

Abstract
Technologies that profile chromatin modifications at single-cell resolution offer enormous promise for functional genomic characterization, but the sparsity of the measurements and integrating multiple binding maps represent substantial challenges. Here we introduce scCUT&Tag-pro, a multimodal assay for profiling protein-DNA interactions coupled with the abundance of surface proteins in single cells. In addition, we introduce scChromHMM, which integrates data from multiple experiments to infer and annotate chromatin states based on combinatorial histone modification patterns. We apply these tools to perform an integrated analysis across nine different molecular modalities in circulating human immune cells. We demonstrate how these two approaches can characterize dynamic changes in the function of individual genomic elements across both discrete cell states and continuous developmental trajectories, nominate associated motifs and regulators that establish chromatin states, and identify extensive and cell type-specific regulatory priming. Finally, we demonstrate how our integrated reference can serve as a scaffold to map and improve the interpretation of additional scCUT&Tag datasets.
Introduction
Technologies that enable unsupervised transcriptomic profiling at single-cell resolution (i.e. scRNA-seq) represent powerful tools not only for the discovery of cell types and states, but also to reveal the structure and function of transcriptional regulatory networks1–6. In the same way, new methods for single-cell chromatin profiling, such as scATAC-seq, can lead to the identification and characterization of individual genomic regulatory regions, and to explore their variation in a heterogeneous population7,8. Assays that profile chromatin accessibility exhibit a largely binarized phenotype - partitioning genomic regions into accessible or inaccessible elements in different tissue and cellular contexts. As a complement to accessibility measurements, genome-wide histone modification profiles offer an exciting opportunity to segment the genome into a more nuanced set of functional elements9–12. The “histone code hypothesis” proposes that the combinatorial presence of multiple histone modifications marking a genomic region serves as a proxy for its regulatory function13,14.
Techniques like ChIP-seq and CUT&Tag have been widely applied to measure the binding profiles of multiple transcription factors and histone modifications15,16. In addition, tailored computational tools such as ChromHMM17 and Segway10 can integrate the combinatorial binding patterns defined by each mark, and assign individual genomic elements to a set of learned functional ‘states’. Recently, multiple studies have demonstrated the ability to measure CUT&Tag profiles in single cells16,18–21. This represents an opportunity to not only identify regions that exhibit cell type-specific accessibility, but also to highlight elements whose acquisition of activating, repressive, or heterochromatic signatures varies within a heterogeneous population.
These advances lay out an exciting challenge for single cell genomics: can the binding profiles of multiple histone modifications be used to infer the function of any individual genomic element, within a single cell? While the availability of scCUT&Tag takes an important step towards this vision, two key challenges remain. First, single-cell chromatin data remains extremely sparse, particularly due to the inherent challenges associated with limited DNA input molecules. Second, scCUT&Tag experiments typically measure only a single histone modification profile in a single cell. While informative, only the integrated combination of multiple histone modifications can be used to annotate functional state, but no current technology can robustly and simultaneously profile many histone modifications in the same cell, particularly when these marks may exhibit overlapping binding patterns. Recent pioneering advances enable paired measurements of cellular transcriptomes and individual CUT&Tag profiles using custom combinatorial-indexing based workflows22,23, and demonstrate how multimodal technologies can facilitate integrative analysis.
Here, we present two techniques aimed to address these key challenges. We first introduce scCUT&Tag-pro, a multimodal single-cell technology, that enables simultaneous profiling of an individual CUT&Tag profile with surface protein abundances and is compatible with the widely used 10x Genomics Chromium system. Second, we introduce a downstream analysis strategy for multimodal datasets: scChromHMM. Our method first integrates data from multiple scCUT&Tag-pro experiments together into a common manifold, computationally generating co-assay profiles for six histone modifications within individual cells. These measurements are used as input to a single-cell extension of the ChromHMM algorithm, in order to return chromatin state annotations for each 200 base pair (bp) genomic window at single-cell resolution.
We apply our tools and technologies to generate 64,876 scCUT&Tag-pro profiles (Supplementary Table 1), and combine this with existing datasets to create an integrated multimodal atlas of human peripheral blood mononuclear cells (PBMCs) that encompasses nine modalities spanning the central dogma. Together, these technologies and analytical tools allow us to explore heterogeneity in genome function at single-cell resolution, identify sequence elements whose presence accompanies changes in chromatin state, and project new datasets onto our multimodal reference. We propose that these tools will help to facilitate the analysis and interpretation of chromatin state heterogeneity in single cells, and lead to a better understanding of its role in establishing and regulating cellular identity.
Results
Simultaneous profiling of surface proteins and scCUT&Tag
The combinatorial pattern of post-translational modifications present on histone proteins correlates with the chromatin structure and regulatory potential of a genomic locus13,14,24,25. We reasoned that multimodal single-cell technologies could help address challenges in scCUT&Tag analysis by coupling robust single-cell measurements of one modality with more sparse measurements from another. For example, CITE-seq/REAP-seq26,27, and ASAP-seq/ICICLE-seq28,29 pair simultaneous measurements of highly expressed and well-characterized cell surface proteins with unsupervised but sparser transcriptomic measurements or chromatin accessibility profiles. We and others have recently demonstrated that the protein information in CITE-seq is highly informative in determining cellular state30–32, while the paired RNA-seq or ATAC-seq measurements enable the characterization of gene regulatory networks. ASAP-seq and ICICLE-seq are enabled by recent optimizations in cell fixation and permeabilization that enable ATAC-seq to be performed on whole cells instead of individual nuclei33.
We propose that a similar multimodal strategy could be applied for genome-wide profiling of protein-DNA interactions as well. We developed scCUT&Tag-pro, an assay that performs CUT&Tag assays16 on whole cells while simultaneously measuring surface protein levels (Figure 1A; Methods). Inspired by ASAP-seq28, our fixation and permeabilization conditions retain the cell membrane and associated proteins, enabling us to simultaneously measure cellular immunophenotypes (Figure 1A). Briefly, cells are first stained with a panel of commercially available oligo-conjugated antibodies. Subsequently, monovalent Fab fragment is added in order to block the potential binding of proteinAG to these antibodies. Cells are then lightly fixed with 0.1% formaldehyde and permeabilized with an isotonic lysis buffer. We found that removing Digitonin from all buffers substantially alleviated cell ‘clumping’ issues which have been previously described18 without diminishing the efficiency of tagmentation (Supplementary Figure 1A). We use an antibody against a histone modification of interest to direct the proteinAG-Tn5 fusion protein to marked genomic sites. Tagmented cells can then be used as input for library preparation using the 10x Genomics scATAC-seq kit, followed by sequencing of both CUT&Tag as well as antibody-derived tag (ADT) libraries. We note that scCUT&Tag-pro is also compatible with antibody-mediated Cell Hashing34, allowing us to increase the throughput of each experiment.
Figure 1: scCUT&Tag-pro enables simultaneous profiling of CUT&Tag and protein levels.
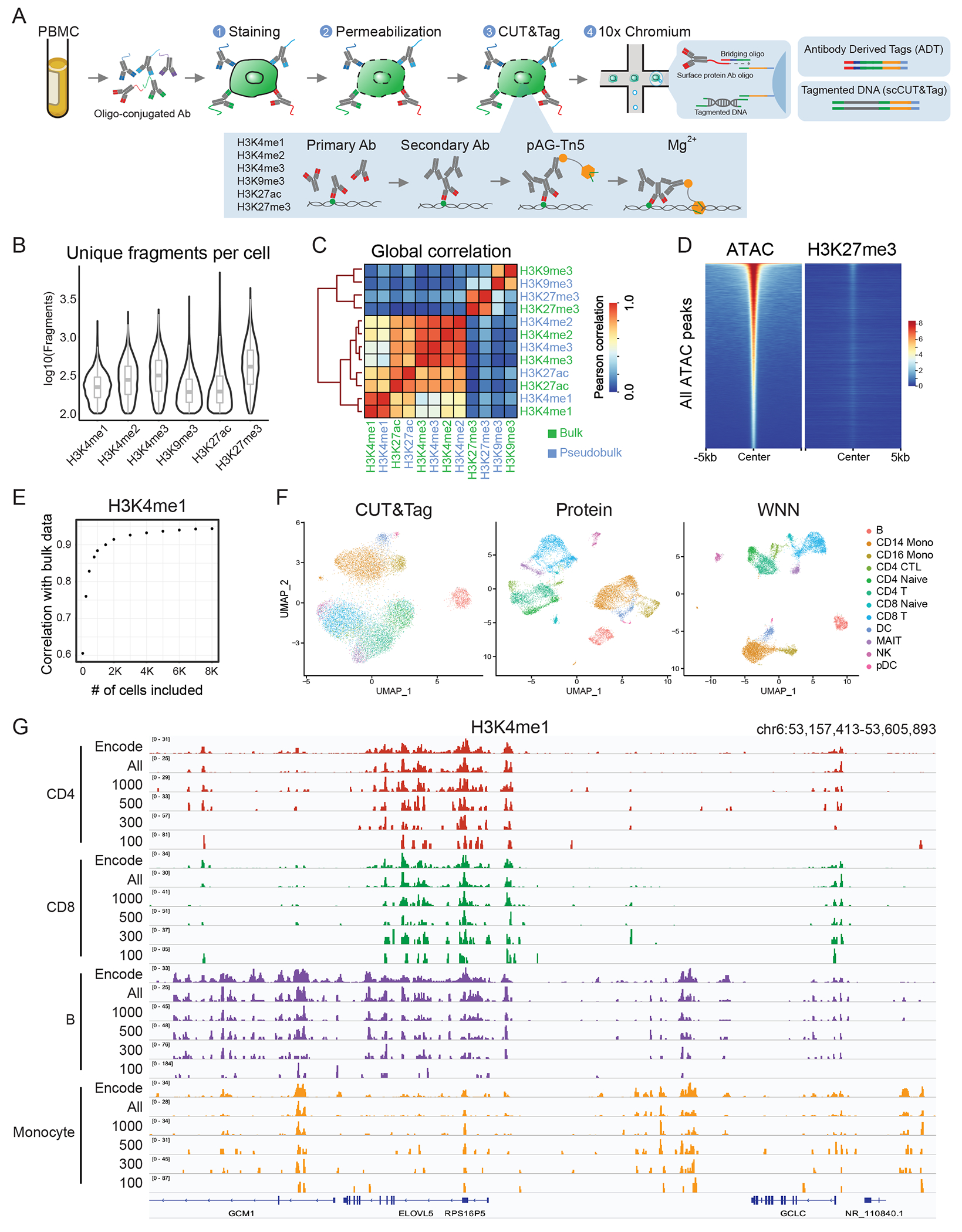
(A) Schematic of experimental workflow, which is compatible with the 10x Chromium system. (B) Distribution of unique fragments obtained per-cell for six histone modifications, profiled in separate PBMC scCUT&Tag-pro experiments (left to right n = 12,770, 9,575, 10,386, 8,304, 15,609 and 8,232 cells). The center, bounds and whiskers of the boxplot show median, quartiles and data points that lie within 1.5× interquartile range of the lower and upper quartiles, respectively. Data beyond the end of the whiskers are plotted individually. (C) Pseudobulk profiles of scCUT&Tag are well-correlated with bulk CUT&Tag profiles of human PBMC. (D) Tornado plots of genomic regions ordered by chromatin accessibility. We observe no enrichment of H3K27me3 in accessible regions, indicating minimal open chromatin bias. (E) Relationship between the number of cells included in a pseudobulk CUT&Tag profile of human PBMC, and the pearson correlation with a bulk experiment. (F) UMAP visualizations of 12,770 single cells profiled with H3K4me1 scCUT&Tag-pro and clustered on the basis of CUT&Tag profiles, cell surface protein levels, and weighted nearest neighbor (WNN) analysis which combines both modalities. Cluster labels are derived from WNN analysis. (G) Comparing pseudobulk CUT&Tag-pro profiles with ChIP-seq data from ENCODE. Including all cells assigned to each cell type results in pseudobulk tracks that closely mirror ENCODE profiles. However, even when downsampling to 300 cells per cluster, cell type-specific patterns can still be observed
In this study, we focused on acquiring scCUT&Tag-pro data from healthy human PBMC, and aimed to study the genome-wide localization patterns for six histone modifications (H3K4me1, H3K4me2, H3K4me3, H3K27ac, H3K27me3, and H3K9me3). In each experiment (Supplementary Table 1), we profiled a single histone modification, alongside an optimized panel of 173 antibodies from the Biolegend TotalSeq-A catalog (Supplementary Table 2). In total, we collected scCUT&Tag-pro from 64,876 individual cells (Supplementary Table 3).
