

Abstract
Previous results indicated poor sugar consumption and early inhibition of metabolism and growth when Clostridium cellulolyticum was cultured on medium containing cellobiose and yeast extract. Changing from complex medium to a synthetic medium had a strong effect on (i) the specific cellobiose consumption, which was increased threefold; and (ii) the electron flow, since the NADH/NAD+ ratios ranged from 0.29 to 2.08 on synthetic medium whereas ratios as high as 42 to 57 on complex medium were observed. These data indicate a better control of the carbon flow on mineral salts medium than on complex medium. By continuous culture, it was shown that the electron flow from glycolysis was balanced by the production of hydrogen gas, ethanol, and lactate. At low levels of carbon flow, pyruvate was preferentially cleaved to acetate and ethanol, enabling the bacteria to maximize ATP formation. A high catabolic rate led to pyruvate overflow and to increased ethanol and lactate production. In vitro, glyceraldehyde-3-phosphate dehydrogenase, lactate dehydrogenase, and ethanol dehydrogenase levels were higher under conditions giving higher in vivo specific production rates. Redox balance is essentially maintained by NADH-ferredoxin reductase-hydrogenase at low levels of carbon flow and by ethanol dehydrogenase and lactate dehydrogenase at high levels of carbon flow. The same maximum growth rate (0.150 h−1) was found in both mineral salts and complex media, proving that the uptake of nutrients or the generation of biosynthetic precursors occurred faster than their utilization. On synthetic medium, cellobiose carbon was converted into cell mass and catabolized to produce ATP, while on complex medium, it served mainly as an energy supply and, if present in excess, led to an accumulation of intracellular metabolites as demonstrated for NADH. Cells grown on synthetic medium and at high levels of carbon flow were able to induce regulatory responses such as the production of ethanol and lactate dehydrogenase.
Cellulolytic clostridia are of cardinal importance in anaerobic environments rich in plant material (4, 23, 42). For many years, the cellulase complex of cellulolytic clostridia and genes encoding cellulases have been the subjects of considerable research, which has led to the cellulosome concept (1–3, 12). The cellulosomes found at the surface of the cells, where they form protuberances, are responsible for the specific adherence of numerous cellulolytic clostridia to cellulose. They contain a multiplicity of enzyme components showing a marked synergism against cellulosic compounds. Thus, enzymes involved in degradation of cellulose and hemicellulose have been well characterized, while few studies have focused on the carbon metabolic pathway. Poor sugar consumption and an early inhibition of metabolism and growth have been documented (7, 10, 15, 18, 19, 27, 37, 38), and Clostridium cellulolyticum, a mesophilic cellulolytic bacterium isolated from compost (35), shows this behavior. Using yeast extract, Casamino Acids, and vitamin supplements, Giallo et al. (15) tried to improve C. cellulolyticum growth, but without success; nevertheless, complex media were used systematically for the cultivation of this organism (4, 7, 13–16, 34). However, the complex metabolism associated with the use of the numerous compounds included in the rich media is such that analyses of metabolism and energy use are difficult to undertake. Moreover, many natural ecosystems are oligotrophic and rarely contain all nutrients in high quantity (22). In light of these considerations, during this investigation a synthetic medium was used to study the behavior of C. cellulolyticum under conditions of nutrient limitation. To permit identification of regulatory responses occasioned by low nutrient concentrations, studies were conducted in chemostats, which can maintain low steady-state nutrient concentrations, using cellobiose as the carbon source, since this disaccharide is the major end product of the cellulose degradation process (32, 39).
MATERIALS AND METHODS
Chemicals.
All chemicals were of highest-purity analytical grade. Unless indicated otherwise, commercial reagents, enzymes, and coenzymes were supplied by Sigma Chemical Co., St. Louis, Mo. All gases used were purchased from Air Liquide, Paris, France.
Organism and medium.
The bacterium used in this study, C. cellulolyticum ATCC 35319, was originally isolated by Petitdemange et al. from decayed grass (35). Stock cultures of C. cellulolyticum were maintained on cellulose and were grown for one transfer in cellobiose before initiation of growth experiments. The anaerobic culture technique used was that proposed by Hungate (20) as modified by Bryant (6).
The defined medium used in all experiments was a modification of the CM3 medium described by Weimer and Zeikus (44), in which 5 g of yeast extract per liter is replaced by oligoelement and vitamin solutions. The composition was (in grams liter−1 unless otherwise indicated): KH2PO4, 1.40; K2HPO4 · 3H2O, 2.90; (NH4)2SO4, 1.00; MgCl2 · 6H2O, 0.10; CaCl2, 0.02; FeSO4 · 7H2O, 9.15% (wt/vol) in 50 mM H2SO4, 25 μl; oligoelement solution, 1.0 ml; vitamin solution, 10 ml; Na2S, 0.50; and resazurin at 0.2% (wt/vol), 0.5 ml. In addition, a constant limited cellobiose concentration (5.84 mM) was added to the feed medium.
The oligoelement solution contained (in grams liter−1 unless otherwise indicated): FeSO4 · 7H2O, 5.00; ZnSO4 · 7H2O, 1.44; MnSO4 · 7H2O, 1.12; CuSO4 · 5H2O, 0.25; Na2B4O7, 0.20; (Mo)7(NH4)6O24 · 4H2O, 1.00; NiCl2, 0.04; CoCl2, 0.02; HBO3, 0.03; Na2SeO3, 0.02; HCl (10 M), 50.0 ml.
The composition of the vitamin solution was (in milligrams per 100 ml of distilled water): d-biotin, 10; para-aminobenzoic acid, 25; nicotinic acid, 15; riboflavin, 25; pantothenic acid, 25; thiamine, 25; and cyanocobalamin, 10. The vitamin in solution was sterilized by filtration through a 0.2-μm-pore-size filter.
Growth conditions.
C. cellulolyticum ATCC 35319 was grown in chemostat culture at varying dilution rates on cellobiose (5.84 mM) as carbon and energy source. Cultures were maintained aseptically in a 2-liter bioreactor (LSL-Biolafitte, St. Germain en Laye, France) with a 1.5-liter working volume. The temperature was maintained at 34°C, and the pH was controlled at 7.2 by the automatic addition of 1 M NaOH. All tubing used was made of Viton (Du Pont Co., Wilmington, Del.) to eliminate oxygen entry. Agitation was kept constant at 50 rpm. Medium was pumped into the fermentor at the appropriate dilution rate. The volume was kept constant at 1.5 liters by automatic regulation of the culture level. The inoculum was 10% by volume and was in the exponential growth phase. The culture was grown in batch for 15 h before the medium flow was started.
