

Abstract
The cytosolic innate immune sensor cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) pathway is crucial for priming adaptive antitumour immunity through antigen-presenting cells (APCs). Natural agonists such as cyclic dinucleotides (CDNs) activate the cGAS-STING pathway, but their clinical translation is impeded by poor cytosolic entry and serum stability, low specificity, and rapid tissue clearance. Here, we developed an ultrasound (US)-guided cancer immunotherapy platform using nanocomplexes composed of 2’3’ cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) electrostatically bound to biocompatible branched cationic biopolymers that are conjugated onto APC-targeting microbubbles (MBs). The nanocomplex-conjugated MBs engaged with APCs and efficiently delivered cGAMP into the cytosol via sonoporation, resulting in activation of cGAS-STING and downstream proinflammatory pathways that efficiently prime antigen-specific T cells. This bridging of innate and adaptive immunity inhibited tumour growth in both localized and metastatic murine cancer models. Our findings demonstrate that targeted local activation of STING in APCs under spatiotemporally US stimulation results in systemic antitumour immunity and improves the therapeutic efficacy of checkpoint blockade, thus paving the way toward novel image-guided strategies for targeted immunotherapy of cancer.
Cancer immunotherapy has generated exciting clinical benefits for treatment-refractory metastatic cancers such as melanoma, non-small cell lung cancer, and renal cell cancer. Unfortunately, only a small subset of patients respond to T cell checkpoint blockade1. Therefore, more effective immunotherapy strategies that can benefit larger numbers of cancer patients, in both localized and metastatic disease settings, are desperately needed2–4. Although the majority of cancer immunotherapies have focused on boosting the adaptive branch of the body’s immune system, there has been a growing realization that both the innate and adaptive branches of the body’s immune system need to be engaged to generate optimal antitumoural immunity5–7. This understanding has led to the development of new immunotherapies that target the regulators of innate immune systems, including the cytosolic DNA sensor cyclic GMP-AMP Synthase-Stimulator of Interferon Genes (cGAS-STING) pathway4,8–10. Activation of STING stimulates type I interferon (IFN) responses that are essential for priming tumour-specific cytotoxic T cells11–15. However, STING is a cytoplasmic protein, and natural agonists such as cyclic GMP-AMP (cGAMP) have poor cytosolic entry16,17. Moreover, non-specific activation of STING can cause widespread inflammatory responses, potentially impeding its translation into clinical applications18,19. Thus, strategies that enable targeted and efficient activation of STING with cGAMP would greatly facilitate its clinical translation for treating human cancers.
Preparation and characterization of MUSIC platform.
Here we report a novel Microbubble-assisted UltraSound-guided Immunotherapy of Cancer (MUSIC) strategy, which uses nanocomplex-conjugated microbubbles (ncMBs) that target antigen-presenting cells (APC) to effectively deliver cGAMP into their cytosol via ultrasound (US)-guided release and activate STING (Fig. 1a,b). Because targeted MBs oscillate when exposed to US, they create transient pores in the plasma membrane when bound to the cell surface, enabling the delivery of payload directly into the cytosol20,21. Although US-responsive MBs have previously been used to create pores in tumour cells to enable entry of plasmid DNAs for transfection purposes14, its utility to deliver more labile nucleic acids has not been demonstrated. Furthermore, how to modify MB surfaces to enhance nucleic acid loading without compromising its functions, remains a challenge. To maximize cGAMP loading, spermine-modified dextran (SpeDex) was synthesized, thiolated and conjugated onto the surface of maleimide-bearing MBs (Fig. 1c, Supplementary Fig. 1a,2a). SpeDex conjugation efficiency onto MBs was quantified using fluorescently labeled SpeDex (Supplementary Fig. 3a–c). To allow MBs to target APCs such as macrophages and dendritic cells (DCs), anti-CD11b antibodies (aCD11b) or isotype IgG (non-targeting control antibody) were thiolated and conjugated onto the surface of the MBs’ maleimide groups (Supplementary Fig. 4a–c). The thiol-maleimide coupling reactions were highly efficient, with more than 99% of MBs conjugated with both SpeDex and aCD11b on their surface, resulting in SpeDex- and aCD11b- conjugated MBs (cMBs) with an average size of 2.6 μm and ~300,000 antibodies per cMB (Fig. 1d). Two strategies were used to load cGAMP and form ncMBs; the first involved adding cGAMP to cMBs, which resulted in highly efficient binding with more than 98% of cMBs carrying cGAMP and an average load of 1.25 × 108 cGAMP molecules per ncMB (Fig. 1e,f). MBs without SpeDex did not have any measurable loading of cGAMP, suggesting that SpeDex is a necessary component for cGAMP loading (Supplementary Table 1). In addition, ncMBs can also be formulated by first forming cGAMP-SpeDex nanocomplexes (average size 169 nm), and then conjugating those nanocomplexes onto MBs to yield ncMBs (Supplementary Fig. 5a–c). To confirm targeting specificity, ncMBs were fluorescently labeled with DiD and incubated with THP-1 human macrophages (CD11b+) or EO771 breast cancer cells (CD11b−). Both bright field and fluorescence microscopy revealed an abundance of cell-ncMB complexes after incubation of ncMBs with THP-1 macrophages but not with EO771 cells (Fig. 1g). To show that ncMBs can efficiently deliver CDNs to the cytosol of APCs, we formulated ncMBs loaded with a fluorescently labeled CDN (DY547-c-diGMP) and incubated them with mouse bone marrow-derived macrophages (BMDMs). The cells were then sonoporated at 1 MHz by using a planar wave transducer (MUSIC treatment). Mean fluorescence intensity (MFI) of the BMDMs revealed that MUSIC treatment increased the uptake of CDNs by ~4 folds relative to uptake of free CDN (Fig. 1h,i, Supplementary Fig. 6a). To assess sonoporation toxicity, cell viabilities of EO771 and THP-1 cells were measured 48 h after MUSIC treatment at 1 W cm−2 which showed no loss of viability in EO771 cells and ~25% viability decrease in THP-1 cells (Supplementary Fig. 6b). These results show that ncMBs have high conjugation efficiency, high cGAMP loading, specific targeting to APCs, improved cytosolic delivery of CDNs into macrophages, and acceptable toxicity.
Fig. 1 |. Nanocomplex-decorated Microbubbles (ncMBs) targeting CD11b on antigen-presenting cells (APCs).
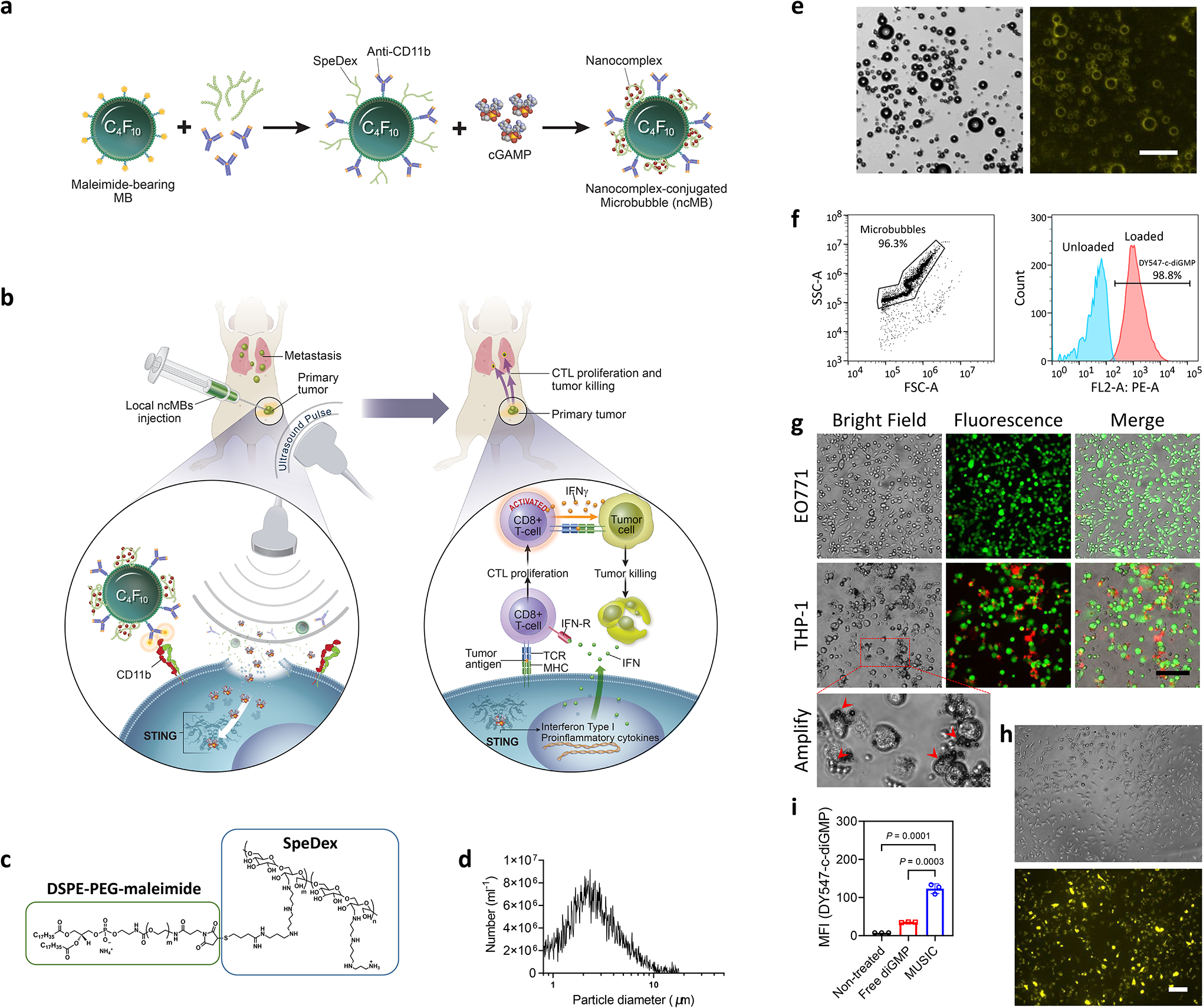
a, ncMBs are obtained by conjugating MBs with anti-CD11b antibodies (aCD11b) and spermine-conjugated dextran (SpeDex) and loading negatively charged cGAMP. b, Upon binding of ncMBs to APCs and under US exposure, cGAMP is delivered directly into the cytosol of the APCs by sonoporation to activate STING and downstream antitumour immunity, a process termed Microbubble-assisted UltraSound-guided Immunotherapy of Cancer (MUSIC). c, The lipid shell of the MB is partly composed of DSPE-PEG-maleimide, which was conjugated with thiolated SpeDex through a thiol-maleimide coupling reaction. d, Coulter counter measurements of SpeDex-aCD11b MBs (cMBs) show a size distribution of 1–10 μm, with a mean size of 2.6 μm. e,f, A fluorescent analog of cGAMP (DY547-c-diGMP) was used to verify binding to cMBs (forming ncMBs). Flow cytometry (e) and fluorescence microscopy (f) confirmed binding of the fluorescent analog to all ncMBs. Scale bar=50 μm. g, DiD-labeled cMBs were added to EO771 murine breast cancer cells and THP-1 human macrophages to confirm CD11b-specific targeting of cMBs. Confocal microscopy confirmed the binding of cMBs to THP-1 cells. h, Fluorescence microscopy of mouse bone marrow-derived macrophages (BMDMs) after sonoporation with DY547-c-diGMP loaded ncMBs indicate cytosolic delivery of the cyclic dinucleotide into all cells. g,h, Scale bar=100 μm. i, Intensity quantification of the DY547-c-diGMP uptake in BMDMs. The data represent mean ± s.d. with n=3 biologically independent samples (i). All data are shown as representative from at least three independently experiments (d-i), and were analysed by one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test (i).
