

Abstract
Toll-like receptors (TLRs) are pattern recognition receptors with a well-documented role in the innate and adaptive immune response. Interestingly, TLR activation has also been linked to several brain functions including neurogenesis and synaptogenesis. Increasing evidence supports TLR involvement in peripheral and central inflammation underlying normal aging and the pathogenesis of clinical conditions characterized by cognitive decline. These include not only major neurodegenerative diseases, but also traumatic brain injuries, surgeries, alcohol consumption- and chemotherapy-induced cognitive impairment. This review will first summarize the physiological roles of TLRs in the nervous system, and then illustrate the emerging involvement of TLRs in cognitive functions, pointing to these receptors as novel enticing pharmacological targets to develop more efficient drugs for the treatment of cognitive impairment.
Keywords: Toll-like receptors, neuroinflammation, cognitive functions, glia, gut-brain axis
TLRs structure, signaling and expression in the brain
Toll-like receptors (TLRs) are pattern recognition receptors (PRRs) with a primary and well-documented role in innate and adaptive immunity. Each TLR consists of a leucine-rich horseshoe-shaped extracellular domain, a single transmembrane helix, and an intracellular toll/interleukin-1 receptor (TIR) domain that interacts with different downstream adaptors (see Glossary) to propagate signal transduction (Figure 1) [1]. Ten functional TLRs have been identified in humans (TLR1–10) [1] and can be classified according to their cellular localization: all TLRs, excepting TLR8, are expressed on the plasma membrane, whereas only TLR3, TLR7, TLR8 and TLR9 are detected on the endosomal membrane [2]. The fact that TLR3, TLR7, and TLR9 may occur both on the cell surface and intracellularly could increase functional responses through the crosstalk with the other TLRs, depending on the tissue and the cell type [2]. Mice, which express twelve TLRs subtypes (TLR1–9 and TLR11–13), lack a functional ortholog for TLR10 [1]. TLR extracellular domains respond to molecules expressed by microbes and invading pathogens that share highly conserved structural motifs (microbe- or pathogen-associated molecular patterns, MAMPs or PAMPs), as well as lipids, nucleic acids and other metabolites endogenously released during an inflammatory response (damage-associated molecular patterns, DAMPs) [1]. Considering the huge variety of ligands recognized and intracellular responses mediated by TLRs, a series of accessory molecules intervene to ensure proper TLRs arrangement on the cell surface and ligand detection as well as to facilitate TLRs signaling (Figure 1) [3]. For example, cluster differentiation-14 (CD14), which can exist both as soluble and as a membrane bound protein, contributes to ligand delivery to different TLRs; lipopolysaccharide-binding protein (LBP) binds to lipopolysaccharide (LPS) and other PAMPs and activates multiple TLRs; myeloid differentiation factor 2 (MD2) is involved in TLR4 conformational changes and LPS binding [3]. Following ligand stimulation, TLRs form both homo- and heterodimers via their lateral faces, triggering the intracellular recruitment of adaptor proteins (Figure 1). Myeloid differentiation primary response 88 (MyD88) is the most common adaptor protein that interacts with the intracellular domain of TLRs [1]. All TLR subtypes, except for TLR3, which uses a different adaptor protein called TIR domain-containing adaptor-inducing interferon-β (TRIF), display MyD88-dependent signaling. TLR4 represents another exception as it is the only TLR that uses both MyD88-dependent and TRIF-dependent pathways [1]. TLR stimulation prompts a complex signaling network that involves the activation of different transcription factors such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), mitogen-activated protein kinase (MAPK) and interferon regulatory factors (IRFs) [1]. Eventually, several inflammatory cascades, including NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome-induced interleukin-1β (IL1β) as well as interferon (IFN)-inducible genes are activated (Figure 1) [4].
Figure 1. TLRs signaling in the brain.