A period of three to four residence times was found to be sufficient to achieve steady-state values of biomass and residual cellobiose concentrations. Generally, these cultures were maintained 28 days. Cultures were carried out at atmospheric pressure, but due to the low qcellobiose values (the specific rates of cellobiose used), the fermentor was sparged with oxygen-free nitrogen (6 ml min−1) to prevent back diffusion of oxygen into the fermentor headspace. If a pink color of resorufin appeared, the culture became unstable and washout occurred.
Analytical procedures.
Bacterial growth was measured spectrophotometrically at 600 nm and calibrated against cell dry weight measurements. Samples (30 ml) were centrifuged for 10 min at 8,000 × g, washed with 0.9% (wt/vol) NaCl, and dried at 65°C to constant weight (48 h). A mean biomass formula of C4H7O2N and an average extracellular protein formula of C16H25O9N6 were determined by elemental analysis (Service Central d’Analyses, CNRS, Vernaison, France) and were used for elemental recovery calculations.
Cell-free supernatants (10,000 × g, 15 min, 4°C) were stored at −80°C. Cellobiose was determined colorimetrically by using dinitrosalicylic reagent (29). Acetate, lactate, ethanol, and succinate were estimated by using the appropriate enzyme kits (Boehringer Mannheim, Meylan, France). Extracellular proteins from the cell-free supernatant were measured by the Bradford dye method (5). The quantity of amino acids present in supernatant was measured by using the procedure of Church et al. (11) and by ion exchange chromatography on a cation-exchange resin with a Beckman 7300 amino acid analyzer. The average elemental amino acid composition was C5H10O2.5N. Exopolysaccharides were determined by using glucose as a standard as described previously (34). Ammonia was assayed by the method of Chaney and Marbach (8).
Gas samples were assayed for hydrogen and carbon dioxide by means of an Intersmat model IGC MB chromatograph equipped with a thermoconductivity detector (80 mA) and a column (2 m by 2 mm) fitted with Carbosieve (120 to 140 mesh size; Supelco). The column temperature was 100°C and the carrier gas was argon (1.2 bar at the column head). The injection temperature was 130°C and the detector temperature was 110°C. One-milliliter samples of the culture gas phase were injected directly into the gas chromatography apparatus with a 1-ml syringe.
Cell lysis was monitored by recording the DNA levels as a proportion of the intracellular cell proteins versus the protein/DNA ratios found in the supernatant. Cell DNA was measured by the method of Giles and Myers (17), and the DNA level in cell-free supernatants was determined after concentration by ethanol precipitation (36).
Assay of glycolytic intermediates in cell extracts.
Dihydroxyacetone phosphate (DHAP), glyceraldehyde-3-phosphate (GAP) and fructose-1,6-biphosphate (FBP) were extracted from a culture broth sample by HClO4 by using the rapid system described by Thomas et al. (40). The average time between cells leaving the fermentor and mixing with HClO4 was ca. 30 ms, and the final HClO4 concentration was 0.6 M. Samples were held in the syringe with the needle closed by a septum for 2 min at room temperature. Extracts were placed in ice for 10 min under N2 atmosphere before addition of 230 mg of K2CO3 and were finally neutralized with 3 M KOH. Extracts were centrifuged (10,000 × g, 4°C, 10 min), and supernatants were stored at −80°C until assayed.
Metabolites were measured by coupling appropriate enzyme assays with fluorimetric determination of the coenzyme NADH. Emission was measured at 459 nm after excitation at 341 nm with a fluorimeter (model F2000; Hitachi, Tokyo, Japan). DHAP, GAP, and FBP concentrations were determined by using an assay mixture containing 50 mM triethanolamine buffer (pH 7.6), 50 mM KCl, 5 mM MgCl2, 12.5 μM NADH, extract, and 4 U of glycerophosphate dehydrogenase (EC 1.1.1.8) to initiate DHAP consumption. After complete depletion of the DHAP in the extract, 100 U of triose phosphate isomerase (EC 5.3.1.1) was added to measure the GAP concentration. Addition of 2.5 U of fructose-1,6-biphosphate aldolase (EC 4.1.2.13) allowed the FBP concentration in the extract to be measured.
Determination of nucleotide pools.
Levels of nucleotides, ATP, ADP, NAD(P)+, and NAD(P)H in the biomass were measured by first extracting the nucleotides from a sample of culture. ATP and ADP were extracted with perchloric acid as described above for glycolytic intermediates.
ATP levels were measured by a luminescence assay employing the luciferin-luciferase system (Microbiol Biomass Test kit; Celsis Lumac, Landgraaf, The Netherlands). ADP was converted to ATP in a reaction mixture containing 2 ml of supernatant, 14 mM phosphocreatine in glycine buffer (0.1 M, pH 9.0), 0.4 mM MgSO4 and 4 U of creatine phosphokinase from rabbit muscle (EC 2.7.3.2) maintained for 15 min at 38°C. The reaction was stopped by heating (100°C) for 3 min, and the mixture was centrifuged for 15 min at 8,000 × g. NAD(P)+ and NAD(P)H were extracted with HCl and KOH, respectively, as described by Wimpenny and Firth (45).
Levels of coenzymes in both extracts were determined by fluorimetric measurements (see above). NAD+ was assayed with NAD(H)-specific alcohol dehydrogenase (EC 1.1.1.1), and NADP+ was assayed with a glucose-6-phosphate dehydrogenase (EC 1.1.1.49) (21, 24). NADH levels were determined by lactate dehydrogenase assay (21). NADPH was measured in a reaction mixture containing 100 mM triethanolamine buffer (pH 6.0), 20 mM α-ketoglutaric acid, and 20 U of NAD(P)H-specific glutamate dehydrogenase from Proteus species (EC 1.4.1.4).
Assay of extracellular pyruvate.
The NADH fluorimetric assay (see above) was adapted for the measurement of extracellular pyruvate by using 0.1 M triethanolamine buffer (pH 7.6) and 12.5 μM NADH. Enzymatic determination of intracellular pyruvate levels was not possible due to significant interference with extracellular pyruvate, which lead to erroneous estimates of intracellular concentrations.
Preparation of cell extracts.
Cell extracts were obtained as described previously (34). Protein concentrations of cell extracts were determined by the Lowry method (26) by using crystalline bovine serum albumin as the standard.