Activations of STING and downstream signaling by MUSIC.
cGAMP, as an endogenous second messenger, activates the cytosolic sensor STING and the downstream type I IFN pathway22. Since we observed excellent MUSIC-mediated cytosolic delivery of CDNs into macrophages, we hypothesized that MUSIC would also enhance STING activation in APCs. As expected, MUSIC produced robust phosphorylation of STING and IFN regulatory factor 3 (IRF3) 6 hours after treatment in both mouse BMDMs and human THP-1 cells (Fig. 2a, Supplementary Fig. 7a). Although we did not observe any STING or IRF-3 phosphorylation in BMDMs treated with cGAMP alone, a small degree of activation of the immune sensors was noted in THP-1 cells, likely due to limited uptake of cGAMP by previously identified transporters on the plasma membrane of human cells23,24. To confirm that IRF3 activation and downstream inflammatory responses are mediated by STING, we treated BMDM from STING−/− mice with MUSIC and did not observe any type I IFN responses, therefore confirming that the immune response generated by MUSIC and the activation of downstream effectors are STING-dependent (Fig. 2b). Finally, to demonstrate that the activation of STING leads to the mobilization of downstream signal cascade and transcriptional activities, we examined the nuclear translocation of phosphorylated IRF3 (pIRF3), which acts as a transcription factor for proinflammatory genes (Fig. 2c, Supplementary Fig. 7b,c). NF-κB, another major downstream component of the STING pathway, was also activated upon MUSIC treatment, as shown by the phosphorylation of IKKα/β, IκBα, and p65 (Fig. 2d, Supplementary Fig. 7d), which was not observed in BMDMs from STING−/− mice (Fig. 2e). The activation of NF-κB signaling is supported by the increased translocation of p65 into the nucleus in both BMDMs and THP-1 cells after MUSIC treatment, where it acts as a transcription factor for proinflammatory cytokine expression (Fig. 2f, Supplementary Fig. 7e,f). Indeed, the expression of IFN genes was higher in MUSIC-treated BMDMs and THP-1 cells compared to cells treated with cGAMP alone, which correlated with a greater production of the proteins (Supplementary Fig. 8a,b). In contrast, APCs from STING−/− mice showed no IFN mRNA and protein expression, supporting that MUSIC’s effects rely on STING signaling (Fig. 2g,h). We repeated these experiments in primary human peripheral blood monocyte derived macrophages and found similar results (Supplementary Fig. 9a–d), thus confirming the specificity and efficacy of MUSIC in activating STING signaling in human primary APCs. In addition to increased cytokine production, MUSIC was also able to enhance the phagocytosis ability of treated macrophage (Supplementary Fig. 10a–c), which is consistent with previous findings that STING activation in macrophages can improve their phagocytosis functions25.
Fig. 2 |. Sonoporation of antigen presenting cells targeted by ncMBs (MUSIC) induces activation of STING-IRF signaling in vitro.
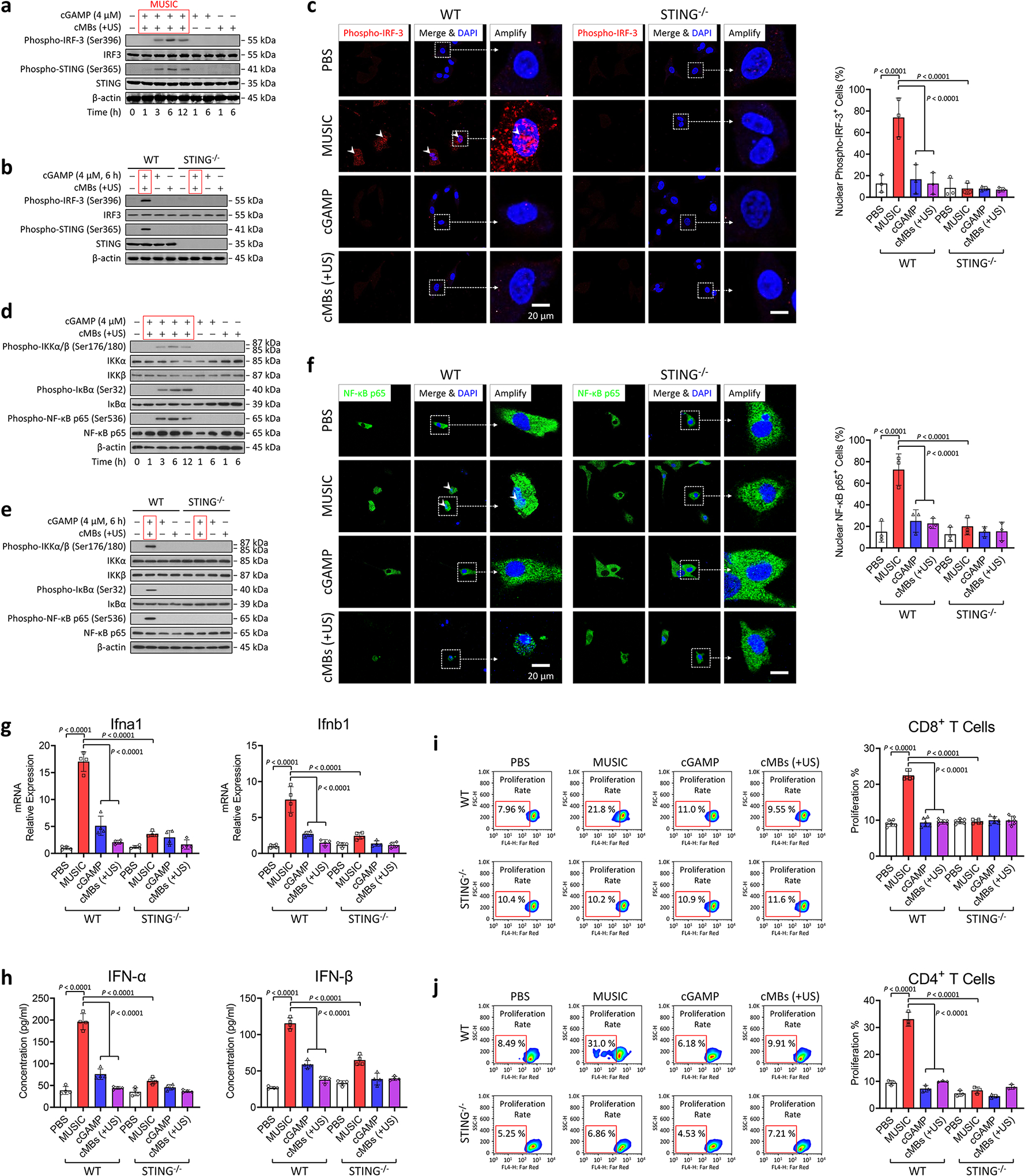
a,b, BMDMs from C57BL/6J mice were treated with MUSIC, cGAMP, or cMBs (+US), respectively. After treatment, the cells were cultured for indicated periods, followed by western blotting of proteins in the STING-IRF-3 pathway. c, Confocal fluorescence microscopy images of the BMDMs nuclear translocation of activated IRF-3 (phosphorylated IRF-3 or pIRF-3) at 6 h after treatment. d,e, Western blotting of NF-κB pathway signaling were performed using same samples from the cells treated as above. The red box in (a,b,d,e) refers specifically to MUSIC treatment. f, Confocal fluorescence microscopy images of the BMDM nuclear translocation of activated NF-κB (p65). c,f, Scale bar=20 μm. Right panels indicate quantification of nuclear fluorescent positive cells done by randomly measuring 500 cells in each group. g, ifna1 and ifnb1 mRNA expression levels in mouse BMDMs treated as above were determined via real-time PCR. h, IFN-α and IFN-β cytokine released in cell culture supernatants from above were measured by ELISA. g,h, n=4 repeats in each group. i,j, CD8+ T cells from OT-I mice and CD4+ T cells from OT-II mice were stained with Far-red. T cell proliferation was measured by flow cytometry after co-culture with MUSIC-activated BMDMs with pulsed OVA peptides for 72 h, respectively. Right panels indicate the quantification of proliferated cells as gated. i, n=6; j, n=3. All data are representative from at least three biologically independent experiments. c,f-j, data are presented as mean ± s.d.; statistical significance was calculated by one-way ANOVA with Tukey’s multiple comparisons test.
