5 |
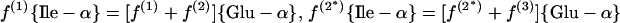 |
6 |
(ii) Biosynthesis of lysine (Fig. 3C).
(a) Diaminopimelate pathways. Pyruvate and aspartate are assessed via Ala-β and Asp-α or Asp-β, respectively.
(a.1) Diaminopimelate-dehydrogenase variant.
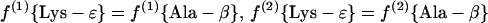 |
7 |
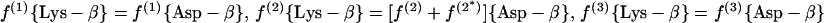 |
8 |
(a.2) Acetylase/succinylase variant.
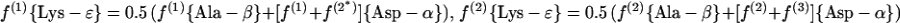 |
9 |
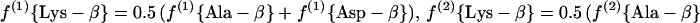 |
10 |
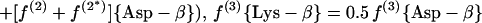 |
(b) α-Aminoadipate pathway. 2-Oxoglutarate is assessed via Glu-γ and Glu-α.
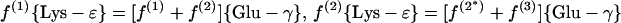 |
11 |
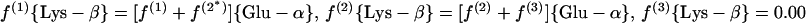 |
12 |
(iii) Biosynthesis of the aromatic ring of tyrosine.
In the biosynthesis of the aromatic ring of tyrosine through the common shikimate pathway (7, 68, 69), the ring is assembled from phosphoenolpyruvate and erythrose-4-phosphate. Phosphoenolpyruvate is assessed via Phe-α and Tyr-α. Erythrose-4-phosphate is assumed to include an intact C2—C3—C4 fragment derived from a first glycerol molecule via the C3 pool of glycolysis and a C1 atom that originates from a second glycerol molecule (see Fig. 1) (50). Due to the symmetry of the aromatic ring of tyrosine, the Tyr-δx and Tyr-ɛx carbons give rise to only one 13C fine structure. Therefore, f(i){Tyr-δ} and f(i){Tyr-ɛ} are calculated as averages of the f values predicted for Tyr-δ1 and Tyr-δ2 or Tyr-ɛ1 and Tyr-ɛ2, respectively (63).
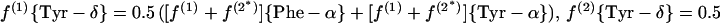 |
13 |
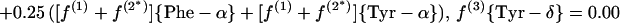 |
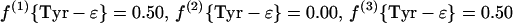 |
14 |
RESULTS
The 2D 13C,1H COSY spectra were analyzed as described by Szyperski (63) (Table 1). Virtually identical 13C scalar-coupling fine structures were observed for samples taken from the mid-exponential growth phase and the early stationary phase, and hence there are no significant differences in the resulting relative abundances of intact carbon fragments, as collected in Table 1. This suggests that there are no major differences in flux ratios through the central metabolic network when the two growth periods are compared (63–66). During hydrolysis, cysteine and tryptophan were oxidized and could thus not be evaluated and asparagine and glutamine were deamidated to aspartate and glutamate (65, 77). The ring carbons of phenylalanine were not evaluated because of strong-coupling effects (63). The evaluation of the observations for all other carbon positions (Table 1) showed that except for isoleucine, lysine, and the aromatic ring of tyrosine, the proteinogenic amino acids in H. hispanica are synthesized according to the pathways commonly found in both bacteria and eucarya (27, 46, 68, 69).
TABLE 1.
Relative intensities of 13C multiplet components and derived relative abundances of intact C2 and C3 fragments in the amino acids
| Observed carbon position | Preparationc | Relative intensity of multiplet componenta
|
Relative abundance of intact carbon fragmentb
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Is | Id | Id* | Idd | It | f(1) | f(2) | f(2*) | f(3) | ||
| Terminal carbons | ||||||||||
| Ala-β | ME | 0.12 | 0.88 | 0.05 | 0.95 | |||||
| ES | 0.11 | 0.89 | 0.05 | 0.95 | ||||||
| Arg-δ | ME | 0.11 | 0.89 | 0.04 | 0.96 | |||||
| ES | 0.11 | 0.89 | 0.05 | 0.95 | ||||||
| Gly-α | ME | 0.09 | 0.91 | 0.02 | 0.98 | |||||
| ES | 0.09 | 0.91 | 0.02 | 0.98 | ||||||
| His-δ2 | ME | 0.08 | 0.92 | 0.00 | 1.00 | |||||
| ES | 0.09 | 0.91 | 0.03 | 0.97 | ||||||
| Ile-γ2 | ME | 0.13 | 0.87 | 0.07 | 0.93 | |||||
| ES | 0.11 | 0.89 | 0.04 | 0.96 | ||||||
| Ile-δ | ME | 0.32 | 0.68 | 0.31 | 0.69 | |||||
| ES | 0.33 | 0.67 | 0.32 | 0.68 | ||||||
| Leu-δ1 | ME | 0.15 | 0.85 | 0.09 | 0.91 | |||||
| ES | 0.11 | 0.89 | 0.04 | 0.96 | ||||||
| Leu-δ2 | ME | 0.87 | 0.13 | 1.00 | 0.00 | |||||
| ES | 0.87 | 0.13 | 0.99 | 0.01 | ||||||
| Lys-ɛ | ME | 0.11 | 0.89 | 0.04 | 0.96 | |||||
| ES | 0.11 | 0.89 | 0.05 | 0.95 | ||||||
| Pro-δ | ME | 0.11 | 0.89 | 0.04 | 0.96 | |||||
| ES | 0.11 | 0.89 | 0.04 | 0.96 | ||||||
| Ser-β | ME | 0.51 | 0.49 | 0.55 | 0.45 | |||||
| ES | 0.54 | 0.46 | 0.58 | 0.42 | ||||||
| Thr-γ2 | ME | 0.48 | 0.52 | 0.51 | 0.49 | |||||
| ES | 0.50 | 0.50 | 0.53 | 0.47 | ||||||
| Val-γ1 | ME | 0.11 | 0.89 | 0.05 | 0.95 | |||||
| ES | 0.11 | 0.89 | 0.04 | 0.96 | ||||||
| Val-γ2 | ME | 0.83 | 0.17 | 0.95 | 0.05 | |||||
| ES | 0.87 | 0.13 | 0.99 | 0.01 | ||||||
| Central carbons in C3 fragments (different scalar couplings) | ||||||||||
| Ala-α | ME | 0.07 | 0.01 | 0.03 | 0.88 | 0.00 | 0.01 | 0.04 | 0.95 | |
| ES | 0.08 | 0.01 | 0.03 | 0.88 | 0.00 | 0.01 | 0.05 | 0.94 | ||
| Asp-α | ME | 0.18 | 0.09 | 0.32 | 0.41 | 0.16 | 0.09 | 0.37 | 0.38 | |
| ES | 0.19 | 0.09 | 0.33 | 0.39 | 0.18 | 0.08 | 0.38 | 0.36 | ||
| Asp-β | ME | 0.18 | 0.34 | 0.31 | 0.17 | 0.16 | 0.39 | 0.36 | 0.09 | |
| ES | 0.19 | 0.32 | 0.32 | 0.17 | 0.18 | 0.37 | 0.37 | 0.08 | ||
| Glu-α | ME | 0.18 | 0.34 | 0.31 | 0.17 | 0.16 | 0.40 | 0.36 | 0.08 | |
| ES | 0.19 | 0.33 | 0.32 | 0.16 | 0.18 | 0.39 | 0.38 | 0.07 | ||
| Glu-γ | ME | 0.09 | 0.02 | 0.77 | 0.12 | 0.04 | 0.00 | 0.96 | 0.00 | |
