

Abstract
Genome sequencing advances have enabled researchers and clinicians to probe vast numbers of human variants to distinguish pathogenic from benign variation. Model organisms have been critical in variant assessment and delineating molecular mechanisms of some of the diseases caused by these variants. The fruit fly, Drosophila melanogaster, has played a valuable role in this endeavor, taking advantage of its genetic technologies and established biological knowledge. In this review, we highlight the utility of the fly in studying the function of genes associated with rare neurological diseases that have led to a better understanding of common disease mechanisms. We emphasize that shared themes emerge among disease mechanisms, including the importance of lipids, in two prominent neurodegenerative diseases: Alzheimer’s disease and Parkinson’s disease.
Keywords: Drosophila, neurodegeneration, mitochondria, lipid, Alzheimer’s disease, Parkinson’s disease
Using flies to study neurological diseases
An ever-increasing trove of human genomic sequences is available but our ability to distinguish disease-causing variants (See Glossary) from benign variation remains a hurdle to utilizing this information for disease diagnosis or for disease prediction. The rate of genetic sequence acquisition far outpaces our ability to characterize the consequences of individual variants [1,2]. Filling this knowledge gap is critical for our understanding of gene function and disease diagnosis. Increasingly better computational tools together with thoughtful and well-controlled genetic studies are helping to fill this gap [3–6]. Efforts to improve genomic analysis in silico together with the addition of hundreds of thousands of reference genome sequences to the public domain have helped make great strides in distinguishing genetic signal from noise. While the use of these bioinformatics tools for disease diagnosis is very valuable, it is not always sufficient. Indeed, sequence analysis may reveal multiple putative disease-causing variants, variants of unknown significance, or variants in a gene of unknown function. In these instances, experimental analysis of the variants becomes necessary and model organisms play an important role in this endeavor [4,7–9].
As one of the premier genetic model organisms, the fruit fly, Drosophila melanogaster, has been at the forefront of genetic research for over a century [10–12]. The advantages of the fly include fewer gene paralogs compared to vertebrates, the observation that most genes that are associated with or cause human diseases have orthologs in the fly genome, and that most cellular and molecular processes are conserved between fly and human [12–16] (Figure 1). Flies are easy to rear, and it is simple to obtain large numbers of animals in a relatively short period of time, bolstering statistical power. Importantly, the fly community continues to develop and refine open-access tools and resources which are aggregated and curated in the online Drosophila database, FlyBase [17]. This unparalleled and growing genetic toolkit includes the use of visible phenotypic markers and balancer chromosomes, allowing researchers to follow mutations through generations without the need for genotyping or extensive stock maintenance [18,19]. The fly also offers practical advantages including a short lifespan, allowing researchers to readily perform aging experiments necessary for the study of neurodegenerative disorders [20,21]. These and other advantages set the fly apart from other animal models as particularly useful in studying a broad range of human diseases (Figure 1).
Figure 1. Advantages of fly biology together with innovative genetic technologies make Drosophila melanogaster a valuable genetic tool.
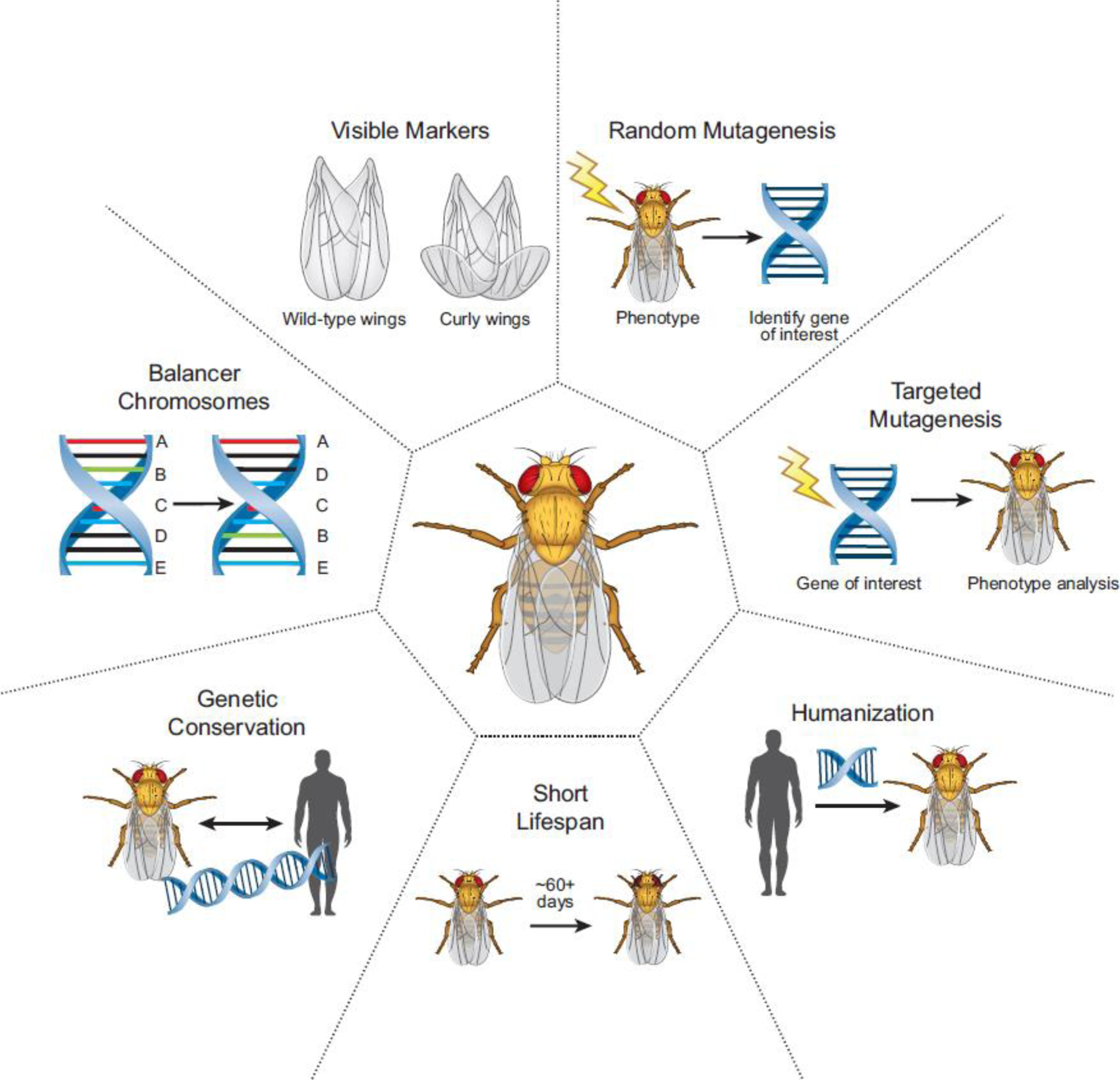
The fly is a valuable screening platform for random or targeted mutagenesis with visible markers and balancer chromosomes to help track mutations without the need for genotyping. The high degree of genetic conservation between human and fly genes together with humanization strategies widely available in the fly provide a useful resource to probe causes of human genetic disease. The biology of the fly, including a short lifespan, allow for simple and rapid assessment of age-dependent phenotypes including neurodegeneration.
Research efforts using model organisms to understand fundamental cellular and molecular mechanisms have significantly advanced our understanding of the pathogenic mechanism of numerous diseases and identified important therapeutic targets [4,7,22–26]. Drosophila has helped elucidate mechanisms of some of the most prevalent neurodegenerative disorders including Alzheimer’s disease (AD) and Parkinson’s disease (PD). Several molecular mechanistic themes have emerged in recent years that are shared between these diseases, including lipid dysregulation and mitochondrial dysfunction [26–31]. In this review, we discuss important advances in rare neurological disease research using the fly that have shed important insights into these two common neurodegenerative diseases, while emphasizing the overlapping disease mechanisms. Finally, we discuss the state of the field and some of the questions that are yet to be answered.
Alzheimer’s disease (AD)
Among all neurodegenerative disorders, Alzheimer’s disease (AD) is the most common in the United States [32], accounting for the majority of dementia cases. While the etiology of AD is complex, it is becoming increasingly clear that lipid dysregulation is a prominent feature of the disease [33]. Glial lipid accumulation in saccules was originally described in patient brains by Alzheimer himself [34,35], but the association of lipid accumulation and lipid dysregulation in AD has only recently become generally appreciated.