Enzyme assays.
Fd-NAD(P)+ reductase, NADH-fd reductase, hydrogenase (EC 1.12.7.1), glyceraldehyde-3-P-dehydrogenase (GAPDH) (EC 1.2.1.12), acetate kinase (EC 2.7.2.1), alcohol dehydrogenase (EC 1.1.1.1) (ADH), and lactate dehydrogenase (EC 1.1.1.27) (LDH), activities were assayed at 34°C as described previously (34). Endo-1,4-β-glucanase (EC 3.2.1.4) activity was determined by the method of Miller et al. (30) with carboxymethylcellulose as the substrate.
Determination of the Km values of LDH and PFO for pyruvate.
Crude extract (5 ml) was dialyzed anaerobically against 5 liters of phosphate buffer (5 mM, pH 7.4) changed three times at 4°C to remove intracellular pyruvate and FBP. LDH activity was measured as described previously (34). To determine Km and Vmax of the LDH, the NADH concentration was held constant at 0.4 mM and the pyruvate concentrations were varied from 0.25 to 30.00 mM. Pyruvate-fd oxidoreductase (PFO) (EC 1.2.7.1) was assayed as described by Meinecke et al. (28) except that 1 mM methyl viologen was used as the artificial electron acceptor. The reduction of methyl viologen was measured at 570 nm, and to determine Km and Vmax of the PFO, the pyruvate concentrations were varied from 0.05 to 2.00 mM.
Calculations.
According to Papoutsakis and Meyer (33), a stoichiometric balance equation for biomass formation from cellobiose can be written in the form:
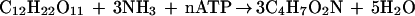 |
where C4H7O2N denotes the elemental composition of the biomass.
The main products of cellobiose fermentation by C. cellulolyticum were acetate, lactate, ethanol, H2, and CO2 (15); because of the very low concentrations of extracellular pyruvate (detected in the medium only at dilution rates from 0.062 to 0.138 h−1), we have omitted this compound in the reactions leading to the formation of the metabolites, the formula for which can be written as follows: cellobiose + 8 ADP + 8 Pi + 4 NAD+ → 4 acetate + 8 ATP + 4 NADH + 4 CO2 + 4 H2
The conversion of cellobiose to lactate is as follows:
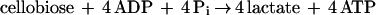 |
The conversion of cellobiose to ethanol can be written as follows: cellobiose + 4 ADP + 4 Pi + 4 NADH → 4 ethanol + 4 ATP + 4 NAD+ + 4 CO2 + 4 H2
NADH is formed by GAPDH, and ATP is formed by phosphoglycerate kinase, pyruvate kinase, and acetate kinase. The specific rates of NADH production and NADH consumption were calculated as follows: qNADH produced = qacetate + qlactate + qethanol, qNADH used by ethanol and lactate production = qlactate + 2 qethanol, and qNADH oxidized by the path NADH-fd-hydrogenase (qNADH-fd) = qNADH produced − qNADH used (qacetate, qlactate, and qethanol are the specific rates of product formation in millimoles per gram of cells per hour). In the following sections, qcellobiose is the specific rate of cellobiose used in millimoles per gram of cells per hour. The specific rate of pyruvate formation (qpyruvate) was determined as follows: qpyruvate = qacetate + qlactate + qethanol. YATP can be calculated (using acetate, lactate, and ethanol concentrations [concn] according to the equations described above) as follows:
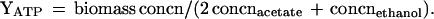 |
YATP is expressed in grams of cells per mole of ATP produced.
Carbon recoveries were calculated from the production of metabolites, biomass, amino acids, proteins, and polysaccharides present in supernatant. CO2 levels were calculated from the concentrations of acetate and ethanol.
RESULTS
Effect of the dilution rate on biomass and metabolite formation.
C. cellulolyticum was grown under cellobiose limitation at proportions of culture volume replaced per hour (D) of 0.016 to 0.138 h−1, the highest D value at which a steady state could be attained (Table 1), since a maximum growth rate (μmax) of 0.150 h−1 was calculated during the early exponential phase of batch growth. At a cellobiose concentration in the feed medium of 5.84 mM and over a wide range of low dilution rates, the residual cellobiose concentrations were low and increased only at a D of 0.138 h−1. These data are typical of a continuous culture carried out under carbon limitation. The dry weight increased between 0.016 and 0.120 h−1 and decreased markedly at 0.138 h−1, corresponding to cellobiose accumulation.
TABLE 1.
Fermentation parameters for continuous steady-state cultures of C. cellulolyticum
| Parametera | Result obtained at a D (h−1) of:
|
||||||
|---|---|---|---|---|---|---|---|
| 0.016 | 0.033 | 0.053 | 0.062 | 0.085 | 0.120 | 0.138 | |
| Biomass concn (g liter−1) | 0.145 ± 0.005 | 0.210 ± 0.007 | 0.229 ± 0.007 | 0.250 ± 0.008 | 0.310 ± 0.012 | 0.370 ± 0.015 | 0.243 ± 0.009 |
| Residual concn (mM)b of cellobiose | 0.15 ± 0.06 | 0.14 ± 0.06 | 0.05 ± 0.02 | 0.15 ± 0.06 | 0.17 ± 0.07 | 0.11 ± 0.05 | 2.35 ± 0.12 |
| qcellobiose (mmol [g of cells]−1 h−1) | 0.63 | 0.89 | 1.34 | 1.41 | 1.55 | 1.86 | 1.98 |
| qpyruvate (mmol [g of cells]−1 h−1) | 1.63 | 2.73 | 2.98 | 3.21 | 3.79 | 4.27 | 4.35 |
| Product yieldc | |||||||
| Acetate | 74.8 | 68.5 | 62.7 | 65.3 | 55.4 | 51.1 | 47.8 |
| Lactate | 2.8 | 2.0 | 2.5 | 4.5 | 6.6 | 11.5 | 8.3 |
| Ethanol | 22.5 | 29.5 | 34.9 | 30.6 | 38.0 | 37.5 | 43.9 |
| Extracellular proteins (mg liter−1) | 41.5 ± 1.5 | 14.0 ± 0.6 | 66.6 ± 2.8 | 42.0 ± 1.7 | 68.9 ± 2.7 | <5.0 | <5.0 |
| Extracellular amino acids (mM) | 1.14 ± 0.06 | 1.20 ± 0.07 | 1.05 ± 0.05 | 0.52 ± 0.03 | 0.98 ± 0.03 | 1.03 ± 0.06 | 0.82 ± 0.03 |
| Polysaccharide (mg liter−1) | 4.5 ± 0.5 | 4.8 ± 0.5 | 5.6 ± 0.6 | 5.0 ± 0.6 | 8.3 ± 0.7 | 8.6 ± 0.7 | 27.7 ± 1.6 |
| YATP (g of cells [mol of ATP]−1) | 5.6 | 7.2 | 10.9 | 10.7 | 14.4 | 18.6 | 21.4 |
| Carbon recovery (%) | 84.2 | 98.2 | 80.5 | 77.9 | 90.7 | 86.8 | 90.0 |
Values are the averages (± standard deviations) from three different chemostats. All other values were determined with an average accuracy of ±10%.