| ES | 0.09 | 0.02 | 0.77 | 0.12 | 0.03 | 0.01 | 0.95 | 0.01 | ||
| His-α | ME | 0.08 | 0.01 | 0.00 | 0.91 | 0.01 | 0.01 | 0.00 | 0.98 | |
| ES | 0.08 | 0.00 | 0.00 | 0.92 | 0.00 | 0.00 | 0.00 | 1.00 | ||
| His-β | ME | 0.07 | 0.80 | 0.01 | 0.12 | 0.01 | 0.99 | 0.00 | 0.00 | |
| ES | 0.08 | 0.79 | 0.01 | 0.12 | 0.02 | 0.98 | 0.00 | 0.00 | ||
| Ile-α | ME | 0.18 | 0.03 | 0.70 | 0.10 | 0.16 | 0.00 | 0.84 | 0.00 | |
| ES | 0.18 | 0.03 | 0.69 | 0.10 | 0.17 | 0.00 | 0.83 | 0.00 | ||
| Leu-α | ME | 0.09 | 0.02 | 0.77 | 0.12 | 0.04 | 0.01 | 0.95 | 0.00 | |
| ES | 0.09 | 0.03 | 0.77 | 0.11 | 0.03 | 0.02 | 0.95 | 0.00 | ||
| Met-α | MEd | |||||||||
| ES | 0.17 | 0.09 | 0.31 | 0.43 | 0.14 | 0.09 | 0.36 | 0.41 | ||
| Phe-α | ME | 0.08 | 0.04 | 0.00 | 0.88 | 0.00 | 0.05 | 0.00 | 0.95 | |
| ES | 0.08 | 0.01 | 0.00 | 0.91 | 0.01 | 0.01 | 0.00 | 0.98 | ||
| Phe-β | ME | 0.07 | 0.81 | 0.01 | 0.12 | 0.00 | 1.00 | 0.00 | 0.00 | |
| ES | 0.07 | 0.79 | 0.02 | 0.12 | 0.01 | 0.98 | 0.01 | 0.00 | ||
| Pro-α | ME | 0.17 | 0.36 | 0.33 | 0.14 | 0.15 | 0.42 | 0.38 | 0.05 | |
| ES | 0.17 | 0.35 | 0.31 | 0.17 | 0.15 | 0.41 | 0.36 | 0.08 | ||
| Ser-α | ME | 0.08 | 0.00 | 0.43 | 0.49 | 0.01 | 0.00 | 0.53 | 0.46 | |
| ES | 0.08 | 0.01 | 0.45 | 0.46 | 0.01 | 0.00 | 0.56 | 0.43 | ||
| Thr-α | ME | 0.17 | 0.10 | 0.32 | 0.41 | 0.15 | 0.10 | 0.37 | 0.38 | |
| ES | 0.18 | 0.10 | 0.32 | 0.40 | 0.16 | 0.10 | 0.37 | 0.37 | ||
| Tyr-α | ME | 0.07 | 0.01 | 0.00 | 0.92 | 0.00 | 0.01 | 0.00 | 0.99 | |
| ES | 0.08 | 0.00 | 0.00 | 0.92 | 0.01 | 0.00 | 0.00 | 0.99 | ||
| Tyr-β | ME | 0.07 | 0.81 | 0.01 | 0.12 | 0.00 | 1.00 | 0.00 | 0.00 | |
| ES | 0.07 | 0.80 | 0.01 | 0.12 | 0.01 | 0.99 | 0.00 | 0.00 | ||
| Val-α | ME | 0.08 | 0.01 | 0.79 | 0.12 | 0.02 | 0.00 | 0.98 | 0.00 | |
| ES | 0.08 | 0.03 | 0.77 | 0.12 | 0.02 | 0.03 | 0.95 | 0.00 | ||
| Central carbons in C3 fragments (equal scalar couplings) | ||||||||||
| Arg-β | ME | 0.41 | 0.52 | 0.07 | 0.50 | 0.50 | 0.00 | |||
| ES | 0.42 | 0.51 | 0.07 | 0.51 | 0.49 | 0.00 | ||||
| Glu-β | ME | 0.42 | 0.51 | 0.07 | 0.51 | 0.49 | 0.00 | |||
| ES | 0.44 | 0.50 | 0.06 | 0.54 | 0.46 | 0.00 | ||||
| Ile-γ1 | ME | 0.30 | 0.60 | 0.10 | 0.33 | 0.65 | 0.02 | |||
| ES | 0.32 | 0.60 | 0.08 | 0.36 | 0.64 | 0.00 | ||||
| Leu-β | ME | 0.75 | 0.22 | 0.03 | 0.99 | 0.00 | 0.01 | |||
| ES | 0.76 | 0.22 | 0.02 | 1.00 | 0.00 | 0.00 | ||||
| Lys-β | ME | 0.17 | 0.67 | 0.16 | 0.15 | 0.78 | 0.07 | |||
| ES | 0.19 | 0.67 | 0.14 | 0.18 | 0.77 | 0.05 | ||||
| Lys-γ | ME | 0.44 | 0.50 | 0.06 | 0.54 | 0.46 | 0.00 | |||
| ES | 0.43 | 0.51 | 0.06 | 0.53 | 0.47 | 0.00 | ||||
| Lys-δ | ME | 0.08 | 0.81 | 0.12 | 0.02 | 0.98 | 0.00 | |||
| ES | 0.08 | 0.81 | 0.12 | 0.02 | 0.98 | 0.00 | ||||
| Pro-β | ME | 0.40 | 0.53 | 0.07 | 0.48 | 0.52 | 0.00 | |||
| ES | 0.41 | 0.52 | 0.07 | 0.50 | 0.50 | 0.00 | ||||
| Pro-γ | ME | 0.07 | 0.77 | 0.16 | 0.01 | 0.94 | 0.05 | |||
| ES | 0.08 | 0.77 | 0.15 | 0.02 | 0.94 | 0.04 | ||||
| Thr-β | ME | 0.19 | 0.67 | 0.14 | 0.18 | 0.77 | 0.05 | |||
| ES | 0.19 | 0.65 | 0.16 | 0.18 | 0.75 | 0.07 | ||||
| Tyr-δx | ME | 0.14 | 0.74 | 0.12 | 0.11 | 0.88 | 0.01 | |||
| ES | 0.15 | 0.74 | 0.11 | 0.12 | 0.87 | 0.01 | ||||
| Tyr-ɛx | ME | 0.28 | 0.25 | 0.47 | 0.30 | 0.22 | 0.48 | |||
| ES | 0.29 | 0.24 | 0.47 | 0.31 | 0.21 | 0.48 | ||||
Is singlet; Id, doublet split by the smaller coupling; Id*, doublet split by the larger coupling; Idd, doublet of doublets; It, triplet.
Experimental error, ±0.02.
ME, sample harvested in the mid-exponential phase; ES, sample harvested in the early stationary phase.
Not evaluated due to an insufficient signal-to-noise ratio.
Amino acid synthesis from glycolytic intermediates.
Identical values for intact carbon fragments (f values) are observed for phenylalanine and tyrosine, where f(3){Phe-α} approximately equals f(3){Tyr-α}, which approximately equals 1 (Table 1). This finding is in agreement with the shikimate/chorismate pathway (7, 68, 69), where the Cβ—Cα—C′ fragments of phenylalanine and tyrosine are both derived from phosphoenolpyruvate, which is itself expected to derive from glycerol without cleavage of carbon-carbon bonds (50). Serine appears to be synthesized from 3-phosphoglycerate, which is also directly derived from glycerol. However, reversible interconversion into glycine and a C1 unit leads to cleavage of Cβ—Cα connectivities, so that f(1){Ser-β} approximately equals 0.55 in both preparations (Table 1). Moreover, the fact that [f(2*) + f(3)]{Ser-α} approximately equals [f(2)]{Gly-α} (Table 1) shows that glycine is nearly exclusively derived from Cα—C′ of serine.
Virtually identical f values are detected for Val-γ1, Leu-δ1, and Ala-β, which provides evidence that valine, leucine, and alanine are derived from pyruvate according to the well-known biosynthetic pathways (68, 69). This is further supported by the following observations: [f(2*) + f(3)]{Ala-α} ≈ [f(2*) + f(3)]{Val-α} and f(1){Leu-β} ≈ f(1){Val-γ2} ≈ f(1){Leu-δ2} ≈ 1 (Table 1) (63). That f(2){Ala-β} approximately equals f(2*){Leu-α} further shows that Leu-α is derived from C2 of acetyl-CoA.
