

Abstract
Cysteine, a thiol-containing amino acid, is critical for the synthesis of sulfur-containing biomolecules that control multiple essential cellular activities. Altered cysteine metabolism has been linked to numerous driver oncoproteins and tumor suppressors, and to malignant traits in cancer. Cysteine can be acquired from extracellular sources or synthesized de novo via the transsulfuration (TSS) pathway. Limited availability of cystine in tumor interstitial fluids raises the possible dependency on de novo cysteine synthesis via TSS. However, the contribution of TSS to cancer metabolism remains highly contentious. Based on recent findings, we provide new perspectives on this critical but understudied metabolic pathway in cancer.
Keywords: Cysteine metabolism, Transsulfuration, Glutathione, Redox homeostasis, Ferroptosis, Cancer
CELLULAR FUNCTIONS OF CYSTEINE AND ITS CRITICAL ROLE IN CANCER
Over the past decade, research in cysteine metabolism has skyrocketed. As described in this review, cysteine has a myriad of cellular functions, including critical roles in cancer cells. Therefore understanding how cells maintain their cysteine pools, either through uptake from the environment or de novo synthesis through the TSS pathway is key for understanding the many functions of this perplexing amino acid. Although most studies in cancer cells have focused on cysteine replenishment by extracellular uptake of its oxidized dipeptide, cystine, recent studies point to de novo cysteine synthesis via TSS as an additional mechanism to retain critical cysteine pools. However, the role of the TSS in cancer cells remains contentious due to a paucity of definitive supportive experimental data. One emerging view is that TSS may have a transient and context-specific role in cancer cell survival and behavior during intermittent periods of extracellular cystine depletion, such in harsh tumor microenvironments. Together, this calls for a reappraisal of the role of the TSS pathway in cancer. In this review, we provide new perspectives on the biology of the TSS pathway and its regulation, and how this might be exploited by transformed cells.
Apart from being a proteogenic amino acid, thiol-containing cysteine is indispensable for synthesis of essential metabolites involved in a myriad of biological processes (Figure 1). Cysteine supplies sulfur for biosynthesis of iron-sulfur (FeS) clusters [1] and coenzyme A (CoA) [2–3], both essential for metabolic enzymes in anabolic and catabolic pathways [1–3]. Cysteine is also a component of the Cys-Gly-Pro-Cys motif of thioredoxins that maintain protein dithiol/disulfide balance across all kingdoms [4–5]. Cysteine is rate-limiting for production of glutathione (GSH), a prominent antioxidant in mammalian cells [1,6–7]. Mitochondrial GSH import is essential for activity of FeS cluster proteins and mitochondrial mRNA translation [8]. GSH dysregulation contributes to many pathological conditions, including neurodegenerative disorders, anemia, aberrant aging, and cancer initiation and progression [7–8]. Cysteine is also a pivotal regulator of ferroptosis, a form of iron-dependent programmed cell death triggered by lipid peroxidation, involved in both health and pathological conditions [9–11], and GSH is a co-factor for the lipid hydroperoxidase, GPX4, which limits ferroptosis [10–11]. Therefore, despite being a conditionally essential amino acid, cysteine is vital for a plethora of pathophysiological processes (Figure 1).
Figure 1.
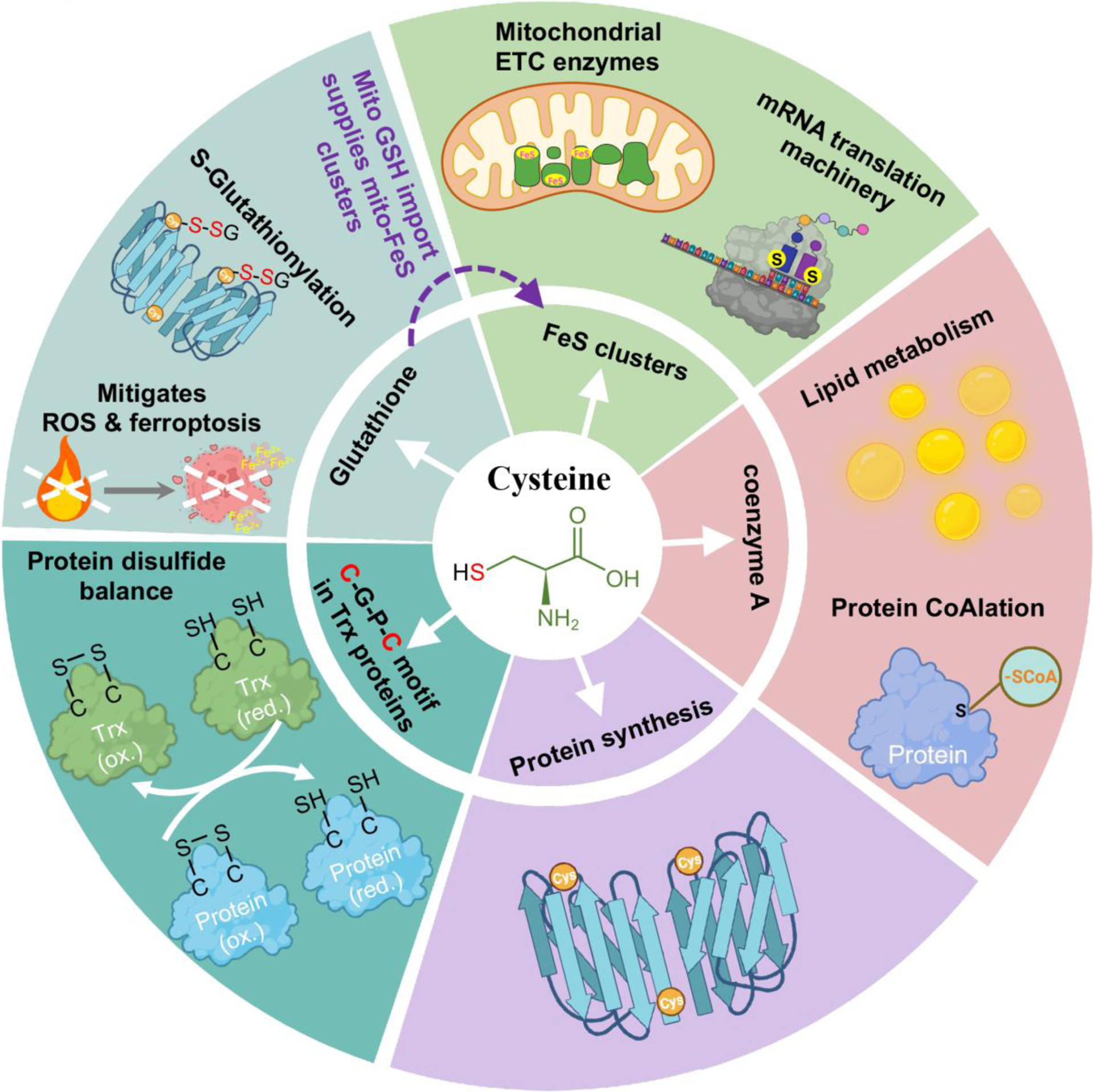
The multifaceted role of cysteine in a myriad of biochemical and biological processes.
Enhanced interest in cysteine metabolism in cancer emanates from landmark discoveries over the past 5–10 years that causally connect oxidative stress and/or GSH biogenesis with cancer biology, including cancer cell stemness [12], anchorage-independent growth [13–14], oncogenesis [15–16], ferroptosis [9,17–18], and metastatic dissemination [19–20]. Several studies have identified augmented uptake of cystine (the oxidized cysteine dimer) in cancer [21–25], directly linking this process to the oncogenic/tumor suppressive functions of oncoproteins such as mutant KRAS and EWS-FLI1, and tumor suppressors such as p53 and BAP1 [21–23,25], as described in Box 1.
Box 1. Augmented cystine uptake in diverse cancers.