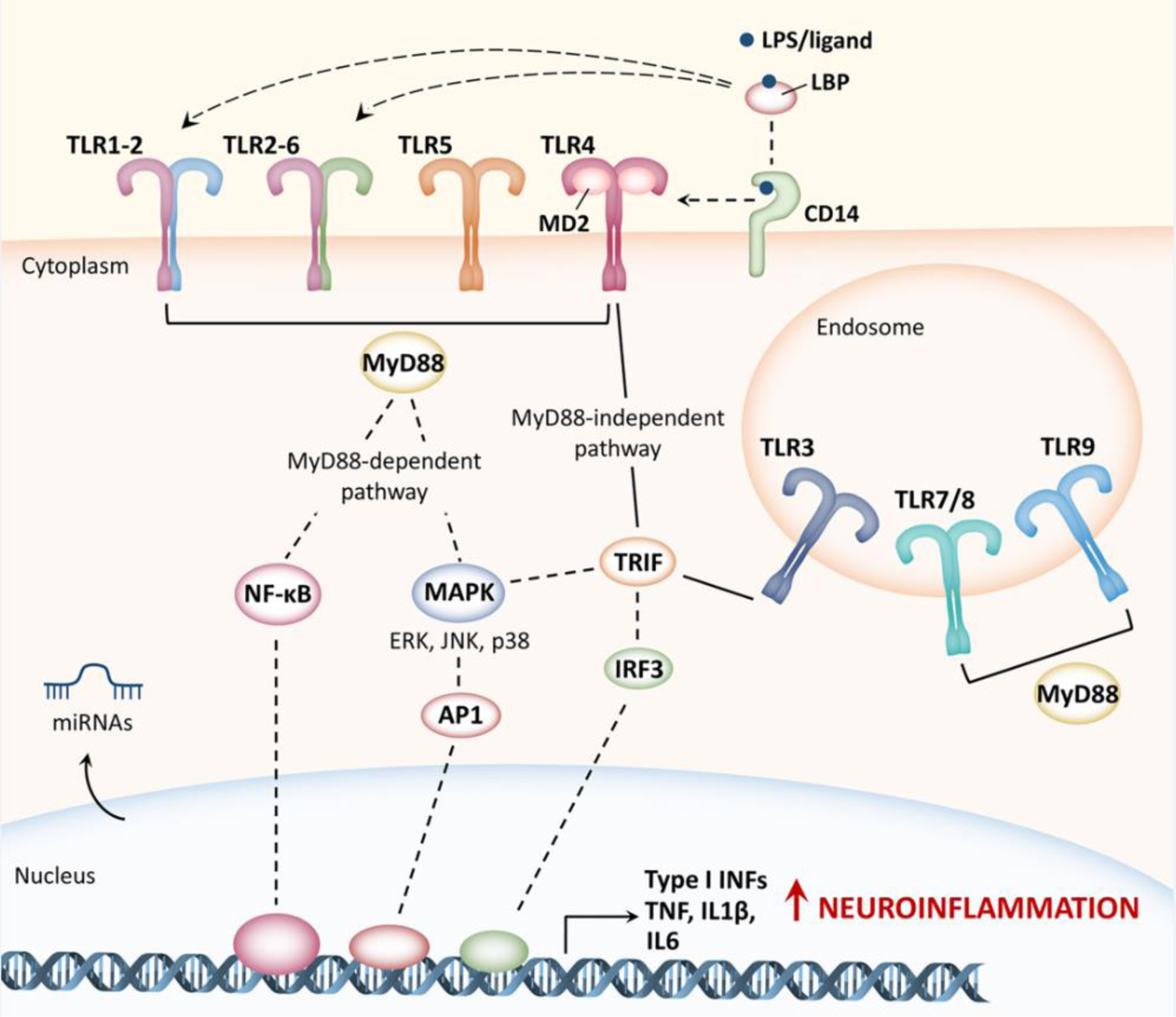
TLRs are expressed on the cell surface as well as in endosomes, and mainly in immune and glial cells. TLRs activation is extremely complex and requires multiple accessory proteins (including cluster differentiation-14, CD14, lipopolysaccharide-binding protein, LBP, and myeloid differentiation factor 2, MD2) that participate in ligand recognition and TLRs conformational changes. Following stimulation, TLRs form both homo- and heterodimers that triggers two main signaling cascades, depending on the adaptor protein involved: the myeloid differentiation primary response protein 88 (MyD88)-dependent pathway) or the TIR domain-containing adaptor-inducing interferon-β (TRIF) MyD88-independent pathway). Downstream signaling networks include the activation of different transcription factors such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), mitogen-activated protein kinase (MAPK) and interferon regulatory factors (IRFs, including IRF3) ultimately leading to the production of inflammatory cytokines (TNF, IL1β, IL6) including Type I interferons (Type I INFs), which are responsible for T lymphocyte stimulation and recruitment. Noteworthy, TLR-mediated inflammatory cascades (in particular TLR2 and TLR4) in both neurons and glia overlap in normal aging and in the onset of different forms of dementia. TLRs activation can also drive the transcription of micro RNAs (miRNAs), which, in turn, can either stimulate or block TLRs activation. This is particularly interesting as multiple miRNAs are implicated in the pathogenesis of different diseases associated with cognitive impairment. ERK, extracellular signal-regulated kinases; JNK, c-Jun N-terminal kinases; AP1, activator protein 1.