The cellobiose and ammonium inputs were 5.84 and 15.13 mM, respectively.
Product yields are expressed as percentages of qpyruvate.
Table 1 summarizes the concentrations of the most important products measured at each steady state as a function of the growth rate. At all D values, the residual ammonium concentrations ranging from 9.5 to 13.7 mM were always in excess. Acetate, ethanol, and lactate were the primary metabolic end products; succinate accumulation was not observed. The rate of cellobiose consumption varied from 0.63 to 1.98 mmol (g of cells)−1 h−1 with increasing growth rate. The ratio of qpyruvate to qcellobiose indicated that 55 to 77% of the consumed cellobiose was converted into end products and the rest was converted into biomass, polysaccharides, proteins, and amino acids.
Whatever the dilution rate, growth on ammonium led to amino acid appearance in the medium varying from 3 to 67 μmol liter−1. Besides the common amino acids, the following other amino compounds accumulated in the medium: phosphoserine (27 μmol liter−1), citrulline (4 μmol liter−1), aminobutyrate (2.5 μmol liter−1), ornithine (3.5 μmol liter−1), and phosphoethanolamine (4 μmol liter−1). There was no apparent correlation between the extent of extracellular amino acids and protein accumulation. Cell lysis was a minor phenomenon since the protein/DNA ratios in cell extracts were found to be approximately 16 to 21, whereas values between 783 and 842 were found in the supernatant. DNA accumulation coming from cell lysis was chiefly observed when cellobiose was depleted. Conversely, carboxymethylcellulase activity was found to be related to the production of extracellular proteins. Extracellular polysaccharide production increased with the carbon flow mainly at a D of 0.138 h−1. The global carbon balance (calculated by taking into account these compounds) was found to be in the range of 77.9 to 98.2%.
Cells growing on limiting concentrations of cellobiose formed hardly any lactate at low growth rates (Table 1); at a D of 0.016 h−1, 74.8% of the qpyruvate representing the cellobiose involved in energy production was converted into acetate, 22.5% was converted into ethanol, and only 2.8% was converted into lactate. The ratio of end products changed with the increase of the carbon flow and was strictly connected with levels of qpyruvate; at a D of 0.138 h−1, the percentage of acetate decreased to 47.8% whereas lactate and ethanol percentages increased to 8.3 and 43.9%, respectively. The specific rate of lactate production increased rapidly when the specific rate of pyruvate production reached approximately 3 mmol (g of cells)−1 h−1 (Fig. 1a). Ethanol production increased linearly with the specific rate of pyruvate production, whereas acetate production showed a biphasic linear increase, slowing down when the specific rates of pyruvate production reached approximately 3 mmol (g of cells)−1 h−1 (Fig. 1b).
FIG. 1.

Influence of the specific rates of pyruvate production in a cellobiose-limited continuous culture of C. cellulolyticum on specific rates of lactate production (▴) and in vitro specific LDH activities (▵) (a) and on specific rates of acetate (○) and ethanol production (●) (b).
Redox balances.
The formation of biomass from cellobiose is stoichiometrically written as follows:
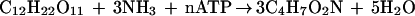 |
where C4H7O2N denotes the elemental biomass composition and corresponds to a molecular weight of 101 g mol−1.
Since YATP values were found to be between 5.6 and 21.4 g mol of ATP−1 in continuous culture (Table 1), an average YATP of 12.7 g mol of ATP−1 is calculated, and the preceding equation can be rewritten as
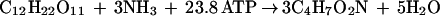 |
From this equation, reducing equivalents NAD(P)H required for biomass synthesis were supplied via the NAD(P)H generated from biomass formation, and no excess of NAD(P)H which could be balanced by the formation of products was formed. Furthermore, we have assumed that little CO2 was produced from general decarboxylation enzymes implicated in biomass synthesis. The pathways to acetic acid, ethanol, and lactic acid generate ATP for synthesis and maintenance of biomass.
The coenzyme balance calculated from the known catabolic pathways producing or consuming reducing equivalents demonstrate an excess of NADH since the qNADH produced/qNADH used ratio was always greater than 1 except at a D of 0.138 h−1, where the NADH used equilibrated the NADH produced (Table 2). In spite of this imbalance, the NADH/NAD+ ratios ranged from 0.25 to 0.41, meaning that the cells contained more NAD+ than NADH except at a D of 0.016 h−1, where a ratio of 2.08 was observed (Table 3). The regeneration of the excess of NADH into NAD+ is due to the path NADH-fd-hydrogenase, since the measured H2/CO2 ratios were found to be greater than 1 (Table 2), although the phosphoroclastic reaction produces 1 mol of CO2 and 1 mol of H2 per mol of pyruvate oxidized (31). It must be noted that the NAD+ + NADH pools increased from 13.16 to 29.84 μmol (g of cells)−1 with the increase of D.
TABLE 2.
Coenzyme balance for continuous steady-state culture of C. cellulolyticum
| Parametera | Result obtained at a D (h−1) of:
|
||||||
|---|---|---|---|---|---|---|---|
| 0.016 | 0.033 | 0.053 | 0.062 | 0.085 | 0.120 | 0.138 | |
| qNADH produced | 1.63 | 2.73 | 2.98 | 3.21 | 3.79 | 4.27 | 4.35 |
| qNADH used | 0.78 | 1.67 | 2.16 | 2.10 | 3.13 | 3.69 | 4.18 |
| qNADH produced/qNADH used | 2.09 | 1.63 | 1.38 | 1.53 | 1.21 | 1.16 | 1.04 |
| H2/CO2 ratio | NDc | 1.69 | 1.37 | ND | 1.33 | 1.29 | ND |
| qNADH-fd from NADHb | 0.85 | 1.06 | 0.82 | 1.11 | 0.66 | 0.58 | 0.17 |
qNADH produced, qNADH used, and qNADH-fd from NADH are the specific rate of NADH production, the specific rate of NADH utilization, and the specific rate of hydrogen production from NADH, respectively (in millimoles per gram of cells per hour).
qNADH-fd from NADH was calculated as qNADH produced − qNADH used.