Due to oxidizing conditions within the extracellular environment, cysteine is mainly present in an oxidized dipeptide form linked via a disulfide bond, i.e. cystine. The cystine/glutamate antiporter xCT (together with the heavy chain subunit CD98/SLC3A2) is critical for the uptake of cystine in exchange for intracellular glutamate, and cystine is rapidly reduced to cysteine by NADPH intracellularly [6,26–27]. The mutant KRAS oncoprotein and tumor suppressors such as p53 and BAP1 activate or repress xCT expression, respectively, which is critical for their biological function during tumorigenesis [21–23]. Expression of xCT is implicated in the oncogenesis and therapeutic resistance of multiple cancers such as triple-negative breast cancer, glioblastoma, non-small cell lung cancer (NSCLC), pancreatic cancer, colorectal cancer, and hepatocellular carcinoma (HCC) [24]. Certain cancers that lack high xCT expression, such as chronic lymphocytic leukaemia (CLL), rely on alanine-serine-cysteine (ASC) transporters to uptake cysteine released by bone marrow stromal cells in the tumor microenvironment [28]. This alternative source of cysteine supply is utilized for GSH synthesis and ROS (reactive oxygen species) detoxification that are important for CLL cell survival, further corroborating the crucial role of cysteine metabolism in malignant cells.
HOW CANCER CELLS COPE WITH DIMINISHED EXTRACELLULAR CYSTINE SUPPLY
Despite enhanced cysteine requirements in many cancers, an unresolved question is, what happens when the extracellular supply chain of cystine is diminished? Cystine levels in plasma and in particular tumor interstitial fluids are limited compared to many other amino acids [29–31]. Moreover, cystine uptake via the cystine/glutamate antiporter xCT, a predominant route for cystine acquisition (see Box 1), reduces intracellular glutamate pools and increases NADPH consumption for cystine-to-cysteine reduction, which can induce redox crisis and metabolic vulnerabilities in cancer cells [27,31–35]. Alternative pathways for cysteine acquisition in tumor cells are known, such as cysteine salvage from GSH degradation, macropinocytosis and protein scavenging, and de novo cysteine synthesis via the TSS pathway [6]. However, cysteine is one of the least abundant amino acid in proteins due to its high reactivity [36], and therefore proteins may not be a major cysteine source. Although extracellular GSH degradation is an important source of cysteine during organismal development, it represents a small fraction of available extracellular cysteine [6], and its long-term role is unclear in cancer. Similarly, cysteine scavenging from intracellular GSH degradation is likely unsustainable because GSH is essential for redox homeostasis [1,6–7] and for mitochondrial function via FeS clusters [8]. The TSS pathway is largely understudied in cancer, and its importance is still under considerable debate. Recent studies point to activation of this pathway in specific cancers, and demonstrate its importance during tumor progression through enhanced de novo cysteine synthesis. New studies have also identified novel rate-limiting factors for TSS in cancer, and the activity or availability of these factors may underlie the contrasting roles of TSS in different cancers. Moreover, multiple GSH-independent cytoprotective mechanisms under cyst(e)ine starvation involving TSS have been uncovered. Together, these findings call for a reappraisal of the role of the TSS pathway in cancer.
THE TSS PATHWAY
Basic biology and functions of the TSS pathway
The TSS pathway is essential for de novo cysteine synthesis in normal cells. For example, TSS is a major source of cysteine in astrocytes [37], and ~half of cysteine molecules used for GSH production are generated through TSS in hepatic cells [38]. In genetically engineered mice with liver-specific ablation of Thioredoxin reductase-1 (Txnrd1) and Glutathione reductase (Gsr), major downstream effectors of NADPH that support antioxidant pathways, methionine-fueled TSS activity can adequately supply cysteine precursors for GSH synthesis, thereby sustaining cytosolic redox homeostasis [39]. The TSS pathway also generates H2S, a gaseous signaling molecule important for protein sulfhydration, which enhances resistance to oxidative stress and increases lifespan in nematodes, fruit flies, mice, and humans [40–42]. The role of H2S has been covered elsewhere [43–44], and this review will mainly focus on the role of the TSS pathway in de novo cysteine synthesis.
As indicated by the name, transsulfuration involves the transfer of sulfur from methionine to serine through multiple reactions (Figure 2; the flow of different moieties is color-coded). Sulfur transfer is indirect, and Hcy (homocysteine), an intermediate metabolite from the methionine cycle, is the sulfur-containing precursor that is channelled to the TSS pathway. The Hcy-to-cysteine conversion is sequentially catalyzed by two enzymes, Cystathionine β-Synthase (CBS) and Cystathionine γ-Lyase (CGL, or Cystathionase, CTH). Specifically, CBS condenses Hcy and serine into cystathionine, which is then cleaved and hydrolyzed by CTH to form cysteine, alpha-ketobutyrate and NH4+ (Figure 2).
Figure 2. Schematic showing the metabolic pathways that contribute to de novo cysteine synthesis.
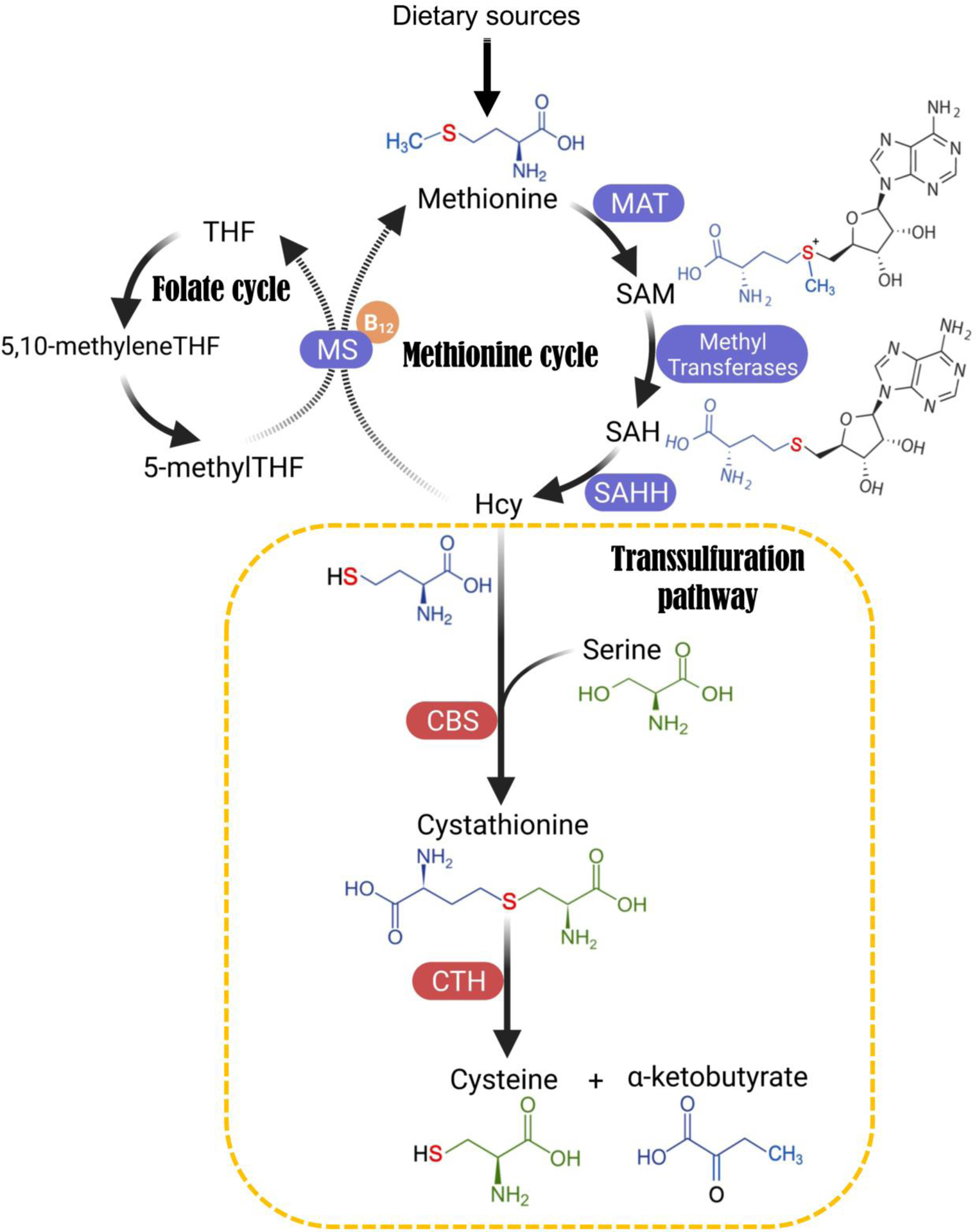
Folate cycle, methionine cycle, and the transsulfuration (TSS) pathway mediate de novo cysteine synthesis. Atoms are colour-coded to indicate their flow in the metabolic reactions. This sulfur transfer process is indirect, and Hcy (homocysteine), an intermediate metabolite from the methionine cycle, is the sulfur-containing precursor that ultimately gets channelled to the TSS pathway. Hcy can either enter the TSS pathway to transfer the sulfur atom to serine, eventually leading to cysteine synthesis, or Hcy is recycled back to methionine using folate cycle metabolite 5-methyl-tetrahydrofolate (5-me-THF) as the methyl donor and vitamin B12 as a cofactor. The Hcy-to-cysteine conversion is again indirect, and is sequentially catalyzed by two enzymes: Cystathionine β-Synthase (CBS) and Cystathionine γ-Lyase (CGL, also named Cystathionase or CTH). Specifically, CBS condenses Hcy and serine into a single molecule called cystathionine, which is then cleaved and hydrolyzed by CTH to form cysteine, alpha-ketobutyrate and NH4+. Abbreviations: B12, Vitamin B12; CBS, Cystathionine beta-synthase; CTH, Cystathionine gamma-lyase; Hcy, Homocysteine; MAT, Methionine adenosyltransferase; MS, Methionine synthase; SAHH, S-Adenosylhomocysteine Hydrolase; SAH, S-Adenosyl homocysteine; SAM, S-Adenosyl methionine; THF, Tetrahydrofolate.