Among all PRRs, TLRs possess the most extensive spectrum of pathogen/damage signal recognition, representing the first line of defense for the organism. In the central nervous system (CNS), TLRs are abundantly expressed on microglia, as well as on astrocytes and oligodendrocytes [5]. Microglia and astrocytes represent the primary innate immune defense in the CNS. In case of injury, TLRs activation on microglia and astrocytes contribute to their shift towards an inflammatory phenotype culminating in the release of cytokines and chemokines, and antigen presentation on the cell surface [6, 7]. Generally immune responses in the CNS are faster than the one mediated by peripheral immune cells, and inflammation has a shorter duration than the one in the periphery [6]. However, chronic glial activation in the CNS, can lead to the engagement and infiltration of peripheral immune cells in the brain that engender sustained immune responses [7].
Even if at a much lower concentration, TLRs have also been found expressed on neural progenitor cells (NPCs) and mature neurons [1, 5]. As neurons do not participate in the immune response, this implies that TLRs have a role in neuronal physiology and functional responses.
This review will first briefly illustrate the roles of TLRs in brain physiology and aging. Then, the most recent findings on TLR involvement in peripheral and central inflammation underpinning the pathogenesis of several clinical conditions characterized by cognitive decline will be discussed.
Roles of TLRs in brain physiology
Using mainly genetic approaches (i.e. mice knockouts), several studies started to unravel TLRs involvement in neuronal development, plasticity, and physiology, all processes that also depend on TLRs temporal and spatial expression in the brain [8–12]. TLR2 absence has a severe impact on brain function and structure: TLR2 knockout mice develop cognitive decline around 7 months of age and show reduced regional cerebral blood flow, inhibited long-term potentiation and increased blood–brain barrier permeability at 12 months [8]. In NPCs, TLR2 activation promotes hippocampal neurogenesis, whereas TLR3 and TLR4 signaling negatively regulate neuronal proliferation and differentiation [1, 9]. Interestingly, recent findings showed sexually dimorphic and region-specific effects of TLR4 on hippocampal neurogenesis, with TLR4 signaling mainly affecting neuronal proliferation in the brain of young adult females [9, 10]. TLR5 was found to promote neural differentiation both during brain development and in the adult brain, via NF-κB and interleukin-6/CREB pathways and the c-Jun N-terminal Kinase (JNK) pathway, respectively [12]. Moreover, TLR5 deficiency was linked to impairment in memory performances in mice [12]. TLR3 was found involved in neuronal maturation and morphogenesis together with TLR7 and TLR8 [11]: 1) all three receptors participate in the reduction of dendritic arborization; 2) TLR3 and TLR7 act as negative regulators of axonal growth; and 3) TLR3 and TLR8 activation lead to increased dendritic spine density [11]. Besides their role during development, these endosomal TLR-mediated mechanisms can also have an important role after brain injury: cell membrane TLRs sense the released DAMPs from the extracellular environment and, through TLR3/7/8 activation, inhibit neurons from forming unproductive or even harmful connections with adjacent dying cells [1]. Besides neuronal cells, TLRs have been also extensively studied in glia. During the early phases of the inflammatory response, TLRs role in mediating phagocytosis of pathogens and misfolded/aggregated proteins is of paramount importance (see below); however, chronic microglial TLRs activation has detrimental effects on neurons (see below) [5, 7]. TLR2/3/4/5 activated microglia trigger neuronal injuries through the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)/NLRP3 inflammasome and PI3K/Akt/mammalian target of rapamycin complex 1 (mTORC1) pathway activation [13, 14]. The resultant increase of nitroxidative stress and inflammatory mediators generates a persistent state of neuroinflammation similar to that observed in the brain during normal aging (Box 1). An aging brain is characterized by a chronic and progressive immune activation and inflammation, mainly TLR-mediated, that over the years can impair memory and processing speed and also increase susceptibility to develop actual dementia and neurocognitive disorders (Box 1).