Macrophages are professional APCs that can prime T cells26. This process can be amplified through activation of STING, resulting in enhanced T cell proliferation and potent antitumour immunity27,28. To assess whether MUSIC can potentiate APCs to generate antitumoural adaptive immune responses, we first evaluated proliferation of CD8+ and CD4+ T cells after co-culture with BMDMs pulsed with OVA peptides and treated with MUSIC. Proliferation of both CD8+ and CD4+ T cells was increased when co-cultured with MUSIC-treated BMDMs relative to other treatment groups. This enhanced T-cell priming effect by MUSIC was absent in STING−/− BMDMs, suggesting that it is a STING-dependent response (Fig. 2i,j, Supplementary Fig. 11a–c). In addition to BMDMs, MUSIC treatment of tumour-associated macrophages (TAMs) from EO771 tumours implanted in wild-type mice also induced IFN protein expression and potentiated T-cell priming but had no effect on TAMs from STING−/− mice, thus supporting the potential of MUSIC to induce antitumour responses in vivo (Supplementary Fig. 12a–d). Since DCs are another major type of professional APC29, we also treated bone marrow-derived dendritic cells (BMDCs) with MUSIC and noted similar robust STING and down-stream IRF3/NF-κB activation (Supplementary Fig. 13a–d), increased type I IFN responses (Supplementary Fig. 14a,b), and enhanced priming of antigen-specific T cells (Supplementary Fig. 15a–f). Together, these findings demonstrated that MUSIC can effectively enhance STING activation in APCs, leading to improved priming of T cell responses.
Intratumoral immune activations.
To assess whether the enhanced STING activation of MUSIC observed in vitro translated into potent antitumour efficacy in vivo, we evaluated its antitumour effect in murine breast cancer models established in syngeneic hosts. Using ncMBs loaded with DY547-c-diGMP, we first assessed the delivery efficiency of CDNs in vivo. We found that the delivery of DY547-c-diGMP in tumour-associated CD11b+ cells was more than 7-fold higher when using ncMBs as compared to non-targeted IgG-ncMBs (Supplementary Fig. 16a–c). Importantly, non-specific uptake of CDNs in CD11b− cells was negligible and 4-fold lower with ncMBs as compared to the IgG-ncMBs. We then showed that MUSIC enabled imaged-guided delivery of CDNs and activation of STING in vivo (Fig. 3a, Supplementary Fig. 17a,b, and Supplementary Video 1). Immunohistochemical staining of tumour sections confirmed that MUSIC prevented Ki67 expression, indicating inhibited tumour cell proliferation, but had no direct effects on other tissues (Supplementary Fig. 18a,b). Evaluation of the tumour immune microenvironment revealed increased phosphorylation of STING in MUSIC-treated tumour tissues, most preferentially in CD11b+ cells (Fig. 3b,c, Supplementary Fig. 19a–e). The increased phosphorylation of STING in CD11b+ cells correlated with increased recruitment of CD8+ and CD4+ T cells into the tumour after MUSIC treatment (Fig. 3d, Supplementary Fig. 20a–d).
Fig. 3 |. MUSIC activates STING signaling and T cell response in primary breast cancer in vivo.
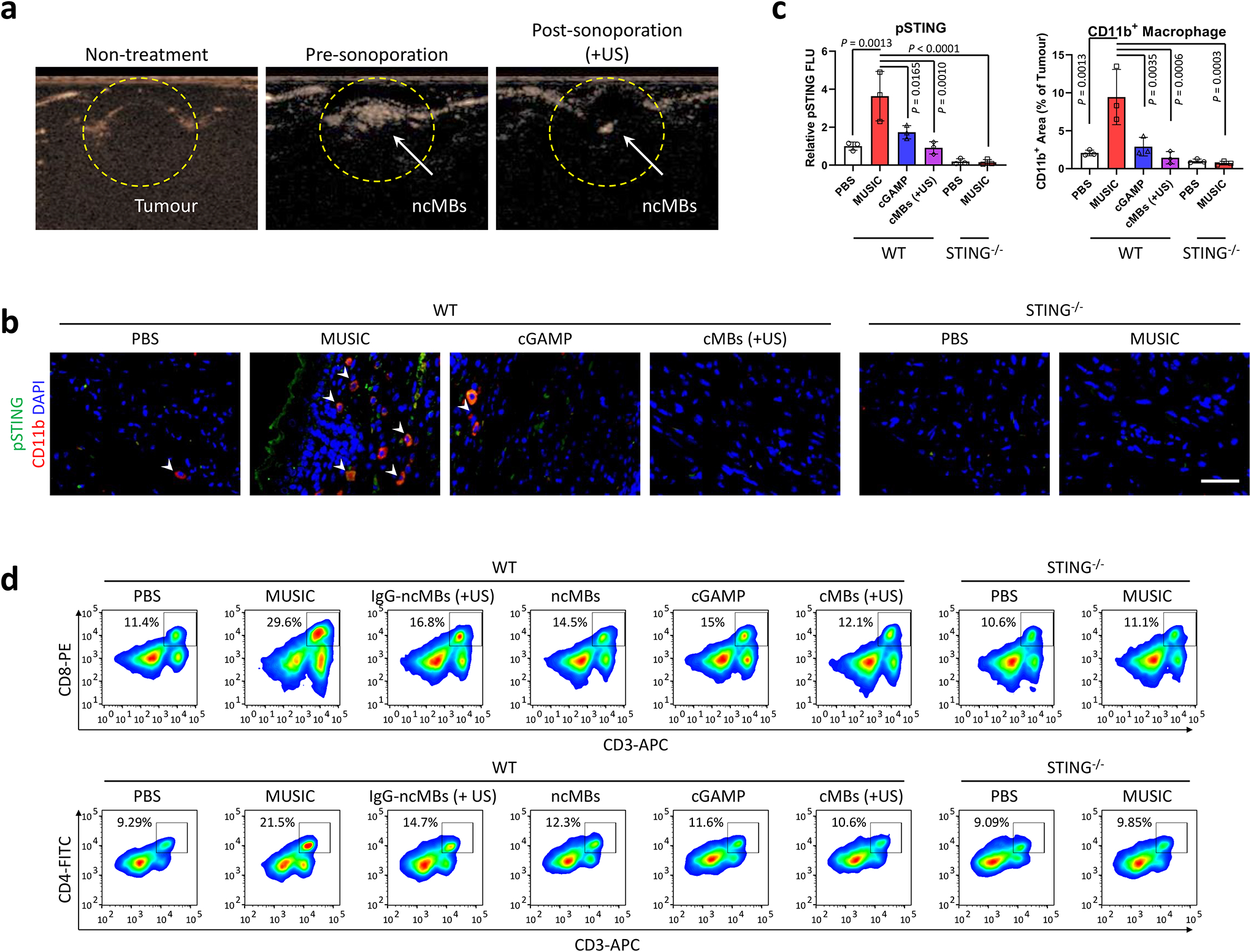
a, Contrast mode ultrasound (US) images of EO771 breast tumours (13 days) in C57BL/6J mice before ncMBs injection (left, Non-treatment), after ncMBs injection (middle, Pre-sonoporation), and after US sonoporation (right, Post-sonoporation). Loss in signal represents bubbles being destroyed after exposure to US. Images are from the same mouse and is representative of 5 randomly treated wild-type (WT) mice. b-d, WT and STING−/− mice were inoculated with EO771 breast tumours, and treated with MUSIC, cGAMP, or cMBs (+US) following the strategy in Supplementary Fig. 17a. b, At 18 days post tumour inoculation, immunostaining by confocal microscopy visualized recruited CD11b+ cells and pSTING+ cells in tumour paraffin section slides. Representative images from random fields of view in one of the three biologically independent mice. Scale bar=50 μm. c, Fluorescence intensity measurements and comparison by ImageJ software from three randomly selected images of three biologically independent mice, analysed by one-way ANOVA with Tukey’s multiple comparisons test. d, Flow cytometry analysis and quantification of CD8+ T or CD4+ T cells in tumours of representative mice at day 18 in each group. Data are representative from three biological independent samples (d) and are shown as mean ± s.d. (c), analysed by one-way ANOVA with Tukey’s multiple comparisons test (c).
MUSIC activates STING mediated antitumour immunity.