Regulation of the TSS pathway
TSS activity is regulated at multiple levels, including bioavailability of precursors and expression and activity of TSS metabolic enzymes, as described below and in Figure 2.
1. CBS and CTH:
CBS and CTH play pivotal roles in TSS, directly executing the formation and splitting of cystathionine, respectively, leading to transfer of sulfur from methionine to serine to form cysteine (Figure 2). Consistent with the major contribution of TSS to cysteine pools (~50%) in liver [38], both CBS and CTH are highly expressed in liver [45–46], which is controlled by the transcription factor HNF4α (Hepatocyte nuclear factor 4A), key for liver development [47]. In addition to transcriptional control, protein stability of CBS is allosterically activated by the TSS pathway precursor, S-adenosyl methionine (SAM) [48], an abundant methionine cycle metabolite in the liver [38]. In support, defects in SAM-generating enzymes, such as of MAT1A in liver cancer cells, substantially impair CBS expression, rendering those cells susceptible to oxidative stress [48]. In other tissues where basal expression of CBS and CTH is relatively low, cells maintain cysteine and redox homeostasis through TSS activation via transcriptional positive feedback of CBS and CTH when exogeneous cystine is depleted [25,49–53]. This has been documented in normal fibroblasts, striatal cells, and various cancer cells [25,49–53]. CTH can also be stimulated by cell stress, including ER stress, Golgi stress, oxidative stress, and mitochondrial respiratory chain dysfunction [54–55]. ATF4, activated by the PERK/GCN2-eIF2α pathway [50,52–54,56], and the master regulator of redox homeostasis, NRF2 [57–59], are key transcription factors for CBS and CTH expression. Both enzymes are physiologically important, based on mice with Cbs or Cth ablation (see Box 2) [60–62].
Box 2. Pathophysiological roles of key TSS enzymes CBS and CTH.
Cbs−/− mice suffer growth retardation and only survive for about two weeks after birth, and mice also display various abnormalities, including severe hepatopathy, vascular abnormalities, and skeletal deformities [62]. In contrast, Cth−/− mice develop normally, but exhibit acute skeletal muscle atrophy when fed a low cyst(e)ine diet, which leads to lethality due to severe paralysis of the extremities [60–61]. Supplementation of cysteine but not NaHS (an H2S donor) in the drinking water of Cth−/− mice restores plasma cysteine and GSH levels, as well as body weight, supporting animal survival on a low cyst(e)ine diet, underlining the physiological importance of de novo cysteine synthesis by TSS [61]. Although de novo cysteine synthesis is impaired in both Cbs−/− and Cth−/− mice, only Cbs−/− mice display deleterious phenotypes with a standard diet, which is likely due to homocystinuria resulted from CBS deficiency, a more severe disorder than cystathioninuria caused by CTH deficiency [60,63].
Loss of Cth mRNA expression has been causally linked to Huntington’s disease [52–53,64], and mutant Huntingtin (Htt), the pathological driver of this disease, sequesters the transcription factor Specificity protein 1 (SP1), leading to reduced Cth expression [64]. This abnormality drives heightened oxidative stress and neurodegeneration, and cysteine supplementation delays these deleterious effects both in cultured cells and in mouse models [64]. Moreover, pharmacological strategies may also be used to rescue CTH deficiency in Huntington disease cells, e.g. a mild Golgi stress response by subtoxic levels of the ionophore monensin stimulates CTH expression via ATF4 upregulation, which protects those cells from cell death elicited by cystine deprivation [53].
2. Serine:
Serine is a non-essential amino acid but is crucial for TSS, as all carbon and nitrogen atoms in newly synthesized cysteine derive from serine (Figure 2). Thus, stable isotope-tracing using 13C and/or 15N labeled serine is often used to quantify de novo cysteine synthesis [25,50,65–66]. Active de novo serine synthesis correlates with high cysteine metabolism in human cancers [67], and disruption of serine synthesis or exogenous serine depletion decreases GSH pools and exacerbates oxidative stress [68–69]. Under serine depletion, de novo synthesized serine is utilized for GSH rather than nucleotide synthesis, which is critical for counteracting oxidative stress [69]. However, previous studies have not elucidated whether serine-to-cysteine flux actually contributes to GSH pools, focusing instead on serine-derived glycine [68–69]. Thus, although serine is essential for TSS, it remains uncertain if aberrant serine metabolism is “rate-limiting” for cysteine dysregulation in cancer.
3. Methionine cycle metabolites and enzymes:
Methionine, an essential amino acid, is an upstream precursor of TSS as it donates the sulfur atom present in newly synthesized cysteine (Figure 2). Recent studies point to certain metabolites of this cycle that are critical for TSS. Studies in both yeast and human cells reveal that capacity to convert SAM to SAH (S-adenosyl homocysteine), generated during SAM-mediated substrate methylation (e.g. on phospholipids, histones, and amino acids), is a major determinant of TSS activity [50,70]. SAH is the predominant precursor of Hcy (homocysteine) which is then funneled into the TSS pathway [71] (Figure 2). In support, in methionine-replete and cystine-depleted cultures, Hcy supplementation can often fully rescue cell survival [25,50,65], highlighting the critical role of downstream metabolites of the methionine cycle rather than methionine itself for TSS activity.
Hydrolysis of SAH by the enzyme SAHH (S-adenosylhomocysteine hydrolase) is the only source of Hcy in mammals [71]. The SAHH gene is frequently amplified in human malignancies, including cervical and colon cancers [72–73]. Enzymatic activity of SAHH is critical for Hcy generation, TSS activity, GSH synthesis, and resistance to ferroptosis induced by the xCT inhibitor, erastin [74]. Hcy sits at the crossroads between entry into the TSS pathway versus recycling back to methionine upon Hcy re-methylation (Figure 2), utilizing folate-dependent or -independent pathways [54]. Hcy utilization in the methionine cycle is linked to generation of SAM, a predominant reactive methyl donor [75]. The balance between those two routes of Hcy usage determines the activity of TSS versus methionine regeneration. In support, deficiency of CBS, the first enzyme in the TSS pathway, tilts the balance toward methionine recycling and is associated with hypermethioninemia [76]. In contrast, preferential Hcy usage in TSS is recently reported in cancer cells [77]. In breast cancer, PIK3CA mutations direct Hcy flux towards TSS to meet cellular cysteine demands, as these mutants transcriptionally suppress xCT expression and inhibit xCT activity via mTORC2-mediated phosphorylation [77–78]. This shifts the balance of Hcy usage away from methionine regeneration, thereby creating a methionine-dependency in these cancer cells [77].