Box 1 -. TLRs in age-related cognitive decline.
As people age, the brain is subjected to profound structural and functional changes. Cerebral blood flow may decrease, impairing the high demand of oxygen and nutrients of the brain, and unrepaired accumulation of DNA damage, naturally occurring in all brain areas, can cause dysregulation of cell metabolism and increased DAMPs release [103]. Such changes result in TLRs-expressing glial cells activation and generate a chronic low-level inflammation (also called “sterile inflammation”, as occurs in the absence of pathogens), that eventually leads to neuronal death and brain networks disruption (Figure I) [103, 104]. Consequently, the brain’s overall size begins to shrink. Volume reduction of the frontal lobe and the hippocampus have been linked to impairment of cognitive functions [105]. Multiple evidence reported the correlation between TLRs overexpression/activation in the brain and age-related cognitive decline. Aged rats showed significantly increased TLR4 levels in the hippocampus compared to younger animals [106], and TLR4 deletion was found to improve cognitive brain function and structure in aged mice [107]. Moreover, in the aging brain, overexpressing-TLR2/TLR4 microglia showed a maladaptive cell response: these cells when stimulated boosted brain neuroinflammation leading to impairments in hippocampal-dependent memory [106]. In a murine model of β2-microglobulin-induced age-related cognitive decline, the TLR4/MyD88/NF-κB signaling in the hippocampus was found 1) to compromise synaptic functions, as demonstrated by reduced levels of synaptophysin and postsynaptic density protein 95, and 2) promote neuronal apoptosis through IL1β and tumor necrosis factor production [108].
Age-related cognitive decline has also been associated with TLRs dysregulation along the gut-brain axis [91, 109]. Besides brain, aged mice showed higher TLR4 expression/activation also in the gut, that correlated with worse cognitive outcomes. Restoring gut microbiota attenuated TLR4/NFκB-induced enteric and central inflammation as well as learning and memory in old mice [91, 109], confirming the importance of gut-brain communication in cognition also during normal aging.
Maladaptive cell responses in an aged brain also promote the uncontrolled neuroinflammation following trauma. For example, overexpression of hippocampal MD2 (an essential cofactor for TLR4 activation) was recently linked to increased susceptibility of old mice to develop post operative cognitive dysfunction (POCD) [110]. Similarly, aging is the primary risk factor for major neurodegenerative diseases, such as vascular dementia, Alzheimer’s, and Parkinson’s disease. With the world population aging rapidly, age-related dementia represents a growing global health concern. Strategies aimed to counteract progressive immune activation and inflammation, such as targeting TLRs, could successfully prevent or improve age-related cognitive decline.
Below, we discuss TLRs-driven neuroinflammation in major neurodegenerative disorders and following injuries to the nervous system, pointing out how pathological pathways often overlap.
Roles of TLRs in major neurodegenerative disorders
The term ‘neurodegenerative disorders’ encompasses a wide range of clinical phenotypes that have in common the progressive loss of neuronal integrity and/or function. The most common symptoms include mood alterations, mild to severe impairment in voluntary and involuntary motor functions and, of course, impaired cognitive abilities. Neuroprotective or neurotoxic effects of TLR activation are disease-stage dependent and also depend on the TLR subtype/s and the cell type involved.