ND, not determined.
TABLE 3.
Nucleotide levels in continuous steady-state cultures of C. cellulolyticum
| Nucleotide | Concentration (μmol [g of cells]−1)a observed at a D (h−1) of:
|
||||||
|---|---|---|---|---|---|---|---|
| 0.016 | 0.033 | 0.053 | 0.062 | 0.085 | 0.120 | 0.138 | |
| NADH | 8.89 ± 1.62 | 3.50 ± 0.67 | 4.06 ± 0.76 | 3.65 ± 0.65 | 6.16 ± 1.19 | 6.73 ± 1.25 | 5.51 ± 1.03 |
| NAD+ | 4.27 ± 1.19 | 10.42 ± 2.71 | 10.35 ± 2.59 | 11.53 ± 3.19 | 15.03 ± 3.35 | 23.11 ± 3.70 | 22.05 ± 3.41 |
| NADH/NAD+ | 2.08 | 0.33 | 0.39 | 0.32 | 0.41 | 0.29 | 0.25 |
| NADH + NAD+ pool | 13.16 | 13.92 | 14.41 | 15.18 | 21.19 | 29.84 | 27.56 |
| NADPH | 8.27 ± 1.30 | 5.66 ± 0.98 | 3.18 ± 0.50 | 2.04 ± 0.37 | 3.87 ± 0.68 | 5.54 ± 0.89 | 6.46 ± 1.11 |
| NADP+ | NDb | ND | ND | ND | ND | 0.04 ± 0.01 | 0.06 ± 0.01 |
Values are the averages (± standard deviations) from three different chemostats.
ND, not detectable.
Intracellular levels of NAD(P)+ and NAD(P)H were measured under different growth conditions (Table 3). Whatever the dilution rate, levels of NADP+ were hardly detectable or were undetectable, whereas the NADPH levels were between 2.04 and 8.27 μmol (g of cells)−1 and were available for biosynthesis reactions.
Enzyme activities.
Specific activities in extracts of pelleted cells were measured at each steady state, and the influence of the growth rate on the specific activities of the enzymes is shown in Table 4. In vitro, GAPDH, lactate dehydrogenase, ethanol dehydrogenase, and acetate kinase activities were higher under conditions giving higher in vivo specific production rates. From the lowest (0.016 h−1) to the highest (0.138 h−1) D values, GAPDH increased 4.4-fold, LDH increased 5.3-fold, ADH increased 7.9-fold, and acetate kinase increased 1.8-fold.
TABLE 4.
Enzymatic activities from cell extracts of continuous cultures of C. cellulolyticum
| Enzyme | Specific activity (μmol min−1 [mg of protein]−1)a observed at a D (h−1) of:
|
||||||
|---|---|---|---|---|---|---|---|
| 0.016 | 0.033 | 0.053 | 0.062 | 0.085 | 0.120 | 0.138 | |
| GAPDH | 1.31 ± 0.17 | 3.34 ± 0.47 | 3.63 ± 0.45 | 3.30 ± 0.43 | 4.90 ± 0.66 | 4.41 ± 0.53 | 5.74 ± 0.70 |
| Hydrogenase | 0.030 ± 0.006 | 0.031 ± 0.004 | 0.042 ± 0.008 | 0.069 ± 0.018 | 0.068 ± 0.012 | 0.145 ± 0.022 | 0.176 ± 0.025 |
| Fd-NADP+ reductase | 0.057 ± 0.015 | 0.070 ± 0.018 | 0.050 ± 0.013 | 0.072 ± 0.017 | 0.051 ± 0.012 | 0.062 ± 0.016 | 0.060 ± 0.014 |
| Fd-NAD+ reductase | 0.047 ± 0.013 | 0.044 ± 0.011 | 0.046 ± 0.011 | 0.054 ± 0.015 | 0.047 ± 0.014 | 0.043 ± 0.010 | 0.054 ± 0.015 |
| NADH-fd reductase | 0.017 ± 0.004 | 0.009 ± 0.003 | 0.019 ± 0.006 | 0.018 ± 0.005 | 0.029 ± 0.009 | 0.018 ± 0.006 | 0.020 ± 0.006 |
| Lactate dehydrogenase | 0.15 ± 0.02 | 0.41 ± 0.05 | 0.43 ± 0.05 | 0.59 ± 0.07 | 0.60 ± 0.06 | 0.69 ± 0.10 | 0.79 ± 0.10 |
| Alcohol dehydrogenase | 0.22 ± 0.02 | 0.32 ± 0.02 | 0.53 ± 0.04 | 0.51 ± 0.06 | 0.58 ± 0.06 | 1.52 ± 0.14 | 1.73 ± 0.19 |
| Acetate kinase | 1.15 ± 0.13 | 1.22 ± 0.18 | 1.15 ± 0.11 | 1.16 ± 0.11 | 1.57 ± 0.06 | 2.34 ± 0.25 | 2.02 ± 0.21 |
One unit of enzyme activity is defined as the amount of enzyme that catalyzes the conversion of 1 μmol of substrate per min. Values are the mean determinations (± standard deviations) from three different chemostats.
A comparison of the specific activities measured with the flows of metabolites suggested that LDH had in vitro specific activities that correlated only weakly with the rate of lactate production (Fig. 1a). This can be explained by variation in the concentration of intracellular compounds which could regulate the lactate dehydrogenase activity. An increase in the extracellular pyruvate which was probably correlated with intracellular pyruvate was also noticed, but this intracellular pyruvate concentration was difficult to measure due to significant interference from extracellular pyruvate (Table 5). Excretion of pyruvate, a partially oxidized metabolic intermediate, must be considered an overflow and means that the PFO could no longer support the flux arriving from cellobiose. This overflow was well correlated with lactate formation (Fig. 2).
TABLE 5.