CYTOPROTECTIVE FUNCTIONS OF THE TSS PATHWAY IN CANCER
In support of the role of cysteine in critical biological processes (Figure 1), dependence on cyst(e)ine supply is supported by distinct tumor models, whereby extracellular cyst(e)ine depletion by cyst(e)inase effectively blocks tumor growth both in vitro and in vivo [2,16]. Moreover, to circumvent the limited supply of cystine in plasma and tumor interstitial fluids in rapidly growing tumors [29–31], tumor cells enhance cystine uptake by upregulating the xCT cystine transporter [21–24, and Box 1]. However, it is reasonable to speculate that by continuously drawing on a limited supply of extracellular cystine, particularly during rapid tumor growth or metastatic dissemination, where cells experience high oxidative stress [19], tumor cells may become increasingly reliant on de novo cysteine synthesis via the TSS pathway. Having said this, the requirement for TSS might be tempered in vivo by reduced cell division rates compared with in vitro cultures, a potential caveat of this notion. As mentioned, while a role for TSS in malignant cells is not universally accepted, several levels of evidence support the importance of TSS, which will now be reviewed.
Activation and cytoprotective functions of the TSS pathway in cancer
Cystine deprivation induces feedback upregulation of CBS and CTH in various tumor types, such as neuroblastoma, glioblastoma, melanoma, fibrosarcoma, Ewing sarcoma, breast cancer, colorectal cancer, pancreatic ductal adenocarcinoma, and ovarian cancer [25,57,79–80,51,65]. In HCC cells, post-translational activation of CBS under TNFα-induced oxidative stress enhances TSS activity, mediated by a proteolytically cleaved and highly active form of CBS, enhancing cystathionine and GSH production [81]. In breast cancer, transcriptional and posttranslational inhibition of xCT by oncogenic PI3KCA enhances TSS activity, promoting de novo cysteine synthesis [77], and recent studies found that CTH is constitutively expressed in clear cell carcinoma of the ovary and endometrium [82–83]. After prolonged treatment the xCT inhibitor erastin [9], ovarian cancer cells upregulate CBS, promoting TSS activity and GSH pools [57]. CBS depletion re-sensitizes the resistant cells to erastin-induced ferroptosis, and ectopic CBS expression protects the parental (erastin sensitive) cells by enhancing TSS flux and blocking ferroptosis [57]. In Ewing sarcoma, IL1RAP (IL1 receptor accessory protein), a cell surface protein transcriptionally induced by driver EWS-FLI1 and EWS-ERG oncoproteins of this disease, not only enhances xCT uptake of cystine, but also promotes CTH transcription and TSS activity, thereby conferring survival under cystine deprivation [25].
Cytoprotection by TSS is further exemplified by supplementation with cystathionine or Hcy, which both restore growth in cystine-free media in diverse tumor types [25,50,65,74,77]. Accordingly, enhanced Hcy supply from the methionine cycle confers ferroptosis resistance during cystine depletion in NSCLC cells [74]. Mechanistically, SAH-to-Hcy conversion mediated by SAHH is augmented by PARK7, which promotes SAHH activity [74]. Moreover, Hcy and cystathionine each increase production of cysteine-containing metabolites such as γ-glutamyl-cysteine and GSH [25,50,65]. Stable isotope tracing using 13C- and/or 15N-serine and 13C-methionine further demonstrates activity of the TSS pathway in neuroblastoma, Ewing sarcoma, breast, and pancreatic cancers [25,50,65,77], which can be blocked by depletion of CBS or CTH [25,50].
Pharmacological inhibition of CBS or CTH induces tumor ferroptosis
Cysteinyl-tRNA Synthetase is a newly identified suppressor of cystine starvation-induced ferroptosis, whereby its depletion upregulates CBS and CTH, as well as cystathionine and cysteine pools, rendering cells ferroptosis resistant [49]. Blocking the TSS pathway by CBS knockdown or the CTH inhibitor (propargylglycine; PAG) each markedly enhance ferroptosis in tumor cells [49]. In colorectal (SW480) and breast cancer (MDA-MB-468) cells that are resistant to cystine starvation, pharmacological inhibition of CTH by beta-cyano-L-Alanine specifically blocked cell growth under cystine starvation, while no effect was observed in complete growth media [65]. A similar observation was made in HeLa cells via CTH inhibition by PAG [84]. Moreover, a newly identified CBS inhibitor (CH004) blocks CBS and TSS activity, leading to Hcy accumulation and reduced H2S formation, inducing ferroptosis in HCC cells [80], highlighting the dependency of these cells on the TSS pathway.
Evidence supporting the role of the TSS pathway in vivo
The importance of TSS in cancer has also been confirmed in vivo. CBS depletion severely blunted neuroblastoma xenograft growth and decreased cystathionine and GSH pools in the tumors that eventually did form [50]. In prostate cancer, high CTH expression correlates with advanced clinical stage and decreased survival in patients, and CTH depletion in tumor xenografts suppressed tumor growth and metastasis to lymph nodes and bone, while overexpression of CTH promoted these malignant traits [79]. In Ewing sarcoma, high CTH expression is associated with poor prognosis, and CTH knockdown reduced primary tumor growth and local invasion, and dramatically reduced spontaneous lung metastasis, which was rescued by CTH re-expression [25]. Collectively, these findings demonstrate the biochemical and functional importance of de novo cysteine synthesis in redox stress adaptation, cell survival, and malignant progression in cancer.
EVIDENCE AGAINST A ROLE FOR THE TSS PATHWAY IN CERTAIN CANCERS
Despite the above evidence, TSS dependency is not a universal feature of transformed cells. In a large panel of NSCLC cell lines, short-term cystine deprivation almost completely depleted intracellular cysteine pools in all lines tested, and most lines underwent ferroptosis, arguing against compensatory de novo cysteine synthesis to maintain cell survival [66]. Moreover, stable isotope tracing of starved cells with 13C3-serine revealed minimal 13C-cysteine (M+3) and 13C-GSH (M+3) pools in both sensitive and resistant cells, arguing against a role for TSS activity in these cells [66]. In colorectal, breast, and pancreatic cancer cell lines, cystine deprivation in vitro also led to ~100% intracellular cysteine depletion in all lines tested, although several lines were resistant to cystine starvation [65]. Thus, exogenous cyst(e)ine supply appears to be essential in these cells. Moreover, pharmacological inhibition of xCT by erastin, Sorafenib, or analogs induced close to 100% cell death in diverse cancer cell lines [51,85–87]. Extracellular cyst(e)ine depletion alone by cyst(e)inase, an engineered enzyme that depletes cyst(e)ine both in vitro cultures and in vivo, was highly effective in inducing cell death and blocking tumor growth in prostate, breast, and pancreatic cancers [2,16]. These findings imply that exogenous cyst(e)ine supply instead of de novo synthesis is critical, and that TSS may not be important in these malignancies. Certain cancer cells with epigenetic silencing of CBS have diminished TSS activity, including gastric and colorectal cancers [88]. CBS induces apoptosis and suppresses HCC tumor growth in vivo, and reduced CBS expression correlates with unfavorable prognosis in HCC patients, although a causative role for CBS loss remains unproven [47,89]. De novo cysteine synthesis did not mediate the tumor suppressor function of CBS in HCC, but instead suppressed IL-6/STAT3 signaling and blocked epithelial-to-mesenchymal transition (EMT) [47,89]. Taken together, these findings starkly contrast with those of the previous section, suggesting that the role of the TSS in tumors is highly context specific.