To assess the effect of MUSIC-mediated STING activation on tumour growth, mice were treated with MUSIC, cGAMP, cMBs (+US), or non-targeted IgG-ncMBs (+US) and monitored over time. We observed that MUSIC given every other day for three treatments led to the most significant tumour growth inhibition and survival benefit (Fig. 4a–d). Furthermore, the necessity of US and CD11b targeting was confirmed by comparing MUSIC with IgG-ncMBs (+US) and ncMBs groups, both of which had lower antitumour effects (Fig. 4c, Supplementary Fig. 21a,b). As expected, no antitumour effect were observed in STING−/− tumour-bearing mice treated with MUSIC (Fig. 4e,f). Additionally, complete tumour eradication was observed in 6 of the 10 MUSIC-treated mice (Supplementary Fig. 17b). Notably, these MUSIC-treated tumour-free mice were resistant to EO771 tumour cell rechallenge, suggesting that MUSIC treatment produced antitumour memory responses (Fig. 4g). To further characterize MUSIC-mediated antitumour memory, we analysed treated tumour tissue samples by flow cytometry and noted a moderate increase in the populations of CD44highCD62Llow effector memory and CD44highCD62Lhigh central memory cells upon MUSIC treatment (Supplementary Fig. 22a–c). In addition, when we collected T cells from spleens of MUSIC-treated tumour-bearing mice and rechallenged them with the EO771 tumour cells in vitro, we observed a robust IFN-γ response, thus confirming that the local MUSIC treatment generated systemic immune memory in vivo (Fig. 4h,i). To further establish the T cell’s role in mediating antitumour response of MUSIC, we depleted CD8+ T cells from tumour-bearing mice using an anti-CD8 antibody prior to MUSIC treatment. The elimination of CD8+ T cells in these animals abrogated the antitumour effect of MUSIC, resulting in half the mice dying at day 21 (Supplementary Fig. 23a–c). Given that activation of STING in APCs leads to type I IFN production, and CD8+ T cells secrete IFN-γ to produce antitumour effects, we next measured type I IFN and IFN-γ levels and found that these cytokines were elevated in tumours and serum upon MUSIC treatment (Supplementary Fig. 24a,b). Since IFN-γ is known to induce the expression of immune checkpoints such as PD-L130, we measured the expression of PD-1 and PD-L1 in T cells and tumour cells respectively. We found that in the MUSIC-treated group, over 95% of intratumoural CD8+ T cells exhibited elevated expression of PD-1 (Supplementary Fig. 25a,b), a marker of cytotoxic T-cell maturation and exhaustion31. Tumour tissue PD-L1 expression also correlated positively with IFN-γ level (Fig. 4j,k). Together, these results support the claim that MUSIC treatment activates both innate and adaptive immune responses via STING-mediated T cell priming by APCs and provide a rationale for the use of MUSIC in combination with immune checkpoint blockade to generate improved antitumour responses.
Fig. 4 |. MUSIC activates STING mediated anti-tumour immunity.
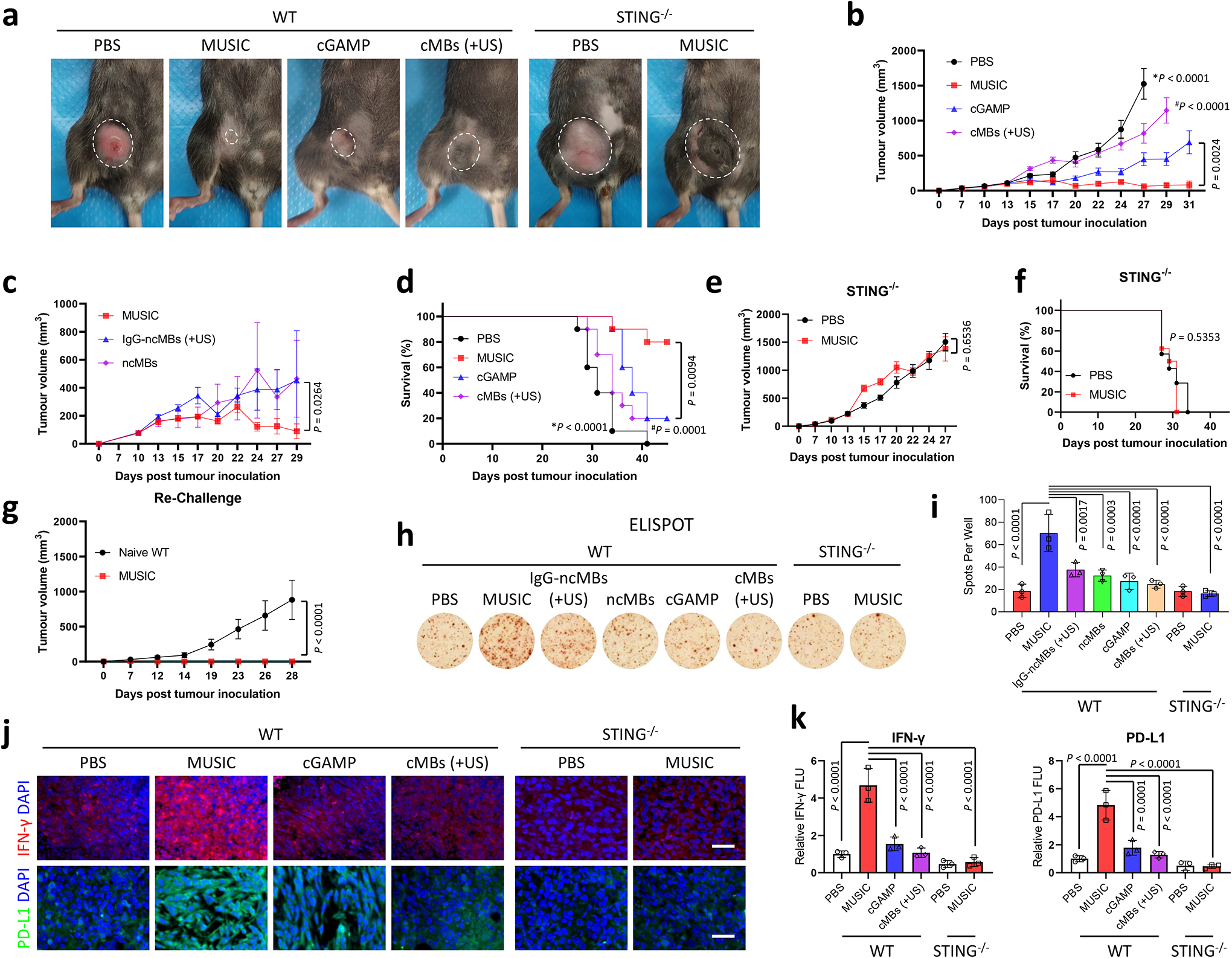
a-g, WT and STING−/− mice were inoculated with EO771 breast tumours, and treated with MUSIC, cGAMP, or cMBs (+US) following the strategy in Supplementary Fig. 17a. a, Representative photographs of mice at 24 days post tumour inoculation. b,c,e, Tumour volumes were monitored and analysed over the indicated periods. d,f, Survival curves for the mice in the different treatment groups. g, The six living tumour-free (TF) mice from the above MUSIC-treated group (b,d) were rechallenged with EO771 cells. Tumour volumes were measured over the following 28 days. n=10 for all WT mice, n=7 (PBS) or 8 (MUSIC) for STING−/− mice (a,b,d-f), n=6 for ncMBs and n=7 for both MUSIC and IgG-ncMBs (+US) (c). a-g, n means biologically independent animals. h,i, Splenic T cells from mice treated 18 days post tumour inoculation were assessed by ELISPOT to further verify immune memory enhancement upon MUSIC treatment. n=3 biologically independent samples. j, IFN-γ and PD-L1 protein expression levels were detected by immunostaining in tumour paraffin section slides. Representative images from random fields of view from one of three biologically independent animals. Scale bar=50 μm. k, Quantification and comparison of fluorescence intensity using ImageJ software from three randomly selected images of 3 biologically independent mice. The data represent mean ± s.e.m. (b,c,e,g) or mean ± s.d. (i,k), analysed by two-sided log-rank (Mantel-Cox) test (d,f), or one-way ANOVA with Tukey’s multiple comparisons test (b,c,e,g,i,k). *P and #P in (b,d) denote the statistical significance relative to the MUSIC group, respectively.
Improved systemic antitumour response by MUSIC.
To test this hypothesis, we next sought to evaluate whether MUSIC could further sensitize poorly immunogenic tumours to PD-1 blockade, particularly those that are highly aggressive and widely metastatic. We treated spontaneously metastatic murine triple negative 4T1 breast tumour-bearing mice with MUSIC in combination with an anti-PD-1 antibody (aPD-1) (Supplementary Fig. 26a). Local MUSIC treatment in combination with systemic aPD-1 administration not only exhibited enhanced primary tumour control, but also significantly decreased systemic disease progression, as compared to either therapy alone (Fig. 5a,b, Supplementary Fig. 26b–e). This improved antitumour response in the combination treatment arm directly translated into a superior survival benefit with a 76% increase in median survival as compared to free cGAMP or aPD-1 alone (Fig. 5c). Macroscopic organ imaging and examination revealed significantly reduced lung disease burden and approximately 60% decrease in pulmonary metastatic nodules (Fig. 5d–f). To assess the effect of the combination treatment on local and systemic immunity, we measured type I IFN levels in both tumour and serum and found out that they were higher in the combination treatment group compared to the control groups (Fig. 5g, Supplementary Fig. 27a). Both CD8+ and CD4+ T cell infiltrations also increased in the combination treatment group (Fig. 5h–k, Supplementary Fig. 27b,c, 28a,b). Finally, the combination treatment showed enhanced phosphorylation of STING and production of IFN-γ (Fig. 5l–o) with a low level of Ki67 expression (Supplementary Fig. 29a,b). To investigate the effect of the combination treatment on memory T cell responses, we collected tumour infiltrating lymphocytes from different treatment groups and found that the combination treatment increased the proportion of CD44highCD62Llow effector memory and CD44highCD62Lhigh central memory cells (Fig. 5p,q). Together, these results provide compelling evidence that MUSIC sensitized poorly immunogenic tumors to PD-1 blockade, and their combination enhanced local and systemic immune activations to produce improved antitumor responses.
Fig. 5 |. MUSIC activates systemic anti-tumour immunity to inhibit breast cancer metastasis.