The f values of His-α, His-β, and His-δ (Table 1) show that the precursor for histidine, ribose-5-phosphate, is synthesized from glyceraldehyde-3-phosphate and fructose-6-phosphate via the nonoxidative part of the pentose phosphate pathway (Fig. 1). The presence of transketolase (EC 2.2.1.1) and transaldolase (EC 2.2.1.2) has been reported for other halophilic archaea (15, 50). The equations f(3){His-α} ≈ f(3){Phe-α} ≈ f(3){Tyr-α} ≈ 1 and f(2){His-δ} ≈ f(2){Phe-β} ≈ f(2){Tyr-β} ≈ 1 indicate that histidine is composed of a Cβ—Cα—C′ fragment and a Cδ2—Cγ fragment from two different glycerol molecules (Fig. 1). This is due to the facts that (i) the glyceraldehyde-3-phosphate pool is almost exclusively derived from glycerol without cleavage of carbon-carbon bonds (see above) and that (ii) fructose-6-phosphate is synthesized from two glycerol molecules via reversal of glycolytic reactions (15, 50), so that it comprises the intact fragments C1—C2—C3 and C4—C5—C6 from two different glycerol molecules.
The f values that characterize pyruvate and phosphoenolpyruvate are slightly different, i.e., pyruvate exhibits about 5% cleavage of C3—C2 connectivities, as evidenced by the equation f(1){Ala-β} ≈ f(1){Val-γ1} ≈ f(1){Leu-δ1} ≈ f(1){Ile-γ2} ≈ 0.05, whereas only intact C3—C2 connectivities are detected for phosphoenolpyruvate (f(2){Phe-α} ≈ f(2){Tyr-α} ≈ 1) (Table 1). This suggests that, in addition to the synthesis of pyruvate from phosphoenolpyruvate via pyruvate kinase (EC 2.7.1.40) (50), the malic enzyme (EC 1.1.1.38, 1.1.1.39, and 1.1.1.40) contributes to pyruvate synthesis via oxalacetate. In fact, malic enzyme activity has been reported for other archaea, such as Halobacterium salinarium (10), Halobacterium cutirubrum (13), and Sulfolobus solfataricus (5).
Amino acid synthesis from intermediates of the tricarboxylic acid cycle.
The f values observed for Asp-α and Asp-β coincide with those observed for Thr-α and Met-α (Asp-α) and Thr-β (Asp-β). Moreover, virtually identical f values were found for Glu-α and Pro-α; for Glu-β, Pro-β, and Arg-β; for Glu-γ and Pro-γ; and for Pro-δ and Arg-δ (Table 1). This is consistent with the well-known pathways in which aspartate, threonine, and methionine are derived from oxalacetate and in which 2-oxoglutarate serves for the biosynthesis of glutamine, proline, and arginine (63). Furthermore, the following observations demonstrate that 2-oxoglutarate is formed by irreversible condensation of oxalacetate with acetyl-CoA in the citric acid cycle (63): (i) the equation f(3){Glu-β} ≈ f(3){Pro-β} ≈ f(2){Glu-γ} ≈ f(3){Glu-γ} ≈ f(3){Pro-γ} ≈ 0 shows that intact C3—C4 connectivities in 2-oxoglutarate are absent; (ii) the f values observed for Asp-β (and also Thr-β) are equal to those observed for Glu-α (and Pro-α), which is in agreement with the fact that the C1—C2—C3 segment of 2-oxoglutarate is derived from C2—C3—C4 of oxalacetate; and (iii) the equation f(2*){Glu-γ} ≈ f(2*){Leu-α} ≈ f(2){Pro-δ} ≈ f(2){Arg-δ} shows that C4—C5 of 2-oxoglutarate is derived from acetyl-CoA (Table 1).
Isoleucine biosynthesis.
A variety of pathways are known for isoleucine biosynthesis in microorganisms. Most common is the threonine pathway, which uses threonine and pyruvate as precursors (1, 68, 69) (Fig. 2 and 3B). In the pyruvate pathway, isoleucine biosynthesis proceeds from pyruvate and acetyl-CoA (14) (Fig. 2 and 3B), whereas in the glutamate pathway, glutamate and pyruvate serve as precursors (36, 48) (Fig. 2 and 3B). In rare cases, isoleucine synthesis has also been found to proceed from homoserine (26, 71), propionate (23, 45, 52), or 2-methylbutyrate (23, 45, 51). In all pathways except the one starting from 2-methylbutyrate, α-ketobutyrate serves as an intermediate and pyruvate yields the Cγ2—Cβ fragment of isoleucine.
In H. hispanica, Ile-γ2 exhibits the same distribution of intact carbon fragments originating from a single molecule of glycerol as Ala-β (Table 1), indicating that the Cγ2—Cβ fragment of isoleucine and the Cβ—Cα fragment of alanine are both derived from pyruvate (Fig. 2 and 3B). However, the relative abundances of intact carbon fragments determined at Ile-α and Ile-δ cannot be explained by a single one of the possible individual pathways (Table 2). A satisfactory fit of the data was obtained with the assumption that the threonine and pyruvate pathways operate simultaneously in a split-pathway fashion. For the early stationary growth phase, the decomposition of the 13C—13C fine structures at both Ile-α and Ile-δ indicates that 56% of isoleucine is synthesized via the threonine pathway and 44% via the pyruvate pathway, and virtually identical values were obtained for the mid-exponential phase (Table 2; Fig. 4).
TABLE 2.
Exploration of isoleucine biosynthesis
| Pathway(s) | Carbon atom analyzed | Relative abundance of indicated intact carbon fragmenta at:
|
|||
|---|---|---|---|---|---|
| Mid-exponential phase
|
Early stationary phase
|
||||
| f(1) | f(2) or f(2*) | f(1) | f(2) or f(2*) | ||
| Experimentalb | Ile-δb | 0.31 | 0.69 | 0.32 | 0.68 |
| Ile-α | 0.16 | 0.84 | 0.17 | 0.83 | |
| Pyruvate and threoninec | Ile-δb | 0.31 | 0.69 | 0.32 | 0.68 |
| Ile-α | 0.16 | 0.84 | 0.17 | 0.83 | |
| Pyruvated (precursors, pyruvate and acetyl-CoA) | Ile-δb | 0.05 | 0.95 | 0.05 | 0.95 |
| Ile-α | 0.05 | 0.95 | 0.05 | 0.95 | |
| Threonine (precursors, threonine and pyruvate)e | Ile-δb | 0.51 | 0.49 | 0.53 | 0.47 |
| Ile-α | 0.25 | 0.75 | 0.26 | 0.74 | |
| Glutamate (precursors, glutamate and pyruvate)f | Ile-δb | 1.00 | 0.00 | 0.98 | 0.02 |
| Ile-α | 0.56 | 0.44 | 0.57 | 0.43 | |
For the definitions of f(1), f(2), and f(2*), see Materials and Methods. f(2) denotes fractions for Ile-δ, and f(2*) denotes fractions for Ile-α. f(2){Ile-α} = f(3){Ile-α} = 0.00 (Table 1; Fig. 3). All values except the experimental values are predictions.
The fractions for Ile-γ1 are not given since those derived from Ile-δ and Ile-γ1 provide the same information (63).
Contributions of the pyruvate and threonine pathways, 44 and 56%, respectively.
Calculated with equations 1 and 2.
Calculated with equations 3 and 4.
Calculated with equations 5 and 6.
FIG. 4.