Studies utilizing Drosophila, initially aimed at studying the role of mitochondrial variants, associated with rare neurological disease [36], have provided insight into the role of mitochondrial function and lipids in these rare diseases as well as in AD [27,37,38]. Leigh syndrome (LS, Phenotype MIM #256000), a rare genetic neurometabolic disorder, is characterized by degenerative features of the central nervous system (CNS) in infants including loss of previously acquired motor skills, and progressive respiratory distress [39]. Genetic causes of Leigh syndrome include mutations in one or several genes encoding proteins that function in metabolic pathways. One cause of LS stems from deleterious variants in the SURF1 gene, which encodes an inner mitochondrial membrane protein involved in the assembly of the of mitochondrial respiratory chain complex IV [39,40]. Silencing the fly homolog, Surf1, in larvae causes severe deficits in all mitochondrial respiratory chain complexes while silencing in adult flies had a selective impairment in cytochrome c oxidase biogenesis, suggesting differential requirements for Surf1 during development and adulthood [41]. Using the genetic tools readily available in the fly, the authors reduced expression of the Surf1 gene specifically in the adult brain and found that this fly mimicked many of the biochemical hallmarks associated with LS, thus providing a suitable animal model in which LS and related mitochondrial disease mechanisms can be probed into the future [42].
Other studies aimed at elucidating mechanisms of LS identified a glial lipid droplet (LD) accumulation phenotype upon loss of genes important for mitochondrial function including the mitochondrial chaperone protein, sicily [27,36,37]. Mutations in the human homolog of sicily, NDUFAF6, are known to cause LS and variants in this gene have recently been identified as risk factors for AD [36,39,43–45]. The studies in the fly demonstrate that neuronal reactive oxygen species (ROS), caused by defective mitochondria, induce lipogenesis and peroxidation of newly synthesized lipids. Peroxidated lipids are shuttled from neurons to glia where they are sequestered in LDs (Figure 2A). The fly model of ROS-induced glial LD formation allowed researchers to probe neuron-glia interactions and the transfer of lipids between these cell types. It was shown that glial LD formation requires the apolipoprotein Glial lazarillo (GLaz). The highest known genetic risk factor for developing AD is a variant in a homologous human lipoprotein, Apolipoprotein E, termed APOE ε4 (APOE4) while other common APOE variants ε2 (APOE2) and ε3 (APOE3) do not confer AD risk [46]. Use of a humanized fly model, in which the expression of the GLaz was replaced with the expression of APOE2, APOE3, and APOE4, demonstrated that expression of APOE2 and APOE3, but not APOE4, rescue glial LD formation, suggesting that APOE4 is a loss-of-function (LOF) mutation in neuron-to-glia lipid transfer. These findings suggest that LD formation and intercellular lipid transport play roles in the pathogenesis of AD, which is bolstered by growing evidence of the importance of lipid regulation in neurodegenerative diseases like AD [29,33,47].
Figure 2. Oxidated lipids produced upon neuronal ROS are shuttled to glia and sequestered in glial LD which protects neurons from damage.
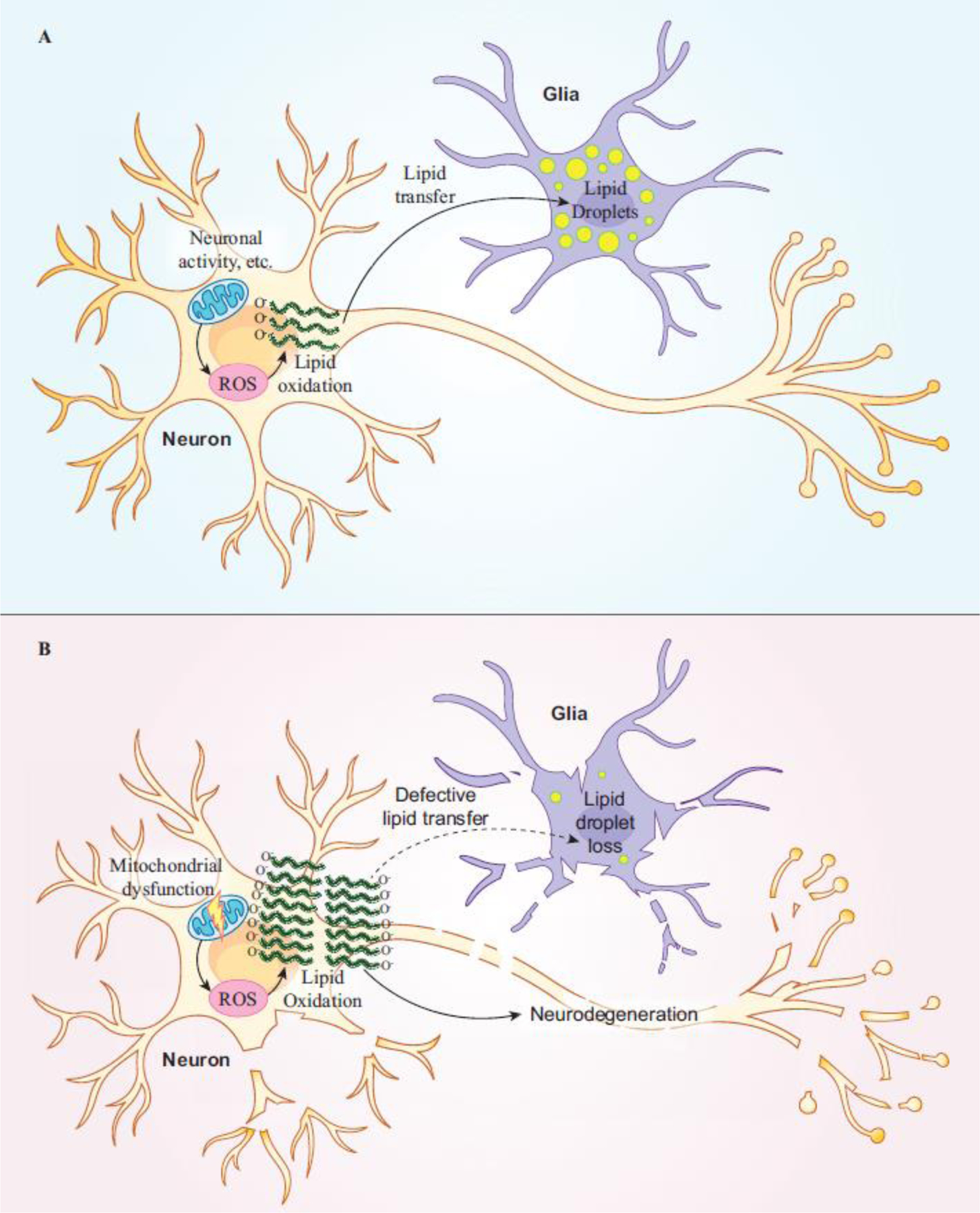
(A) Glial LD formation defends against neurotoxicity caused by oxidative stress in the form of ROS. Neuronal ROS production from mitochondria induces lipogenesis and lipid oxidation. Oxidated lipids are shuttled from neurons to glia where they become sequestered in LDs.
(B) Loss of glial LD formation leads to neurodegeneration. Elevated ROS (due to aging, genetic risk, etc.) together with defects in neuron-to-glia lipid transfer leads to the accumulation of oxidated lipids in neurons and in the extracellular space where they induce neurodegeneration. Genes important for lipid transfer overlap with AD risk factors suggesting an important role for glial LD formation in disease.
A recent study utilizing the fly model of ROS-induced glial LD accumulation identified a tissue-specific role for several known AD risk genes identified in GWAS and other studies [44,45,48–50], in neuron-to-glia lipid transport and LD accumulation [38]. This study showed that AD risk genes that function in APOE lipidation are required in neurons, while genes important for APOE uptake are required in glia for LD formation. These findings suggest that AD risk may be conferred by altered gene function in neurons or glia and that multiple insults synergize to cause disease (Figure 2B).