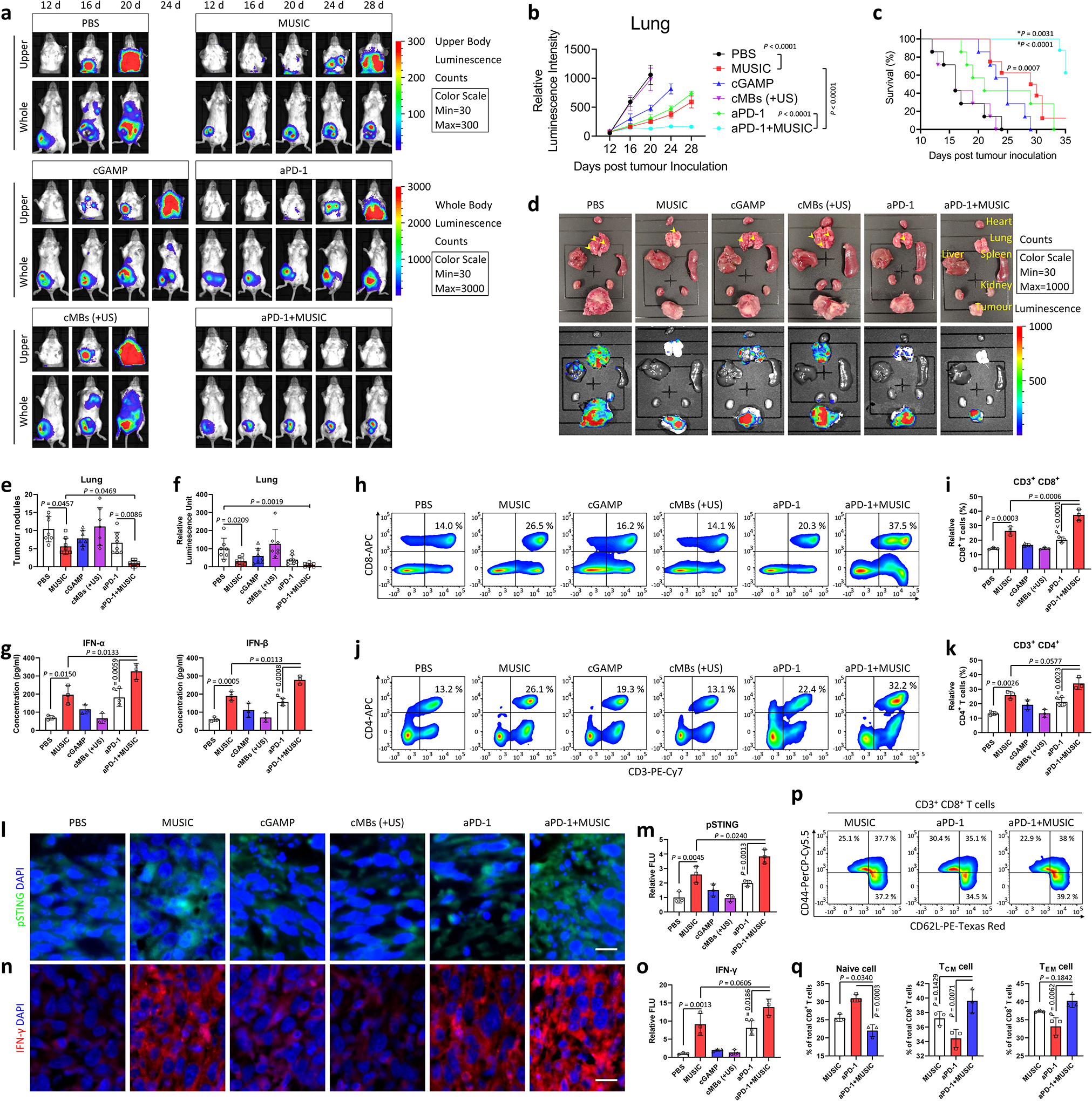
a-f, BALB/cJ mice were inoculated with luciferase-expressing 4T1 (Luc-4T1) tumours and treated as indicated in Supplementary Fig. 26a. n=7 for PBS, cGAMP, and cMBs (+US); n=8 for MUSIC, aPD-1, and aPD-1+MUSIC. Representative IVIS spectrum images (a) and quantified signal intensity (b) showing metastases of Luc-4T1 breast tumours at 28 days. c, Survival curves were plotted and analysed for the mice in each treatment arm. d, Representative photographs and bioluminescence images of organs ex vivo after euthanasia show the presence of tumour metastases. e, Quantification of tumour nodules in lungs from (d). f, Quantified bioluminescence signals in lungs from (d). g, Accumulation of IFN-α or IFN-β in tumours (determined using ELISA) at 21 days post tumour inoculation. h-k, Flow cytometry analysis and quantification of CD8+ T or CD4+ T cells in tumours at day 21 in each group. i,k Proportions of CD8+ or CD4+ T cells. l-o, Levels of activated (phosphorylated) STING and IFN-γ protein in tumour paraffin section slides detected by immunostaining. l,n, Images were collected in a blinded fashion and represent three biologically independent experiments. Scale bar=20 μm. m,o, Quantification and comparisons of fluorescence intensity using ImageJ software from three randomly selected images. p,q, Flow cytometry of tumour lymphocytes demonstrate that 4T1 tumour-bearing mice treated with aPD-1+MUSIC experienced a shift from naive to memory CD8+ T cells at 21 days post tumour inoculation. q, Proportions of CD62L+/− or CD44+/− T cells. The data represent mean ± s.e.m. (b) or mean ± s.d. (e-g,i,k,m,o,q) from representative experiments of three independent experiments with n=3 (g,i,k,m,o,q), analysed by two-sided log-rank (Mantel-Cox) test (c), or one-way ANOVA with Tukey’s multiple comparisons test (b,e,g,i,k,m,o,q), or one-way ANOVA with Dunnett’s multiple comparisons test, PBS group as control (f). P, *P, and #P in (c) denote the statistical significance relative to the MUSIC vs PBS group, aPD-1+MUSIC vs MUSIC group, or aPD-1+MUSIC vs aPD-1 group, respectively.
Conclusions
In summary, inefficient cytosolic entry and concerns about systemic toxicity have been major limitations for the clinical translation of STING agonists. The MUSIC platform can overcome these limitations by providing a robust strategy to enhance STING activation in a targeted manner by using clinically approved technologies. By investigating the mechanisms of action of MUSIC in producing robust STING activation, type I IFN responses, and more efficient priming of antigen-specific T cells in primary tumours, we identified a new strategy to activate innate and adaptive antitumour immune responses with minimal toxicity effects. MUSIC is the first image-guided cancer immunotherapy strategy that uses antibody guided targeting to activate STING in APCs through delivery of molecular drugs14. Unlike previous studies, which relied on the MBs solely to produce passages for molecules to enter cells, MUSIC integrates MBs with its cargos to enhance the stability, loading and delivery efficiency of the incorporated nucleic acids, resulting in targeted and localized immune activation. This local activation in turn generated systemic antitumour immunity by bridging the innate and adaptive immune responses. Although the MUSIC technology employs a microbubble platform, the same concept and design principle could be readily translated to nanoscale systems for targeted delivery and activation of innate immune sensors under image guidance for cancer immunotherapy applications. As such, the MUSIC platform provides a new framework for developing image-guided immunotherapy by using acoustically responsive biomaterials to enable efficient, targeted and robust immune activation to produce potent antitumour effects while minimizing systemic toxicity. The versatility of the MUSIC platform also enables it to be applied to targeted delivery of other immune-stimulating agents such as nucleotide-based vaccines as well as gene therapies for multiple human diseases.
Methods
Synthesis of SpeDex and Conjugation onto MBs.
Spermine-modified dextran (SpeDex) polymer was synthesized as previously reported32. Briefly Potassium periodate (6.25 mmol) was added to a solution of Dextran 40k (6.25 mmol of glucose monomers) in milliQ water (20 ml). The reaction was vigorously stirred in the dark for 7 h at room temperature and then spin filtered 2 × using Amicon spin filters (Molecular Weight Cut Off [MWCO]=10 kDa, Sigma) at 4,000 g for 10 min at 4 °C with water washing. The resulting retentate was dialyzed for 24 h against water using a regenerated cellulose semi permeable membrane (MWCO 3.5–5 kDa) and then added to a solution of spermine (2.96 mmol) in borate buffer (19 ml, 0.1 M, pH=11) over 5 h via syringe pump. The resulting solution was gently stirred for 24 h at room temperature followed by the addition of NaBH4 (9.48 mmol) under ice bath and stirring for 48 h at room temperature. An additional portion of NaBH4 (9.48 mmol) was then added and stirring continued for 24 h under the same conditions. Crude product was dialyzed against water (MWCO=3.5–5 kDa) for 48 h followed by lyophilization for 48 h to yield SpeDex. For thiolation, SpeDex (3.55 μmol NH2) was dissolved with phosphate-buffered saline (PBS) 1 ×, 5 mmol l−1 ethylenediaminetetraacetic acid (EDTA, 0.5 ml). To this a 1 mg ml−1 aqueous solution of 2-iminothiolane HCl (35.45 μmol) was added dropwise with vigorous stirring. The resulting mixture was stirred for 1 h, dialyzed for 48 h (MWCO=3.5 kDa), and lyophilized for 48 h. Fluorescent labeling of SpeDex polymers was done using amine reactive 5/6-carboxyfluorescein succinimidyl ester (NHS-fluorescein). Briefly, SpeDex polymer (5 mg) was dissolved in 1 ml of 1 × borate buffer (50 mmol l−1, pH=8.5). NHS-fluorescein (5 mg, 10.562 μmol) was dissolved in DMF (0.5 ml) and added dropwise to SpeDex solution with vigorous stirring. The reaction was stirred for 1 h, dialyzed for 48 h (MWCO=3.5 kDa), and lyophilized for 48 h. MBs were formulated as previously reported33. Briefly, Lipid films containing a mixture of 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-Distearoyl-sn-Glycero-3-Phosphoethanolamine-N-[Methoxy(Polyethylene glycol)-2000] (DSPE-PEG2k), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N[maleimide(polyethylene glycol)5000] (DSPE-PEG(5000)-mal) with a 90:5:5 molar ratio were prepared. These films were prepared by dissolving DSPC, DSPE-PEG(2000), and DSPE-PEG(5000)-mal in 100 μl of chloroform and slowly evaporating the mixture with a rotary evaporator (Büchi Rotavapor R-100) until mostly dry. The resulting films were then dried overnight under vacuum and stored at −20 °C for later use. The lipid films were solvated in a mixture of PBS 1 ×/propylene glycol/glycerol (80:10:10 v/v/v, 2 ml total) and sonicated at 70 °C until clear or for 15 min. Perfluorobutane (PFB) vapor was then introduced in the solution, and the resulting mixture was was tip sonicated at 70% amplitude for 5 seconds before being cooled down in an ice bath. The resulting MB formulation was washed with PBS 1 × pH 6.5 plus 1 mmol l−1 EDTA using centrifugation (300 g, 3 min) to yield PEGylated MBs with terminal maleimide functions (mal-MBs). Thiolated SpeDex was then dissolved in the same PBS solution at 10 mg ml−1 and added to mal-MBs at a 1:20 maleimide:SpeDex molar ratio. The solution was rotated end-over-end for 4 h then washed (300g, 3 min) and characterized using a Coulter Counter (Beckman Coulter Multisizer 4) to afford SpeDex MBs.