We first evaluated the sensitivity and specificity of our CUT&Tag profiles. For example, in our H3K27me3 dataset we observed, on average, 501 unique fragments per cell with an 84.1% uniquely mapping rate (Supplementary Figure 1B), comparable to but slightly below a recent pioneering study19 introducing scCUT&Tag on single nuclei from the same system (802 average unique fragments/cell, Figure 1B, Supplementary Figure 1C). For all marks, we observed that a pseudobulk profile generated from our single-cell data closely mirrored simultaneously generated bulk CUT&Tag profiles from PBMC, exhibited minimal overlap with accessible regions identified in a scATAC-seq dataset28 (Figure 1C, D). Through downsampling analysis, we found that the quantitative accuracy of each histone modification pseudobulk profile was dependent on the number of included single cells, but began to saturate between 500-1,000 cells (Figure 1E). Moreover, we found that unsupervised clustering of each scCUT&Tag dataset using an LSI-based dimensional reduction workflow (Supplementary Methods) was capable of identifying major cell groups that define the human immune system (Figure 1F), but did not recapitulate the high-resolution identification of cell states as observed in scRNA-seq or CITE-seq data30.
Surface protein levels facilitate integration across experiments
We therefore examined the antibody-derived tags (ADT) for 173 cell surface proteins, measured simultaneously in each experiment (Supplementary Figure 1D). For example, in the H3K4me1 dataset, ADT expression patterns were fully concordant with cell types determined from chromatin-based clustering (i.e. uniform expression of CD3 in T cells, CD14 in monocytes, and CD19 on B cells; Supplementary Figure 1E). However, the protein modality revealed additional substructure, including mutual exclusivity of CD4 and CD8 in T cells, a division of monocytes into classical CD14+ and nonclassical CD16+ subgroups, and canonical marker expression denoting Mucosal Associated Invariant T (MAIT) (Figure 1F, Supplementary Figure 1E). We used our recently introduced Weighted Nearest Neighbor (WNN) analysis30 (Supplementary Methods) to simultaneously cluster scCUT&Tag-pro based on a weighted combination of modalities (Figure 1F, Supplementary Figure 2A, B). For each experiment, we found that the protein modality substantially enhanced our ability to separate distinct cell types, echoing our previous findings in CITE-seq and ASAP-seq analysis26,28 (Supplementary Figure 2C).
We compared the results of our single-cell analysis with FACS-sorted H3K4me1 ChIP-seq profiles from ENCODE35 (Supplementary Methods). We observed that pseudobulk profiles generated from our WNN-derived scCUT&Tag-pro clusters clearly recapitulated the cell type-specific ENCODE profiles (Figure 1G). We also profiled biological replicates for one of the histone modifications (H3K4me1) and observed that these profiles were reproducible across replicates as well (Supplementary Figure 2D). Moreover, downsampling our single-cell clusters to as low as 300 cells/profile still retained substantial cell type-specificity in the ensuing pseudobulk tracks (Figure 1G). Taken together, these analyses demonstrate that scCUT&Tag-pro datasets are comparable in sensitivity to existing technologies, exhibit low background, contain sufficient information to identify granular cell types and states, and can be used to generate pseudobulk profiles that reflect high-quality bulk data when a sufficient number of cells are included.
As an alternative to fully unsupervised analysis, we found that single-cell protein measurements also facilitated high-resolution supervised annotation using reference datasets. For example, we recently introduced a carefully annotated reference dataset of 58 major and minor cell types and states in the circulating human immune system, using a CITE-seq dataset of 161,764 cells and 228 surface proteins30. As our protein panel in this study largely overlaps with the reference dataset, we ‘mapped’ the cells from each scCUT&Tag-pro experiment onto our reference dataset (Supplementary Methods), and repeated the procedure for our published ASAP-seq dataset28 as well. Performing query-to-reference mapping using shared protein markers enables joint visualization of all datasets (Figure 2A, B), and also transfers a consistent set of annotations to each cell at multiple levels of resolution. This enabled us to partition each dataset into broad (level 1), granular (level 2), and fine-grained (level 3) classifications (Supplementary Figure 3A). We also used this approach to infer a unified pseudotime trajectory (Supplementary Methods) that modeled CD8 T cell transitions from naive to effector states, and confirmed the accuracy and resolution of our trajectory by comparing the developmental dynamics of key markers across experiments (Figure 2C, Supplementary Figure 3B).
Figure 2: Protein measurements facilitate integrated analysis across modalities.
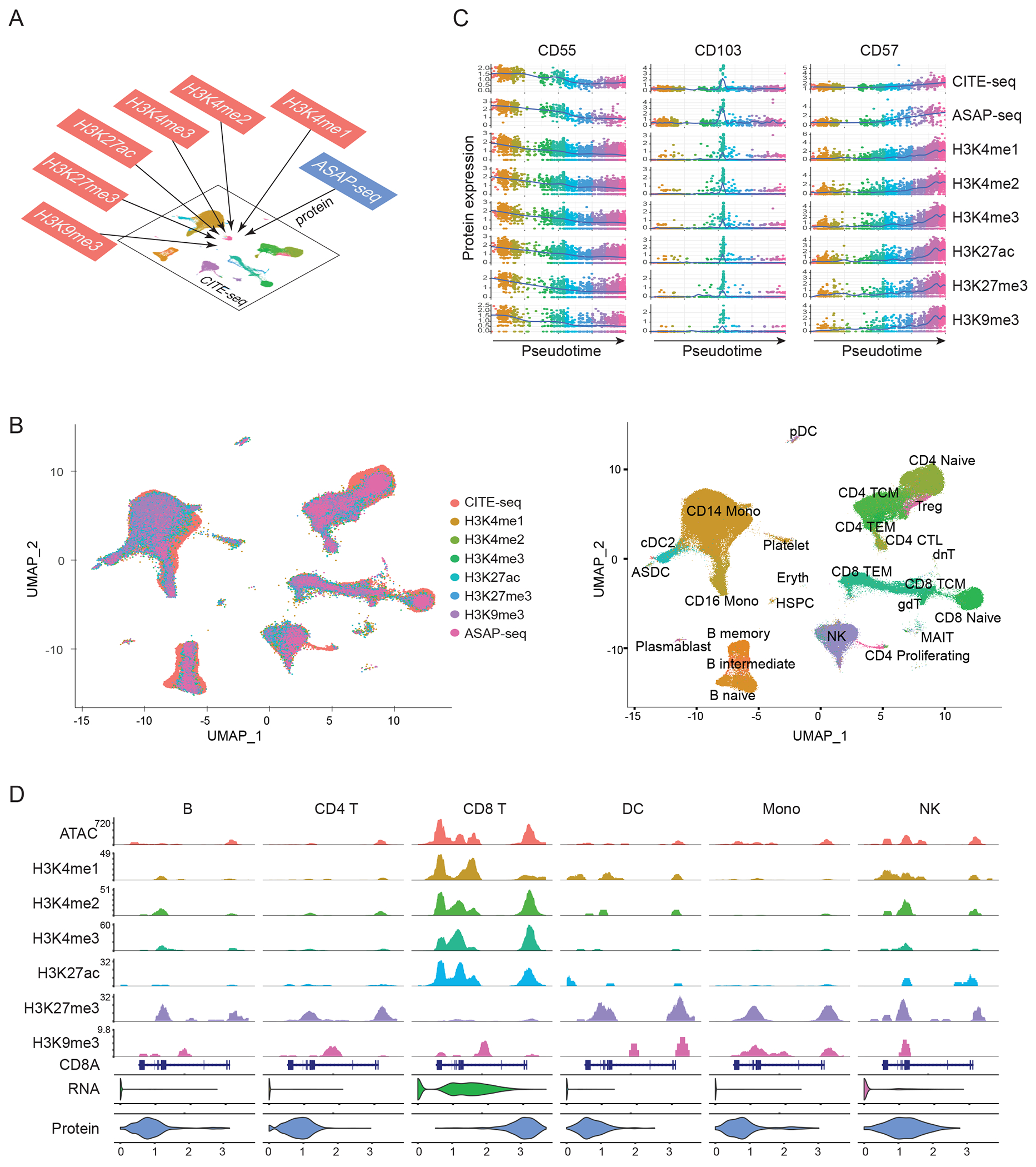
(A) Schematic workflow for integrated analysis. Datasets produced by scCUT&Tag-pro, ASAP-seq, and CITE-seq are integrated together on the basis of a shared panel of cell surface protein measurements. (B) Left: UMAP visualization of 230,597 total cells projected onto the reference dataset from (Hao et al, Cell 2021). Right: In addition to a harmonized visualization, cells from all experiments are annotated with a unified set of labels. (C) We learned a unified pseudotime trajectory based on all experiments, representing the CD8 T cell transition from naive to effector states. We observe identical molecular dynamics for naive (CD55), memory (CD103), and effector (CD57) markers across all experiments, demonstrating that integrative analysis accurately identifies cells in matched biological states across experiments. (D) Visualization of nine molecular modalities at the CD8A locus in B cell (B), CD4 T cell (CD4 T), CD8 T cell (CD8 T), dendritic cell (DC), monocyte (Mono) and natural killer cell (NK) groups.