The link between elevated ROS levels in neurons and LD accumulation in glia is not confined to genes that cause LS. Rather, ROS and LD formation is a shared phenotype resulting from mutations in proteins important for mitochondrial function which are associated with rare neurological diseases. Examples of this include a mitofusin protein encoded by the MFN2 gene (Marf in flies), which is associated with Charcot-Marie-Tooth Disease Type 2A2A/2A2B (Phenotype MIM#609260 and 617087), and a mitochondrial Methionyl-tRNA synthetase encoded by the MARS2 gene (MetRS-m or Aats-met in flies), which is associated with autosomal recessive spastic ataxia with leukoencephalopathy (ARSAL, Phenotype MIM #611390) [27,51,52]. Additionally, one study aimed at probing the mechanistic underpinnings of a rare form of hereditary spastic paraplegia (Spastic Paraplegia 39 [SPG39], Phenotype MIM #612020) and other rare syndromes [53], revealed links between mitochondrial abnormalities, ROS accumulation, and LD formation in the fly brain. SPG39 is caused by variants in the gene patatin like phospholipase domain containing 6, PNPLA6, which encodes an enzyme that facilitates the breakdown of phosphatidylcholine in membranes [54]. The fly ortholog of PNPLA6, swiss cheese (sws), shares high conservation with human PNPLA6 [53]. Loss of sws in the fly leads to a dramatic loss of neurons in the brain, causing behavioral abnormalities, memory deficits, and shortened lifespan. Moreover, knockdown of sws specifically in neurons or in glia activates the production of neuronal ROS and triggers the upregulation of antioxidant genes [53,55]. Consistent with previous reports [27,37,38], loss of sws and the concomitant activation of ROS in neurons leads to enhanced LD formation. Overall, the links between ROS, LD formation, and neurodegeneration in this study are striking and have implications for the etiology of SPG39. The cell-specific effects observed in this case and other studies highlights the complexities of both rare and common neurological disease. Certainly, studies that utilize tissue-specific inquiry for neurological disease research will be key to understanding disease as well as therapeutic discovery.
Additional support for the links between ROS, LD formation, and neurological dysfunction related to rare neurological disease and AD have been discovered using the fly [37,56,57]. Intercellular transport of lipids and glial LD formation is part of normal brain development [56,58,59] as well as a consequence of cell stress [27,38,60,61]. This transfer relies on apolipoproteins [37,38,62–64]. As APOE4 is a risk-factor for AD, these and other findings [65–68] suggest that a better understanding of AD will require careful probing of tissue-specific consequences of genetic variants required for LD formation. Furthermore, these studies implicate mitochondrial dysfunction and subsequent ROS production in glial LD accumulation, suggesting that therapeutic interventions aimed at reducing the effects of ROS may prove effective at preventing neurodegeneration and dysregulation of lipids in the brain. It should be emphasized that antioxidant therapies for AD and neurological disorders utilize a potent blood-brain-barrier (BBB) penetrating compound that can be administered prior to the onset of disease which is safe to administer over a long period of time. One compound that has these features is N-acetylcysteine amide (NACA) [69]. In flies, this compound alleviates ROS-mediated neurodegeneration and relieves glial LD accumulation [27,37,38] as well as strongly suppresses neurodegenerative phenotypes in a fly model of ACOX1 deficiency [70]. Hence, further exploration of NACA or similar compounds in AD and other ROS-associated disorders is warranted. A better understanding of cell-specific lipid biology in the brain will be critical to understand the etiology of neurodegenerative diseases and to identify therapeutic strategies for these illnesses.
Parkinson’s disease (PD)
Parkinson’s disease (PD) is a common neurodegenerative movement disorder characterized by chronic loss of dopaminergic neurons in the substantia nigra (SN) and the presence of abnormal protein inclusions called Lewy bodies [71]. α-synuclein (α-Syn), one of the primary protein components of Lewy bodies, forms aggregates in neurons and can also be found in glia of PD patients [72–74]. Indeed, SNCA, the α-Syn encoding gene, is one of the strongest risk factors of sporadic PD [75–77] and mutations as well as copy-number variants in SNCA cause familial PD [78]. During the past decades, multiple cellular pathways have been associated with PD pathology, however the mechanisms underlying PD are not yet fully understood. Recent findings reveal that lipids play a key role in the pathogenesis of PD and here we discuss several fly studies that indicate lipid metabolism is a common mechanism in PD and related rare diseases.
Dysregulated ceramide metabolism as an emerging mechanism shared by Parkinson’s disease and PLA2G6-associated neurodegeneration
The phospholipase A2 Group 6 (PLA2G6) gene encodes an enzyme that catalyzes the release of fatty acids from phospholipids [79]. Studies carried out in in vitro systems and cultured cell lines have shown that this group of genes play critical roles in maintaining homeostasis of cell membranes and lipid signaling [80]. Dysfunction of PLA2G6 has been observed to cause dysregulated lipid metabolism in the CNS of a knock-out mouse model, and has been reported to be associated with several human neurological diseases [81,82]. Autosomal recessive mutations in PLA2G6 have been identified in a group of rare diseases collectively termed PLA2G6-associated neurodegeneration (PLAN). PLAN disorders have a broad clinical spectrum and can be classified into three major types including infantile neuroaxonal dystrophy (INAD, Phenotype MIM #256600), atypical neuroaxonal dystrophy (aNAD, Phenotype MIM #610217), and PLA2G6-related dystonia-parkinsonism (Phenotype MIM #612953). The age of onset and severity of disease vary greatly between the three [83–85]. Possible explanations for this clinical heterogeneity include distinct tissue distribution of PLA2G6 isoforms, derived via alternative splicing [79,86], and different pathogenic alterations that may impose selective effects on protein function [87]. Variants in PLA2G6 have been associated with NBIA (neurodegeneration with brain iron accumulation) disorder [88,89]. However, elevated levels of iron are only observed in a subgroup of INAD and in aNAD patients but not in PLA2G6-related dystonia-parkinsonism patients [89]. Hence, the precise mechanisms that underlie the pathogenesis of PLAN are still not fully understood.
A study in flies identified phenotypes associated with loss of the fly homolog of PLA2G6, iPLA2-VIA, including sleep disturbances and loss of dopaminergic neurons [90]. The authors documented a change in phospholipid composition in the brain of iPLA2-VIA−/− flies as they contain shortened phospholipid acyl chains when compared to wild-type controls. These lipid alterations reduce the binding affinity of α-Syn and phospholipids, which in turn promotes α-Syn aggregation. Another study in flies demonstrated that loss of iPLA2-VIA causes an excessive ceramide buildup in neurons, impaired retromer function, lysosomal dysfunction, and neurodegeneration [83]. iPLA2-VIA interacts with retromer proteins, including VPS35 and VPS26, and loss of iPLA2-VIA leads to a reduction of these proteins, causing excessive trafficking of protein and membrane lipids to lysosomes, resulting in lysosomal expansion and dysfunction. These observations have been confirmed by studies in many other tissue culture and model systems including mice and rats, and are also consistent with observations that variants in VPS35 cause familial PD [91,92]. The authors proposed that excessive ceramide levels leads to stiffened cellular membranes, based on biophysical data (reviewed in [93]), and documented that ceramide-reducing drugs are able to significantly rescue observed neurodegenerative phenotypes (Figure 3). Another link between ceramide accumulation and PD comes from observations of excessive ceramide as well as a retromer dysfunction in fly PD models in which α-Syn is overexpressed [83]. These phenotypes are strongly suppressed by feeding flies with food containing drugs that lower the ceramide levels. Together, these studies connect variations in PLA2G6/iPLA2-VIA with rare neurological diseases as well as PD. Indeed, dysregulations of ceramides are found to be associated with PD progression. The plasma ceramide levels are increased in PD patients [94,95], especially in those who have cognitive impairment [96]. In the postmortem PD brains and cerebrospinal fluid, lipidomic analyses also revealed an unbalanced ratio of sphingolipid metabolites [97–99]. However, these observations in human samples do not address the causal relationship between disrupted ceramide metabolism and PD pathogenesis. Luckily, studies in flies provide new evidence for a possible underlying mechanism. Fly pink1 is the ortholog of human PTEN-induced kinase 1 (PINK1), which is associated with an autosomal recessive familial form of early-onset PD. PINK1 is involved in mitochondrial quality control, and loss of PINK1 leads to dysfunction in mitochondrial activities as well as defective clearance of damaged mitochondria [100,101]. In a recent study, lipidomic analyses of mitochondria isolated from pink1 deficient flies also documented and increase ceramide levels [102]. Genetic or pharmacological reduction of ceramide levels in flies rescued the mitochondrial phenotypes of pink1 mutants and restored normal flight behavior. Hence, ceramide reduction may be a potential therapeutic strategy in the treatment of PD [83,103].