Alzheimer’s disease
An estimated 6.2 million people with Alzheimer’s disease (AD) are currently living in the US, and this number is expecting to grow to 13.8 million by 2060 [15]. The role of TLRs in AD has been studied for more than 15 years [16, 17]. In human and mouse brain, both TLR2 and TLR4 are up-regulated in the microglia surrounding amyloid-β (Aβ) plaques [18, 19]. During the early phases of AD, fibrillar Aβ-induced TLR2 and TLR4 activation on microglia increases microglia recruitment and phagocytosis of Aβ deposits with consequent preservation of cognitive functions in mice [20, 21]. Moreover, repeated systemic administration of low doses of LPS results in a chronic mild TLR4 stimulation that attenuates AD-related tauopathy and improves synaptic impairments and cognitive performance [22]. However, as the disease progresses, sustained TLR2 and TLR4 activation induces extensive neuroinflammation, leading to severe neurotoxic effects [5, 23, 24]. Both TLR2 and TLR4 are involved in the microglial switch towards the inflammatory M1 phenotype, characterized by the production and release of high levels of inflammatory cytokines that ultimately lead to neuronal death [25–28]. In rodents, pharmacological strategies that directly (intravenous injection of anti-TLR2 antibody) or indirectly (oral administration of the dual cholinesterase inhibitor DL0410) target TLR2 and TLR4 signaling attenuate neuroinflammation and improve cognitive functions [29, 30]. TLR4 signaling activation is also implicated as a potential link between diabetes mellitus and the onset of AD [31]. TLR3 is increased in the human phagocytic microglia surrounding Aβ deposits, and its up-regulation correlates with the progression of pathology [32]. However, in vitro experiments demonstrate that fibrillar Aβ is not responsible for increased TLR3 expression on microglia, suggesting that TLR3 may be involved in other aspects of AD pathogenesis [32]. TLR5 has been proposed as a relevant target gene for AD treatment [33], as a TLR5 decoy receptor-based strategy counteracts Aβ aggregation [34]. The cerebrospinal fluid of AD patients contains high levels of neurotoxic microRNAs belonging to the let-7 family: in particular, let-7b was found to activate endosomal TLR7 triggering caspase-3-mediated apoptosis in CNS neurons [35, 36]. Interestingly, in two triple-transgenic mouse model of AD (3xTg-AD and Tg-SwDI), the first in vivo evidence for the beneficial effects of TLR9 agonism was reported [37, 38]. TLR9 specifically binds to DNA sequences containing unmethylated cytosine-guanosine (CpG) motifs; systemic administration of type B CpG oligodeoxynucleotides (CpG ODNs) ameliorates cognitive performance, reducing all the major pathological hallmarks of AD while increasing plasma levels of the neuroprotective cytokine IL10 [37, 38].
Vascular dementia
Vascular dementia (VD) is the second most common neurodegenerative disorder [39]. Strokes can cause a progressive a decrease in blood supply to the brain, triggering inflammatory TLR4/MyD88/NF-κB pathway-driven neural death and cognitive impairment [39]. Recently, low-intensity focused ultrasound stimulation was found to ameliorate working memory dysfunctions in VD through the inhibition of the TLR4/NF-κB pathway in rats prefrontal cortex [40]. The microRNA-93 is implicated in the pathogenesis of VD; it was found to activate the TLR4/MyD88/NF-κB pathway in hippocampal microglia contributing to memory deficits [41].