Our reference-mapping workflow enables us to explore the relationships between nine different modalities by harmonizing them into a common space, and providing a consistent set of annotations (Figure 2B). The modalities span the central dogma and range from measurements of chromatin accessibility, protein-DNA interactions (six targets), gene expression, and protein abundances. While the individual modalities are collected in separate experiments, each originates from a multimodal technology (ASAP-seq, scCUT&Tag-pro, CITE-seq), that is paired with a large and comprehensive panel of surface protein measurements to facilitate integration. For example, in Figure 2D we visualize data from each modality collected at the CD8A locus. As expected, we find robust gene and protein expression (as measured by CITE-seq), enriched chromatin accessibility (as measured by ASAP-seq), and the presence of activating histone modifications (as measured by scCUT&Tag-pro) in CD8 T cells. In myeloid cell types, the locus is characterized by inaccessible chromatin and the presence of repressive histone modifications, which results in undetectable expression of CD8 RNA and protein.
scChromHMM characterizes chromatin state at single-cell resolution
We next aimed to use our dataset to explore heterogeneity in chromatin state within the human immune system. We first considered an analysis strategy exclusively based on tools that have been previously developed for bulk chromatin analysis, and provide 25 pseudobulk CUT&Tag tracks (based on level-2 reference derived annotations) as input. For example, ChromHMM12,17 trains a hidden markov model (HMM) on the concatenated histone modification profiles for all cell types, and applies the Baum-Welch training algorithm36 to identify a set of possible hidden ‘chromatin states’. ChromHMM then applies the forward-backward algorithm36 individually on each pseudobulk track, in order to calculate the posterior probability that each 200bp genomic window in each cell type is assigned to a particular chromatin state.
After running the Baum-Welch step (Supplementary Methods), ChromHMM obtained 12 states (Figure 3A), which could be broadly grouped into active promoter (enriched for H3K4me3/H3K4me2/H3K27ac), active enhancer (enriched for H3K4me1/H3K27ac), repressed (enriched for H3K27me3), and heterochromatic (enriched for H3K9me3) states. We also observed more subtle heterogeneity in chromatin state within these broad categories, likely corresponding to strong/weak states as has been previously described17. We identified a very similar set of chromatin states when running the Baum-Welch on pseudobulk tracks derived from level-3 annotations (Supplementary Figure 4A, B). These results suggest that the Baum-Welch step of ChromHMM can be successfully run on pseudobulk tracks generated from scCUT&Tag-pro, returning a functionally interpretable set of chromatin states.
Figure 3: scChromHMM annotates chromatin states at single-cell resolution.

(A) Chromatin states returned by ChromHMM, which was run on 25 pseudobulk tracks for six histone marks. States can be broadly grouped into five categories. (B) Correlation comparing cell type-specific pseudobulk profiles of H3K4me1 in CD14 monocytes, generated from the original experiment, or from the interpolated values. Each point corresponds to a 200bp genomic window (Supplementary Methods) (C) For each cell type, the interpolated and original profiles are highly correlated and clustered together. (D) scChromHMM outputs at the PAX5 locus. (Top) Pseudobulk profiles for six chromatin marks in three cell types. Yellow bar highlights a 200bp genomic window near the TSS. (Bottom) scChromHMM posterior probabilities representing the annotation for the highlighted window in each cell. The region is uniformly annotated with a promoter state in B cells where PAX5 is transcriptionally active, and as a repressive state in other cell types. (E) Metaplots exhibiting the enrichment of chromatin accessibility and histone modifications at functional regions identified by chromHMM (left) and scChromHMM (right) in CD14 monocytes.
However, when attempting to associate these states with individual genomic elements in each cell type, we identified a limitation associated with the application of bulk methods to single-cell datasets. In particular, we found that in contrast to the initial Baum-Welch step for state identification, the final outputs of ChromHMM were highly dependent on the level of granularity used when generating pseudobulk profiles. For example, when we considered genomic windows assigned to enhancer states in CD8+ T cells (level 1), 16.4% were not annotated as enhancers in any of the more granular T cell subsets when we repeated the analysis using a higher-resolution subset of cell labels (level 2). Especially when analyzing developmental tissues, or systems characterized by continuous sources of heterogeneity, it may be detrimental to condition all downstream analyses on a fixed set of discrete cell type labels37.
We therefore devised an alternative approach, scChromHMM, where chromatin states are assigned at single-cell resolution. In order to do so, we first generate single-cell profiles with simultaneous measurements of six histone marks. While it is not currently feasible to experimentally generate these data, we used our previously described anchoring workflow38 as a computational alternative. We recently demonstrated the use of this workflow to ‘transfer’ modalities across experiments38, for example, using a CITE-seq reference to accurately and robustly predict cell surface protein levels in an scRNA-seq dataset of human bone marrow. We apply a similar procedure in this study (Supplementary Methods), in order to interpolate 20,000 single-cell profiles, each of which consists of quantitative and genome-wide profiles for all six histone modifications and chromatin accessibility, alongside RNA and protein measurements. As in our previous studies, interpolated profiles represent a weighted average of ‘anchor’ cells, where the identification of anchors denotes a ‘matching’ of biological states across experiments based on shared protein expression patterns.
We assessed the accuracy of our interpolated profiles by comparing them to the original measurements that were obtained in separate experiments. For example, we computed the pseudobulk profile for H3K4me1 measurements based on our interpolated profiles, or the original measured values, and observed high quantitative concordance (Figure 3B; R=0.95). For each cell type, pseudobulk profiles of interpolated measurements clustered specifically with the pseudobulk profiles from the original measurements (Figure 3C, additional histone marks shown in Supplementary Figure 5A and cross comparison across marks show in Supplementary Figure 5B), demonstrating that our procedure retains cell type-specific variation. Finally, we repeated the interpolation procedure to generate an independent set of 20,000 profiles, and observed high reproducibility (R=0.98) between runs (Supplementary Figure 5C). While our procedure cannot capture stochastic fluctuations, it does represent a form of data ‘denoising’ by averaging histone modification signals across cells in similar biological states, and therefore alleviates the sparsity limitations associated with scCUT&Tag measurements. We emphasize that the acquisition of protein measurements is essential for this strategy, as the protein modality enables the accurate identification of anchors across diverse datasets.
Having acquired interpolated chromatin profiles for six histone modifications at single-cell resolution, we next aimed to annotate the chromatin state of each 200bp genomic window, in each cell. To achieve this, we ran the forward-backward algorithm12,36 individually on each cell (Supplementary Methods), using our interpolated profiles for six histone modifications and our previously calculated set of emission and transition probabilities as input. We note that data binarization is a requirement for this procedure and found that the frequency of our interpolated profiles across genomic windows exhibited clear evidence of bimodality, facilitating this process (Supplementary figure 6A). The output of scChromHMM represents, for each genomic window in each cell, the posterior probability distribution across 12 chromatin states. For example, in Figure 3D, we visualize the posterior probabilities (promoter) of a genomic window located at the PAX5 transcriptional start site (TSS), at single-cell resolution.
Regions classified by scChromHMM as active promoters were enriched near the TSS of actively transcribed genes, active enhancer regions were distal to the TSS but enriched for accessible chromatin, and heterochromatic regions largely overlapped (76.3%) with a set of repetitive elements annotated by RepeatMasker39 (Figure 3E, Supplementary Methods). We also found that putatively functional regions identified by scChromHMM exhibited increased accuracy and robustness compared to regions identified by ChromHMM run on pseudobulk profiles. Regions classified by scChromHMM consistently exhibited stronger enrichment for key histone modifications (Figure 3E) and RNA PolII (Supplementary Figure 6B) compared to active promoters identified by ChromHMM. We conclude that scChromHMM enables chromatin state predictions at single-cell resolution, and also improves the accuracy of state prediction.