Antibody Thiolation and Preparation of ncMBs.
Anti-CD11b (aCD11b, clone M1/70, BioLegend) or IgG was first thiolated to allow for conjugation onto maleimide-bearing SpeDex MBs. Briefly, a 2 mg ml−1 solution of Traut’s reagent in PBS pH 8.0 with 5 mmol l−1 EDTA was added at a 600:1 molar ratio to a solution of aCD11b or IgG in PBS with 50 mmol l−1 EDTA. This solution was rotated for 2 h before having its buffer exchanged with PBS pH 6.5 with 1 mmol l−1 EDTA by using a Zeba Spin desalting column (7 kDa MWCO). The extent of thiolation was determined with the Measure-IT assay (ThermoFisher) and was reported to be 1.67 thiols per antibody. SpeDex MBs were then added to the solution of aCD11b or IgG at a ratio of 0.345 equivalents of antibody per maleimide and rotated end over end for 15 h at 4 °C to allow for conjugation. Afterwards, the solution was washed 3 times with PBS at 300 g for 3 min to afford SpeDex-aCD11b conjugated MBs (cMBs). To validate targeting, cMBs were incubated with cGAMP at a nitrogen:phosphate (N:P) ratio of 1:34 and the resulting MBs conjugated with aCD11b and cGAMP/SpeDex nanocomplexes (ncMBs) were added to 12-well plates containing either 300,000 THP-1 macrophages or EO771 murine breast cancer cells. The wells were washed three times, filled with perfluorobutane-saturated PBS, and imaged with bright field and fluorescence microscopy.
Formation of SpeDex-cGAMP nanocomplexes and conjugation onto MBs.
A solution of SpeDex polymer was added to a solution of cGAMP at equal volumes and at a N:P ratio of 1:10. The mixture was vortexed and then incubated for 30 min. Nanocomplexes size was determined to be 168.8 ± 9.4 nm using nanoparticle tracking analysis (Particle Metrix, ZetaView). Nanocomplexes were then conjugated onto maleimide-bearing MBs at a 1:20 maleimide:SpeDex molar ratio and rotated for 4 h followed by three washes with PBS to afford ncMBs. ncMBs were characterized using a Coulter Counter (Beckman Coulter Multisizer 4).
Mice.
C57BL/6J, C57BL/6J-Sting1gt/J, BALB/cJ, OT-I (C57BL/6-Tg(TcraTcrb)1100Mjb/J) and OT-II (C57BL/6-Tg(TcraTcrb)425Cbn/J) mice were purchased from The Jackson Laboratory and maintained at the animal facility of The University of Texas Southwestern Medical Center or The University of Texas MD Anderson Cancer Center in a specific pathogen free (SPF) environment with an ambient temperature of 22 °C and a relative humidity of 50%. All the mice were maintained on a standard diet and water in a 12 h:12 h light-dark cycle. Tumours were inoculated into the mammary fat pad of 6 to 8 week-old female mice. For CD8 depletion, 300 μg/mouse CD8 antibody were i.p. injected 24 h prior to other treatments. Inject every 72 h until the end of the experiment. All animal experiments were approved by and performed in accordance with the Institutional Animal Care and Use Committee of The University of Texas Southwestern Medical Center and The University of Texas MD Anderson Cancer Center.
Cell culture.
Human monocyte THP-1 and mouse mammary breast carcinoma 4T1 cell lines were obtained from the American Type Culture Collection (ATCC). The Luc-4T1 cell line was constructed with the red-shifted firefly luciferase (Red-FLuc) gene (Luciola Italica), and its luminescence was measured with an in vivo imaging system to evaluate the luminescence intensity. Six days before treatment, THP-1 monocytes were spun down and re-suspended in medium. Phorbol 12-myristate 13-acetate (PMA) was added to obtain a final concentration of 200 nmol l−1 to prompt differentiation of the monocytes into macrophages. Primary human peripheral blood monocytes derived macrophages were induced differentiation using CD11b+ cells sorted from PBMC (STEMCELL Technologies). Briefly, cells were cultured with macrophage colony-stimulating factor (M-CSF, 50 ng ml−1) for 7 days. The mouse mammary carcinoma cell line EO771 was obtained from CH3 Biosystems. Mouse bone marrow-derived macrophages (BMDMs) and bone marrow-derived dendritic cells (BMDCs) were isolated from the hind leg femur bone marrow of C57BL/6J mice and were cultured or activated with macrophage colony-stimulating factor (M-CSF, 50 ng ml−1) or granulocyte macrophage colony-stimulating factor (GM-CSF, 20 ng ml−1), according to the standardized procedures34,35. The purity of the induced cells was assessed by flow cytometry as follows BMDM, CD45+CD11b+; and BMDCs: CD45+CD11c+. THP-1, 4T1, and EO771 cells were cultured in RPMI 1640 medium, and BMDM and BMDCs were maintained in Dulbecco’s minimum essential medium (DMEM). All media were supplemented with 10 mmol l−1 HEPES, 10% fetal bovine serum (FBS), 1% sodium pyruvate and 100 U ml−1 of penicillin G sodium and 100 μg ml−1 of streptomycin sulfate. Cells were incubated at 37 °C under humidified conditions equilibrated with 5% CO2. All the cell lines were tested and found to be free of mycoplasma contamination by using a biochemical method with Hoechst staining.
Treatment of THP-1 cells, BMDMs, Human Monocyte Derived Macrophages, and BMDCs with ncMBs and US (MUSIC).
One day before treatment, cells were plated at 400,000 cells/well in 12-well plates. On the day of treatment, cMBs (5 MBs/cell) were rotated with 10 nmol cGAMP for 15 min to allow binding to yield ncMBs. Cells were washed once, the medium was aspirated off, and the entire ncMBs volume was added to the wells. Plates were flipped upside down and left to incubate for 10 min at 37 °C. Cells were diluted to 2.5 ml with perfluorobutane-saturated medium, placed on top of a water bath set to 35 °C, then sonoporated at 1 W cm−2, 20% duty cycle (DC), for 60 seconds using a planar wave transducer (Bulldog-Bio, Sonitron GTS Sonoporation System). cGAMP only (diluted in sterile PBS, hereafter cGAMP), cMBs+(US) (SpeDex-aCD11b MBs, no cGAMP loading, hereafter cMBs), and PBS were used as controls. Cells were collected at various times after treatment for quantitative reverse transcription polymerase chain reaction (qRT-PCR), enzyme-linked immunosorbent assay (ELISA), western blotting, and other immunostaining experiments. For delivery experiments (Fig. 1h,i, Supplementary Fig. 6a), DY547-c-diGMP was used in place of cGAMP at the same molar ratio. At different time points after sonoporation, cells were washed 3 times with PBS before being imaged using a Zeiss Axio A1 upright microscope connected with an ORCA-Spark Digital CMOS camera and Olympus TRITC filter cube (Exposure time = 0.3 s, Gain = 240).
Cell viability assays
Propidium Iodide staining cell viability assay (ThermoFisher) was performed following the manufactures’ protocols. Briefly, Propidium Iodide Ready Flow™ Reagent was added to MUSIC-treated cells after 48 hours of incubation. Flow cytometry assays were performed for distinguishing dead cells from live cells. Fluorescence associated with cell viability was measured.
Macrophage phagocytosis assay
BMDMs were treated with MUSIC, cGAMP, cMBs (+US), ncMBs, or non-targeted IgG-ncMBs (+US), respectively. After treatment, the cells were incubated for 6 h. EO771 cells were plated in 12 well plate at the density of 0.5 million cells/well, and stained with Far-Red (2 μM) following the vendor’s protocol. Then add macrophage to EO771 cells with a ratio of 1:1 for another 4 h. The mixed cells were collected and fixed by 4% PFA. Flow cytometry was performed to analyse the attachment of EO771 cells to macrophages.
Western blotting.
Cells were lysed with RIPA Lysis and Extraction Buffer (ThermoFisher) with 1% proteinase inhibitor cocktail (ThermoFisher, Cat #78429) according to the manufacturer’s instructions. Protein samples were collected from the supernatants and concentrations quantified by BCA assay. Samples were boiled for 10 min, and equal amounts of each sample were applied to 10% SDS-polyacrylamide minigels and subjected to electrophoresis (80 V in stacking gel and 120 V in separating gel). The proteins in the gel were then transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad) and blocked in 5% nonfat milk blocking buffer for 1 h at room temperature. Membranes were then incubated overnight at 4 °C with primary antibodies and then secondary antibodies according to the manufacturer’s (Cell Signaling Technology) instructions (NF-κB Pathway Sampler Kit #9936, Human-Reactive STING Pathway Antibody Sampler Kit #38866, and Mouse-Reactive STING Pathway Antibody Sampler Kit #16029). Signals were detected using X-ray film to determine the band intensity. β-actin (Mouse mAb, #3700, Cell Signaling Technology) was used as a loading control.