Decomposition of the experimental 13C—13C scalar-coupling fine structures for the Ile-α and Ile-δ carbons into contributions from the threonine and pyruvate pathways (Fig. 2; Table 2). The stick diagrams represent the fine structures that would be expected if only a single pathway were operational, and the experimental cross sections on the right were taken along ω1(13C) from the 2D 13C,1H COSY spectrum recorded with biomass that was harvested in the early stationary phase (see the text). Fifty-six and 44% of isoleucine are synthesized via the threonine and pyruvate pathways, respectively. The carbon chemical shifts are relative to those of DSS (2,2-dimethyl-2-silapentane-5-sulfonate sodium salt).
To verify that the threonine pathway, which has so far not been described for archaea, is operational in H. hispanica, we performed an additional labeling experiment using [13C4] threonine instead of [13C3]glycerol (see Materials and Methods). Consistent with the presence of the threonine pathway, the 13C—13C scalar-coupling fine structures observed at Ile-δ, Ile-γ1, and Ile-α are dominated by a doublet (Fig. 5), which proves that intact Cδ—Cγ1 and Cα—C′ fragments originate from [13C4]threonine (Fig. 3). The poor signal-to-noise ratio of the γ2-carbon cross peaks indicates that this carbon position is only sparsely enriched with 13C. Consistently, the γ2 carbon exhibits a fine structure that is also observed at Ala-β. This is due to the fact that the Cγ2—Cβ fragment is derived from pyruvate in both the threonine and the pyruvate pathways (note that pyruvate serves for alanine biosynthesis). Long-range 3JCαCδ couplings of 3.1 Hz are observed as additional splittings of the doublet lines at Ile-α and Ile-δ, and 3JCγ2C′ couplings of 1.7 Hz (see reference 35) in threonine are observed as broadenings of the doublet lines at Thr-γ2 (Fig. 5). The observation of these long-range carbon-carbon couplings confirms that both of the Cδ—Cγ1 and Cα—C′ fragments in isoleucine are derived from the same single threonine molecule via α-ketobutyrate.
FIG. 5.

Cross sections taken along ω1(13C) from the 2D 13C,1H COSY spectrum recorded with the biomass labeled with [13C4]threonine. (A to C) 13C—13C scalar-coupling fine structures of the precursors used for isoleucine biosynthesis via the threonine pathway (Fig. 2), i.e., with Thr-α, Thr-γ2, and Ala-β representing C3 of pyruvate. (D to F) 13C—13C fine structures observed at Ile-α, Ile-δ, and Ile-γ2 (Ile-γ1 yields the same information as Ile-δ [63]). The fine structures of Ile-α and Ile-δ are dominated by a doublet, which proves that the fragments Cδ—Cγ1 and Cα—C′ originate from [13C4]threonine. The γ2 carbon exhibits a 13C—13C fine structure similar to that of Ala-β, which is consistent with the fact that the Cγ2—Cβ fragments of Ile are derived from pyruvate. The additional small splittings of the doublet components of Ile-α and Ile-δ (D and E) and the broadening of the Thr-γ2 doublet lines (B) arise from the vicinal scalar couplings 3JCαCδ in isoleucine and 3JCγ2C′ in threonine (35) (see the text). The carbon chemical shifts are relative to those of DSS. The asterisk in panel E indicates an impurity.
Alternative combinations of multiple biosynthesis pathways are not supported by the experimental data. For example, decomposition of the isoleucine 13C—13C fine structures into fractions stemming from the pyruvate and glutamate pathways leads to different contributions for the two pathways when they are derived either from Ile-α or from Ile-δ, both for the mid-exponential and the early stationary phase (Table 2). Decomposition into the threonine and the glutamate pathways yields negative contributions for one of the pathways. Finally, if one assumes simultaneous operation of all three pathways of Fig. 2, one finds that the contribution of the glutamate pathway would be below 10% while the relative contributions of the pyruvate and threonine pathways would be similar to those obtained when only those two pathways are assumed to be active (see above and Fig. 4).
Lysine biosynthesis.
Lysine biosynthesis via the “diaminopimelate pathway” starts from aspartate and pyruvate, whereas the “α-aminoadipate pathway” relies on 2-oxoglutarate and acetyl-CoA as precursors (9, 54) (Fig. 3C and 6A). Two variants of the diaminopimelate pathway differ in the reaction sequence used to convert l-Δ1-piperidine-2,6-dicarboxylate to dl-diaminopimelate (Fig. 6B). In the dehydrogenase variant, l-Δ1-piperidine-2,6-dicarboxylate is directly converted to dl-diaminopimelate by diaminopimelate dehydrogenase (EC 1.4.1.16) (42, 43, 74). In the acetylase/succinylase variant (8, 32, 62), l-Δ1-piperidine-2,6-dicarboxylate is first acetylated or succinylated and then converted to the symmetric intermediate ll-diaminopimelate, which is finally epimerized to dl-diaminopimelate. The dehydrogenase variant can readily be distinguished from the acetylase/succinylase variants in the fractional labeling experiment, because the involvement of the symmetric intermediate ll-diaminopimelate implies symmetrization of the 13C-labeling pattern about the Cβ—Cγ bond (Fig. 3C).
FIG. 6.

Lysine biosynthesis. (A) Synthesis via the α-aminoadipate pathway (9, 54); (B) synthesis via the diaminopimelate pathway. In the acetylase/succinylase variant, l-Δ1-piperidine-2,6-dicarboxylate is converted to dl-diaminopimelate via the symmetric intermediate ll-diaminopimelate (route 1) (8, 32, 62). In the dehydrogenase variant, l-Δ1-piperidine-2,6-dicarboxylate is directly converted to dl-diaminopimelate by diaminopimelate dehydrogenase (route 2) (42, 43, 74).
For H. hispanica our study indicates that biosynthesis during the mid-exponential as well as the early stationary growth phase proceeds via the dehydrogenase variant of the diaminopimelate pathway (Fig. 3C and 6B; Table 3). The relative abundances of intact carbon fragments observed at Lys-δ and Lys-ɛ correspond to the values observed at Ala-β (Table 1), which is expected if the fragment Cɛ—Cδ comes entirely from the fragment C3—C2 of pyruvate when lysine is synthesized via diaminopimelate dehydrogenase (Fig. 3C). The data in Table 3 show that there are no significant contributions to lysine biosynthesis either from the acetylase/succinylase variant of the diaminopimelate pathway or from the α-aminoadipate pathway. This result is of special interest since simultaneous operation of both variants of the diaminopimelate pathway has previously been documented for the bacterium Corynebacterium glutamicum (29, 56).
TABLE 3.
Exploration of lysine biosynthesis
| Pathway | Carbon atom analyzed | Relative abundance of indicated intact carbon fragmenta at:
|
|||||
|---|---|---|---|---|---|---|---|
| Mid-exponential phase
|
Early stationary phase
|
||||||
| f(1) | f(2) | f(3) | f(1) | f(2) | f(3) | ||
| Experimental | Lys-ɛb | 0.04 | 0.96 | 0.05 | 0.95 | ||
| Lys-β | 0.15 | 0.78 | 0.07 | 0.18 | 0.77 | 0.05 | |
| Diaminopimelate-dehydrogenase variant of the diaminopimelate pathwayc | Lys-ɛ | 0.05 | 0.95 | 0.05 | 0.95 | ||
| Lys-β | 0.16 | 0.75 | 0.09 | 0.18 | 0.74 | 0.08 | |
| Acetylase/succinylase variant of the diaminopimelate pathwayd | Lys-ɛ | 0.29 | 0.71 | 0.30 | 0.70 | ||
| Lys-β | 0.11 | 0.85 | 0.04 | 0.12 | 0.84 | 0.04 | |
| α-Aminoadipatee | Lys-ɛ | 0.04 | 0.96 | 0.04 | 0.96 | ||
| Lys-β | 0.52 | 0.48 | 0.00 | 0.55 | 0.45 | 0.00 | |
For the definitions of f(1), f(2), and f(3), see Materials and Methods. All values except the experimental values are expected.