Heterogeneity in repressive chromatin suggests regulatory priming
We used the output of scChromHMM to explore the dynamics of chromatin state changes across a trajectory of CD8 T cell maturation from naive to effector states. For example, we identified 14,585 genomic windows that acquired or lost a repressive chromatin state (Supplementary Methods), indicating that extensive remodeling of repressive chromatin is associated with this cellular process, particularly at the sharp transition from naive to memory cells (Figure 4A). In order to explore motifs that were associated with these transitions, we modified the chromVAR algorithm40 to identify motifs whose presence was correlated with an increased posterior probability across the learned chromatin states (Supplementary Methods).
Figure 4: Extensive heterogeneity in repressive chromatin encodes cellular identity.

(A) Remodeling of repressive chromatin during CD8 T cell maturation. Heatmap shows the posterior probabilities (repressive state) in single cells for 14,585 genomic loci, as returned by scChromHMM. Cells are ordered by their progression along pseudotime (Figure 2C). (B) ChromVar deviation scores for the TBX21 and LEF1 motifs in single cells, ordered by their progression along pseudotime. We used the scChromHMM-derived posterior probabilities as input to ChromVar, instead of chromatin accessibility levels. (C) Unsupervised analysis of scChromHMM-derived probabilities (repressive state) separates granular cell types. (D) Single-cell correlation matrix based on repressive chromatin at TSS (Supplementary Methods) when using all TSS (left heatmap), or after excluding the top 3,000 transcriptionally variable genes (right heatmap). In each case, the observed correlation structure is fully consistent with cell type labels, suggesting that there is extensive heterogeneity in repressive chromatin even for genes that do not vary transcriptionally. (E) Scatter plot showing average gene expression levels for all genes in CD14 monocytes (x-axis) and other cell types (y-axis). Colored points represent 1,597 loci where we detect changes in repressive chromatin for monocytes (Supplementary Methods). Blue points represent 1,340 loci where we do not detect an accompanying transcriptional change. Red points represent 257 genes where we detect a transcriptional shift. TPM: Transcripts Per Kilobase Million. (F) Four representative examples of individual genes shown as blue points in (E).
Our analyses revealed that genomic windows containing the motif bound by the transcription factor TBX21 (T-bet), which is a crucial regulator of T cell responses41,42, exhibit highly dynamic chromatin states during T cell maturation (Figure 4B). In particular, as cells transition from naive to memory and effector states, genomic windows containing TBX21 motifs acquire an increased probability of entering an active promoter or enhancer state. Conversely, genomic windows containing motifs bound by the critical T cell regulator LEF143 tend to exist in active chromatin states at the onset of the trajectory, but the same windows transition to acquire a repressive state as cells progress through the trajectory (Figure 4B). More generally, when aggregating cells into granular subgroups, we observed substantial cellular heterogeneity and cell type-specificity for motif activity scores computed based on scChromHMM-determined probabilities (Supplementary Figure 6C). This analysis nominated a suite of regulators for each cell type that were also differentially expressed at the transcriptional level (Supplementary Figure 6C). Taken together, our results demonstrate the potential for integrated analyses of scCUT&Tag data to identify DNA sequence motifs that correlate with diverse chromatin states.
We next asked if cellular heterogeneity in repressive chromatin state was fully reflected in the cellular transcriptome. We first considered the scChromHMM derived probabilities for the repressive state at genomic windows overlapping TSS. We found that granular cellular states could be separated based on unsupervised clustering of these measurements (Supplementary Methods), indicating that cellular identity is encoded not only in transcriptional output or immunophenotypes, but also in genome-wide profiles of repressive chromatin state (Figure 4C). To our surprise, we found that granular cellular identity remained encoded in repressive chromatin probabilities at TSS, even after excluding the 3,000 most variable genes in the transcriptome (Figure 4D). This suggested that even for genes whose expression did not vary across cell types, there may be substantial heterogeneity in chromatin state.
We therefore considered the set of loci where we observed cell type-specific changes in repressive chromatin state (Supplementary Methods), and calculated their overlap with transcriptional changes as well. For example, we observed 1,597 TSS loci whose posterior probability (repressive chromatin state) and H3K27me3 signal was enriched or depleted in CD14 monocytes. Within the associated genes for these loci, we detected monocyte-specific transcriptional shifts in only 257 (16.1%) (Figure 4E, F). When we did observe both transcriptional and chromatin-based shifts, they were highly concordant (i.e. the acquisition of a repressive state was associated with a decrease in gene expression). However, when we identified heterogeneity in repressive chromatin state in the absence of transcriptional variation, gene expression was either absent or at very low levels in all cell types (Figure 4E). When considering their pseudobulk expression, the median expression of the set of 1,340 genes in PBMC was below 0.5 transcripts per million. These genes are not informative when determining cellular state from scRNA-seq data, yet their repressive chromatin landscape exhibits clear patterns of cell type-specificity (Figure 4F).
We note that some genes exhibiting this pattern may in fact be differentially expressed across cell types but cannot be accurately quantified by scRNA-seq. To address this, we analyzed a publicly available transcriptome atlas dataset44, with high-quality bulk total RNA-seq profiles for sorted PBMC subsets. Of the 1,340 genes where we detected monocyte-specific changes in repressive chromatin but not transcription, 1,081 (81%) failed to show evidence of differential expression in bulk data as well (DESeq2 adjusted p-value > 0.01), demonstrating that our findings are not driven by artifacts driven by scRNA-seq.
Gene Ontology ontology analysis45 of these gene sets was enriched for DNA-binding transcription factors, and transmembrane and channel proteins whose activity may regulate the activation of downstream signaling pathways and immune responses (Supplementary Figure 6D). In a meta-analysis of these TSS, monocytes exhibited enrichment for H3K27me3, weak enrichment of H3K4me3 and H3K4me2, and the absence of H3K27ac (Supplementary Figure 6E). We therefore hypothesize that these chromatin shifts may represent a form of regulatory ‘poising’, that is not yet reflected in the current transcriptional output of each cell but may be relevant to its future behavior or potential. Our findings are consistent with previous studies of the epigenetic landscape in human T cells, which have shown that the promoters of many silent genes were enriched for active histone modifications, and could be rapidly induced in response to environmental stimuli46. Moreover, studies in differentiating T cells have found that distinct and lineage-specific chromatin states can be present even for non-expressed genes47–49.
Projecting scCUT&Tag datasets onto a multimodal atlas
Our analyses demonstrate the utility of projecting data from diverse molecular modalities into a harmonized manifold, creating a multimodal atlas that spans CITE-seq, ASAP-seq, and scCUT&Tag-pro experiments. However, it is not always possible to measure cell surface protein levels when performing scCUT&Tag, particularly when performing analysis of single nuclei. We therefore considered how to project unimodal scCUT&Tag profiles into our reference dataset.
We have recently released a framework, Azimuth30, which addresses a similar problem of projecting scRNA-seq datasets onto a CITE-seq defined human PBMC reference. In Azimuth, we use a semi-supervised dimensionality reduction approach, supervised PCA50, which identifies the best transcriptomically-defined vectors that separate CITE-seq defined cell types. We pursued a similar strategy to map a recently published H3K27me3 scCUT&Tag dataset of human PBMC nuclei19 onto our multimodal reference atlas. Using our scCUT&Tag-pro dataset as a reference, we calculated a modified LSI (Supplementary Methods) which transforms the histone modification profiles into a low-dimensional space that retains separation of our multimodally defined cell types. We use this transformation to identify anchors and ‘map’ the query dataset into our reference dataset (Supplementary Methods).
We observe that by performing reference-mapping we could substantially improve the interpretation of the query dataset, and in particular, the ability to resolve granular cell types. For example, unsupervised clustering of the query dataset19 was consistent with the previously published analysis and revealed broad clusters of immune subsets, including Monocytes, B, and T/NK subroups (Figure 5A). After reference mapping, monocytes could be segregated into CD14 and CD16+ subsets, T/NK subclusters could be further resolved into CD4 T, CD8 T, and NK groups with additional heterogeneity for naive and effector states, and B cells separated into different developmental stages (Figure 5B). We found that we could classify (prediction score >0.5) 88% of cells at level-1 resolution, and 73% of cells at level-2 resolution. We find high concordance when comparing patterns of cell type-specific H3K27me3 binding compared in both the reference and query datasets (Figure 5C–E), verifying the accuracy of our predictions. We note that while we transfer discrete annotations in this example, it is also possible to transfer continuous data, which would enable the prediction of one histone modification based on the measurement of another. Therefore, we propose that our multimodal atlas represents a broad scaffold for the circulating human immune system that can be used to map datasets from a wide variety of cutting edge single-cell technologies.
Figure 5: Supervised mapping of scCUT&Tag datasets.