Extracellular pyruvate levels and intracellular metabolite in continuous steady-state cultures of C. cellulolyticum
| D (h−1) | Extracellular pyruvate (μM)a | Intracellular metabolite (μmol [g of cells]−1)a
|
||
|---|---|---|---|---|
| DHAP | GAP | FBP | ||
| 0.033 | NDb | 7.9 ± 0.9 | 3.8 ± 0.7 | 1.0 ± 0.1 |
| 0.053 | ND | 8.9 ± 1.0 | 7.4 ± 1.4 | 2.1 ± 0.2 |
| 0.062 | 79.0 ± 4.3 | 15.5 ± 1.6 | 7.1 ± 1.2 | 2.1 ± 0.2 |
| 0.085 | 138.7 ± 7.7 | 21.9 ± 2.3 | 6.0 ± 1.0 | 3.4 ± 0.5 |
| 0.120 | 147.3 ± 8.0 | 30.2 ± 2.9 | 6.4 ± 1.2 | 2.4 ± 0.3 |
| 0.138 | 70.2 ± 3.5 | 32.7 ± 3.2 | 7.4 ± 1.3 | 3.2 ± 0.4 |
Values are the averages of three determinations (± standard deviations).
ND, not detectable.
FIG. 2.

Relation between the specific rates of extracellular pyruvate diffusion and the specific rates of pyruvate production (●) coming from cellobiose catabolism and the specific rates of lactate production (○).
The apparent Km and Vmax values for pyruvate catalyzed by the LDH and the PFO were calculated from standard Lineweaver-Burk plots (data not shown), and Kmvalues were found to be 4.5 and 0.57 mM, respectively. These two enzymes show markedly different Vmax/Km ratios for pyruvate, 0.182 × 10−3 min−1 mg−1 in the case of LDH and 5.02 × 10−3 min−1 mg−1 for the PFO, explaining the pyruvate overflow. As a consequence of the PFO saturation and the high apparent Km value of the LDH for pyruvate, an increase of the intracellular pool of triose-phosphate occurred, namely, the DHAP, whereas GAP remained constant (Table 5). An increase of the intracellular FBP was also noticed, this compound being a facultative activator of the LDH activity. By assuming an internal volume of 1.67 ml (g of cell)−1 (41), the steady-state internal FBP concentration was 0.60 to 2.04 mM (Table 5). LDH activity assayed in dialyzed cell extracts was increased 2.2-fold and 2.8-fold in presence of 1.00 mM and 2.00 mM FBP, respectively, and 1.8-fold by prior incubation with 4.50 mM pyruvate.
At each steady state, no NADPH-fd reductase activity was detected but a high level of fd-NADP+ reductase activity was measured (Table 4), which suggests the involvement of NADPH-fd oxidoreductase in the production of NADPH, particularly since no glucose-6-phosphate dehydrogenase and transhydrogenase activities were detected.
With high specific activities of fd-NAD+ reductase and of NADH-fd reductase, the NADH-fd oxidoreductase can function reversibly. However, as product formation coincided with an excess of NADH produced (Table 2), the NADH-fd reductase activity combined with hydrogenase activity could function in NADH oxidation and H2 production, explaining how H2/CO2 ratios greater than 1 were obtained (Table 2).
DISCUSSION
The results presented in this article give consistent information on the mechanisms used by C. cellulolyticum to regulate cellobiose catabolism. Previous work carried out by using complex medium have led to a bottleneck, since carbon flow was stopped by a high level of intracellular NADH (34). This study shows that changing from complex to synthetic medium had strong effects on the following: (i) specific cellobiose consumption, which was increased from 0.68 (for the complex medium) to 1.98 mmol (g of cells)−1 h−1 (for the synthetic medium), whereas the highest dilution rate at which a steady state could be attained was increased from 0.120 (for the complex medium) to 0.138 h−1 (for the synthetic medium) (in complex medium at a growth rate [μ] of 0.120 h−1, the dilution rate approaches the washout point since the dry weight of cells decreased by 78% of the maximal biomass [34] whereas in synthetic medium at a μ of 0.138 h−1, the biomass decreased by 35%); (ii) electron flow, since the NADH/NAD+ ratios were in the range 0.29 to 2.08 whereas ratios as high as 42 to 57 were observed on complex media (34). Clearly, these data indicate a better control of cellobiose catabolism by C. cellulolyticum on mineral salts medium than on complex medium.
When C. cellulolyticum was grown on complex medium, regardless of the dilution rate, cellobiose catabolism showed the same pattern, i.e., acetate was the main product, whereas the biosynthesis of ethanol and lactate was low (34). Conversely, on synthetic medium, this study reveals that formation of end products and their ratios can change within broad limits. These changes were not due to iron-limited culture media, which could have effects on the biosynthesis of iron proteins such as ferredoxin (25), PFO (43), and hydrogenase (9) and hence on the cellobiose metabolism, since we found that the growth of C. cellulolyticum was limited when the iron added to the medium was less than 0.2 mg liter−1 (data not shown).
With low D values (Fig. 3), when cellobiose flow and hence ATP synthesis were limited, pyruvate was preferentially cleaved to acetate and ethanol. This enabled the bacteria to maximize formation of ATP. The high percentage of acetate formation versus the low ethanol production found at low growth rates under cellobiose limitation proved that C. cellulolyticum maintained its redox balance via the efficient NADH-fd reductase activity, as corroborated by an H2/CO2 ratio greater than 1. At increased D values, there was a large effect on ethanol and lactate production, which approximately coincided with pyruvate accumulation (Fig. 4). The level of lactate production increased sharply when the qcellobiose reached approximately 1.34 mmol (g of cells)−1 h−1, values which were never obtained on the complex medium, since the highest value reported was 0.68 mmol (g of cells)−1 h−1 (34).
FIG. 3.

Scheme of the carbon and electron flow distribution at a D of 0.033 h−1. Only the major flows are indicated in the direction of the arrows. Carbon and electron flows are symbolized by thick and thin lines, respectively. Enzymes (indicated by circled numbers): 1, GAPDH; 2, PFO; 3, phosphotransacetylase; 4, acetate kinase; 5, acetaldehyde dehydrogenase; 6, ADH; 7, hydrogenase; 8, Fd-NADP+ reductase; 9, NADH-fd reductase.
FIG. 4.

Scheme of the carbon and electron flows distribution at a D of 0.138 h−1. Notation is the same as in the legend for Fig. 3, except that the lactate dehydrogenase is numbered 10.