Figure 3. The involvement of rare disease-associated genes in lipid metabolism as a common mechanism in PD.
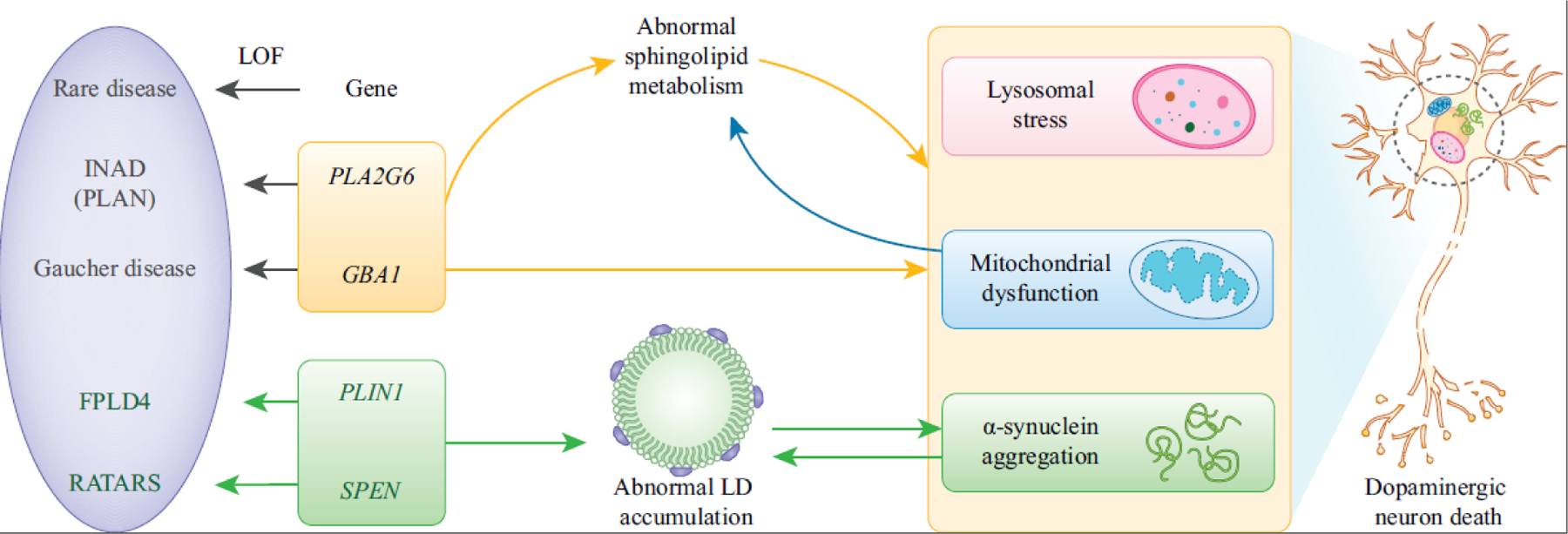
Autosomal recessive mutations in PLA2G6 are associated with INAD (PLAN). Autosomal recessive LOF alleles of GBA1 cause Gaucher disease (GD). Loss of PLA2G6 or GBA1 causes abnormal sphingolipid metabolism. Dysfunction of PLA2G6 or GBA1 causes lysosomal stress, mitochondrial defects and α-Syn aggregation, leading to the dopaminergic neuron death in PD. In addition, mitochondrial stress causes ceramide accumulation.
PLIN1 regulates the homeostasis of LDs as a lipid store gatekeeper. Heterozygous frameshift mutations in PLIN1 are associated with FPLD4. Heterozygous truncating mutations in SPEN are associated with RATARS. Disruption of PLIN1 or SPEN causes accumulation of LDs, which contributes to α-Syn aggregation in the progression of PD. In addition, overexpression of α-Syn also causes LD accumulation.
Dysregulated ceramide metabolism shared by lysosomal storage disease and Parkinson’s disease
Additional insight into the role for ceramide dysregulation in Parkinson’s disease has come from studies using the fly aimed at defining the molecular mechanisms of Gaucher disease (GD, Phenotype MIM#230800 Type I, #230900 Type II, #231000 Type III), a lysosomal storage disorder. Variants in the lysosomal acid β-glucocerebrosidase (GCase) encoded by Glucocerebrosidase (GBA1) lead to accumulation of glucosylceramides (GluCer) in phagocytic ‘Gaucher cells’ [104]. While autosomal recessive LOF alleles of GBA1 cause GD, heterozygous mutant alleles of GBA1 are genetic risk factors for PD and dementia with Lewy bodies (Phenotype MIM #127750) [105]. GD patients and carriers of GD-associated mutations have a higher propensity to develop PD, and develop a faster progression of both motor and cognitive deficits associated with disease [106,107]. How GBA1 mutations influence PD etiology, including α-Syn aggregation, is under intensive investigation and still remains to be fully elucidated (reviewed in [108,109]).
Several GBA1 mutant fly models have been generated by different groups to elucidate the signaling pathways and cellular processes underling disease pathogenesis [110–115]. Flies have two orthologues of GBA1, Gba1a and Gba1b, and it is the loss of Gba1b that accounts for the phenotypes resembling symptoms observed in PD patients [104,105,110,114,115]. Gba1b is expressed in the brain and fat body, consistent with its role in neurological function and lipid metabolism [112]. Proteomic analyses of Gba1b null mutants revealed dysregulation of proteins involved in extracellular vesicle (EV) biology, and GCase deficiency was shown to promote protein aggregate spread between cells and tissues via dysregulated EVs [115]. Gba1b deficiency affects a specific subset of ceramides that are critical for EVs and alters the cargo components of EVs. The pathogenic effects of ceramide dysregulation were also observed in preformed α-synuclein fibrils (PFF) treated SH-SY5Y cell line, and treating the cells with myriocin, a ceramide synthesis inhibitor, rescued the cellular phenotypes associated with PD [116]. These and other studies (reviewed in [29,117]) implicate ceramide homeostasis as an important candidate for PD therapeutic intervention (Figure 3). Although the working models of the interaction between GBA1 and PD are complex and have not been well-tested in different cellular contexts [113,118–120], the links between GBA1 and PD identify new therapeutic targets and strengthen evidence for PD being a lipidopathy (reviewed in [121–123]).
Lipid droplet biology as a functional node in PD and rare diseases
There is also growing evidence that PD pathology is associated with the presence of LDs. In vitro data show that α-Syn can accumulate on LD surfaces and may affect the turnover of LDs, but this mechanism has not been confirmed in vivo. LD membranes contain perilipin proteins, and perilipin 1 (PLIN1) is the major scaffold protein that encircles the LDs in adipocytes [124,125]. PLIN1 regulates the homeostasis of LDs as a lipid store gatekeeper for the coordinated assembly and disassembly of lipolytic complexes upon phosphorylation and dephosphorylation [125,126]. PLIN1 is associated with familial partial lipodystrophy type 4 (FPLD4, Phenotype MIM #613877), characterized by childhood or young adult onset lipodystrophy, severe dyslipidemia and insulin-resistant diabetes mellitus [127]. In Drosophila, there are two genes encoding fly perilipins, Lipid storage droplet-1 (Lsd-1) and Lsd-2. The ortholog of human PLIN1, 2, 3 and 5 is Lsd-2 [128] and it is ubiquitously expressed. As in vertebrates, Lsd-2 is considered to be the guardian of LDs by limiting the access of lipases [129–131]. The fly eye provides an in vivo model to study the relationship of α-Syn and LDs and an accumulation of LDs can be promoted by expression of Lsd-2 as well as Lsd-1 in photoreceptor neurons [132]. Interestingly, the overexpression of α-Syn initiates LD accumulation. Moreover, α-Syn aggregation is enhanced in the presence of LDs (Figure 3). Hence, these data provide evidence that LDs contribute to α-Syn aggregation in the progression of PD pathology.