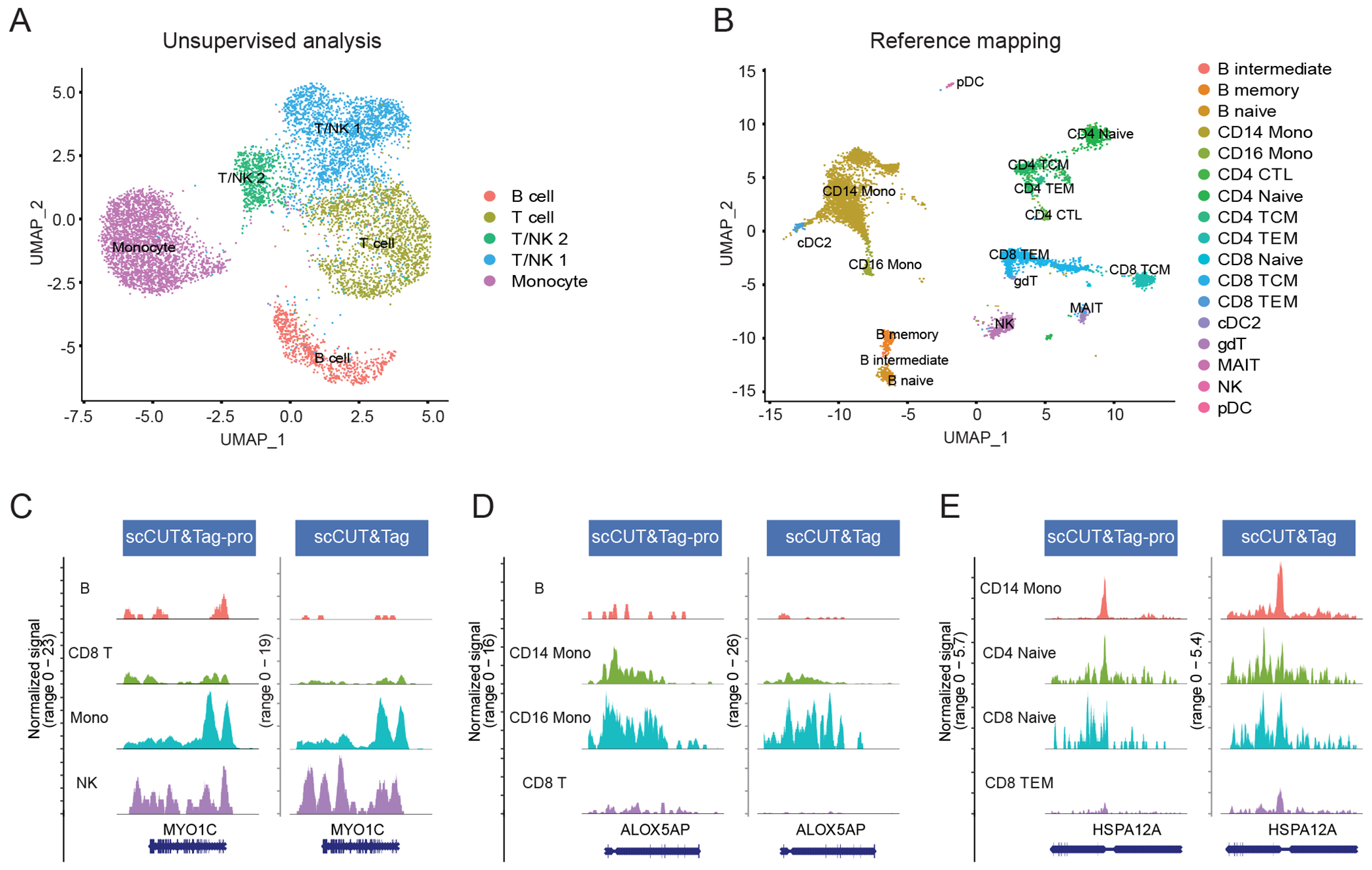
(A) UMAP visualization of 8,362 H3K27me3 scCUT&Tag profiles of human PBMC, from (Wu et al, 2021), based on an unsupervised analysis and clustering. (B) Same cells as in (A), but after mapping to the multimodal reference defined in this paper. Cells are colored by their reference-derived level 2 annotations. (C)-(E) Coverage plots showing the cell type-specific binding patterns of H3K27me3 at three loci. Plots are shown for our dataset (reference), as well as the scCUT&Tag profiles from the query dataset (query, Wu et al, 2021). Cells in the query dataset are grouped by their predicted labels. We observe highly concordant patterns across datasets for all loci, supporting the accuracy of our predictions. Four representative cell types are shown at each locus. TEM: T effector memory.
Discussion
One of the key goals of multi-omic single-cell studies is to explore the relationships across different molecular modalities, and to explore how changes in one modality affect variation in another. Here, we pursue an integrative analysis where nine different molecular modalities are each measured in separate experiments. We subsequently integrate these data and derive what we colloquially refer to as single-cell ‘mega-omic’ profiles, each representing an individual cell containing measured or interpolated measurements for many modalities. In addition, we introduce scChromHMM, which enables the exploration of heterogeneity in chromatin state across both discrete cell types and continuous trajectories.
Substantial recent progress has been made in simultaneous profiling of multiple modalities in single cells22,23,51,52, but currently it is not feasible to simultaneously profile the genome-wide binding patterns of six different histone modifications in the same cell, particularly when there is a partial overlap in their localization. Additionally, future extensions of CUT&Tag enabling simultaneous measurements of RNA and protein levels are likely to result in decreased per-modality data quality, as has been previously described28,29. Our approach therefore represents a feasible and broadly applicable alternative, and we demonstrate that our ‘mega-omic’ profiles are highly quantitative, sensitive, and robust. However, interpolated modality predictions cannot capture stochastic technical or biological variation or be used to detect associations between multiple histone modifications within the same cell.
The successful integration of CITE-seq, ASAP-seq, and scCUT&Tag-pro datasets originates from a shared panel of cell surface protein levels that are collected in each experiment. We emphasize that cell surface protein levels represent one of multiple potential modalities that can be used for ‘mega-omics’ integration. For example, two recent pioneering approaches (Paired-Tag22, and CoTECH23) introduced combinatorial indexing-based approaches for simultaneous scCUT&Tag and scRNA-seq in single cells, enabling the integration of additional multi-omic technologies (i.e. Paired-Seq) via transcriptomic measurements. Our scCUT&Tag-pro technology represents a complementary solution that is compatible with the 10x Genomics Chromium system, and will be particularly valuable for profiling immune cell types that are well-defined by their surface protein landscape.
We found substantial heterogeneity in chromatin state even at genes that did not exhibit transcriptional variation. In particular, we observed cell type-specific repression, as demarcated by the presence of H3K27me3, even for genes that were expressed at similarly low levels across all cell types. While it is possible that this heterogeneity in repressive chromatin state has minimal functional consequence, cellular variation in chromatin state may also underlie future cell type-specific transcriptional responses. In addition to previous findings in human immune cells46–49, Mellis et al. found that cell type specificity was encoded not only in steady-state transcriptional output, but also in future responses to environmental perturbations53. Similarly, Shim et al. found that genes with heterogeneous H3K27me3 landscapes across distinct organ systems were enriched for key developmental, regulatory, and morphological functions54. We propose that scCUT&Tag-pro represents a sensitive technique for identifying loci exhibiting heterogeneous repressive chromatin within an organ system as well. Future experiments profiling chromatin state in the bone marrow will help to elucidate the developmental dynamics that establish this heterogeneity.
Moving forward, we expect that integrated analyses across multiple modalities will not only help to understand the molecular state of single cells, but also will reveal new insights on fundamental questions in gene regulation. For example, the activation of gene expression is correlated with changes in chromatin accessibility, the acquisition and loss of histone modifications, the binding of transcription factors and RNA polymerase, and the formation of new DNA contacts. However, the regulatory relationships and temporal ordering between these steps remains poorly understood. Motivated by the ability of RNA velocity analysis to use known temporal relationships between unspliced and spliced reads in order to infer the direction of developmental trajectories55, we envision that single-cell multi-omic analysis will help to order the coordinated molecular events that collectively establish cellular heterogeneity.
Methods:
Protocols
A step-by-step online protocol for scCUT&Tag-pro is available on the protocols.io platform, here: https://www.protocols.io/private/57E45A034D5311ECA43B0A58A9FEAC02.
PBMC acquisition and processing
Cryopreserved healthy donor PBMCs were purchased from AllCells. After thawing into DMEM with 10% FBS, the cells were spun down at 4°C for 5 min at 400 g and washed twice with PBS with 2% BSA. After centrifugation, the cell pellet is resuspended in staining buffer (2% BSA and 0.01% Tween in PBS).
scCUT&Tag antibodies
The antibodies used were H3K4me1 (1:100, Abcam, ab8895), H3K4me2 (1:100, Abcam, ab32356), H3K4me3 (1:100, Abcam, ab213224), H3K27ac (1:100, Abcam, ab177178), H3K27me3 (1:100, Cell Signaling Technology, 9733), H3K9me3 (1:100, Abcam, ab8898), Phospho-Rpb1 CTD (Ser2/Ser5) (1:50, Cell Signaling, 13546) and guinea pig anti-rabbit (1:100, Novus Biologicals, NBP1-72763). TotalSeq-A conjugated antibodies and panels were obtained from BioLegend (399907, see Supplementary Table 2 for a list of antibodies, clones and barcodes).
scCUT&Tag-pro experimental workflow
Surface Protein Antibody staining
5 million thawed PBMC were resuspended in 200 µL staining buffer (2% BSA and 0.01% Tween in PBS) and incubated for 15 min with 10 µL Fc receptor block (TruStain FcX, BioLegend) on ice. The samples were evenly distributed into 5 tubes and 2 µL hashing antibody was added separately. Cells were then washed three times with 1 mL staining buffer and pooled together. 2 million PBMC were used for each histone modification. After cell hashing, the panel of oligo-conjguated antibodies (one test is sufficient for 2 million cells) was added to the cells to incubate for 30 min on ice. After staining, cells were washed three times with 1 mL staining buffer and resuspended in 100 µL staining buffer. Subsequently, 4 µL Fab Fragment Goat Anti-Mouse IgG (Jackson ImmunoResearch, 115-007-003) was added for incubation on ice for 15 min. The cells were then washed three times with 1 mL staining buffer. After the final wash, cells were resuspended in 200 μl PBS and ready for fixation.
Fixation and permeabilization
1.25 µL of 16% Methanol-free formaldehyde (Thermo Fisher Scientific, PI28906) was added for fixation (final concentration: 0.1%) at room temperature for 5 min. The cross-linking reaction was stopped by addition of 12 µL 1.25M glycine solution. Subsequently, cells were washed twice with PBS. The permeabilization was performed by adding isotonic lysis buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 3 mM MgCl2, 0.1% NP-40, 0.1% Tween-20, 1% BSA, 1× protease inhibitors) on ice for 7 min. Subsequently, 1 mL of cold wash buffer (20 mM HEPES pH 7.6, 150 mM NaCl, 0.5 mM spermidine, 1× protease inhibitors) was added, and cells were centrifuged at 800g for 5 min at 4°C.