Values coincide with those observed for Ala-β.
Calculated with equations 7 and 8.
Calculated with equations 9 and 10.
Calculated with equations 11 and 12.
Threonine cleavage.
In the labeling experiment with [13C4]threonine, the fine structure of Leu-α (representing C2 of acetyl-CoA [see Fig. 2 in reference 63]) contains a higher proportion of the doublet component than the fine structure of Ala-β (representing C3 of pyruvate) and a large proportion of 13C—13C′ fragments is also found for Gly-α (Fig. 7). This provides evidence that the exogenously supplied threonine is cleaved into glycine and acetaldehyde, which would be compatible with the assumptions that there is threonine aldolase activity in H. hispanica and subsequent conversion of acetaldehyde into acetyl-CoA (69). This indication of threonine aldolase activity in a halophilic archaeon complements data obtained with other organisms: l-threonine aldolases (EC 4.1.2.5) from Saccharomyces cerevisiae (GLY1 [37, 44]), Escherichia coli (ltaE [39]), and Pseudomonas sp. strain NCIMB 10558 (ltaP [38]) have been cloned and expressed, and l-threonine aldolase activity has been demonstrated for serine hydroxymethyltransferase (EC 2.1.2.1) in E. coli (55), although this enzyme serves primarily for the cleavage of serine to glycine and a C1 unit. The serine hydroxymethyltransferase from the thermophilic archaeon Sulfolobus solfataricus has been shown to possess allo-l-threonine aldolase activity (18). Genome sequencing showed that serine hydroxymethyltransferase is also present in Methanobacterium thermoautotrophicum (60), in Methanococcus jannaschii (12, 57), and in the hyperthermophilic, sulfate-reducing archaeon Archaeoglobus fulgidus (34).
FIG. 7.

Evidence for threonine cleavage in H. hispanica derived from the labeling experiment with [13C4]threonine. The 13C—13C scalar-coupling fine structure of Leu-α (A), which represents C2 of acetyl-CoA, exhibits a significantly more intense doublet component than that of Ala-β (B), which represents C3 of pyruvate. A strong doublet is also detected for Gly-α (C). These data indicate that a fraction of the exogenously supplied threonine was cleaved into glycine and acetaldehyde, with subsequent conversion of acetaldehyde into acetyl-CoA (69). The carbon chemical shifts are relative to those of DSS.
Analysis of the 13C-labeling pattern in tyrosine indicates yet unknown biosynthetic pathways.
Commonly, erythrose-4-phosphate and phosphoenolpyruvate serve for the biosynthesis of the aromatic ring of tyrosine via the shikimate pathway (7, 68, 69). To evaluate the 13C-labeling pattern of the tyrosine ring in light of this pathway, f values for this pathway were predicted with the assumption that histidine biosynthesis proceeds via ribose-5-phosphate (68, 69). The pentose pool must then be composed of molecules carrying intact C1—C2—C3 and C4—C5 fragments (Fig. 1) (see above for f values obtained for His), so that erythrose-4-phosphate would be expected to bear a C2—C3—C4 fragment from one source molecule and a C1 carbon from a second glycerol molecule (Fig. 1). Significant deviations between the experimental data and the thus calculated values for a “pure” shikimate pathway (Table 4) cannot be explained within the framework of commonly known pathways. In particular, biosynthesis of the aromatic ring from 2-keto-3-deoxyarabino-heptulosonate-7-phosphate (DAHP), which itself arises from erythrose-4-phosphate and phosphoenolpyruvate via DAHP synthase (EC 4.1.2.15), predicts that 50% of the ɛ carbons must not be connected to carbon atoms that arise from the same source molecule, i.e., f(1){Tyr-ɛ} equals 0.5 and f(2){Tyr-ɛ} equals 0 (Fig. 8). Hence, the detection of intact Cδ—Cɛ fragments (f(2){Tyr-ɛ} ≈ 0.21) (Table 4) excludes the sole operation of the standard shikimate pathway. This result appears to be in line with data obtained previously for the methanogen Methanococcus maripaludis with a different 13C-labeling approach, where the standard shikimate pathway (67) could not account for all the observations. (Note that the operation of the oxidative part of the pentose phosphate cycle has not yet been observed for halophilic archaebacteria. In cell extracts of Haloferax mediterranei and Haloarcula vallismortis, 6-phosphogluconate dehydrogenase (EC 1.1.1.44) was not active at physiological salt concentrations whereas the observed transketolase and transaldolase activities ensured the formation of pentoses from hexoses (50). However, even if pentose biosynthesis in H. hispanica occurred via glucose oxidation, the same relative abundances of intact carbon fragments would be predicted for the erythrose-4-phosphate pool.
TABLE 4.
Analysis of aromatic 13C fine structures of tyrosine
| Nature of values | Carbon atom analyzed | Relative abundance of indicated intact carbon fragmenta at:
|
|||||
|---|---|---|---|---|---|---|---|
| Mid-exponential phase
|
Early stationary phase
|
||||||
| f(1) | f(2) | f(3) | f(1) | f(2) | f(3) | ||
| Calculatedb | Tyr-δ | 0.00 | 1.00 | 0.00 | 0.01 | 0.99 | 0.00 |
| Observed | 0.11 | 0.88 | 0.01 | 0.12 | 0.87 | 0.01 | |
| Calculatedc | Tyr-ɛ | 0.50 | 0.00 | 0.50 | 0.50 | 0.00 | 0.50 |
| Observed | 0.30 | 0.22d | 0.48 | 0.31 | 0.21d | 0.48 | |
For the definitions of f(1), f(2), and f(3), see Materials and Methods.
Calculated with equation 13.
Calculated with equation 14.
Denotes the relative abundance of tyrosine molecules carrying Cɛ—Cδ fragments originating from a single molecule of glycerol.
FIG. 8.

Scheme indicating the transfer of intact C2 and C3 fragments of glycerol via DAHP (2-keto-3-deoxyarabino-heptulosonate-7-phosphate) to the aromatic ring of tyrosine. Thick lines indicate carbon-carbon connectivities arising from a single molecule of glycerol. DAHP was assumed to be synthesized from erythrose-4-phosphate and phosphoenolpyruvate (compare with Fig. 1). One of the ɛ carbons of tyrosine must then be bound to two carbons that originate from different glycerol molecules. The experimental detection of Cδ—Cɛ fragments thus excludes the possibility that the standard shikimate pathway is the only pathway leading to tyrosine biosynthesis in this system (see the text and Table 4). The double line indicates the cyclization site of the Tyr ring. C1 of DAHP is lost as CO2.
DISCUSSION
In perspective with current knowledge on amino acid biosynthesis in bacteria and eucarya, the present data on isoleucine and lysine biosynthesis are particularly intriguing and expand knowledge accumulated for other organisms in the domain of the archaea. Thus, the identification of a split threonine/pyruvate pathway for isoleucine biosynthesis in H. hispanica is a novel finding, since the threonine pathway has not previously been reported for archaea. In archaebacterial species, such as methanobacteria (21–25, 61) and the thermophilic archaeon Thermoproteus neutrophilus (54), the pyruvate pathway is used. In methanogens, isoleucine synthesis can also proceed from propionate or 2-methylbutyrate (23). Notably, the threonine pathway is common in eucaryotic microorganisms and bacteria, although the pyruvate pathway has also been reported for such species (20, 33, 71, 73). However, constitutive, simultaneous operation of the threonine and pyruvate pathways has so far been observed only in a very few organisms, e.g., spirochetes of the genus Leptospira (73). In addition, split isoleucine synthesis pathways have been reported for eucaryotic and procaryotic microorganisms such as Serratia marcescens (threonine/pyruvate pathways) (33), E. coli Crookes and K-12 (threonine/glutamate pathways) (36, 48), Rhodopseudomonas sphaeroides (threonine/glutamate pathways) (16), and S. cerevisiae (threonine/pyruvate pathways or synthesis from homoserine) (71), but with these organisms a genomic mutation or special growth conditions are required to activate routes other than the threonine pathway.