Pyruvate overflow means that PFO could no longer support the flow arriving at pyruvate from the catabolism of cellobiose. Under these conditions, electron flow from glycolysis was balanced chiefly by ethanol and lactate production; the NADH-fd reductase was not active since the H2/CO2 ratio was near 1. This indicates that the NADH-fd reductase activity under low rates of cellobiose consumption (Fig. 3) and ADH and LDH activities under high rates of cellobiose consumption (Fig. 4) were able to maintain the redox balance of C. cellulolyticum; the increase in the NAD+ and NADH content with the D values could be correlated with the increase in the levels of the three NAD+ oxidoreductases GAPDH, ADH, and LDH.
We must also take into account the fact that on synthetic medium, cellobiose carbon can be converted into cell mass and used in catabolism to produce ATP, while on complex medium, yeast extract carried many cell building blocks and cellobiose carbon served mainly as an energy supply. Since the same μmax of 0.150 h−1 was found in both media, it is clear that the uptake of nutrients or the generation of biosynthetic precursors occurs faster than the utilization of these precursors for biomass production. This interpretation is confirmed by the fact that numerous precursors were found in the synthetic culture medium. The fact that lactate production occurred only in conditions of pyruvate overflow avoids competition between the lactate pathway and (i) PFO, which maximizes ATP formation via acetate production, and (ii) anabolism, since pyruvate is also the precursor of compounds such as alanine, valine, and leucine.
We can suppose that in natural environments, C. cellulolyticum will rarely find nutrient substances in high concentrations and that the complex medium with high concentrations of substrate may be unfavorable to C. cellulolyticum, which was unable to deal with a surfeit of substrates; under these conditions, the nutrients or products of its metabolism, such as were demonstrated for NADH (34), may accumulate intracellularly to toxic levels. It can be argued that during the course of evolution, C. cellulolyticum has evolved to optimize cellobiose catabolism and nitrogen anabolism under nutrient-poor conditions.
This study of synthetic medium with cellobiose has permitted the identification of regulatory responses of C. cellulolyticum and serves as a basis for additional work using bacteria growing on cellulose. Much remains to be learned about the physiology of these bacteria on insoluble substrates.
ACKNOWLEDGMENTS
This work was supported by the Commission of European Communities FAIR program (contract CT 95-0191 [DG 12 SSMA]).
We thank M. Young for a critical reading of the manuscript. The technical assistance of Guy Raval and Cynthia Rousselot was greatly appreciated.
REFERENCES
- 1.Bayer E A, Shoham Y, Tormo J, Lamed R. The cellulosome: a cell surface organelle for the adhesion to and degradation of cellulose. In: Fletcher M, editor. Bacterial adhesion molecular and ecological diversity. New York, N.Y: Wiley-Liss; 1996. pp. 155–182. [Google Scholar]
- 2.Bayer E A, Morag E, Lamed R. The cellulosome—a treasure-trove for biotechnology. Trends Biotechnol. 1994;12:379–386. doi: 10.1016/0167-7799(94)90039-6. [DOI] [PubMed] [Google Scholar]
- 3.Béguin P, Aubert J P. The biological degradation of cellulose. FEMS Microbiol Rev. 1994;13:25–28. doi: 10.1111/j.1574-6976.1994.tb00033.x. [DOI] [PubMed] [Google Scholar]
- 4.Benoit L, Cailliez C, Petitdemange E, Gitton J. Isolation of cellulolytic mesophilic clostridia from a municipal solid waste digestor. Microb Ecol. 1992;23:117–125. doi: 10.1007/BF00172634. [DOI] [PubMed] [Google Scholar]
- 5.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 6.Bryant M P. Commentary on the Hungate technique for culture of anaerobic bacteria. Am J Clin Nutr. 1972;25:1324–1328. doi: 10.1093/ajcn/25.12.1324. [DOI] [PubMed] [Google Scholar]
- 7.Cailliez C, Benoit L, Thirion J P, Petitdemange H. Characterization of 10 mesophilic cellulolytic clostridia isolated from a municipal solid waste digestor. Curr Microbiol. 1992;25:105–112. [Google Scholar]
- 8.Chaney A L, Marbach E P. Modified reagents for determination of urea and ammonia. Clin Chem. 1962;8:130–132. [PubMed] [Google Scholar]
- 9.Chen J S, Mortenson L E. Purification and properties of hydrogenase from Clostridium pasteurianum W5. Biochim Biophys Acta. 1974;371:283–298. doi: 10.1016/0005-2795(74)90025-7. [DOI] [PubMed] [Google Scholar]
- 10.Chung K T. Inhibitory effects of H2 on growth of Clostridium cellobioparum. Appl Environ Microbiol. 1976;31:342–348. doi: 10.1128/aem.31.3.342-348.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Church F C, David H P, Catagniani G L, Swaigsgood H G. An O-phthalaldehyde spectrometric assay for proteinase. Anal Biochem. 1985;146:343–348. doi: 10.1016/0003-2697(85)90549-4. [DOI] [PubMed] [Google Scholar]
- 12.Felix C R, Ljungdahl L G. The cellulosome: the exocellular organelle of Clostridium. Annu Rev Microbiol. 1993;47:791–819. doi: 10.1146/annurev.mi.47.100193.004043. [DOI] [PubMed] [Google Scholar]
- 13.Gelhaye E, Gehin A, Petitdemange H. Colonization of crystalline cellulose by Clostridium cellulolyticum ATCC 35319. Appl Environ Microbiol. 1993;59:3154–3156. doi: 10.1128/aem.59.9.3154-3156.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gelhaye E, Petitdemange H, Gay R. Adhesion and growth rate of Clostridium cellulolyticum ATCC 35319 on crystalline cellulose. J Bacteriol. 1993;175:3452–3458. doi: 10.1128/jb.175.11.3452-3458.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giallo J, Gaudin C, Belaich J P, Petitdemange E, Caillet-Mangin F. Metabolism of glucose and cellobiose by cellulolytic mesophilic Clostridium sp. strain H10. Appl Environ Microbiol. 1983;45:843–849. doi: 10.1128/aem.45.3.843-849.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giallo J, Gaudin C, Belaich J P. Metabolism and solubilization of cellulose by Clostridium cellulolyticum H10. Appl Environ Microbiol. 1985;45:1216–1221. doi: 10.1128/aem.49.5.1216-1221.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giles K W, Myers A. An improved diphenylamine method for the estimation of deoxyribonucleic acid. Nature. 1965;206:93. [Google Scholar]