Tagmentation
Permeabilized cells were directly resuspended with 150 µL antibody buffer (20 mM HEPES pH 7.6, 150 mM NaCl, 2 mM EDTA, 0.5 mM spermidine, 1% BSA, 1× protease inhibitors) with primary antibody (for example, anti-H3K4me1) and incubated overnight on a rotator at 4°C. The next day, cells were washed once with 150 μl wash buffer and centrifuged for 5 min at 800g. After removing the supernatant, the cells were resuspended in 150 μL of wash buffer with secondary antibody and incubated for 1h at room temperature on rotator. The cells were washed twice with 150 μL wash buffer to remove excess remaining antibodies. The cells were then resuspended in 150 μL high salt wash buffer (20 mM HEPES pH 7.6, 300 mM NaCl, 0.5 mM spermidine, 1× protease inhibitors) with 7.5 µL pAG-Tn5 (EpiCypher, 15-1017) and incubated for 1 h on rotator at room temperature. The cells were then washed twice with high salt wash buffer and resuspended in 100 μL tagmentation buffer (20 mM HEPES pH 7.6, 300 mM NaCl, 0.5 mM spermidine, 10 mM MgCl2, 1× protease inhibitors). The samples were placed on PCR machine to incubate for 1h at 37 °C. When tagmentation was done, the reaction was stopped by adding 4 μL of 0.5 M EDTA. Tagmentation steps were performed in 0.2 mL tubes in order to minimize cell loss.
Single-cell encapsulation, PCR, and library construction
After tagmentation, cells were centrifuged for 5 min at 1000g and the supernatant was discarded. Cells were resuspended with 30 µL 1× Diluted Nuclei Buffer (10x Genomics), counted, and diluted to a concentration based on the targeted cell number. The transposed cell mix was prepared as following: 7 µL of ATAC buffer and 8 µL cells in 1× Diluted Nuclei Buffer. All remaining steps were performed according to the 10x Chromium single-cell ATAC protocol and the library construction method was adapted from ASAP-seq28. Briefly, 0.5 μL of 1 μM bridge oligo A (TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGNNNNNNNNNVTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT/3InvdT/) was added to the barcoding mix. Linear amplification was performing using the following PCR program: (40 °C for 5 min, 72 °C for 5 min, 98 °C for 30 s; 12 cycles of 98 °C for 10 s, 59 °C for 30 s and 72 °C for 1 min; ending with hold at 15 °C). The remaining steps were performed according to the 10x scATAC-seq protocol (v1.1), with the following additional modifications:
Antibody-derived tags: During silane bead elution (Step 3.1o), beads were eluted in 43.5 μL of Elution Solution I. The extra 3 μL was used for the surface protein tags library. During SPRI cleanup (Step 3.2d), the supernatant was saved and the short DNA derived from antibody oligos was purified with 2.0× SPRI beads. The eluted DNA was combined with the 3 µL left aside after the silane purification to be used as input for protein tag amplification. PCR reactions were set up to generate the protein tag library with KAPA Hifi Master Mix (P5 and RPI-x primers): 95 °C for 3 min; 14–16 cycles of 95 °C for 20 s, 60 °C for 30 s and 72 °C for 20 s; followed by 72 °C for 5 min and ending with hold at 4 °C.
P5 primer: AATGATACGGCGACCACCGAGATCTACAC
RPI-x primer: CAAGCAGAAGACGGCATACGAGATxxxxxxxxGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA.
Sequencing
The final libraries were sequenced on NextSeq 550 with the following recipe: i5:16bp, i7: 8bp, read1: 34bp, read2: 34bp.
scCUT&Tag-pro data preprocessing
Antibody-derived tags and Cell Hashing libraries
We use salmon alevin 1.4.056 for the alignment and quantification of single-cell antibody-derived tags (ADT) and hashtag oligos (HTO), using a custom index that is constructed only from known ADT and HTO barcode sequences. We apply a threshold of 10 protein and 10 hashing antibody counts. Cells that pass these thresholds are used as input to the HTODemux() function in Seurat30 for demultiplexing and doublet removal. Antibody data was normalized using the centered log ratio transformation. In order to visualize cells on the basis of their ADT profiles, we performed PCA using all features as input, and retained the first 25 principal components to construct a nearest-neighbor graph and perform UMAP visualization.
pAG-Tn5 tagmented libraries (i.e. scCUT&Tag)
We use cellranger-atac v1.2 pipeline with default settings to align reads to the hg38 human reference annotation and generate fragment files. We filter the fragment file to keep the cellular barcodes (CBs) that were present in the filtered ADT and HTO libraries, and also contained at least 100 fragments. We use the AggregateTiles function from the Signac package57 to quantify the fragment coverage based on 5,000bp genomic windows, remove windows with less than 5 counts, and then merge adjacent windows. We next perform modified TD-IDF normalization57, which corrects for differences in cellular sequencing depth and weights each window based on its average fragment abundance across all cells. We next run singular value decomposition (SVD) on the TF-IDF matrix to generate the low dimensional representation of the data. We use dimensions 2:30 for UMAP visualization, as the first dimension is highly correlated with sequencing depth.
We use the Seurat v4 Weighted Nearest Neighbor (WNN) framework30 to generate a multimodal representation of the scCUT&Tag-pro datasets. We use the FindMultiModalNeighbors function to generate a WNN graph, using dimensions 1:25 (ADT modality) and dimensions 2:30 (scCUT&Tag modality).
Reference mapping
We integrate each of our scCUT&Tag-pro datasets with a previously published reference CITE-seq dataset30. In previous work, we apply ‘supervised PCA’ (sPCA) to identify a linear projection of the scRNA-seq measurements in the CITE-seq dataset that maximally retains the information captured in both modalities. This transformation can be applied to new scRNA-seq query datasets in order to identify anchors with the CITE-seq reference. These anchors are used for annotation and visualization of the query cells. Our framework for query-to-reference mapping is implemented as part of our Azimuth tool, accessible at azimuth.hubmapconsortium.org and fully described in our previous paper30.
Here, we employ a similar strategy, but perform sPCA on the protein measurements from the CITE-seq dataset. As input to sPCA, we use all 128 protein features that were measured both in this study as well as in the reference dataset. This procedure identifies a linear transformation of cell surface protein measurements that can be applied to scCUT&Tag-pro measurements. We use the FindTransferAnchors anchors (reduction = ‘spca’, dims = 1:30) in Seurat v4 to identify anchors between reference and query datasets, and the MapQuery function (default parameters) in order to annotate query cells at multiple levels of resolution, and to visualize these cells in a 2D UMAP. After all datasets have been mapped, we subset all CD8 T cells (which encompass memory, naive, and effector states). We use monocle3 with default parameters58 to identify an integrated developmental trajectory that orders cells from all experiments.
Interpolating multiple histone modification profiles in single cells
In order to perform an integrated analysis of chromatin state at single-cell resolution, we first generate a set of single-cell profiles that comprise interpolated, genome-wide, and quantitative measurements of six histone modifications. In previous work38, we demonstrated how to use anchors between reference and query datasets to transfer not only discrete information (i.e. cell type annotations), but also quantitative measurements. For example, we demonstrated how to accurately interpolate cell surface protein measurements for a scRNA-seq dataset of 274,599 human bone marrow mononuclear cells generated by the Human Cell Atlas, based on a CITE-seq dataset of 35,543 cells from the same system.
We apply a similar workflow for this study (schematized in Figure 2A). First, we consider a CITE-seq dataset30, where each cell contains transcriptomic measurements alongside quantifications for a panel of 228 surface proteins. While the original dataset contains 161,764 cells, interpolating genome-wide profiles for multiple modalities at this scale creates memory and storage constraints, and we therefore select a random subset of 20,000 cells for downstream analysis. In Supplementary Figure 3B, we demonstrate that we obtain highly concordant results when repeating the procedure on a second random downsample of 20,000 cells.
We then find anchors between each scCUT&Tag-pro dataset, and the 20,000 CITE-seq cells. Specifically, the ‘anchors’ in this procedure represent pairwise correspondences of cells: a CITE-seq cell that lacks the histone modification profile of interest, and an scCUT&Tag-pro cell in a matched biological state, but where the histone modification was directly measured. Anchors are determined using the FindTransferAnchors function, and use the same protein sPCA transformation as described above (‘Reference mapping’). We use these anchors to interpolate (i.e. ‘impute’) the number of fragments for each histone modification, in each 200bp genomic window, at single-cell resolution (see Stuart et al. 201938, ‘Feature Imputation’). For example, in each of the 20,000 CITE-seq cells, the interpolated levels of H3K4me1 represent a weighted average of the anchor-forming cells in the H3K4me1 scCUT&Tag-pro. We repeat this procedure for each of the six modifications, as well as the ASAP-seq dataset28. Therefore, each of the 20,000 cells contains information for nine modalities, encompassing both the original measurements (RNA + protein), as well as seven interpolated modalities representing histone modifications and chromatin accessibility.
Based on our pseudobulk saturation analysis (Figure 1E), we used a weighted average of 500 anchors when interpolating values for each cell. We highlight that this weighting prioritizes anchors that are most similar to the query cell, enabling us to accurately interpolate histone modification profiles even for cell types present at moderate abundance (Figure 3C, Supplementary Figure 3A). However, for rare cell types, there is a possibility that an insufficient number of anchors may reduce the accuracy of these predictions. We therefore removed cell types that were present at <2.5% frequency in our reference dataset from further analysis.
In Figure 3B we plot the correlation between interpolated and original values for the H3K4me1 histone modification. Each point represents a 200bp genomic window. To avoid overplotting, we only plot windows on chr22 (n=254,093).