FACTORS INFLUENCING EXPERIMENTAL ASSESSMENT OF THE TSS PATHWAY
Several factors may contribute to the apparent dichotomy described in the above sections. Complicating the assessment of TSS activity, acute cystine depletion can markedly increase ROS accumulation [25,50,52,65]. High ROS in turn can oxidize cysteine residues of multiple core components of the mRNA translation machinery to block global translation [90], which may impair proper feedback activation of the TSS pathway. Thus, we speculate that under high ROS load during experimental acute cystine deprivation in vitro (e.g. 0μM cystine), rapidly proliferating cancer cells may not have sufficient time or fitness to execute meaningful feedback activation of the TSS pathway. Therefore, measuring de novo cysteine synthesis under complete cystine starvation might be prone to artificially negative results. To circumvent these challenges, culture media supplemented with β-mercaptoethanol is often used to preserve redox homeostasis during de novo cysteine analysis in cystine-free cultures [25,50]. This strategy has facilitated the evaluation of TSS activities in other cancers, demonstrating CBS- and CTH-dependent TSS activity as validated by gene depletion or rescue of these enzymes [25,50]. However, it should be noted that the use of β-mercaptoethanol has its own caveats, including potential disruption of disulfides within the cell, release of lysosomal cystine as cysteine into the cystosol, and loss of persulfides from proteins. Thus, isotope tracing such as with 13C-serine is necessary to properly assess de novo cysteine synthesis [25,50] and increase the reliability of results, if β-mercaptoethanol is used to overcome cystine deprivation-induced high ROS.
GSH-INDEPENDENT CYTOPROTECTIVE MECHANISMS
Several other newly described mechanisms can also explain why some cancer cells do not rely on TSS during cyst(e)ine depletion, including several TSS-independent mechanisms that confer resistance versus sensitivity to cystine starvation [65–66]. Firstly, a study characterized a GSH-independent mechanism that promotes cell survival under acute cystine deficiency. They found that glutamate accumulation (and resulting glutamate stress) leads to cell death upon cystine starvation [66]. Glutamate consumption via γ-glutamyl-peptide synthesis (rather than a lack of GSH synthesis due to cysteine deficiency) protects cells from cystine starvation-induced ferroptosis [66] (Figure 3). Non-canonical activity of the Glutamate-Cysteine Ligase Catalytic subunit (GCLC), which generates γ-glutamyl-cysteine during GSH synthesis, mediates γ-glutamyl-peptide synthesis to alleviate glutamate stress during cystine starvation [66]. Secondly, another study identified that ROS generated by the polyamine synthesis pathway downstream of the methionine cycle triggers cell death upon cystine starvation, even though methionine can donate also sulfur atoms to the TSS pathway [65] (Figure 3). Blocking key enzymes of during spermidine and spermine synthesis, such as Adenosylmethionine decarboxylase 1 (AMD1), Polyamine oxidase (PAOX), and Spermine oxidase (SMOX), alleviated ROS levels and protected cells from cystine starvation-induced cell death [65] (Figure 3). This study concludes that this mechanism, rather than a requirement for the TSS activity, correlates with sensitivity to cystine starvation in cancer cells.
Figure 3. Mechanisms in the TSS pathway that regulate cystine deprivation-induced ferroptosis independent of de novo cysteine synthesis and GSH.

A non-canonical function of GCLC, an enzyme that is known to catalyze γ-glutamyl-cysteine formation during GSH synthesis, mediates γ-glutamyl-peptide synthesis upon cystine deprivation. This non-canonical mechanism alleviates glutamate stress and prevents cystine starvation-induced ferroptosis in cancer cells. Genetic or pharmacological blockade of GCLC sensitizes cancer cells to cystine deficiency-induced ferroptosis. In contrast, activation of the polyamine synthesis pathway downstream of methionine promotes ROS (H2O2) production, which triggers cell death upon cystine starvation. Blocking each of the key enzymes in the polyamine synthesis pathway, such as AMD1, PAOX and SMOX, alleviates ROS levels and protects cells from cystine starvation-induced ferroptosis. Abbreviations: GCLC, Glutamate-cysteine ligase catalytic subunit; GSS, Glutathione synthetase; AMD1, Adenosylmethionine decarboxylase 1; PAOX, Polyamine oxidase; SMOX, Spermine oxidase; dcSAM, decarboxylated S-adenosylmethionine.
Other studies have identified mechanisms harnessing coenzyme Q10 (CoQ10) antioxidants to prevent lipid peroxidation and thus ferroptosis when the GSH system is compromised [91–93] (Figure 4). Specifically, apoptosis-inducing factor mitochondria-associated 2 (AIFM2/FSP1) [91–92] and dihydroorotate dehydrogenase (DHODH) [93] reduce ubiquinol (CoQ10) in the cytosol and mitochondria respectively, reducing lipid peroxidation and ferroptosis when the GSH-GPX4 defense system is inhibited (Figure 4). Genetic screens identified the tetrahydrobiopterin (BH4) biosynthesis pathway as another mechanism preventing ferroptosis when the GSH-GPX4 system is blocked [94]. By trapping radicals, BH4 protects lipid membranes from autoxidation, and enzymes promoting BH4 production including GCH1, PTS, and SPR are important cytoprotective enzymes in this context [94] (Figure 4). Phospholipase A2 group VI (iPLA2β) suppresses ferroptosis when the GSH/GPX4 system is blocked [95–96], mediated by hydrolysis of peroxidized phospholipids that trigger ferroptosis (Figure 4). Finally, genetic and pharmacologic screens revealed that deubiquitinating enzymes (DUBs) act as cytoprotective factors when GSH is depleted in breast cancer cells. Mechanistically, DUBs mitigate GSH depletion-induced buildup of ubiquitinated proteins that cause proteotoxic and ER stress, increasing tolerance to GSH depletion in some cancer cell types [97]. Therefore, although not all cancer cells express high levels of some of these enzymes, or produce BH4, we speculate that cells that can utilize these mechanisms might bypass TSS pathway dependency to withstand cystine starvation or diminished GSH production (Figure 4). Together, these mechanisms and accompanying factors may explain the lack of contribution of the TSS pathway in certain cancers such as NSCLC and pancreatic cancers [65–66].
Figure 4. Alternative cytoprotective mechanisms exploited by cancer cells when cyst(e)ine-mediated GSH antioxidant system is compromised.