- 18.Huang L, Forsberg C W. Cellulose digestion and cellulase regulation and distribution in Fibrobacter succinogenes subsp. succinogenes S85. Appl Environ Microbiol. 1990;56:1221–1228. doi: 10.1128/aem.56.5.1221-1228.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hungate R E. The anaerobic mesophilic cellulolytic bacteria. Bacteriol Rev. 1950;14:1–49. doi: 10.1128/br.14.1.1-49.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hungate R E. A roll tube method for cultivation of strict anaerobes. Methods Microbiol. 1969;33:117–132. [Google Scholar]
- 21.Klingenberg M. Nicotinamide-adenine dinucleotides (NAD+, NADP+, NADH, NADPH). Spectrophotometric and fluorimetric methods. In: Bergmeyer H Y, editor. Methods in enzymatic analysis. New York, N.Y: Academic Press, Inc.; 1965. pp. 2045–2059. [Google Scholar]
- 22.Koch A L. Microbial physiology and ecology of slow growth. Microbiol Mol Biol Rev. 1997;61:305–318. doi: 10.1128/mmbr.61.3.305-318.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leschine S B. Cellulose degradation in anaerobic environments. Annu Rev Microbiol. 1995;49:399–426. doi: 10.1146/annurev.mi.49.100195.002151. [DOI] [PubMed] [Google Scholar]
- 24.London J, Knight M. Concentrations of nicotinamide nucleotide coenzymes in micro-organisms. J Gen Microbiol. 1966;44:241–254. doi: 10.1099/00221287-44-2-241. [DOI] [PubMed] [Google Scholar]
- 25.Lovenberg W, Buchanan B B, Rabinowitz J C. Studies on the chemical nature of clostridial ferredoxin. J Biol Chem. 1963;238:3899–3913. [PubMed] [Google Scholar]
- 26.Lowry O H, Rosebrough N J, Farr A L, Randall R J. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 27.Maglione G, Russel J B. The adverse effect of nitrogen limitation and excess cellobiose on Fibrobacter succinogenes S85. Appl Microbiol Biotechnol. 1997;48:720–725. [Google Scholar]
- 28.Meinecke B, Bertram J, Gottschalk G. Purification and characterization of the pyruvate-ferredoxin oxidoreductase from Clostridium acetobutylicum. Arch Microbiol. 1989;152:244–250. doi: 10.1007/BF00409658. [DOI] [PubMed] [Google Scholar]
- 29.Miller G L. Use of dinitrosalicylic acid reagent for determination of reducing sugars. Anal Chem. 1959;31:426–428. [Google Scholar]
- 30.Miller G L, Blum R, Glennon W, Burton A L. Measurement of carboxymethylcellulase activity. Anal Chem. 1960;138:481–487. [Google Scholar]
- 31.Mortenson L E, Valentine R C, Carnahan J E. Ferredoxin in the phosphoroclastic reaction of pyruvic acid and its relation to nitrogen fixation in Clostridium pasteurianum. J Biol Chem. 1963;238:794–800. [Google Scholar]
- 32.Ng T, Zeikus J G. Differential metabolism of cellobiose and glucose by Clostridium thermocellum and Clostridium thermohydrosulfuricum. J Bacteriol. 1982;150:1391–1399. doi: 10.1128/jb.150.3.1391-1399.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Papoutsakis E T, Meyer C L. Equations and calculations of product yields and preferred pathways for butanediol and mixed-acid fermentations. Biotechnol Bioeng. 1985;27:50–66. doi: 10.1002/bit.260270108. [DOI] [PubMed] [Google Scholar]
- 34.Payot S, Guedon E, Cailliez C, Gelhaye E, Petitdemange H. Metabolism of cellobiose by Clostridium cellulolyticum growing in continuous culture: evidence for decreased NADH reoxidation as a factor limiting growth. Microbiology. 1998;144:375–384. doi: 10.1099/00221287-144-2-375. [DOI] [PubMed] [Google Scholar]
- 35.Petitdemange E, Caillet F, Giallo J, Gaudin C. Clostridium cellulolyticum sp. nov., a cellulolytic mesophilic species from decayed grass. Int J Syst Bacteriol. 1984;34:155–159. [Google Scholar]
- 36.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 37.Shi Y, Weimer P J. Utilization of individual cellodextrins by three predominant ruminal cellulolytic bacteria. Appl Environ Microbiol. 1996;62:1084–1088. doi: 10.1128/aem.62.3.1084-1088.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sleat R, Mah R, Robinson R. Isolation and characterization of an anaerobic, cellulolytic bacterium, Clostridium cellulovorans sp. nov. Appl Environ Microbiol. 1984;48:88–93. doi: 10.1128/aem.48.1.88-93.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Strobel H J, Caldwell F C, Dawson K A. Carbohydrate transport by the anaerobic thermophile Clostridium thermocellum LQRI. Appl Environ Microbiol. 1995;61:4012–4015. doi: 10.1128/aem.61.11.4012-4015.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomas T D, Elwood D C, Longyear M C. Change from homo- to heterolactic fermentation by Streptococcus lactis resulting from glucose limitation in anaerobic chemostat cultures. J Bacteriol. 1979;138:109–117. doi: 10.1128/jb.138.1.109-117.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thompson J, Thomas T D. Phosphoenolpyruvate and 2-phosphoglycerate: endogenous energy source(s) for sugar accumulation by starved cells of Streptococcus lactis. J Bacteriol. 1977;130:583–595. doi: 10.1128/jb.130.2.583-595.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tomme P, Warren R A J, Gilkes N R. Cellulose hydrolysis by bacteria and fungi. Adv Microb Physiol. 1995;37:1–81. doi: 10.1016/s0065-2911(08)60143-5. [DOI] [PubMed] [Google Scholar]
- 43.Uyeda K, Rabinowitz J C. Pyruvate-ferredoxin oxidoreductase. Purification and properties of the enzyme. J Biol Chem. 1971;246:3111–3119. [PubMed] [Google Scholar]
- 44.Weimer P J, Zeikus J G. Fermentation of cellulose and cellobiose by Clostridium thermocellum in the absence and presence of Methanobacterium thermoautotrophicum. Appl Environ Microbiol. 1977;33:289–297. doi: 10.1128/aem.33.2.289-297.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wimpenny J W T, Firth A. Levels of nicotinamide adenine dinucleotide and reduced nicotinamide adenine dinucleotide in facultative bacteria and the effect of oxygen. J Bacteriol. 1972;111:24–32. doi: 10.1128/jb.111.1.24-32.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]