Additional studies have shed light on the role that the protein Split Ends (SPEN), an RNA-binding protein, plays in lipid regulation in PD [133]. Heterozygous truncating mutations in SPEN are associated with a rare neurodevelopmental disorder called Radio-Tartaglia syndrome (RATARS, Phenotype MIM #619312), characterized by global developmental delay and variable behavioral abnormalities [134]. Recently, SPEN was also found to be associated with PD using microarray analysis from the substantia nigra of PD patients [61] (Figure 3). In Drosophila, loss of spen leads to the inhibition of fat catabolism, which delays larval development, indicative of a failure to generate energy to fuel proper development [135]. spen also regulates LD number, size, and localization in glia. Knockdown of spen in glia leads to increased PLIN2 levels and a dramatic accumulation of LDs in the Drosophila brain [61]. Moreover, glial-specific knockdown of spen causes enhanced sensitivity to paraquat, a model of PD, while overexpression of spen in glia protects against paraquat-induced neurotoxicity [61]. Together, these data support an important role for SPEN in the control of lipid metabolism, and implicate SPEN and lipid dysregulation in neurodegeneration associated with PD.
These and other studies [136–138] demonstrate that lipid dysregulation in the nervous system is a key component of PD and related neurological disorders. Therapeutic interventions aimed at restoring proper balanced lipid production and preventing buildup of toxic lipid species may be important in PD prevention.
Concluding Remarks
In the pursuit to delineate cell biological mechanisms, including those involved in human diseases, it is critical that researchers have access to genetic models with a history of translational success. There are numerous recent examples of successful rare disease studies using Drosophila that have not only shed light into the rare disease diagnosis and pathogenesis, but have also led to insights into common disease [70,139–142]. Continued emphasis on model-organism-based research will fuel insights into rare and common disease mechanisms as well as shed light on therapeutic strategies and identify targets of therapeutic value. Obviously, as with any model system, Drosophila has limitations which need to be carefully considered [143–145], but studies in flies balance utility and translatability.
As researchers continue to probe the etiology of both rare and common diseases, common themes emerge. For AD, PD, and related disorders, a key role for mitochondria, ROS, and lipid biology emerges as driving forces for disease, with mitochondrial dysfunction driving lipid dyshomeostasis and peroxidation, leading to neurodegeneration. While general mechanistic themes of disease etiology emerge, the detailed differences of cellular changes as well as the nature of affected lipids distinguish one neurodegenerative disease from another. Future investigation will yield specific molecules for which focused therapeutic intervention will likely aid the prevention or cure of these diseases. A broader therapeutic strategy that aims to prevent general lipid dysregulation and mitochondrial dysfunction might also prove effective at preventing disease with common mechanisms. One such therapeutic strategy is the administration of antioxidants. While this intervention has been tried with mixed success, it is possible that a better knowledge of the genetic make-up of individuals will allow administration of potent, BBB-penetrating antioxidants prior to the onset of disease symptoms for AD. Similarly, lowering ceramide levels may have beneficial effects in individuals that carry variants associated with PD. While many outstanding questions remain (see outstanding questions box), the future of neurodegenerative disease research as well as therapeutic interventions aimed to prevent them will inevitably be led by the use of model organisms, including the fly.
Outstanding Questions Box.
What is the prevalence of lipid dysregulation in neurodegenerative diseases beyond AD and PD?
Are there signatures of alterations in lipid accumulation and metabolism that can be used as biomarkers and diagnostic tools?
What is the role of different brain cell types in neurodegenerative diseases? Which cell types are affected first?
Which organelles are relevant to neurodegenerative diseases? Which organelles are affected first?
Can therapeutics be generated that selectively target specific proteins and lipids in different cell types?
How can researchers more readily translate observations in fly studies to human disease?
Highlights.
The study of rare neurological disease using the fruit fly, Drosophila melanogaster, has shed valuable insight into common neurodegenerative diseases including Alzheimer’s disease and Parkinson’s disease.
Mitochondrial dysfunction and lipid dysregulation are shared features of many rare neurological disorders and common neurodegenerative diseases.
Studies aimed at delineating mechanisms in flies have shed light on disease pathogenesis and identified novel therapeutic targets for the treatment of neurological disease.
Acknowledgments
We are grateful to Liping Wang, Guang Lin and Yiming Zheng for their insight and feedback on this review. We are also thankful to the reviewers and editors for their comments and efforts to improve our manuscript. This work was supported by grants from The Huffington Foundation, CureINAD, The National Institute on Aging of The National Institutes of Health (NIH) under the award numbers R01 AG07326 and U01 AG072439, The Office of Strategic Coordination/Office of the NIH Director under the award numbers U54 NS093793 and R01 HG011795, The Office of Research Infrastructure Programs of the NIH under the award numbers R24 OD022005 and R24 OD031447. Support also comes from the Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital. M.J.M. is supported by the Brain Disorders & Development Fellowship Training Grant from the NIH under the award number T32 NS043124–19.
Glossary Box
- Apolipoproteins
Proteins that are associated with lipids to form lipoproteins. Apolipoproteins stabilize lipoprotein complexes and aid in lipid solubilization
- Balancer chromosomes
Genetically engineered chromosomes that carry a recessive lethal or sterile allele as well as a dominant allele that causes a visible phenotype (marker). Balancers also contain multiple chromosomal inversions that inhibit the formation of viable recombinant chromosomes, therefore allowing maintenance of the desired mutation(s) or transgene(s) in future generations
- Benign variation
One or more genetic alterations that do not induce any obvious phenotype or cause disease
- Disease-causing variants
One or more genetic alterations that cause or are associated with a particular disease
- Extracellular vesicle (EV)
Particles released from cells surrounded by a phospholipid bilayer to transport cargo proteins, lipids, etc
- Fat body
The fly tissue containing adipocytes which facilitate lipid metabolism and storage as well as protein synthesis and secretion
- Gaucher cells
Monocyte-macrophage cells that accumulate unprocessed glucocerebrosidase. These cells are typically characterized by their large size, small and eccentrically placed nuclei, spindle or rod-shaped membrane-bound inclusion bodies, and numerous small tubules in the cytoplasm
- Lewy bodies
Inclusion bodies in which abnormal protein aggregations (composed of the protein α-synuclein in association with ubiquitin, neurofilament protein, alpha B crystalline, etc.) form and which serve as a pathological marker for PD
- Lipid droplet (LD)
A phospholipid monolayer-bound organelle containing lipids (esp. di- and tri-acylglycerols and sterol esters) together with embedded proteins. LDs serve as lipid stores and are most prevalent in adipose tissue
- Lipogenesis
The process of fatty acid synthesis. Lipogenesis occurs predominantly in the liver, adipose tissue, and small intestine
- Mitochondrial respiratory chain
Five enzymatic protein complexes (I-V) critical for the production of energy in the form of adenosine triphosphate (ATP)
- Neurometabolic disorders
Genetic disorders that result from abnormalities of enzymes that cause a deficiency or an accumulation of certain essential metabolites or cause the accumulation of toxic compounds, impairing the development or function of the nervous system
- Peroxidation
The process of chemical degradation of lipids by oxidants. Polyunsaturated fatty acids, containing multiple carbon-carbon double bonds, are particularly susceptible to peroxidation
- Phospholipid
A type of amphiphilic lipid that is composed of two fatty acids (hydrophobic “tails”), a phosphate group (hydrophilic “head”), and a glycerol molecule. Phospholipids comprise the key components of cell membranes often forming a bilayer with oppositely oriented molecules (heads out)
- Reactive oxygen species (ROS)
Radical oxygen-containing molecules that readily oxygenate and alter cellular molecules
- Substantia nigra (SN)