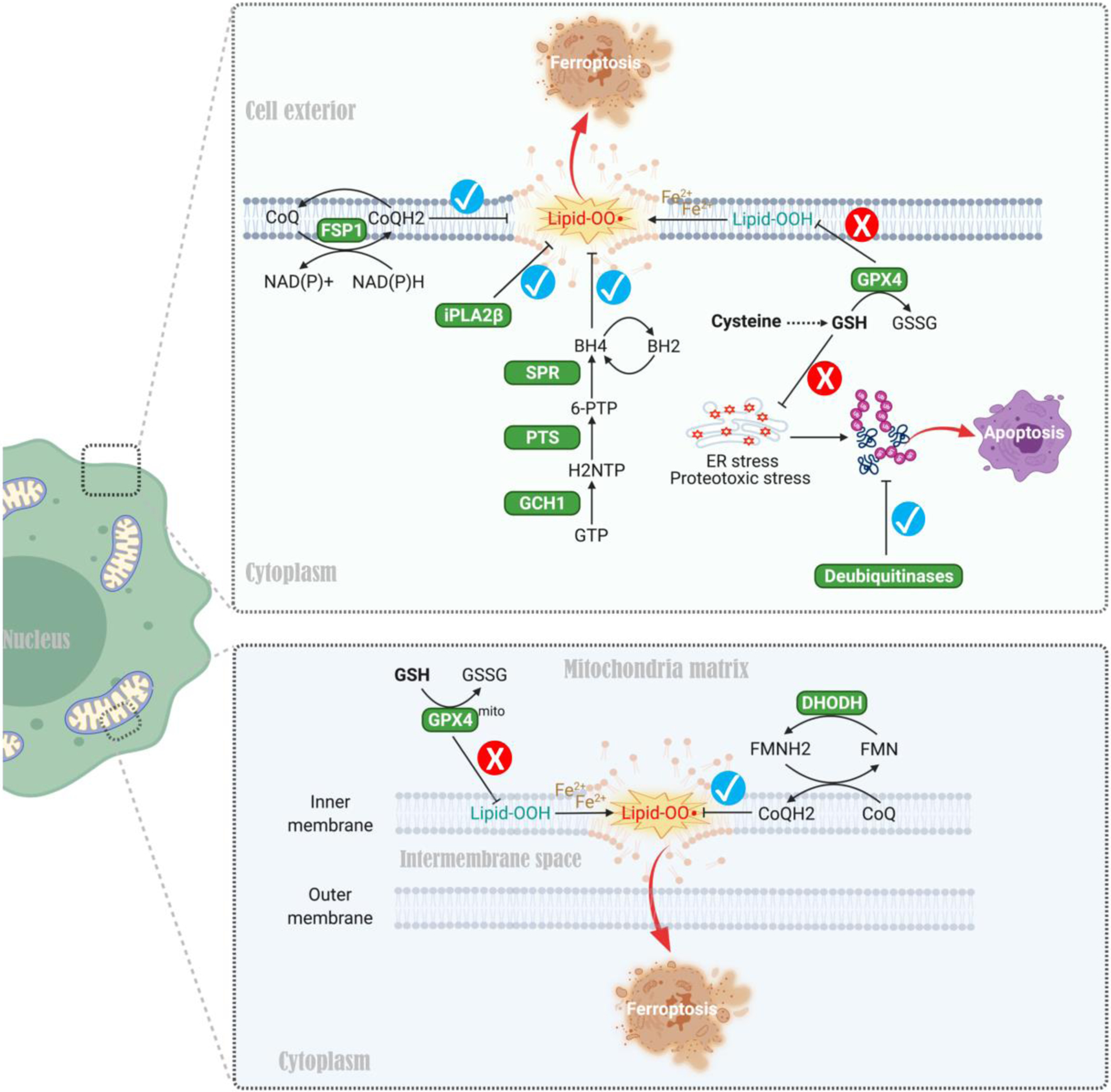
First, apoptosis-inducing factor mitochondria-associated 2 (AIFM2/FSP1) and dihydroorotate dehydrogenase (DHODH) promote reduction of ubiquinone to ubiquinol (two forms of CoQ10) in the cytosol and mitochondria respectively, thereby mitigating lipid peroxidation and ferroptosis when the GSH-GPX4 system is inhibited. Second, radical-trapping antioxidant BH4 (tetrahydrobiopterin) protects lipid membranes from autoxidation, and enzymes promoting BH4 production such as GCH1, PTS, and SPR are exploited by cancer cells for survival when the cytoprotective GSH-GPX4 axis is blocked. Third, phospholipase A2 group VI (iPLA2β) promotes the hydrolysis of peroxidized phospholipids, thereby preventing ferroptosis when the GSH/GPX4 system is disabled. Finally, deubiquitinating enzymes (DUBs) mitigate GSH depletion-induced buildup of ubiquitinated proteins that cause proteotoxic stress and ER stress, thereby increasing tolerance to GSH depletion in various cancers. These mechanisms may facilitate cancer cells bypassing the TSS pathway to withstand cystine starvation or diminished GSH production. Abbreviations: FSP1, ferroptosis suppressor protein 1; DHODH, dihydroorotate dehydrogenase; CoQ, coenzyme Q; GPX4, Glutathione peroxidase 4; FMN, flavin mononucleotide; FMNH2, 1,5-dihydro flavin mononucleotide (reduced form); GCH1, GTP cyclohydrolase I; PTS, 6-pyruvoyltetrahydropterin synthase; SPR, sepiapterin reductase; iPLA2β, phospholipase A2 group VI; BH4, tetrahydrobiopterin; BH2, oxidized tetrahydrobiopterin; NH2TP, 7,8-dihydroneopterin triphosphate; 6-PTP, 6-pyruvoyltetrahydropterin.
DE NOVO CYSTEINE SYNTHESIS: NECESSARY BUT INSUFFICIENT FOR CANCER CELL GROWTH?
Although some cancer cell lines can survive under cystine starvation, to our knowledge, none have been shown to proliferate robustly under such conditions, even when TSS activity is functionally linked to cellular viability [25,50,57,65]. Although the TSS pathway is critical for cell survival in neuroblastoma, Ewing sarcoma, and erastin-resistant ovarian cancer cells when extracellular cystine is limited, proliferation was not observed under these conditions [25,50,57]. In Ewing sarcoma cells, despite the crucial role of CTH-mediated de novo cysteine synthesis in ferroptosis resistance during partial cystine deprivation, complete cystine starvation was still highly cytotoxic [25]. These observations argue that while de novo cysteine synthesis via TSS is necessary for cell survival in some cancers during cysteine crisis, it is insufficient for sustaining growth on its own. Supporting this notion, the TSS substrate SAH, generated by the GNMT enzyme, is reduced in some cancers [50], which may limit de novo cysteine synthesis. Indeed, overexpressing GNMT to promote SAH production, or supplementing with Hcy, each restored cell survival and proliferation upon cystine starvation [25,50,65]. Moreover, PARK7, a redox-sensitive chaperone and oxidative stress sensor that augments SAHH activity, is critical for TSS activity and ferroptosis resistance in erastin-treated NSCLC cells [74]. These experiments were performed under methionine and serine replete conditions, each key for de novo cysteine synthesis, further corroborating that intermediary metabolites such as SAH and Hcy (and corresponding enzymes), are likely rate-limiting for TSS capacity, at least in some cancer cells.
Taken together, the above studies suggest that de novo cysteine synthesis is likely contextual as a transient but highly plastic emergency stress response program in some cancer cell types, such as when extracellular cystine levels become limited or uptake is blocked. This further suggests that such tumor cells might be able to switch back and forth between cystine import and TSS activity for maintaining cysteine pools, depending on acute cystine availability. It is tempting to speculate that cancer cells that are able to exploit this form of TSS-based plasticity, despite being unsustainable long-term, have a survival advantage during the intermittent stresses imposed by the metastatic cascade or in nutrient-scarce microenvironments during tumor progression in vivo, which is likely more variable than the all-or-none cystine starvation imposed in vitro.
CONCLUDING REMARKS
The role of de novo cysteine synthesis via TSS in cancer remains controversial and understudied. Pre-clinical studies demonstrate the potent effectiveness of targeting cysteine metabolism in various cancer models, including systemic cysteine depletion using an engineered cyst(e)inase [2,16], or pharmacological blockade of xCT with erastin and analogs, sulfasalazine, and a newly identified agent HG106 [9,51,98–100]. These findings support the dominant role of cystine uptake in maintaining tumor growth, and suggest that de novo cysteine synthesis cannot fully compensate for loss of exogenous cyst(e)ine in some cancers. Nevertheless, accumulating evidence in other cancer types indicates that TSS enzymes are highly responsive to cystine deprivation and are functionally important for oncogenesis in vivo. Methods currently used to evaluate TSS activity (i.e. under cystine depletion which induces high ROS) are problematic. β-mercaptoethanol supplementation to facilitate redox homeostasis, although with its own caveats, offers a partial solution [25,50]. While isotope tracing can increase the reliability of such assays [25,50], an exploration of alternative approaches is warranted for future studies of de novo cysteine synthesis. Despite the demonstrated role of TSS in certain cancers, de novo synthesis alone does not appear to support long term tumor cell proliferation. This, as stated in the Outstanding questions, further studies are needed to understand if TSS represents a transient and highly plastic mechanism for cytoprotection under short-term cystine limitation and oxidative stress. This mechanism may function as a “backup plan” for maintaining cysteine pools and redox homeostasis in harsh tumor microenvironments, where nutrient availability can be unstable, or during specific phases of the metastatic cascade during which high oxidative stress can occur [19]. Efforts are also warranted to determine if other cytoprotective mechanisms that are independent of GSH (which is highly dependent on cysteine supply), drive the apparent lack of contribution of TSS in certain cancers. Finally, it should be noted that despite the remarkable efficacy of reducing exogeneous cyst(e)ine with cyst(e)inase in tumor models, tumor growth was not fully curbed in these studies [2,16,98–100]. Further studies are necessary to determine if TSS-mediated de novo cysteine synthesis drove the residual (and sometimes considerable) tumor growth when the cyst(e)ine supply was limited in these models. In conclusion, addressing remaining gaps in knowledge is critical for effectively targeting cysteine metabolism including TSS in cancer and possibly other disorders, as a deeper understanding of this pathway is crucial for unravelling effective therapeutic avenues.