Parkinson’s disease
Parkinson’s disease (PD) is another extremely common neurodegenerative disease, counting about one million patients in US alonei. It is characterized by pathological aggregates of the synaptic protein α-synuclein that spread across the brain along anatomically connected areas, causing neuronal dysfunction and death [42]. The progressive loss of dopaminergic neurons within the substantia nigra negatively impacts the function of the whole basal ganglia network, leading to motor and cognitive impairment in patients [42]. Similar to what happens in AD, TLR4 activation and overexpression in PD was reported as both neuroprotective and neurotoxic: on one hand, its signaling had an important role in microglia-mediated clearance of extracellular α-synuclein that prevented neuronal degeneration and improves behavioral responses [43, 44]; on the other hand, the prolonged exposure of TLR4 to α-synuclein caused the neurotoxic activation of both astrocytes and microglia [45]. In mice, genetic ablation of TLR4 as well as oral administration of Salidroside, an anti-inflammatory glucoside that indirectly targets TLR4 prevents striatal dopaminergic neurons loss and α-synuclein-induced increased glial activation, oxidative stress and inhibits the NLRP3 inflammasome pathway [46–48]. As observed for TLR4, sustained TLR2 activation on glial cells increases neuroinflammation (TNF and IL1β release), whereas neuronal TLR2 activation impairs α-synuclein clearance through an AKT/mTOR-dependent mechanism, establishing a positive loop that increases α-synuclein accumulation and neurodegeneration [49, 50]. Interestingly, two peptides targeting the TLR2 cascade were recently developed: the first, called wtTIDM, blocks TLR2 interaction with MyD88; the second one, called wtNBD, acts downstream to prevent TLR2-mediated NF-κB activation [51]. Both peptides reduce microglia-derived inflammatory mediators, decrease α-synuclein accumulation and spreading, and protect dopaminergic neurons [51]. Recent work provided the first evidence of TLR5 involvement in α-synuclein-induced microglial NLRP3/caspase1/IL1β signaling activation [52]. Neutralizing TLR5 with an anti-TLR5 antibody attenuates α-synuclein-mediated IL1β and CXCL2 release from microglia [52]. TLR7 and TLR8 were also recently associated with PD pathogenesis [53]: TLR7, TLR8 and combined TLR7/8 deletion significantly reduce α-synuclein aggregation, astrogliosis, microgliosis and T-cell activation, preventing the loss of dopaminergic neurons and the onset of motor and non-motor PD symptoms [53]. The role of TLR9 in PD is less investigated, but it has been reported that the downregulation of microglia glucocorticoid receptors, which leads to dopaminergic neurodegeneration in parkinsonism [54], involves TLR9 activation [55].
Multiple sclerosis
Multiple sclerosis is a chronic, demyelinating disease, where cognitive impairment represents a common, but sometimes neglected, symptom. Activation of both microglial and astrocytic TLR2 and TLR4 is involved in the pathogenesis of MS with similar signaling cascades observed in other major neurodegenerative disorders [56]. Human endogenous retrovirus W (HERV-W) is a group of endogenous retroviruses that have been linked to MS pathogenesis: HERV-W envelope protein impairs oligodendrocyte precursor cells (OPCs) maturation and differentiation, thus negatively affecting the myelination process [57, 58]. Recently, an in vitro study demonstrated that blocking TLR4 expressed on oligodendrocytes with L48H37 (a curcumin analog) or C20 (a small molecule component of many natural flavonoids and isoflavonoids) rescues their HERV-W-induced maturation arrest and their myelination capacity by reducing inflammatory cytokine expression and nitroxidative stress [57]. Interestingly, TLR3 signaling was shown to be neuroprotective as it suppressed relapsing demyelination in a mouse model of MS [59].
The pathogenesis of neurodegenerative diseases is multifactorial and most underlying causes remain unknown, but increasing findings encourage strategies aimed at targeting TLR-induced neuroinflammation.
Targeting TLR signaling after injury to the nervous system
Nervous system injury is associated with the onset of cognitive deficits. Besides normal aging, extremely common life events, such as traumatic brain injury (TBI), alcohol consumption, chronic pain states, cancer and chemotherapy treatment, have been acknowledged as risk factors for the development of dementia both immediately after the event, but also years later. Although these occurrences are often unpredictable, early interventions aimed at blocking neuroinflammation have proven to be effective in preventing or attenuating the onset of cognitive impairment.