Real-time qPCR.
RNA was extracted from cells by using TRIzol (Invitrogen) reagent according to the manufacturer’s instructions. To detect target mRNA, the RNA strand was reverse-transcribed to cDNA with a cDNA Synthesis Supermix & Kits (Bio-Rad). The resulting cDNA was amplified and analysed with real-time PCR supermix kits (Bio-Rad) by CFX Manager Software. Expression of each gene was normalized against that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression. The human and mouse IFNs and GAPDH primers were purchased from Bio-Rad (mouse Gapdh, qMmuCED0027497; mouse Ifna1, qMmuCED0041680; mouse Ifnb1, qMmuCED0050444; human IFNA1, qHsaCED0020782; human IFNB1, qHsaCED0019234; and human GAPDH, qHsaCED0038674).
Cytokine assay.
Blood samples (~500 μl) were collected from the orbital sinus of mice anesthetized with isoflurane. Mice were euthanized after blood collection. Samples were then centrifuged, and the serum was collected and diluted with an equal volume of PBS. For collection of cell culture supernatants, samples were collected at various times after treatment and concentrated under vacuum. For collection of tissue samples, tumours were digested with EasyPep™ Lysis Buffer (ThermoFisher), 100 mg tissue in 1 ml buffer. Cytokines were measured with the mouse IFN-α (InvivoGen), IFN-β (BioLegend), IFN-γ (BioLegend), human IFN-α2 (BioLegend), or IFN-β (R&D Systems) ELISA kits according to the manufacturer’s instructions.
Tumour models.
Orthotopic mammary fat pad tumours were established by injecting mouse breast cancer cells (EO771 in C57BL/6J mice, for primary tumour model; 4T1 in BALB/cJ mice, for metastatic tumour model) at 1 × 106 cells in 50 μl PBS per mouse. Tumours were measured with calipers and tumour volumes were calculated according to an ellipsoid formula (1/2 × length × width2). Mice with palpable tumours of similar size were randomized into different groups at 13 days (EO771-C57BL/6J tumour models) or 12 days (4T1- BALB/cJ tumour models) post tumour inoculation. Each group included 10 mice for the EO771-C57BL/6J tumour models, and 7 or 8 mice for the 4T1-BALB/cJ tumour models. For the EO771-C57BL/6J tumour model, 20–30 μL of all treatments were given intratumourally on days 13, 15, 17, respectively. MUSIC, IgG-ncMBs, and cMBs groups all received US. MUSIC, IgG-ncMBs, and ncMBs were all loaded with 100 μg cGAMP (same amount as cGAMP only group). For the 4T1-BALB/cJ tumour model, all groups were treated intratumourally on days 12, 14, 16. The aPD-1 and aPD-1+MUSIC groups were administered anti-PD-1 antibody (200 μg per mouse) intraperitoneally on days 12, 14, 16, 18, 20, and 22. MUSIC, aPD-1+MUSIC, and cMBs groups all received US. MUSIC and aPD-1+ MUSIC were all loaded with 100 μg cGAMP (same amount as cGAMP only group).Procedures in tumour-growth experiments were consistent across all of the tumour models.
Ultrasound-guided in vivo STING Activation using MUSIC.
Murine breast cancer models were used to test the effectiveness of the MUSIC platform in activating the STING pathway in vivo as follows. EO771 or 4T1 tumours were grown for indicated days before mice were randomized into several groups based on tumour size. The average tumour volume was 100 mm3, and any differences between the means were determined to be insignificant by analysis of variance. The mice were injected intratumourally with 20–30 μl ncMBs suspension infused at 1 μl s−1 with a syringe pump; 100 μg of cGAMP was used for all cGAMP samples, and ~2.7e7 cMBs were used for all MBs samples. US was applied by using acoustic coupling gel and a 1-MHz plane wave transducer operating at 4 W cm−2 for 60 seconds and a 50% duty cycle given to opposite sides of the tumour for a total treatment time of 120 seconds. Mice were treated every other day for a total of three treatments, and tumour size was monitored every other day thereafter. Mice were sacrificed if tumour ulceration appeared or if tumour volume reached 2,000 mm3. This was monitored by vet techs that were blinded to the treatment and control groups of the study. cGAMP-loaded SpeDex-aCD11b MBs are ncMBs; ncMBs with US used to sonoporate cells or mice is MUSIC; Non-targeted ncMBs are IgG-ncMBs; Non-targeted ncMBs with US used to sonoporate cells or mice are IgG-ncMBs (+US); Blank SpeDex-aCD11b MBs are cMBs (control MBs); Blank SpeDex-aCD11b MBs with US used to sonoporate cells or mice are cMBs (+US).
T cell priming.
To isolate T cells from the various mouse models (OT-II mice for CD4+ T cells, OT-I mice for CD8+ T cells, C57BL/6J mice for Pan T cells), spleens were carefully removed and washed twice with sterile PBS. Tissue was dissociated through a 70 μm Nylon cell strainer by mechanical force. Splenocytes were centrifuged at 300 g for 5 min to pellet the cells, after which sterile red blood cell lysis buffer was added and the mixture was incubated at room temperature for 10 min. Cells were washed twice with cold PBS, filtered and counted in dilutions of at least 20 × (around 1 × 108 cells/spleen). Mouse naive CD8a+ T cells, naive CD4+ T cells, and Pan T cells were isolated according to the manufacturer’s instructions (Miltenyi Biotec, Cat #130-096-543, 130-104-453, 130-095-130). BMDMs or BMDCs were treated with MUSIC, cGAMP alone, or cMBs (+US) as described above and then incubated with OVA peptide that bind either MHC-I (OVA 257–264; detect a strong CD8+ cytolytic T cell response) or MHC-II (OVA 323–339; to detect CD4+ T-cell activation) for 6 h to allow antigen reorganization and presentation. Afterwards, cells were washed and then incubated for 72 h with OT-I cells or OT-II cells, respectively, and the OT cells were then analysed by flow cytometry to determine the amount of proliferation. Pan T cells were directly restimulated with EO771 cells.
Flow cytometry.
The treated mice from each group were euthanized on the indicated days post tumour inoculation, and the perfused tumour or spleen tissues were collected and digested in 200 U ml−1 collagenase D, 1.6 U ml−1 dispase and 15 U ml−1 DNase I in 10 mmol l−1 HEPES buffer at 37 °C for 60 min to obtain the cell suspensions. The dissociated cells were then filtered through a 70-μm nylon cell strainer and collected for the following analyses. Tumour-associated macrophages (TAMs), CD8+ T cells, CD4+ T cells, and Pan T cells were isolated for flow cytometry analysis as follows. Cells were fixed and permeated to allow entry of the fluorescent probes for flow cytometry. Briefly, cells were first blocked with anti-CD16/CD32 (Bio X Cell, Cat # BE0307, dilution of 1:200) for 15 min to avoid nonspecific binding to the Fc receptor. For CD8+ or CD4+ T-cell proliferation analysis, cells were prestained with CellTrace™ Far Red Cell Proliferation Kit (Invitrogen). For other analyses, cells were stained separately with different antibodies (anti-CD8a-PE, dilution of 1:200; anti-CD8a-APC, dilution of 1:300; anti-CD4-FITC, dilution of 1:300; anti-CD3-PE-Cy7, dilution of 1:200; anti-CD11b-PE, dilution 1:200; anti-CD11b-PerCP-Cy5.5, dilution 1:200; anti-CD68-APC, dilution 1:200; anti-IL-10-PE, dilution 1:200; anti-CD62L-PE-Texas Red, dilution of 1:200; anti-CD44-PerCPCy5.5, dilution of 1:200) according to the manufacturer’s instructions. Both CD8+ T and CD4+ T cells are also gated as CD3 positive.
IFN-γ-secreting T cell ELISPOT assay.
IFN-γ released from stimulated T cell (splenic T cells from EO771 tumour bearing mice) upon sub co-culture in the immediate EO771 cellular environment were measured using IFN-γ ELISPOT assay kit (Affymetrix e Bioscience) according to the manufacturer’s protocol. Briefly, a 96-well assay plate (Millipore) was first pre-coated with anti-IFN-γ antibody at 4 °C overnight. A total of 1 × 107 T cells were co-cultured with EO771 cells at a ratio of 10:1 overnight at incubated at 37 °C in humidified conditions equilibrated with 5% CO2. After washing, the biotinylated detection antibody was added followed by washing and incubation with avidin-HRP reagent. After development with AEC (3-amino-9-ethylcarbazole) substrate solution, IFN-γ spots were viewed by using a dissecting microscope and recorded using an AID vSpot Reader Spectrum.
Histologic analysis.
All slides were deparaffinized in xylene for 10 min, three times, and rehydrated with graded concentrations of ethanol. Antigen retrieval and immunofluorescence staining (IF) or immunohistochemical staining (IHC) were done according to previously described processes and manufacturers’ protocols. For IF staining: green, target protein stained with primary antibody and then FITC-conjugated secondary antibody; red, target protein stained with primary antibody and then Alexa Fluor 555-conjugated secondary antibody; blue, cell nuclei stained with 4,6-diamidino-2-phenylindole (DAPI). Microscopy images from the same group of experiments were acquired using the same microscope settings.
In vivo luminescence imaging.
BALB/cJ mice were used to study the growth and metastasis of Luc-4T1 tumours in vivo as follows. At 12 days post tumour implantation, mice were intraperitoneally injected with 1.5 mg D-luciferin (Syd Labs) per 10 g body weight; at 10 min after injection, the presence and metastases of the tumours were monitored by bioluminescence imaging (BLI) using an In Vivo Imaging System (PerkinElmer IVIS® Lumina III). Data were analysed and presented with Aura Imaging software. All images were adjusted to the same color scale and display threshold. The IVIS images of the upper body were used to detect luminescence signals in the lung. After the live imaging, mice that met the criteria for early euthanasia were sacrificed36, and major organs including heart, liver, spleen, lung, kidneys, and tumour were collected for additional IVIS imaging to analyse luminescence signals in these organs.