Lysine biosynthesis via the dehydrogenase variant of the diaminopimelate pathway has previously been identified, for example, in Bacillus sphaericus (74) and in Corynebacterium glutamicum (29, 56, 72), and here we now present direct evidence that this pathway also exists within the domain of the archaea. It has previously been shown that lysine biosynthesis in the methanogens Methanospirillum hungatei (22), Methanococcus voltae (24), Methanothrix concilii (25), and Methanobacterium thermoautotrophicum (3) occurs via the diaminopimelate pathway, but the experiments performed in these earlier studies did not allow us to distinguish between the two variant pathways of Fig. 6B. For Methanobacterium thermoautotrophicum the enzymes that were assayed, i.e., dihydrodipicolinate synthase (EC 4.2.1.52) and diaminopimelate decarboxylase (EC 4.1.1.20), catalyze reactions that are common to both variants of the pathway. For Methanospirillum hungatei (22), Methanococcus voltae (24), Methanococcus jannaschii (61), and Methanothrix concilii (25), 13C-labeling experiments with [1-13C]acetate, [2-13C]acetate, or 13CO2 were used in biosynthetic studies, but with these specifically labeled precursors both variants of the diaminopimelate pathway (Fig. 6B) yielded the same positional 13C enrichments in lysine (15). Indications for the operation of the acetylase/succinylase variant in methanogens and Archaeoglobus fulgidus were obtained from genome sequencing. The gene for ll-diaminopimelate epimerase (EC 5.1.1.7), which catalyzes the conversion of ll-diaminopimelate to meso-diaminopimelate in the acetylase/succinylase variant (Fig. 6B), was identified in Methanobacterium thermoautotrophicum (60), Methanococcus jannaschii (12, 57), and Archaeoglobus fulgidus (34), and the succinyl-diaminopimelate desuccinylase gene (EC 3.5.1.18) was found in Methanococcus jannaschii (12, 57) and Archaeoglobus fulgidus (34). The α-aminoadipate pathway for lysine synthesis (Fig. 6A), which has previously been identified solely for lower eucarya such as fungi, algae, and yeast (9, 70), operates in the thermophilic archaeon Thermoproteus neutrophilus (54). Overall, with the new data presented in this paper the implication is that all currently known pathways for lysine biosynthesis in bacteria and eucarya exist also in the domain of archaea.
Organisms living under extreme environmental conditions, such as archaebacteria, have considerable interest for applications in biotechnology (17). Elucidation of their bioreaction networks is thus of interest as a basis for metabolic engineering (2). In this context the present investigation is another telling illustration of the power of biosynthetically directed fractional 13C labeling in combination with 2D 13C,1H NMR techniques (58, 65) for support of flux analysis in central metabolism (53, 63, 64, 66). This approach revealed a remarkable diversity of pathways employed for isoleucine and lysine biosynthesis in a halophilic archaeon and showed that with the sole exception of the aromatic ring of tyrosine (and possibly phenylalanine), the proteinogenic amino acids are synthesized according to the common pathways (68, 69). Comparison with the amino acid metabolisms of eucarya and bacteria then also supports the notion that pathways for synthesis of proteinogenic amino acids were probably largely established before the divergence of these three domains (76).
ACKNOWLEDGMENTS
Financial support was obtained from the Swiss Priority Program in Biotechnology and the Fonds der chemischen Industrie.
We thank Brigitte Kessler for excellent technical support.
REFERENCES
- 1.Abelson P H. Amino acid biosynthesis in Escherichia coli: isotope competition with 14C-glucose. J Biol Chem. 1954;206:335–343. [PubMed] [Google Scholar]
- 2.Bailey J E. Towards a science of metabolic engineering. Science. 1991;252:1668–1675. doi: 10.1126/science.2047876. [DOI] [PubMed] [Google Scholar]
- 3.Bakhiet N, Forney F W, Stahly D P, Daniels L. Lysine biosynthesis in Methanobacterium thermoautotrophicum is by the diaminopimelic acid pathway. Curr Microbiol. 1984;10:195–198. [Google Scholar]
- 4.Bartels C, Xia T, Billeter M, Güntert P, Wüthrich K. The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J Biomol NMR. 1995;6:1–10. doi: 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
- 5.Bartolucci S, Rella R, Guagliardi A, Raia C A, Gambacorta A, De Rosa M, Rossi M. Malic enzyme from archaebacterium Sulfolobus solfataricus. J Biol Chem. 1987;262:7725–7731. [PubMed] [Google Scholar]
- 6.Bax A, Pochapsky S. Optimized recording of heteronuclear multidimensional NMR spectra using pulsed field gradients. J Magnet Reson. 1992;99:638–643. [Google Scholar]
- 7.Bentley R. The shikimate pathway—a metabolic tree with many branches. Crit Rev Biochem Mol Biol. 1990;25:307–384. doi: 10.3109/10409239009090615. [DOI] [PubMed] [Google Scholar]
- 8.Berges D A, DeWolf W E, Dunn G L, Newman D J, Schmidt S J, Taggart J J, Gilvarg C. Studies on the active site of succinyl-CoA:tetrahydrodipicolinate N-succinyltransferase. J Biol Chem. 1986;261:6160–6167. [PubMed] [Google Scholar]
- 9.Bhattacharjee J K. α-Aminoadipate pathway for the biosynthesis of lysine in lower eucaryotes. Crit Rev Microbiol. 1985;12:131–151. doi: 10.3109/10408418509104427. [DOI] [PubMed] [Google Scholar]
- 10.Bhaumik S R, Sonawat H M. Pyruvate metabolism in Halobacterium salinarium studied by intracellular 13C nuclear magnetic resonance spectroscopy. J Bacteriol. 1994;176:2172–2176. doi: 10.1128/jb.176.8.2172-2176.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bodenhausen G, Ruben D. Natural abundance nitrogen-15 NMR by enhanced heteronuclear spectroscopy. Chem Phys Lett. 1980;69:185–188. [Google Scholar]
- 12.Bult C J, et al. Complete genome sequence of the methanogenic archaeon Methanococcus jannaschii. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- 13.Cazzulo J J, Vidal M C. Effect of monovalent cations on the malic enzyme from the extreme halophile Halobacterium cutirubrum. J Bacteriol. 1972;109:437–439. doi: 10.1128/jb.109.1.437-439.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Charon N W, Johnson R C, Peterson D. Amino acid biosynthesis in the spirochete Leptospira: evidence for a novel pathway of isoleucine biosynthesis. J Bacteriol. 1974;117:203–211. doi: 10.1128/jb.117.1.203-211.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Danson M J. Central metabolism of the archaea. In: Kates M, et al., editors. The biochemistry of the archaea—1993. Amsterdam, The Netherlands: Elsevier Science Publishers B.V.; 1993. pp. 1–24. [Google Scholar]
- 16.Datta P. Biosynthesis of amino acids. In: Clayton R K, Sistrom W R, editors. The photosynthetic bacteria—1978. New York, N.Y: Plenum Press; 1978. pp. 787–788. [Google Scholar]
- 17.Davis M C. Making a living at the extremes. Trends Biotechnol. 1998;16:102–104. [Google Scholar]