Assigning chromatin states at single-cell resolution
In order to integrate information from multiple histone modifications we use the multivariate Hidden Markov Model introduced in ChromHMM12. For clarity, we reintroduce the key components of the model here. The model assumes that each 200bp genomic window (‘genomic interval’) can be represented based on one of K hidden states, where each state is defined by the combinatorial presence or absence of multiple histone modifications. The full likelihood of the model is:
Where v represents the observed data, such that is ‘1’ if mark m is present at genomic interval and ‘0’ otherwise. represents the probability that mark m is present at a genomic interval in state k (‘emission probability’), represents the probability of the markov chain transitioning from state i to state k (‘transition probability’), and represents the ‘hidden state’ associated with each at genomic interval .
The first step of the procedure is to learn a set of possible chromatin states, and their associated emission and transition probabilities. We use ChromHMM’s variant of the Baum Welch algorithm12 as implemented in the LearnModel command, setting the number of states to 12, and the maximum number of iterations to 1,000. As input to ChromHMM17, we provide the histone modification profiles for all cell types, where cell type identity was determined after reference mapping (level 2 resolution). The 12 learned states and associated emission probabilities are shown in Figure 3A.
After learning a set of chromatin states, we next infer the path of hidden states (hidden state sequence) through each chromosome, at single-cell resolution. To do so, we extend the ChromHMM method in order to calculate the posterior distribution of state assignments for each 200bp genomic interval in each cell. We refer to the following procedure as scChromHMM:
The input to scChromHMM is the set of interpolated histone modifications in single cells, as described above
As ChromHMM models multiple chromatin marks as an independent set of Bernoulli random variables, we must binarize the interpolated values present in each cell. As shown in Supplementary Figure 4A, the distribution of abundances for each histone modification exhibited clear bimodality. We assigned binarization thresholds via manual inspection (red dashed line), which were congruent with the results of Hartigan’s dip test. Binarization thresholds were set independently for each mark, but kept constant across single cells. After thresholding, each genomic interval in each cell is represented by the presence or absence of six histone modifications.
We run the forward-backward algorithm on each cell independently, a dynamic programming algorithm to learn the posterior probabilities of hidden state variables in a hidden markov model36. The output of this procedure is a posterior probability distribution over all 12 states for each genomic interval in each cell.
Comparison of scChromHMM and ChromHMM results
In addition to scChromHMM, we also annotated genomic intervals using the standard ChromHMM workflow17. While this approach does not return state predictions in individual cells, it does return posterior probability distributions for each cell type (level 2 resolution). We therefore compared the predictions from ChromHMM17 with the predictions from scChromHMM after grouping by celltype. In Figure 3E we perform a meta-analysis across all regions assigned to promoter, enhancer, repressor, and heterochromatic states, and explore the level of enrichment for each modification in a 10kb window centered on these regions. We combined the posterior probabilities for functionally related states (promoters: state 1 and 2; enhancers: state 3 and 4; repressor: state 9 and 10).
We compare results on CD14 monocytes, as this is the most abundant cell type in our dataset. For ChromHMM17, we selected all regions with >75% probability posterior probability in CD14 monocytes for meta-analysis, and for scChromHMM we selected all regions where the majority of CD14 monocytes had >75% posterior probability. In Supplementary Figure 4B, we also compare average PolII fragment abundance as measured by scCUT&Tag-pro on promoter regions as identified by either ChromHMM17 or scChromHMM.
Identifying heterogeneity in chromatin state across a continuous trajectory
In previous work38, we used the Moran’s I statistic measure of spatial autocorrelation to identify genes whose expression varied as a function of spatial location. Here, we use a similar approach to identify 14,585 genomic intervals whose posterior probability (repressive state) varies across a continuous trajectory spanning Naive to Effector states in CD8 T cells (Figure 4A). We extract 3,157 CD8 T cells where we have calculated posterior state probabilities using scChromHMM, and where we also have obtained pseudotime estimates (see ‘Reference Mapping’). We use the pseudotime values to construct a Gaussian similarity kernel with unit bandwidth, which defines the spatial weights associated with the Moran’s I calculation. We calculate Moran’s I for each genomic interval as previously described38, and acknowledge the Trapnell Lab Monocle 3 tutorials for suggesting the use of Moran’s I to estimate spatial autocorrelation in single-cell data. We considered all intervals with Moran’s I > 0.15 as varying across the trajectory. In Figure 4A, we visualize the posterior probabilities of these regions as a function of pseudotime, after applying a moving average filter (k=21).
Using chromVAR to identify associations between motifs and functional states
The chromVAR package40 identifies associations between transcription factors and chromatin accessibility by examining deviations (i.e. gains/losses) in the accessibility of peaks that share the same DNA sequence motif while carefully controlling for technical biases. Here, we aim to detect associations between transcription factors and the acquisition of distinct chromatin states. We therefore ran the chromVAR algorithm40, but instead of passing single-cell chromatin accessibility profiles as input, we used our single-cell posterior state probabilities. We repeat this process separately for promoter, enhancer, and repressor states. Using the promoter state as an example, chromVAR calculates - for each motif and each cell - the difference between the observed promoter posterior probabilities for peaks containing the motif, and the expected posterior probability (based on an average across all cells). chromVAR next performs a similar calculation for a matched background peak set in order to calculate bias-corrected deviation scores (Figure 4B, Supplementary Figure 4C).
Characterizing regulatory priming based on repressive chromatin state
In Figure 4, we analyze cells based on their posterior probability (repressive state) at 19,306 transcriptional start sites (TSS). Based on a previous TSS definition54, we consider all genomic intervals that overlap with a window starting 2,500bp upstream of the TSS, and extending 500bp downstream. In each cell, we quantify the level of repression at each TSS as the average of the repressive state posterior probability for each of these windows. We generate a repressive score matrix that represents the level of repression at each cell (rows), at each TSS (columns). We apply TF-IDF normalization57 to this matrix followed by SVD, and use singular vectors 2:30 as input to UMAP visualization in Figure 4C.
In Figure 4D, we visualize cell-cell correlations based on the repressive score matrix. In the left heatmap, we use all 19,306 TSS as input to LSI, and use dimensions 2:30 to calculate a correlation matrix. In the right heatmap, we perform the same procedure, but exclude the top 3,000 TSS that were identified as highly variable based on gene expression levels, as determined by SCTransform59. In both heatmaps, cells are grouped based on their predicted annotation, but the order of cells within an individual group is random.
In Figure 4E, we consider the overlap between heterogeneity in repressive chromatin state and transcriptional variation. Using the FindMarkers command in Seurat (default parameters), which performs a wilcox sum rank test, we compared CD14 Monocytes to all other cells. We performed tests for scChromHMM-derived posterior probabilities (repressive state), H3K27me3 levels (as measured by scCUT&Tag-pro), and gene expression (as measured by CITE-seq), and kept all results with adjusted p-value < 0.01. To ensure that we robustly identified regions varying in their repressive state, we intersected the lists of TSS identified as differential by scChromHMM as well as H3K27me3, resulting in 1,597 loci. Of the associated genes, 257 were identified as differentially expressed in the transcriptome.
Comparative analysis to detect differentially expressed genes in the bulk RNA-seq dataset
In figure 4E, we show gene loci where we observed cell type specific changes in repressive chromatin states, and calculated their overlap with transcriptional changes as well. We identified 1,340 loci where we observed shifts in repressive chromatin when comparing CD14 monocytes to remaining cell types, but did not detect transcriptional differences between these cell types in scRNA-seq data. We further validate the lack in transcriptional shifts of the 1,340 loci using bulk RNA-seq data. We quantify bulk RNA-seq sorted PBMC dataset using salmon 1.4.056 for four major cell-types CD14 Monocyte (3 replicates), B (2 replicates), CD8 T (2 replicates) and Natural killer (3 replicates) cells. We perform differential expression analysis on the bulk RNA-seq data comparing the TPM estimates of the CD14 monocytes with other cell-types using DESeq260 1.30.1. Of the 1,340 loci- 1,081 (81%) did not show evidence of differential expression in the bulk RNA-seq dataset (DESeq260 adjusted p-value > 0.01). Of the remaining 257 loci that did show transcriptional differences in scRNA-seq data, 74% did show evidence (adjusted p-value <0.01) in bulk data as well.
Mapping scCUT&Tag datasets to a multimodal reference
In Seurat v430, we use ‘supervised PCA’ (sPCA) to integrate multimodal (CITE-seq) and unimodal (scRNA-seq) datasets. The sPCA reduction aims to project transcriptomic measurements into a low-dimensional space that most closely resembles the WNN graph. We construct a weighted nearest neighbor graph that identifies, for each cell, a set of neighbor cells in the most similar molecular state based on a weighted combination of RNA and protein modalities. This graph represents a linear kernel, L, that can be used to supervise the dimensional reduction process for transcriptomic measurements.
As described in (Barshan et al, 2011)50, there is a closed form solution to sPCA based on a data matrix X, kernel L, and centering matrix H. The projection matrix U is represented by the eigenvectors of matrix . We apply the resulting projection matrix () to new scRNA-seq datasets, in order to identify anchors between query and reference datasets. We can repeat the same process using the protein modality, in order to calculate ().
Here, we apply a conceptually similar procedure to the integration of multimodal (H3K27me3 scCUT&Tag-pro, from this study) and unimodal datasets (H3K27me3 scCUT&Tag from Wu et.al.)19. For unsupervised analysis of the scCUT&Tag dataset, we downloaded fragment files from Gene Expression Omnibus with accession code GSE157910. We filtered cells with less than 250 fragments or more than 5,000 fragments, and repeated the LSI-based analysis workflow to perform unsupervised analysis (Figure 5A).