Meaning “black substance,” the SN is a part of the basal ganglia in the midbrain which looks like a darkened streak in the unstained brain. The SN itself is made up of two anatomically and functionally distinct portions: the substantia nigra pars compacta (SNpc), mainly consisting of dopaminergic neurons, and the substantia nigra pars reticulata (SNpr), mainly consisting of GABAergic neurons
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chong JX et al. (2015) The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities. Am. J. Hum. Genet 97, 199–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moulton MJ and Letsou A (2016) Modeling congenital disease and inborn errors of development in Drosophila melanogaster. DMM Dis. Model. Mech 9, 253–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karczewski KJ et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baldridge D et al. (2021) Model organisms contribute to diagnosis and discovery in the undiagnosed diseases network: Current state and a future vision. Orphanet J. Rare Dis 16, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang J et al. (2017) MARRVEL: Integration of Human and Model Organism Genetic Resources to Facilitate Functional Annotation of the Human Genome. Am. J. Hum. Genet 100, 843–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu P et al. (2019) Reanalysis of Clinical Exome Sequencing Data. N. Engl. J. Med 380, 2478–2480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wangler MF et al. (2017) Model organisms facilitate rare disease diagnosis and therapeutic research. Genetics 207, 9–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wangler MF et al. (2017) Drosophila and genome-wide association studies: A review and resource for the functional dissection of human complex traits. DMM Dis. Model. Mech 10, 77–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sonoshita M and Cagan RL (2017) Modeling Human Cancers in Drosophila. Curr. Top. Dev. Biol 121, 287–309 [DOI] [PubMed] [Google Scholar]
- 10.Stephenson R and Metcalfe NH (2013) Drosophila melanogaster: A fly through its history and current use. J. R. Coll. Physicians Edinb 43, 70–75 [DOI] [PubMed] [Google Scholar]
- 11.Bellen HJ et al. (2019) The fruit fly at the interface of diagnosis and pathogenic mechanisms of rare and common human diseases. Hum. Mol. Genet 28, R207–R214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pandey UB and Nichols CD (2011) Human disease models in drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol. Rev 63, 411–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rajan A and Perrimon N (2013) Of flies and men: Insights on organismal metabolism from fruit flies. BMC Biol 11, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bellen HJ and Yamamoto S (2015) Morgan’s Legacy: Fruit Flies and the Functional Annotation of Conserved Genes. Cell 163, 12–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reiter LT et al. (2001) A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res 11, 1114–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shih J et al. (2015) Comparison of inter- and intraspecies variation in humans and fruit flies. Genomics Data 3, 49–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Larkin A et al. (2021) FlyBase: Updates to the Drosophila melanogaster knowledge base. Nucleic Acids Res 49, D899–D907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller DE et al. (2019) The joy of balancers. PLOS Genet 15, e1008421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.St Johnston D (2002) The art and design of genetic screens: Drosophila melanogaster. Nat. Rev. Genet 3, 176–188 [DOI] [PubMed] [Google Scholar]
- 20.Linford NJ et al. (2013) Measurement of lifespan in Drosophila melanogaster. J. Vis. Exp 1, e50068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Piper MDW and Partridge L (2018) Drosophila as a model for ageing. Biochim. Biophys. Acta - Mol. Basis Dis 1864, 2707–2717 [DOI] [PubMed] [Google Scholar]
- 22.Scearce-Levie K et al. (2020) Leveraging preclinical models for the development of Alzheimer disease therapeutics. Nat. Rev. Drug Discov 19, 447–462 [DOI] [PubMed] [Google Scholar]
- 23.Duarte-Jurado AP et al. (2021) Antioxidant therapeutics in parkinson’s disease: Current challenges and opportunities. Antioxidants 10, 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rai SN and Singh P (2020) Advancement in the modelling and therapeutics of Parkinson’s disease. J. Chem. Neuroanat 104, 101752. [DOI] [PubMed] [Google Scholar]
- 25.Metaxakis A et al. (2020) Molecular Interventions towards Multiple Sclerosis Treatment. Brain Sci. 2020, Vol. 10, Page 299 10, 299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tavassolifar MJ et al. (2020) The Influence of Reactive Oxygen Species in the Immune System and Pathogenesis of Multiple Sclerosis. Autoimmune Dis 2020, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu L et al. (2015) Glial Lipid Droplets and ROS Induced by Mitochondrial Defects Promote Neurodegeneration. Cell 160, 177–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kolodkin N, et al A (2020) ROS networks: designs, aging, Parkinson’s disease and precision therapies. npj Syst. Biol. Appl. 2020 61 6, 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin G et al. (2019) Sphingolipids in the Pathogenesis of Parkinson’s Disease and Parkinsonism. Trends Endocrinol. Metab 30, 106–117 [DOI] [PubMed] [Google Scholar]
- 30.Farmer BC et al. (2020) Lipid Droplets in Neurodegenerative Disorders. Front. Neurosci 14, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monzio Compagnoni G et al. (2020) The Role of Mitochondria in Neurodegenerative Diseases: the Lesson from Alzheimer’s Disease and Parkinson’s Disease. Mol. Neurobiol 57, 2959–2980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murray MJ and Friederich Murray C (2007) Alzheimer’s Disease. In Complications in Anesthesia pp. 493–495, W.B. Saunders [Google Scholar]
- 33.Chew H et al. (2020) Involvement of Lipids in Alzheimer’s Disease Pathology and Potential Therapies. Front. Physiol 11, 1–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Graeber MB et al. (1997) Rediscovery of the case described by Alois Alzheimer in 1911: Historical, histological and molecular genetic analysis. Neurogenetics 1, 73–80 [DOI] [PubMed] [Google Scholar]
- 35.Alzheimer A (1907) Uber eine eigenartige Erkrankung der Hirnrinde. Allg. Zeitschrift Rsychiatrie Psych. Medizine 64, 146–148 [Google Scholar]
- 36.Zhang K et al. (2013) The C8ORF38 homologue Sicily is a cytosolic chaperone for a mitochondrial complex I subunit. J. Cell Biol 200, 807–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu L et al. (2017) The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cell Metab 26, 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moulton MJ et al. (2021) Neuronal ROS-induced glial lipid droplet formation is altered by loss of Alzheimer’s disease–associated genes. Proc. Natl. Acad. Sci 118, e2112095118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schubert MB and Vilarinho L (2020) Molecular basis of Leigh syndrome: A current look. Orphanet J. Rare Dis 15, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alves CAPF et al. (2020) Pediatric Leigh Syndrome: Neuroimaging Features and Genetic Correlations. Ann. Neurol 88, 218–232 [DOI] [PubMed] [Google Scholar]
- 41.Da-Rè C et al. (2014) Leigh Syndrome in Drosophila melanogaster morphological and biochemical characterization of Surf1 post-transcriptional silencing. J. Biol. Chem 289, 29235–29246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bakare AB et al. (2021) Leigh Syndrome: A Tale of Two Genomes. Front. Physiol 12, 1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moreno-Grau S et al. (2019) Genome-wide association analysis of dementia and its clinical endophenotypes reveal novel loci associated with Alzheimer’s disease and three causality networks: The GR@ACE project. Alzheimer’s Dement 15, 1333–1347 [DOI] [PubMed] [Google Scholar]
- 44.Kunkle BW et al. (2019) Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet 51, 414–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bellenguez C et al. (2020) New insights on the genetic etiology of Alzheimer’s and related dementia. medRxiv DOI: 10.1101/2020.10.01.20200659 [DOI]
- 46.Yamazaki Y et al. (2019) Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol. 2019 159 15, 501–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shamim A et al. (2018) Lipids: An insight into the neurodegenerative disorders. Clin. Nutr. Exp 20, 1–19 [Google Scholar]