- 18.Delle Fratte S, White R H, Maras B, Bossa F, Schirch V. Purification and properties of serine hydroxymethyltransferase from Sulfolobus solfataricus. J Bacteriol. 1997;179:7456–7461. doi: 10.1128/jb.179.23.7456-7461.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DeMarco A, Wüthrich K. Digital filtering with a sinusoidal window function: an alternative technique for resolution enhancement in FT NMR. J Magnet Reson. 1976;24:201–204. [Google Scholar]
- 20.Dunstan R H, Whatley F R, Greenaway W. Growth of Paracoccus denitrificans on [2,3-13C]-succinate and [1,4-13C]-succinate. II. Isoleucine biosynthesis. Proc R Soc Lond B. 1987;231:349–358. doi: 10.1098/rspb.1987.0048. [DOI] [PubMed] [Google Scholar]
- 21.Eikmanns B, Linder D, Thauer R K. Unusual pathway of isoleucine biosynthesis in Methanobacterium thermoautotrophicum. Arch Microbiol. 1983;136:111–113. [Google Scholar]
- 22.Ekiel I, Smith I C P, Sprott G D. Biosynthetic pathways in Methanospirillum hungatei as determined by 13C nuclear magnetic resonance. J Bacteriol. 1983;156:316–326. doi: 10.1128/jb.156.1.316-326.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ekiel I, Smith I C P, Sprott G D. Biosynthesis of isoleucine in methanogenic bacteria: a 13C NMR study. Biochemistry. 1984;23:1683–1687. [Google Scholar]
- 24.Ekiel I, Jarrell K F, Sprott G D. Amino acid biosynthesis and sodium dependent transport in Methanococcus voltae, as revealed by 13C NMR. Eur J Biochem. 1985;149:437–444. doi: 10.1111/j.1432-1033.1985.tb08944.x. [DOI] [PubMed] [Google Scholar]
- 25.Ekiel I, Sprott G D, Patel G B. Acetate and CO2 assimilation by Methanothrix concilii. J Bacteriol. 1985;162:905–908. doi: 10.1128/jb.162.3.905-908.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flavin M, Segal A. Purification and properties of the cystathionine γ-cleavage enzyme of Neurospora. J Biol Chem. 1964;239:2220–2227. [PubMed] [Google Scholar]
- 27.Gottschalk G. Bacterial metabolism. 2nd ed. New York, N.Y: Springer-Verlag; 1986. [Google Scholar]
- 28.Güntert P, Dötsch V, Wider G, Wüthrich K. Processing of multi-dimensional NMR data with the new software PROSA. J Biomol NMR. 1992;2:619–629. [Google Scholar]
- 29.Ishino S, Yamaguchi K, Shirahata K, Araki K. Involvement of meso-diaminopimelate d-dehydrogenase in lysine biosynthesis in Corynebacterium glutamicum. Agric Biol Chem. 1984;48:2557–2560. [Google Scholar]
- 30.IUPAC-IUB Commission on Biochemical Nomenclature. Abbreviations and symbols for the description of the conformation of polypeptide chains. Biochemistry. 1970;9:3471–3479. doi: 10.1021/bi00820a001. [DOI] [PubMed] [Google Scholar]
- 31.Juez G, Rodriguez-Valera F, Ventosa A, Kushner D J. Haloarcula hispanica spec. nov. and Haloferax gibbonsii spec. nov., two new species of extremely halophilic archaebacteria. Syst Appl Microbiol. 1986;8:75–79. [Google Scholar]
- 32.Kindler S H, Gilvarg C. N-Succinyl-l-α,ɛ-diaminopimelate acid deacylase. J Biol Chem. 1960;235:3532–3535. [PubMed] [Google Scholar]
- 33.Kisumi M, Komatsubara S, Chibata I. Pathway for isoleucine formation from pyruvate by leucine biosynthetic enzymes in leucine-accumulating isoleucine revertants of Serratia marcescens. J Biochem. 1977;82:95–103. doi: 10.1093/oxfordjournals.jbchem.a131698. [DOI] [PubMed] [Google Scholar]
- 34.Klenk H-P, et al. The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus. Nature. 1997;390:364–370. doi: 10.1038/37052. [DOI] [PubMed] [Google Scholar]
- 35.Krivdin L B, Della E W. Spin-spin coupling constants between carbons separated by more than one bond. Prog NMR Spectrosc. 1991;23:301–610. [Google Scholar]
- 36.LeMaster D M, Cronan J E., Jr Biosynthetic production of 13C-labeled amino acids with site-specific enrichment. J Biol Chem. 1982;257:1224–1230. [PubMed] [Google Scholar]
- 37.Liu J Q, Nagata S, Dairi T, Misono H, Shimizu S, Yamada H. The GLY1 gene of Saccharomyces cerevisiae encodes a low-specific l-threonine aldolase that catalyzes cleavage of l-allo-threonine and l-threonine to glycine. Expression of the gene in Escherichia coli and purification and characterization of the enzyme. Eur J Biochem. 1997;245:289–293. doi: 10.1111/j.1432-1033.1997.00289.x. [DOI] [PubMed] [Google Scholar]
- 38.Liu J Q, Ito S, Dairi T, Itoh N, Kataoka M, Shimizu S, Yamada H. Gene cloning, nucleotide sequencing, and purification and characterization of the low-specificity l-threonine aldolase from Pseudomonas sp. strain NCIMB 10558. Appl Environ Microbiol. 1998;64:549–554. doi: 10.1128/aem.64.2.549-554.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu J Q, Dairi T, Itoh N, Kataoka M, Shimizu S, Yamada H. Gene cloning, biochemical characterization and physiological role of a thermostable low-specificity l-threonine aldolase from Escherichia coli. Eur J Biochem. 1998;255:220–226. doi: 10.1046/j.1432-1327.1998.2550220.x. [DOI] [PubMed] [Google Scholar]
- 40.Lodwick D, Ross H N M, Walker J A, Almond J W, Grant W D. Nucleotide sequence of the 16S ribosomal RNA gene from the haloalkaliphilic archaeon Natronobacterium magadii, and the phylogeny of Halobacteria. Syst Appl Microbiol. 1991;14:352–357. [Google Scholar]
- 41.Marion D, Ikura M, Tschudin R, Bax A. Rapid recording of 2D NMR spectra without phase cycling. Application to the study of hydrogen exchange in proteins. J Magnet Reson. 1989;85:393–399. [Google Scholar]
- 42.Misono H, Soda K. Properties of meso-α,ɛ-diaminopimelate dehydrogenase from Bacillus sphaericus. J Biol Chem. 1980;255:10599–10605. [PubMed] [Google Scholar]
- 43.Misono H, Togawa H, Yamamoto T, Soda K. Meso-α,ɛ-diaminopimelate d-dehydrogenase: distribution and the reaction product. J Bacteriol. 1979;137:22–27. doi: 10.1128/jb.137.1.22-27.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Monschau N, Stahlmann K P, Sahm H, McNeil J B, Bognar A L. Identification of Saccharomyces cerevisiae GLY1 as a threonine aldolase: a key enzyme in glycine biosynthesis. FEMS Microbiol Lett. 1997;150:55–60. doi: 10.1111/j.1574-6968.1997.tb10349.x. [DOI] [PubMed] [Google Scholar]
- 45.Monticello D J, Hadioetomo R S, Costilow R N. Isoleucine synthesis by Clostridium sporogenes from propionate or α-methylbutyrate. J Gen Microbiol. 1984;130:309–318. doi: 10.1099/00221287-130-2-309. [DOI] [PubMed] [Google Scholar]
- 46.Neidhardt F C, Curtiss III R, Ingraham J L, Lin E C C, Low K B, Magasamik B, Reznikoff W S, Riley M, Schaechter M, Umbarger H E, editors. Escherichia coli and Salmonella: cellular and molecular biology. 2nd ed. Washington, D.C: American Society for Microbiology; 1996. [Google Scholar]
- 47.Otting G, Wüthrich K. Efficient purging scheme for proton-detected heteronuclear two-dimensional NMR. J Magnet Reson. 1988;76:569–574. [Google Scholar]