For each scCUT&Tag-pro dataset, the reference mapping procedure described above uses the protein measurements to map each cell into a reference-defined space. We construct a k-nearest neighbor graph (k=30) in this space. This defines a kernel matrix L, where if cells i, j are neighbors and 0 otherwise. Our goal is to compute a singular value decomposition of the matrix XL, where X is the TF-IDF normalized peak barcode matrix representing the quantified scCUT&Tag profiles in the multimodal experiment. Unlike scRNA data, the large number of features (peaks) in the scCUT&Tag profiles makes it computationally expensive to perform SVD. We circumvent this problem by performing eigendecomposition of the matrix , where the dimensionality is determined by the number of cells, but returns the same result. The result is a modified LSI that represents a low-dimensional transformation of the chromatin profiles, but has been supervised by the protein measurements. We use the FindTransferAnchors (reduction = ‘lsiproject’, reference.reduction = ‘SLSI’, dims = 1:30, k.anchor = 20), to identify anchors between our scCUT&Tag-pro dataset and the unimodal query. We use these anchors to annotate cell types in the query dataset and assign prediction scores.
Lastly, we also use these anchors to interpolate protein values for 173 surface proteins in the scCUT&Tag query dataset. We then apply the previously computed transformation to map these cells onto the original CITE-seq reference30 and we filter cells with prediction score < 0.5. We perform this step for visualization purposes only, as it allows us to view the query scCUT&Tag dataset in the same UMAP visualization as Figure 2B, which represents all datasets in this study (Figure 2B).
Supplementary Material
Acknowledgements:
The authors would like to thank all the members of the Satija Lab for thoughtful discussions related to this work. B.Z. is a postdoctoral fellow of the Jane Coffin Childs Memorial Fund for Medical Research. This investigation has been aided by a grant from the Jane Coffin Childs Memorial Fund for Medical Research. This work was supported by the Chan Zuckerberg Initiative (EOSS-0000000082, HCA-A-1704- 01895 to R.S.), and the NIH (K99CA267677-01 to A.S; K99HG011489-01 to T.S; RM1HG011014-02, 1OT2OD026673- 01, DP2HG009623-01 to R.S).
Footnotes
Code Availability:
Seurat, Signac, and scChromHMM are freely available as open-source software packages:
https://github.com/satijalab/seurat
Reporting Summary:
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Competing interests:
In the past three years, R.S. has worked as a consultant for Bristol-Myers Squibb, Regeneron, and Kallyope and served as an SAB member for ImmunAI, Resolve Biosciences, Nanostring, and the NYC Pandemic Response Lab. P.S. is co-inventor of a patent related to this work. The other authors declare no competing interests.
Data availability:
Data generated for this manuscript are deposited in the Gene Expression Omnibus with accession code: GSE195725. The processed datasets are available as open-access downloads at: https://zenodo.org/record/5504061. The publicly datasets were used in this study: GSM4732109 and GSE164378 (Figure 2, 3, 4 and Supplementary Figure 3); GSM4732123 (Figure 1D); GSM1220567, GSM1220569, GSM1027296 and GSM1102793 (Figure 1G); GSM5034342 and GSM5034344 (Supplementary Figure 1C and Figure 5); Datasets used in DE analysis with the bulk total RNA-seq: http://r2platform.com/rna_atlas and https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE138734
References:
- 1.Tang F et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Jaitin DA et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 343, 776–779 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Macosko EZ et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hashimshony T, Wagner F, Sher N & Yanai I CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2, 666–673 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Picelli S et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 10, 1096–1098 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Shalek AK et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 498, 236–240 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buenrostro JD et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cusanovich DA et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.ENCODE Project Consortium. A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 9, e1001046 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoffman MM et al. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat. Methods 9, 473–476 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bannister AJ & Kouzarides T Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ernst J & Kellis M Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 28, 817–825 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strahl BD & Allis CD The language of covalent histone modifications. Nature 403, 41–45 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Jenuwein T & Allis CD Translating the histone code. Science 293, 1074–1080 (2001). [DOI] [PubMed] [Google Scholar]
- 15.Park PJ ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 10, 669–680 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaya-Okur HS et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 10, 1930 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ernst J & Kellis M ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bartosovic M, Kabbe M & Castelo-Branco G Single-cell CUT&Tag profiles histone modifications and transcription factors in complex tissues. Nat. Biotechnol. 39, 825–835 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu SJ et al. Single-cell CUT&Tag analysis of chromatin modifications in differentiation and tumor progression. Nat. Biotechnol. 39, 819–824 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Q et al. CoBATCH for High-Throughput Single-Cell Epigenomic Profiling. Mol. Cell 76, 206–216.e7 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Carter B et al. Mapping histone modifications in low cell number and single cells using antibody-guided chromatin tagmentation (ACT-seq). Nat. Commun. 10, 3747 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu C et al. Joint profiling of histone modifications and transcriptome in single cells from mouse brain. Nat. Methods (2021) doi: 10.1038/s41592-021-01060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiong H, Luo Y, Wang Q, Yu X & He A Single-cell joint detection of chromatin occupancy and transcriptome enables higher-dimensional epigenomic reconstructions. Nat. Methods (2021) doi: 10.1038/s41592-021-01129-z. [DOI] [PubMed] [Google Scholar]
- 24.Zhou VW, Goren A & Bernstein BE Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 12, 7–18 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Suganuma T & Workman JL Signals and combinatorial functions of histone modifications. Annu. Rev. Biochem. 80, 473–499 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Stoeckius M et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peterson VM et al. Multiplexed quantification of proteins and transcripts in single cells. Nat. Biotechnol. 35, 936–939 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Mimitou EP et al. Scalable, multimodal profiling of chromatin accessibility, gene expression and protein levels in single cells. Nat. Biotechnol. 1–13 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swanson E et al. Simultaneous trimodal single-cell measurement of transcripts, epitopes, and chromatin accessibility using TEA-seq. Elife 10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hao Y et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nathan A et al. Multimodally profiling memory T cells from a tuberculosis cohort identifies cell state associations with demographics, environment and disease. Nat. Immunol. 22, 781–793 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pombo Antunes AR et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 24, 595–610 (2021). [DOI] [PubMed] [Google Scholar]
- 33.Lareau CA et al. Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat. Biotechnol. 37, 916–924 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stoeckius M et al. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 19, 224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bernstein BE et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat. Biotechnol. 28, 1045–1048 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Durbin R, Eddy SR, Krogh A & Mitchison G Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. (Cambridge University Press, 1998). [Google Scholar]
- 37.Dann E, Henderson NC, Teichmann SA, Morgan MD & Marioni JC Milo: differential abundance testing on single-cell data using k-NN graphs. bioRxiv 2020.11.23.393769 (2020) doi: 10.1101/2020.11.23.393769. [DOI] [PubMed] [Google Scholar]
- 38.Stuart T et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smit AFA, Hubley R & Green P RepeatMasker Open-4.0. http://www.repeatmasker.org (2013).
- 40.Schep AN, Wu B, Buenrostro JD & Greenleaf WJ chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szabo SJ et al. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100, 655–669 (2000). [DOI] [PubMed] [Google Scholar]
- 42.Kallies A & Good-Jacobson KL Transcription Factor T-bet Orchestrates Lineage Development and Function in the Immune System. Trends Immunol. 38, 287–297 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Zhao X, Shan Q & Xue H-H TCF1 in T cell immunity: a broadened frontier. Nat. Rev. Immunol. (2021) doi: 10.1038/s41577-021-00563-6. [DOI] [PubMed] [Google Scholar]
- 44.Lorenzi L et al. The RNA Atlas expands the catalog of human non-coding RNAs. Nat. Biotechnol. 39, 1453–1465 (2021). [DOI] [PubMed] [Google Scholar]
- 45.Ashburner M et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roh T-Y, Cuddapah S, Cui K & Zhao K The genomic landscape of histone modifications in human T cells. Proc. Natl. Acad. Sci. U. S. A 103, 15782–15787 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wei G et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30, 155–167 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schoenborn JR et al. Comprehensive epigenetic profiling identifies multiple distal regulatory elements directing transcription of the gene encoding interferon-gamma. Nat. Immunol. 8, 732–742 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cui K et al. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell 4, 80–93 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barshan E, Ghodsi A, Azimifar Z & Zolghadri Jahromi M Supervised principal component analysis: Visualization, classification and regression on subspaces and submanifolds. Pattern Recognit. 44, 1357–1371 (2011). [Google Scholar]
- 51.Gopalan S, Wang Y, Harper NW, Garber M & Fazzio TG Simultaneous profiling of multiple chromatin proteins in the same cells. Mol. Cell (2021) doi: 10.1016/j.molcel.2021.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meers MP, Janssens DH & Henikoff S Multifactorial chromatin regulatory landscapes at single cell resolution. bioRxiv 2021.07.08.451691 (2021) doi: 10.1101/2021.07.08.451691. [DOI] [Google Scholar]
- 53.Mellis IA et al. Responsiveness to perturbations is a hallmark of transcription factors that maintain cell identity in vitro. Cell Syst (2021) doi: 10.1016/j.cels.2021.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shim WJ et al. Conserved Epigenetic Regulatory Logic Infers Genes Governing Cell Identity. Cell Syst 11, 625–639.e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.La Manno G et al. RNA velocity of single cells. Nature 560, 494–498 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Srivastava A, Malik L, Smith T, Sudbery I & Patro R Alevin efficiently estimates accurate gene abundances from dscRNA-seq data. Genome Biol. 20, 65 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stuart T, Srivastava A, Lareau C & Satija R Multimodal single-cell chromatin analysis with Signac. BioRxiv (2020). [Google Scholar]
- 58.Cao J et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hafemeister C & Satija R Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data generated for this manuscript are deposited in the Gene Expression Omnibus with accession code: GSE195725. The processed datasets are available as open-access downloads at: https://zenodo.org/record/5504061. The publicly datasets were used in this study: GSM4732109 and GSE164378 (Figure 2, 3, 4 and Supplementary Figure 3); GSM4732123 (Figure 1D); GSM1220567, GSM1220569, GSM1027296 and GSM1102793 (Figure 1G); GSM5034342 and GSM5034344 (Supplementary Figure 1C and Figure 5); Datasets used in DE analysis with the bulk total RNA-seq: http://r2platform.com/rna_atlas and https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE138734