- 48.Lambert J-C et al. (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet 45, 1452–1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stocker H et al. (2018) The genetic risk of Alzheimer’s disease beyond APOE ε4: systematic review of Alzheimer’s genetic risk scores. Transl. Psychiatry 2018 81 8, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pimenova AA et al. (2018) Untangling Genetic Risk for Alzheimer’s Disease. Biol. Psychiatry 83, 300–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bayat V et al. (2012) Mutations in the mitochondrial methionyl-tRNA synthetase cause a neurodegenerative phenotype in flies and a recessive ataxia (ARSAL) in humans. PLoS Biol 10, e1001288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dorn GW and II (2020) Mitofusin 2 Dysfunction and Disease in Mice and Men. Front. Physiol 11, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Melentev PA et al. (2021) Loss of swiss cheese in neurons contributes to neurodegeneration with mitochondria abnormalities, reactive oxygen species acceleration and accumulation of lipid droplets in Drosophila brain. Int. J. Mol. Sci 22, 8275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Synofzik M et al. (2014) PNPLA6 mutations cause Boucher-Neuhäuser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain 137, 69–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ryabova EV et al. (2021) Morpho‐functional consequences of swiss cheese knockdown in glia of drosophila melanogaster. Cells 10, 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Van Den Brink DM et al. (2018) Physiological and pathological roles of FATP-mediated lipid droplets in Drosophila and mice retina. PLOS Genet 14, 1–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Muliyil S et al. (2020) ADAM 17‐triggered TNF signalling protects the ageing Drosophila retina from lipid droplet‐mediated degeneration. EMBO J 39, e104415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kis V et al. (2015) Specialized cortex glial cells accumulate lipid droplets in drosophila melanogaster. PLoS One 10, e0131250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Welte MA and Gould AP (2017) Lipid droplet functions beyond energy storage. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 1862, 1260–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ioannou MS et al. (2019) Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 177, 1522–1535.e14 [DOI] [PubMed] [Google Scholar]
- 61.Girard V et al. (2020) Spen modulates lipid droplet content in adult Drosophila glial cells and protects against paraquat toxicity. Sci. Rep 10, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qi G et al. (2020) APOE4 impairs neuron‐astrocyte coupling of fatty acid metabolism. Alzheimer’s Dement 16, e045251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Farmer B et al. (2019) Apolipoprotein E4 Alters Astrocyte Fatty Acid Metabolism and Lipid Droplet Formation. Cells 8, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yin J et al. (2021) Brain-specific lipoprotein receptors interact with astrocyte derived apolipoprotein and mediate neuron-glia lipid shuttling. Nat. Commun 12, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sims R et al. (2020) The multiplex model of the genetics of Alzheimer’s disease. Nat. Neurosci 23, 311–322 [DOI] [PubMed] [Google Scholar]
- 66.Gerring ZF et al. (2020) An analysis of genetically regulated gene expression across multiple tissues implicates novel gene candidates in Alzheimer’s disease. Alzheimer’s Res. Ther 12, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cabirol-Pol M-J et al. (2017) Glial lipid droplets and neurodegeneration in a Drosophila model of complex I deficiency. Glia 66, 874–888 [DOI] [PubMed] [Google Scholar]
- 68.Goodman LD and Bellen HJ (2021) Recent insights into the role of glia and oxidative stress in Alzheimer’s disease gained from Drosophila. Curr. Opin. Neurobiol 72, 32–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Atlas D (2021) Emerging therapeutic opportunities of novel thiol-amides, NAC-amide (AD4/NACA) and thioredoxin mimetics (TXM-Peptides) for neurodegenerative-related disorders. Free Radic. Biol. Med 176, 120–141 [DOI] [PubMed] [Google Scholar]
- 70.Chung H lok et al. (2020) Loss- or Gain-of-Function Mutations in ACOX1 Cause Axonal Loss via Different Mechanisms. Neuron 106, 589–606.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goedert M et al. (2013) 100 years of Lewy pathology. Nat. Rev. Neurol 9, 13–24 [DOI] [PubMed] [Google Scholar]
- 72.Braak H et al. (2007) Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol 114, 231–241 [DOI] [PubMed] [Google Scholar]
- 73.Booth HDE et al. (2017) The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci 40, 358–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lu SZ et al. (2019) Suppression of astrocytic autophagy by αb-crystallin contributes to α-synuclein inclusion formation 11 Medical and Health Sciences 1109 Neurosciences 06 Biological Sciences 0601 Biochemistry and Cell Biology. Transl. Neurodegener 8, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nalls MA et al. (2014) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet 46, 989–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Soldner F et al. (2016) Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 533, 95–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lu S and Zhou J (2017) Finding the “Guilty” Gene Variant of Sporadic Parkinson’s Disease Via CRISPR/Cas9. Neurosci. Bull 33, 115–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Miller DW et al. (2004) Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology 62, 1835–1838 [DOI] [PubMed] [Google Scholar]
- 79.Larsson PKA et al. (1998) Multiple splice variants of the human calcium-independent phospholipase A2 and their effect on enzyme activity. J. Biol. Chem 273, 207–214 [DOI] [PubMed] [Google Scholar]
- 80.Brown WJ et al. (2003) Phospholipase A2 (PLA2) enzymes in membrane trafficking: Mediators of membrane shape and function. Traffic 4, 214–221 [DOI] [PubMed] [Google Scholar]
- 81.Beck G et al. (2011) Neuroaxonal dystrophy in calcium-independent phospholipase A2β deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J. Neurosci 31, 11411–11420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Adibhatla RM and Hatcher JF (2008) Phospholipase A2, reactive oxygen species, and lipid peroxidation in CNS pathologies. J. Biochem. Mol. Biol 41, 560–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lin G et al. (2018) Phospholipase PLA2G6, a Parkinsonism-Associated Gene, Affects Vps26 and Vps35, Retromer Function, and Ceramide Levels, Similar to α-Synuclein Gain. Cell Metab 28, 605–618.e6 [DOI] [PubMed] [Google Scholar]
- 84.Iodice A et al. (2017) Infantile neuroaxonal dystrophy and PLA2G6-associated neurodegeneration: An update for the diagnosis. Brain Dev 39, 93–100 [DOI] [PubMed] [Google Scholar]
- 85.Spatola M and Wider C (2014) Genetics of Parkinson’s disease: The yield. Park. Relat. Disord 20, S35–S38 [DOI] [PubMed] [Google Scholar]
- 86.Larsson Forsell PKA et al. (1999) The human calcium-independent phospholipase A2 gene: Multiple enzymes with distinct properties from a single gene. Eur. J. Biochem 262, 575–585 [DOI] [PubMed] [Google Scholar]
- 87.Engel LA et al. (2010) Catalytic function of PLA2G6 is impaired by mutations associated with infantile neuroaxonal dystrophy but not dystonia-parkinsonism. PLoS One 5, e12897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ferese R et al. (2018) Heterozygous PLA2G6 mutation leads to iron accumulation within basal ganglia and Parkinson’s disease. Front. Neurol 9, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Morgan NV et al. (2006) PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat. Genet 38, 752–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mori A et al. (2019) Parkinson’s disease-associated iPLA2-VIA/PLA2G6 regulates neuronal functions and α-synuclein stability through membrane remodeling. Proc. Natl. Acad. Sci. U. S. A 116, 20689–20699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Williams ET et al. (2017) VPS35, the Retromer Complex and Parkinson’s Disease. J. Parkinsons. Dis 7, 219–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rahman AA and Morrison BE (2019) Contributions of VPS35 Mutations to Parkinson’s Disease. Neuroscience 401, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Alonso A and Goñi FM (2018) The Physical Properties of Ceramides in Membranes. Annu. Rev. Biophys 47, 633–654 [DOI] [PubMed] [Google Scholar]
- 94.Guedes LC et al. (2017) Serum lipid alterations in GBA-associated Parkinson’s disease. Park. Relat. Disord 44, 58–65 [DOI] [PubMed] [Google Scholar]
- 95.Klatt-Schreiner K et al. (2020) High Glucosylceramides and Low Anandamide Contribute to Sensory Loss and Pain in Parkinson’s Disease. Mov. Disord 35, 1822–1833 [DOI] [PubMed] [Google Scholar]
- 96.Mielke MM et al. (2013) Plasma ceramide and glucosylceramide metabolism is altered in sporadic Parkinson’s disease and associated with cognitive impairment: a pilot study. PLoS One 8, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cheng D et al. (2011) Lipid pathway alterations in Parkinson’s disease primary visual cortex. PLoS One 6, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Beger AW et al. (2022) Human Brain Lipidomics: Pilot Analysis of the Basal Ganglia Sphingolipidome in Parkinson’s Disease and Lewy Body Disease. Metabolites 12, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fernández-Irigoyen J et al. (2021) Alteration in the Cerebrospinal Fluid Lipidome in Parkinson’s Disease: A Post-Mortem Pilot Study. Biomedicines 9, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ge P et al. (2020) PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Mol. Neurodegener 15, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bayne AN and Trempe JF (2019) Mechanisms of PINK1, ubiquitin and Parkin interactions in mitochondrial quality control and beyond. Cell. Mol. Life Sci 76, 4589–4611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vos M and Klein C (2022) Ceramide-induced mitophagy impairs ß-oxidation-linked energy production in PINK1 deficiency. Autophagy DOI: 10.1080/15548627.2022.2027193 [DOI] [PMC free article] [PubMed]