- 48.Phillips A T, Nuss J I, Moosic J, Foshay C. Alternate pathway for isoleucine biosynthesis in Escherichia coli. J Bacteriol. 1972;109:714–719. doi: 10.1128/jb.109.2.714-719.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rangaswamy V, Altekar W. Ketohexokinase (ATP:d-fructose 1-phosphotransferase) from a halophilic archaebacterium, Haloarcula vallismortis: purification and properties. J Bacteriol. 1994;176:5505–5512. doi: 10.1128/jb.176.17.5505-5512.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rawal N, Kelkar S M, Altekar W. Alternative routes of carbohydrate metabolism in halophilic archaebacteria. Indian J Biochem Biophys. 1988;25:674–686. [PubMed] [Google Scholar]
- 51.Robinson J M, Allison M J. Isoleucine biosynthesis from 2-methylbutyric acid by anaerobic bacteria from the rumen. J Bacteriol. 1969;97:1220–1226. doi: 10.1128/jb.97.3.1220-1226.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sauer F D, Erfle J D, Mahadevan S. Amino acid biosynthesis in mixed rumen cultures. Biochem J. 1975;150:357–372. doi: 10.1042/bj1500357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sauer U, Hatzimanikatis V, Bailey J E, Hochuli M, Szyperski T, Wüthrich K. Metabolic fluxes in riboflavin-producing Bacillus subtilis. Nat Biotechnol. 1997;15:448–452. doi: 10.1038/nbt0597-448. [DOI] [PubMed] [Google Scholar]
- 54.Schäfer S, Paalme T, Vilu R, Fuchs G. 13C-NMR study of acetate assimilation in Thermoproteus neutrophilus. Eur J Biochem. 1989;186:695–700. doi: 10.1111/j.1432-1033.1989.tb15262.x. [DOI] [PubMed] [Google Scholar]
- 55.Schirch V, Hopkins S, Villar E, Angelaccio S. Serine hydroxymethyltransferase from Escherichia coli: purification and properties. J Bacteriol. 1985;163:1–7. doi: 10.1128/jb.163.1.1-7.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schrumpf B, Schwarzer A, Kalinowski J, Pühler A, Eggeling L, Sahm H. A functionally split pathway for lysine synthesis in Corynebacterium glutamicum. J Bacteriol. 1991;173:4510–4516. doi: 10.1128/jb.173.14.4510-4516.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Selkov E, Maltsev N, Olsen G J, Overbeek R, Whitman W B. A reconstruction of the metabolism of Methanococcus jannaschii from sequence data. Gene. 1997;197:11–26. doi: 10.1016/s0378-1119(97)00307-7. [DOI] [PubMed] [Google Scholar]
- 58.Senn H, Werner B, Messerle B A, Weber C, Traber R, Wüthrich K. Stereospecific assignment of the methyl 1H-NMR lines of valine and leucine in polypeptides by nonrandom 13C labeling. FEBS Lett. 1989;249:113–118. [Google Scholar]
- 59.Shaka A J, Barker P B, Freeman R. Computer-optimized decoupling scheme for wideband applications and low-level operation. J Magnet Reson. 1985;64:547–552. [Google Scholar]
- 60.Smith D R, Doucette-Stamm L A, Deloughery C, Lee H, Dubois J, Aldredge T, Bashirzadeh R, Blakely D, Cook R, Gilbert K, Harrison D, Hoang L, Keagle P, Lumm W, Pothier B, Qiu D, Spadafora R, Vicaire R, Wang Y, Wierzbowski J, Gibson R, Jiwani N, Caruso A, Bush D, Safer H, Patwell D, Prabhakar S, McDougall S, Shimer G, Goyal A, Pietrokovski S, Church G M, Daniels C J, Mao J-I, Rice P, Nölling J, Reeve J N. Complete genome sequence of Methanobacterium thermoautotrophicum ΔH: functional analysis and comparative genomics. J Bacteriol. 1997;179:7135–7155. doi: 10.1128/jb.179.22.7135-7155.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sprott G D, Ekiel I, Patel G B. Metabolic pathways in Methanococcus jannaschii and other methanogenic bacteria. Appl Environ Microbiol. 1993;59:1092–1098. doi: 10.1128/aem.59.4.1092-1098.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sundharadas G, Gilvarg C. Biosynthesis of α,ɛ-diaminopimelic acid in Bacillus megaterium. J Biol Chem. 1967;242:3983–3988. [PubMed] [Google Scholar]
- 63.Szyperski T. Biosynthetically directed fractional 13C-labeling of proteinogenic amino acids. An efficient tool to investigate intermediary metabolism. Eur J Biochem. 1995;232:433–448. doi: 10.1111/j.1432-1033.1995.tb20829.x. [DOI] [PubMed] [Google Scholar]
- 64.Szyperski T. 13C-NMR, MS and metabolic flux balancing in biotechnology research. Q Rev Biophys. 1998;31:41–106. doi: 10.1017/s0033583598003412. [DOI] [PubMed] [Google Scholar]
- 65.Szyperski T, Neri D, Leiting B, Otting G, Wüthrich K. Support of 1H-NMR assignments in proteins by biosynthetically directed fractional 13C-labeling. J Biomol NMR. 1992;2:323–334. doi: 10.1007/BF01874811. [DOI] [PubMed] [Google Scholar]
- 66.Szyperski T, Bailey J E, Wüthrich K. Detecting and dissecting metabolic fluxes using biosynthetic fractional 13C labeling and two-dimensional NMR spectroscopy. Trends Biotechnol. 1996;14:453–459. [Google Scholar]
- 67.Tumbula D L, Teng Q, Bartlett M G, Whitman W B. Ribose biosynthesis and evidence for an alternative first step in the common aromatic amino acid pathway in Methanococcus maripaludis. J Bacteriol. 1997;179:6010–6013. doi: 10.1128/jb.179.19.6010-6013.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Umbarger H E. Amino acid biosynthesis and its regulation. Annu Rev Biochem. 1978;47:533–606. doi: 10.1146/annurev.bi.47.070178.002533. [DOI] [PubMed] [Google Scholar]
- 69.Voet D, Voet J G. Biochemistry. 2nd ed. New York, N.Y: John Wiley & Sons, Inc.; 1995. Amino acid metabolism; pp. 727–784. [Google Scholar]
- 70.Vogel H J. Two modes of lysine synthesis among lower fungi: evolutionary significance. Biochim Biophys Acta. 1960;41:172–174. [Google Scholar]
- 71.Vollbrecht D. Three pathways of isoleucine biosynthesis in mutant strains of Saccharomyces cerevisiae. Biochim Biophys Acta. 1974;362:382–389. doi: 10.1016/0304-4165(74)90231-1. [DOI] [PubMed] [Google Scholar]
- 72.Wehrmann A, Phillipp B, Sahm H, Eggeling L. Different modes of diaminopimelate synthesis and their role in cell wall integrity: a study with Corynebacterium glutamicum. J Bacteriol. 1998;180:3159–3165. doi: 10.1128/jb.180.12.3159-3165.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Westfall H N, Charon N W, Peterson D E. Multiple pathways for isoleucine biosynthesis in the spirochete Leptospira. J Bacteriol. 1983;154:846–853. doi: 10.1128/jb.154.2.846-853.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.White P J. The essential role of diaminopimelate dehydrogenase in the biosynthesis of lysine by Bacillus sphaericus. J Gen Microbiol. 1983;129:739–749. [Google Scholar]
- 75.Wider G, Wüthrich K. A simple experimental scheme using pulsed field gradients for coherence pathway rejection and solvent suppression in phase-sensitive heteronuclear correlation spectra. J Magnet Reson. 1993;102:239–241. [Google Scholar]
- 76.Woese C R, Kandler O, Wheelis M L. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wüthrich K, Szyperski T, Leiting B, Otting G. Biosynthetic pathways of the common proteinogenic amino acids investigated by fractional 13C-labeling and NMR spectroscopy. In: Takai K, editor. Frontiers and new horizons in amino acid research: proceedings of the First Biennial International Conference on Amino Acid Research, Frontiers and New Horizons. Amsterdam, The Netherlands: Elsevier; 1992. pp. 41–48. [Google Scholar]