Statistical analyses.
All data are shown as mean ± standard deviation (s.d.) or mean ±standard error of mean (s.e.m.) from at least triplicate conditions unless otherwise indicated. Each experiment was repeated independently at least three times unless otherwise indicated. Statistical analyses included two-sided unpaired Student t-test and one-way ANOVA with Tukey’s or Dunnett’s multiple comparisons test, as appropriate. Survival was determined for mice in every group by the Kaplan-Meier method and compared by the two-sided log-rank (Mantel-Cox) test. The P values of less than 0.05 were considered to indicate statistical significance and are labeled in the results. Statistical analyses were done with GraphPad Prism 7 and Microsoft Excel 2013 software. No animals were excluded from the analyses.
Supplementary Material
Acknowledgements
This work was supported in part by the Cancer Prevention and Research Institute of Texas (CPRIT) Grants RR150010, RP210199 and RP19023, the Department of Defense grants W81XWH-21-1-0332/0333 and W81XWH-17-1-0401, the Susan G. Komen Foundation Career Catalyst Research Grant CCR19605871, and the National Cancer Institute grant 1K08CA241070. R.F.M. is a CPRIT Established Investigator. Research reported in this publication was also supported by the Children’s Cancer Fund Comprehensive Center for Pediatric Oncology Research. The authors acknowledge Siemens Medical Solutions USA. Inc. for the Siemens Sequoia ultrasound scanner loan. The authors acknowledge the UT Southwestern Harold C. Simmons Cancer Center Support Grant P30 CA142543 for the support provided by the Small Animal Imaging shared resource. The authors also would like to acknowledge Erin Moore (Creatives Services, Department of Radiology, The University of Texas Southwestern Medical Center) for the illustrations in Fig. 1a,b, Supplementary Fig. 5a, and Christine Wogan from MD Anderson Cancer Center for editorial help.
Footnotes
Competing interests
A provisional patent application based on the technology described in the manuscript has been filed by The University of Texas Southwestern Medical Center, with Sina Khorsandi, Jacques Lux and Wen Jiang as inventors, application number 63/173,956. All other authors declare no competing interests.
Data availability
The authors declare that data supporting the findings of this study are available within the article and its Supplementary Information. The datasets generated and analysed during the study are publicly available at https://osf.io/phcmx. Source data are provided with this paper.
References
- 1.Jerby-Arnon L et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175, 984–997 e924, doi: 10.1016/j.cell.2018.09.006 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alsaiari SK et al. Sustained and targeted delivery of checkpoint inhibitors by metal-organic frameworks for cancer immunotherapy. Sci Adv 7, doi: 10.1126/sciadv.abe7174 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Topalian SL, Taube JM & Pardoll DM Neoadjuvant checkpoint blockade for cancer immunotherapy. Science 367, doi: 10.1126/science.aax0182 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun X et al. Amplifying STING activation by cyclic dinucleotide-manganese particles for local and systemic cancer metalloimmunotherapy. Nat Nanotechnol 16, 1260–1270, doi: 10.1038/s41565-021-00962-9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bendickova K & Fric J Roles of IL-2 in bridging adaptive and innate immunity, and as a tool for cellular immunotherapy. Journal of leukocyte biology 108, 427–437, doi: 10.1002/JLB.5MIR0420-055R (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wen L et al. An Efficient Combination Immunotherapy for Primary Liver Cancer by Harmonized Activation of Innate and Adaptive Immunity in Mice. Hepatology 69, 2518–2532, doi: 10.1002/hep.30528 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee D, Huntoon K, Wang Y, Jiang W & Kim BY Harnessing Innate Immunity Using Biomaterials for Cancer Immunotherapy. Advanced Materials, 2007576 (2021). [DOI] [PubMed] [Google Scholar]
- 8.Reislander T, Groelly FJ & Tarsounas M DNA Damage and Cancer Immunotherapy: A STING in the Tale. Molecular cell 80, 21–28, doi: 10.1016/j.molcel.2020.07.026 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Luo M et al. A STING-activating nanovaccine for cancer immunotherapy. Nat Nanotechnol 12, 648–654, doi: 10.1038/nnano.2017.52 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shae D et al. Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy. Nat Nanotechnol 14, 269–278, doi: 10.1038/s41565-018-0342-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cousin C et al. Persistence of Integrase-Deficient Lentiviral Vectors Correlates with the Induction of STING-Independent CD8(+) T Cell Responses. Cell reports 26, 1242–1257 e1247, doi: 10.1016/j.celrep.2019.01.025 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu N et al. STING agonist promotes CAR T cell trafficking and persistence in breast cancer. The Journal of experimental medicine 218, doi: 10.1084/jem.20200844 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miao L et al. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat Biotechnol 37, 1174–1185, doi: 10.1038/s41587-019-0247-3 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Ilovitsh T et al. Low-frequency ultrasound-mediated cytokine transfection enhances T cell recruitment at local and distant tumor sites. Proceedings of the National Academy of Sciences of the United States of America 117, 12674–12685, doi: 10.1073/pnas.1914906117 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng M et al. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nature Reviews Cancer 19, 568–586 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gutjahr A et al. The STING ligand cGAMP potentiates the efficacy of vaccine-induced CD8+ T cells. JCI insight 4, doi: 10.1172/jci.insight.125107 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chin EN et al. Antitumor activity of a systemic STING-activating non-nucleotide cGAMP mimetic. Science 369, 993–999, doi: 10.1126/science.abb4255 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Decout A, Katz JD, Venkatraman S & Ablasser A The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nature reviews. Immunology, doi: 10.1038/s41577-021-00524-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y et al. TMEM203 is a binding partner and regulator of STING-mediated inflammatory signaling in macrophages. Proceedings of the National Academy of Sciences of the United States of America 116, 16479–16488, doi: 10.1073/pnas.1901090116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo Y et al. Single-cell analysis reveals effective siRNA delivery in brain tumors with microbubble-enhanced ultrasound and cationic nanoparticles. Sci Adv 7, doi: 10.1126/sciadv.abf7390 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dwivedi P et al. Magnetic Targeting and Ultrasound Activation of Liposome-Microbubble Conjugate for Enhanced Delivery of Anticancer Therapies. ACS applied materials & interfaces 12, 23737–23751, doi: 10.1021/acsami.0c05308 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Takahashi M et al. The tumor suppressor kinase DAPK3 drives tumor-intrinsic immunity through the STING-IFN-beta pathway. Nature immunology 22, 485–496, doi: 10.1038/s41590-021-00896-3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lahey LJ et al. LRRC8A:C/E Heteromeric Channels Are Ubiquitous Transporters of cGAMP. Molecular cell 80, 578–591 e575, doi: 10.1016/j.molcel.2020.10.021 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ritchie C, Cordova AF, Hess GT, Bassik MC & Li L SLC19A1 Is an Importer of the Immunotransmitter cGAMP. Molecular cell 75, 372–381 e375, doi: 10.1016/j.molcel.2019.05.006 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahn J, Xia T, Capote AR, Betancourt D & Barber GN Extrinsic phagocyte-dependent STING signaling dictates the immunogenicity of dying cells. Cancer cell 33, 862–873. e865 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li X et al. Lyn Delivers Bacteria to Lysosomes for Eradication through TLR2-Initiated Autophagy Related Phagocytosis. PLoS pathogens 12, e1005363, doi: 10.1371/journal.ppat.1005363 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu J, Deng H & Xu Z Targeting macrophage priming by polyphyllin VII triggers anti-tumor immunity via STING-governed cytotoxic T-cell infiltration in lung cancer. Scientific reports 10, 21360, doi: 10.1038/s41598-020-77800-w (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.von Roemeling CA et al. Therapeutic modulation of phagocytosis in glioblastoma can activate both innate and adaptive antitumour immunity. Nature communications 11, 1–12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Mingo Pulido A et al. The inhibitory receptor TIM-3 limits activation of the cGAS-STING pathway in intra-tumoral dendritic cells by suppressing extracellular DNA uptake. Immunity 54, 1154–1167 e1157, doi: 10.1016/j.immuni.2021.04.019 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lucas ED et al. Type 1 IFN and PD-L1 Coordinate Lymphatic Endothelial Cell Expansion and Contraction during an Inflammatory Immune Response. Journal of immunology 201, 1735–1747, doi: 10.4049/jimmunol.1800271 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ambler R et al. PD-1 suppresses the maintenance of cell couples between cytotoxic T cells and target tumor cells within the tumor. Science signaling 13, doi: 10.1126/scisignal.aau4518 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Azzam T et al. Polysaccharide-oligoamine based conjugates for gene delivery. Journal of medicinal chemistry 45, 1817–1824, doi: 10.1021/jm0105528 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Lux J et al. Thrombin-activatable microbubbles as potential ultrasound contrast agents for the detection of acute thrombosis. ACS applied materials & interfaces 9, 37587–37596 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Assouvie A, Daley-Bauer LP & Rousselet G Growing Murine Bone Marrow-Derived Macrophages. Methods Mol Biol 1784, 29–33, doi: 10.1007/978-1-4939-7837-3_3 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Roney K Bone Marrow-Derived Dendritic Cells. Methods Mol Biol 1960, 57–62, doi: 10.1007/978-1-4939-9167-9_4 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Underwood W & Anthony R AVMA guidelines for the euthanasia of animals: 2020 edition. Retrieved on March 2013, 2020–2021 (2020). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that data supporting the findings of this study are available within the article and its Supplementary Information. The datasets generated and analysed during the study are publicly available at https://osf.io/phcmx. Source data are provided with this paper.