- 103.Custodia A et al. (2021) Ceramide Metabolism and Parkinson’s Disease-Therapeutic Targets. Biomolecules 11, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pastores GM (1997) Gaucher’s Disease. Pathological features. Baillieres. Clin. Haematol 10, 739–749 [DOI] [PubMed] [Google Scholar]
- 105.Riboldi GM and Di Fonzo AB (2019) GBA, Gaucher Disease, and Parkinson’s Disease: From Genetic to Clinic to New Therapeutic Approaches. Cells 8, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Blandini F et al. (2019) Glucocerebrosidase mutations and synucleinopathies: Toward a model of precision medicine. Mov. Disord 34, 9–21 [DOI] [PubMed] [Google Scholar]
- 107.Brockmann K et al. (2015) GBA-associated Parkinson’s disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov. Disord 30, 407–411 [DOI] [PubMed] [Google Scholar]
- 108.Behl T et al. (2021) Cross-talks among GBA mutations, glucocerebrosidase, and α-synuclein in GBA-associated Parkinson’s disease and their targeted therapeutic approaches: a comprehensive review. Transl. Neurodegener 10, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vos M and Klein C (2021) The Importance of Drosophila melanogaster Research to UnCover Cellular Pathways Underlying Parkinson’s Disease. Cells 10, 579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cabasso O et al. (2019) Drosophila melanogaster Mutated in its GBA1b Ortholog Recapitulates Neuronopathic Gaucher Disease. J. Clin. Med 8, 1–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Maor G et al. (2016) The contribution of mutant GBA to the development of Parkinson disease in Drosophila. Hum. Mol. Genet 25, 2712–2727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kinghorn KJ et al. (2016) A Drosophila Model of Neuronopathic Gaucher Disease Demonstrates Lysosomal-Autophagic Defects and Altered mTOR Signalling and Is Functionally Rescued by Rapamycin. J. Neurosci 36, 11654–11670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Suzuki M et al. (2015) Glucocerebrosidase deficiency accelerates the accumulation of proteinase K-resistant α-synuclein and aggravates neurodegeneration in a Drosophila model of Parkinson’s disease. Hum. Mol. Genet 24, 6675–6686 [DOI] [PubMed] [Google Scholar]
- 114.Kawasaki H et al. (2017) Minos-insertion mutant of the Drosophila GBA gene homologue showed abnormal phenotypes of climbing ability, sleep and life span with accumulation of hydroxy-glucocerebroside. Gene 614, 49–55 [DOI] [PubMed] [Google Scholar]
- 115.Jewett KA et al. (2021) Glucocerebrosidase reduces the spread of protein aggregation in a Drosophila melanogaster model of neurodegeneration by regulating proteins trafficked by extracellular vesicles. PLoS Genet 17, e1008859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mingione A et al. (2021) Inhibition of ceramide synthesis reduces α‐synuclein proteinopathy in a cellular model of parkinson’s disease. Int. J. Mol. Sci 22, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Plotegher N et al. (2019) Ceramides in Parkinson’s Disease: From Recent Evidence to New Hypotheses. Front. Neurosci 13, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Davis MY et al. (2016) Glucocerebrosidase Deficiency in Drosophila Results in α-Synuclein-Independent Protein Aggregation and Neurodegeneration. PLoS Genet 12, e1005944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Khair SBA et al. (2018) Silencing of glucocerebrosidase gene in Drosophila enhances the aggregation of Parkinson’s disease associated α-synuclein mutant A53T and affects locomotor activity. Front. Neurosci 12, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Maor G et al. (2019) The effect of mutant GBA1 on accumulation and aggregation of a synuclein. Hum. Mol. Genet 28, 1768–1781 [DOI] [PubMed] [Google Scholar]
- 121.Fanning S et al. (2020) Parkinson’s disease: proteinopathy or lipidopathy? npj Park. Dis 6, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Xicoy H et al. (2019) The Role of Lipids in Parkinson’s Disease. Cells 8, 1–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Brockmann K (2020) GBA-Associated Synucleinopathies: Prime Candidates for Alpha-Synuclein Targeting Compounds. Front. Cell Dev. Biol 8, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Greenberg AS et al. (1993) Isolation of cDNAs for perilipins A and B: sequence and expression of lipid droplet-associated proteins of adipocytes. Proc. Natl. Acad. Sci. U. S. A 90, 12035–12039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kimmel AR and Sztalryd C (2016) The Perilipins: Major Cytosolic Lipid Droplet-Associated Proteins and Their Roles in Cellular Lipid Storage, Mobilization, and Systemic Homeostasis. Annu. Rev. Nutr 36, 471–509 [DOI] [PubMed] [Google Scholar]
- 126.Brasaemle DL (2007) Thematic review series: adipocyte biology. The perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. J. Lipid Res 48, 2547–2559 [DOI] [PubMed] [Google Scholar]
- 127.Gandotra S et al. (2011) Perilipin deficiency and autosomal dominant partial lipodystrophy. N. Engl. J. Med 364, 740–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hu Y et al. (2011) An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics 12, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Beller M et al. (2010) PERILIPIN-dependent control of lipid droplet structure and fat storage in Drosophila. Cell Metab 12, 521–532 [DOI] [PubMed] [Google Scholar]
- 130.Beller M et al. (2006) Characterization of the Drosophila lipid droplet subproteome. Mol. Cell. Proteomics 5, 1082–1094 [DOI] [PubMed] [Google Scholar]
- 131.Bi J et al. (2012) Opposite and redundant roles of the two Drosophila perilipins in lipid mobilization. J. Cell Sci 125, 3568–3577 [DOI] [PubMed] [Google Scholar]
- 132.Girard V et al. (2021) Abnormal accumulation of lipid droplets in neurons induces the conversion of alpha-Synuclein to proteolytic resistant forms in a Drosophila model of Parkinson’s disease. PLoS Genet 17, e1009921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hazegh KE et al. (2017) An autonomous metabolic role for Spen. PLoS Genet 13, e1006859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Radio FC et al. (2021) SPEN haploinsufficiency causes a neurodevelopmental disorder overlapping proximal 1p36 deletion syndrome with an episignature of X chromosomes in females. Am. J. Hum. Genet 108, 502–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gillette CM et al. (2020) Gene-diet interactions: Dietary rescue of metabolic defects in spen-depleted drosophila melanogaster. Genetics 214, 961–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Guo JF et al. (2018) Coding mutations in NUS1 contribute to Parkinson’s disease. Proc. Natl. Acad. Sci. U. S. A 115, 11567–11572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.De Lazzari F et al. (2020) Antioxidant Therapy in Parkinson’s Disease: Insights from Drosophila melanogaster. Antioxidants 9, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Valadas JS et al. (2018) ER Lipid Defects in Neuropeptidergic Neurons Impair Sleep Patterns in Parkinson’s Disease. Neuron 98, 1155–1169.e6 [DOI] [PubMed] [Google Scholar]
- 139.Marcogliese PC et al. (2018) IRF2BPL Is Associated with Neurological Phenotypes. Am. J. Hum. Genet 103, 245–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Marcogliese PC et al. (2021) Drosophila functional screening of de novo variants in autism uncovers deleterious variants and facilitates discovery of rare neurodevelopmental diseases. bioRxiv 13, 2020.12.30.424813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Link N et al. (2019) Mutations in ANKLE2, a ZIKA Virus Target, Disrupt an Asymmetric Cell Division Pathway in Drosophila Neuroblasts to Cause Microcephaly. Dev. Cell 51, 713–729.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Link N and Bellen HJ (2020) Using Drosophila to drive the diagnosis and understand the mechanisms of rare human diseases. Development 147, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gitler AD et al. (2017) Neurodegenerative disease: models, mechanisms, and a new hope. Dis. Model. Mech 10, 499–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hunter P (2008) The paradox of model organisms. The use of model organisms in research will continue despite their shortcomings. EMBO Rep 9, 717–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lambrechts R et al. (2017) Modelling in miniature: Using Drosophila melanogaster to study human neurodegeneration. Drug Discov. Today Dis. Model 25–26, 3–10 [Google Scholar]