Outstanding Questions.
Is de novo cysteine synthesis via the TSS pathway a minor player or a critical but underappreciated process in cancer? Is this tumor type specific or does it depend on the genetic/metabolic state of tumors?
Is de novo cysteine synthesis necessary but insufficient for cell growth in cancer? The current experimental evidence suggests that the TSS pathway is likely a transient but highly plastic emergency stress response program under short-term cystine crisis (and the associated oxidative stress).
What are the relative contributions of de novo cysteine synthesis versus cyst(e)ine uptake from exogenous sources, e.g. tumor interstitial fluids and stromal or other cells in the tumor microenvironment?
Does de novo cysteine synthesis via the TSS pathway drive the residual (and sometimes considerable) tumor growth upon limiting the cyst(e)ine supply in reported various tumor models? If so, can simultaneous inhibition of cyst(e)ine supply and de novo cysteine synthesis synergistically eradicate tumors?
Under cyst(e)ine starvation and the high oxidative stress associated with this condition, cancer cells can exploit other cytoprotective mechanisms independent of antioxidant GSH (that is highly dependent on cysteine supply), do these mechanisms drive the lack of contribution of the TSS pathway in certain cancers?
Highlights.
Cysteine is critical for synthesis of sulfur-containing biomolecules that control essential cellular activities, such as redox homeostasis, mitochondrial electron transport chain reactions, and the mRNA translation machinery.
During oncogenesis, cystine uptake by the xCT transporter is controlled by driver oncoproteins and tumor suppressors. However, cyst(e)ine levels are limited in tumor interstitial fluids, creating a potential dependency on de novo cysteine synthesis via the TSS pathway.
The contribution of de novo cysteine synthesis in cancer cells is highly contentious.
Novel rate-limiting factors for the TSS pathway and GSH-independent cytoprotective mechanisms have been identified in cancer cells under cystine deprivation.
De novo cysteine synthesis is likely a transient but highly plastic emergency stress response mechanism in cancer cells that encounter intermittent cystine deficiency in vivo.
Acknowledgements:
We thank the funding support from the NIH U54 Pediatric Immunotherapy Discovery and Development Network (1U54CA232568-01), a Stand Up 2 Cancer Phillip A. Sharp Innovation in Collaboration Award (SU2C-AACR-PS-33), and a St. Baldrick’s Foundation/Martha’s Better Ewing Sarcoma Treatment (BEST) Grant (#663113). Stand Up 2 Cancer (SU2C) is a division of the Entertainment Industry Foundation, and research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. H.-F. Zhang is funded by a fellowship from the Canadian Institutes of Health Research (#415377) and a trainee award from the Michael Smith Foundation for Health Research partnered with the Lotte and John Hecht Memorial Foundation (#18569).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: The authors declare no competing interests.
REFERENCES
- 1.Poltorack CD and Dixon SJ (2022) Understanding the role of cysteine in ferroptosis: progress & paradoxes. FEBS J 289, 374–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Badgley MA et al. (2020) Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368, 85–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gout I (2018) Coenzyme A, protein CoAlation and redox regulation in mammalian cells. Biochem. Soc. Trans 46, 721–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghareeb H et al. (2020) The Thioredoxin System: A Promising Target for Cancer Drug Development. Chemistry 26, 10175–10184 [DOI] [PubMed] [Google Scholar]
- 5.Harris IS et al. (2015) Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27, 211–22 [DOI] [PubMed] [Google Scholar]
- 6.Combs JA, and DeNicola GM (2019) The Non-Essential Amino Acid Cysteine Becomes Essential for Tumor Proliferation and Survival. Cancers (Basel) 11,678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allocati N et al. (2018) Glutathione transferases: substrates, inihibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis 7, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y et al. (2021) SLC25A39 is necessary for mitochondrial glutathione import in mammalian cells. Nature 599, 136–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dixon SJ et al. (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stockwell BR et al. (2020) Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol 30, 478–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen X et al. (2021) Organelle-specific regulation of ferroptosis. Cell Death Differ 28, 2843–2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diehn M et al. (2009) Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458, 780–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schafer ZT et al. (2009) Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461, 109–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang L et al. (2016) Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 532,255–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeNicola GM et al. (2011) Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475,106–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cramer SL et al. (2017) Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med 23, 120–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alvarez SW et al. (2017) NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 551, 639–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang WS et al. (2014) Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piskounova E et al. (2015) Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le Gal K et al. (2015) Antioxidants can increase melanoma metastasis in mice. Sci Transl Med 7, 308re8. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y et al. (2018) BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat. Cell Biol 20, 1181–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang L et al. (2015) Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim JKM et al. (2019) Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc. Natl. Acad. Sci. U. S. A 116, 9433–9442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J et al. (2020) xCT: A Critical Molecule That Links Cancer Metabolism to Redox Signaling. Mol Ther 28, 2358–2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang H-F. et al. (2021) Proteomic screens for suppressors of anoikis identify IL1RAP as a promising surface target in Ewing sarcoma. Cancer Discov 11, 2884–2903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Conrad M and Sato H (2012) The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-): cystine supplier and beyond. Amino Acids 42, 231–46 [DOI] [PubMed] [Google Scholar]
- 27.Liu X et al. (2020) Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat. Cell Biol 22, 476–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W et al. (2012) Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat. Cell Biol 14, 276–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muir A et al. (2017) Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife 6, e27713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kamphorst JJ et al. (2015) Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res 75, 544–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sullivan MR et al. (2019) Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 8, e44235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shin CS et al. (2017) The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nat. Commun 8, 15074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koppula P et al. (2017) The glutamate/cystine antiporter SLC7A11/xCT enhances cancer cell dependency on glucose by exporting glutamate. J. Biol. Chem 292, 14240–14249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sayin VI et al. (2017) Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. Elife 6, e28083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.LeBoeuf SE et al. (2020) Activation of Oxidative Stress Response in Cancer Generates a Druggable Dependency on Exogenous Non-essential Amino Acids. Cell Metab 31, 339–350.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bak DW et al. (2019) Cysteine reactivity across the subcellular universe. Curr. Opin. Chem. Biol 48, 96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McBean GJ (2017) Cysteine, Glutathione, and Thiol Redox Balance in Astrocytes. Antioxidants (Basel) 6, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mosharov E et al. (2000) The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by the transsulfuration pathway and its regulation by redox changes. Biochemistry 39, 13005–11 [DOI] [PubMed] [Google Scholar]
- 39.Eriksson S et al. (2015) Dietary methionine can sustain cytosolic redox homeostasis in the mouse liver. Nat. Commun 6, 6479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kabil H et al. (2011) Increased transsulfuration mediates longevity and dietary restriction in Drosophila. Proc. Natl. Acad. Sci. U. S. A 108, 16831–16836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hine C et al. (2015) Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell 160, 132–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zivanovic J et al. (2019) Selective Persulfide Detection Reveals Evolutionarily Conserved Antiaging Effects of S-Sulfhydration. Cell Metab 30, 1152–1170.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Filipovic MR et al. (2018) Chemical Biology of H(2)S Signaling through Persulfidation. Chem. Rev 118, 1253–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kimura H (2020) Signalling by hydrogen sulfide and polysulfides via protein S-sulfuration. Br J Pharmacol 177, 720–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ishii I et al. (2004) Murine cystathionine gamma-lyase: complete cDNA and genomic sequences, promoter activity, tissue distribution and developmental expression. Biochem. J 381, 113–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Namekata K et al. (2004) Abnormal lipid metabolism in cystathionine beta-synthase-deficient mice, an animal model for hyperhomocysteinemia. J Biol. Chem 279, 52961–9 [DOI] [PubMed] [Google Scholar]
- 47.Xu Q et al. (2020) HNF4alpha regulates sulfur amino acid metabolism and confers sensitivity to methionine restriction in liver cancer. Nat. Commun 11, 3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prudova A et al. (2006) S-adenosylmethionine stabilizes cystathionine beta-synthase and modulates redox capacity. Proc. Natl. Acad. Sci. U. S. A 103, 6489–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hayano M et al. (2016) Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ 23, 270–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu J et al. (2019) Transsulfuration Activity Can Support Cell Growth upon Extracellular Cysteine Limitation. Cell Metab 30, 865–876.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dixon SJ et al. (2014) Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3, e02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sbodio JI et al. (2016) Transcriptional control of amino acid homeostasis is disrupted in Huntington’s disease. Proc. Natl. Acad. Sci. U. S. A 113, 8843–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sbodio JI et al. (2018) Golgi stress response reprograms cysteine metabolism to confer cytoprotection in Huntington’s disease. Proc. Natl. Acad. Sci. U. S. A 115, 780–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sbodio JI et al. (2019) Regulators of the transsulfuration pathway. Br. J. Pharmacol 176, 583–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bao XR et al. (2016) Mitochondrial dysfunction remodels one-carbon metabolism in human cells. Elife 5, e10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dickhout JG et al. (2012) Integrated stress response modulates cellular redox state via induction of cystathionine γ-lyase: cross-talk between integrated stress response and thiol metabolism. J. Biol. Chem 287, 7603–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu N et al. (2020) Activation of the reverse transsulfuration pathway through NRF2/CBS confers erastin-induced ferroptosis resistance. Br. J. Cancer 122, 279–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martín JA et al. (2007) Oxidative stress as a signal to up-regulate gamma-cystathionase in the fetal-to-neonatal transition in rats. Cell Mol. Biol. (Noisy-le-grand) 53, Suppl:OL1010–7 [PubMed] [Google Scholar]
- 59.Hassan MI et al. (2012) Platelet-derived growth factor-BB induces cystathionine γ-lyase expression in rat mesangial cells via a redox-dependent mechanism. Br. J. Pharmacol 166, 2231–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ishii I et al. (2010) Cystathionine gamma-Lyase-deficient mice require dietary cysteine to protect against acute lethal myopathy and oxidative injury. J. Biol. Chem 285, 26358–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mani S et al. (2011) A critical life-supporting role for cystathionine gamma-lyase in the absence of dietary cysteine supply. Free Radic. Biol. Med 50, 1280–7 [DOI] [PubMed] [Google Scholar]
- 62.Kruger WD (2017) Cystathionine β-synthase deficiency: of mice and men. Mol. Genet. Metab 121, 199–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zaric BL et al. (2019) Homocysteine and Hyperhomocysteinaemia. Curr. Med. Chem 26, 2948–2961 [DOI] [PubMed] [Google Scholar]
- 64.Paul BD et al. (2014) Cystathionine γ-lyase deficiency mediates neurodegeneration in Huntington’s disease. Nature 509, 96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang T et al. (2020) Polyamine pathway activity promotes cysteine essentiality in cancer cells. Nat. Metab 2, 1062–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kang YP et al. (2021) Non-canonical Glutamate-Cysteine Ligase Activity Protects against Ferroptosis. Cell Metab 33, 174–189.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mehrmohamadi M et al. (2014) Characterization of the usage of the serine metabolic network in human cancer. Cell Rep 9, 1507–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang T et al. (2018) Disruption of De Novo Serine Synthesis in Muller Cells Induced Mitochondrial Dysfunction and Aggravated Oxidative Damage. Mol. Neurobiol 55, 7025–7037 [DOI] [PubMed] [Google Scholar]
- 69.Maddocks OD et al. (2013) Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493, 542–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ye C et al. (2017) A Metabolic Function for Phospholipid and Histone Methylation. Mol Cell 66, 180–193.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chiang PK et al. (1996) S-Adenosylmethionine and methylation. FASEB J 10, 471–80 [PubMed] [Google Scholar]
- 72.Scotto L et al. (2008) Identification of copy number gain and overexpressed genes on chromosome arm 20q by an integrative genomic approach in cervical cancer: potential role in progression. Genes Chromosomes Cancer 47, 755–65 [DOI] [PubMed] [Google Scholar]
- 73.Loo LW et al. (2013) Integrated analysis of genome-wide copy number alterations and gene expression in microsatellite stable, CpG island methylator phenotype-negative colon cancer. Genes Chromosomes Cancer 52, 450–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cao J et al. (2020) DJ-1 suppresses ferroptosis through preserving the activity of S-adenosyl homocysteine hydrolase. Nat. Commun 11, 1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ducker GS and Rabinowitz JD (2017) One-Carbon Metabolism in Health and Disease. Cell Metab 10, 27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stabler SP et al. (2002) Elevated plasma total homocysteine in severe methionine adenosyltransferase I/III deficiency. Metabolism 51, 881–8 [DOI] [PubMed] [Google Scholar]
- 77.Lien EC et al. (2017) Oncogenic PI3K promotes methionine dependency in breast cancer cells through the cystine-glutamate antiporter xCT. Sci. Signal 10, eaao6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gu Y et al. (2017) mTORC2 Regulates Amino Acid Metabolism in Cancer by Phosphorylation of the Cystine-Glutamate Antiporter xCT. Mol. Cell 67, 128–138.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang YH et al. (2019) Dysregulation of cystathionine gamma-lyase promotes prostate cancer progression and metastasis. EMBO Rep 20, e45986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang L et al. (2018) A pharmacological probe identifies cystathionine β-synthase as a new negative regulator for ferroptosis. Cell Death Dis 9, 1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zou GG and Banerjee R (2003) Tumor necrosis factor-alpha-induced targeted proteolysis of cystathionine beta-synthase modulates redox homeostasis. J. Biol. Chem 278, 16802–8 [DOI] [PubMed] [Google Scholar]
- 82.Cochrane DR et al. (2017) Clear cell and endometrioid carcinomas: are their differences attributable to distinct cells of origin? J. Pathol 243, 26–36 [DOI] [PubMed] [Google Scholar]
- 83.Cochrane DR et al. (2020) Single cell transcriptomes of normal endometrial derived organoids uncover novel cell type markers and cryptic differentiation of primary tumours. J. Pathol 252, 201–214 [DOI] [PubMed] [Google Scholar]
- 84.Alborzinia H et al. (2018) Golgi stress mediates redox imbalance and ferroptosis in human cells. Commun. Biol 1, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wu J et al. (2019) Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 572, 402–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zille M et al. (2019) Ferroptosis in Neurons and Cancer Cells Is Similar But Differentially Regulated by Histone Deacetylase Inhibitors. eNeuro 6, ENEURO.0263–18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kwon OS et al. (2020) Systematic identification of a nuclear receptor-enriched predictive signature for erastin-induced ferroptosis. Redox Biol 37, 101719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhao H et al. (2012) Frequent epigenetic silencing of the folate-metabolising gene cystathionine-beta-synthase in gastrointestinal cancer. PLoS One 7, e49683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhou YF et al. (2021) Cystathionine beta-synthase mediated PRRX2/IL-6/STAT3 inactivation suppresses Tregs infiltration and induces apoptosis to inhibit HCC carcinogenesis. J. Immunother. Cancer 9, e003031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chio IIC et al. (2016) NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell 166, 963–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Doll S et al. (2019) FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698 [DOI] [PubMed] [Google Scholar]
- 92.Bersuker K et al. (2019) The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mao C et al. (2021) DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593, 586–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Soula M et al. (2020) Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat. Chem. Biol 16, 1351–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen D et al. (2021) iPLA2beta-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat. Commun 12, 3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sun WY et al. (2021) Phospholipase iPLA2beta averts ferroptosis by eliminating a redox lipid death signal. Nat. Chem. Biol 17, 465–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Harris IS et al. (2019) Deubiquitinases Maintain Protein Homeostasis and Survival of Cancer Cells upon Glutathione Depletion. Cell Metab 29, 1166–1181.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang Y et al. (2019) Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem. Biol 26, 623–633.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hu K et al. (2020) Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J. Clin. Invest 130, 1752–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ghoochani A et al. (2021) Ferroptosis Inducers Are a Novel Therapeutic Approach for Advanced Prostate Cancer. Cancer Res 81, 1583–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]