Traumatic injuries
Trauma (e.g. from accidental falls, construction and car accidents, violence, sport) results in brain edema, hypoxia/ischemia and neuroinflammation (referred as secondary injury) that lead to long-lasting deficits in memory, attention and executive functions in more than 50% of patients [60]. TLR2 and TLR4 are up-regulated in the injured cortex and promote 1) M1 neurotoxic phenotypes through the NF-κB-p65/pERK/NLRP3 pathway [61] and 2) the recruitment of reactive astrocytes at the injury site with detrimental effects on synapses and neurological outcomes [62]. Interestingly, TLR2/TLR4 activation also interferes with oligodendrocyte progenitor cell (OPC) survival, impairing the remyelination of injured axons [63].
Targeting TLR4 signaling with the selective inhibitor TAK-242 (resatorvid), within hours of the injury reduces neuronal apoptosis and improves neurobehavioral impairments in rodents [64–66]. Aside from its contribution to neuroinflammation, TLR2 activation is involved in the endogenous neurogenesis after TBI: it promotes neuronal stem cell (NSC) proliferation and differentiation, contributing to the repair process in the hippocampus [67], confirming its beneficial role in hippocampal neurogenesis [68] also in pathological conditions.
Alcohol consumption
Chronic alcohol abuse impairs CNS homeostasis and contributes to more than 200 chronic diseases, including severe forms of dementia [69]. Following ethanol intake, TLR2, TLR3, TLR4 and TLR7 expression increases in the mouse prefrontal cortex, another important area involved in cognitive function [70]. Ethanol-induced TLR3 activation increases the expression of inflammatory IL1β, IL6 and TNF in the hippocampus [71], as well as voluntary alcohol intake [72, 73]. TLR2 and TLR4 knockout rodents are protected from ethanol-induced neuroinflammation [74], myelin and synaptic dysfunctions, and long-term cognitive alterations [75, 76]. Analysis of postmortem human alcoholic brains revealed an increased TLR7 expression and microglial activation in the hippocampus [77]. The same study demonstrated that ethanol enhances microglia-derived neurotoxic let-7b release that contributes to hippocampal neurodegeneration in rats through TLR7 [77].
Postoperative cognitive dysfunction
Postoperative cognitive dysfunction (POCD) is a recognized clinical condition characterized by the appearance of cognitive impairment in the acute and/or late stage after anesthesia and surgical operations. Following surgery, TLR2, together with TLR4, was found involved in NLRP3- and MAPK-mediated neurological damage [78], as well as in the increased IL1β and IL6 expression in the cerebral cortex and hippocampus, with consequent impairment of learning and memory [79]. Postsurgical treatment with morphine, widely used to alleviate moderate-to-severe postoperative pain, can increase the persistence of POCD through TLR4-induced NLRP3-mediated hippocampal neuroinflammation in aged rats [80]. Moreover, increased TLR3 expression and activation was proven to have a role in POCD, as TLR3 knockout mice show improved hippocampal apoptosis, inflammation and cognitive performances following surgery [81].
TLR-mediated neuroinflammation in the enteric nervous system as early event in cognitive decline
When studying cognitive impairment, the CNS has always been considered as the main area of investigation. However, mounting evidence suggests that signs of neuronal dysfunction (protein misfolding and aggregates, altered neurotransmission, reactive gliosis) in the enteric nervous system (ENS) can be detected long before neurodegeneration in the CNS and the appearance of dementia [82]. For this reason, investigating the ENS has gained attention in studies of neurocognitive disease pathogenesis. The enteric nervous system (ENS) consists of about 500 million neurons (five times the number present in the spinal cord) organized in thousands of ganglia distributed within two main plexuses, the myenteric (Auerbach’s) and submucosal (Meissner’s) (Figure 2) [83]. The bidirectional communication between ENS and CNS occurs through the parasympathetic (Vagus nerve) and sympathetic (i.e. via the prevertebral ganglia) system, known as the gut-brain axis [83]. Interestingly, neurons within the ENS mainly communicate through neurotransmitters similar to the CNS, including dopamine, serotonin and acetylcholine, and it has been shown that the ENS can also operate independently of the CNS [84]. Moreover, enteric glial cells (EGCs) share several structural and functional characteristics with the central astrocytes. Thereby, the ENS is often referred to as the “second brain” [83, 85].
Figure 2. TLR-mediated neuroinflammation in the ENS as an early event in the development of cognitive dysfunctions.
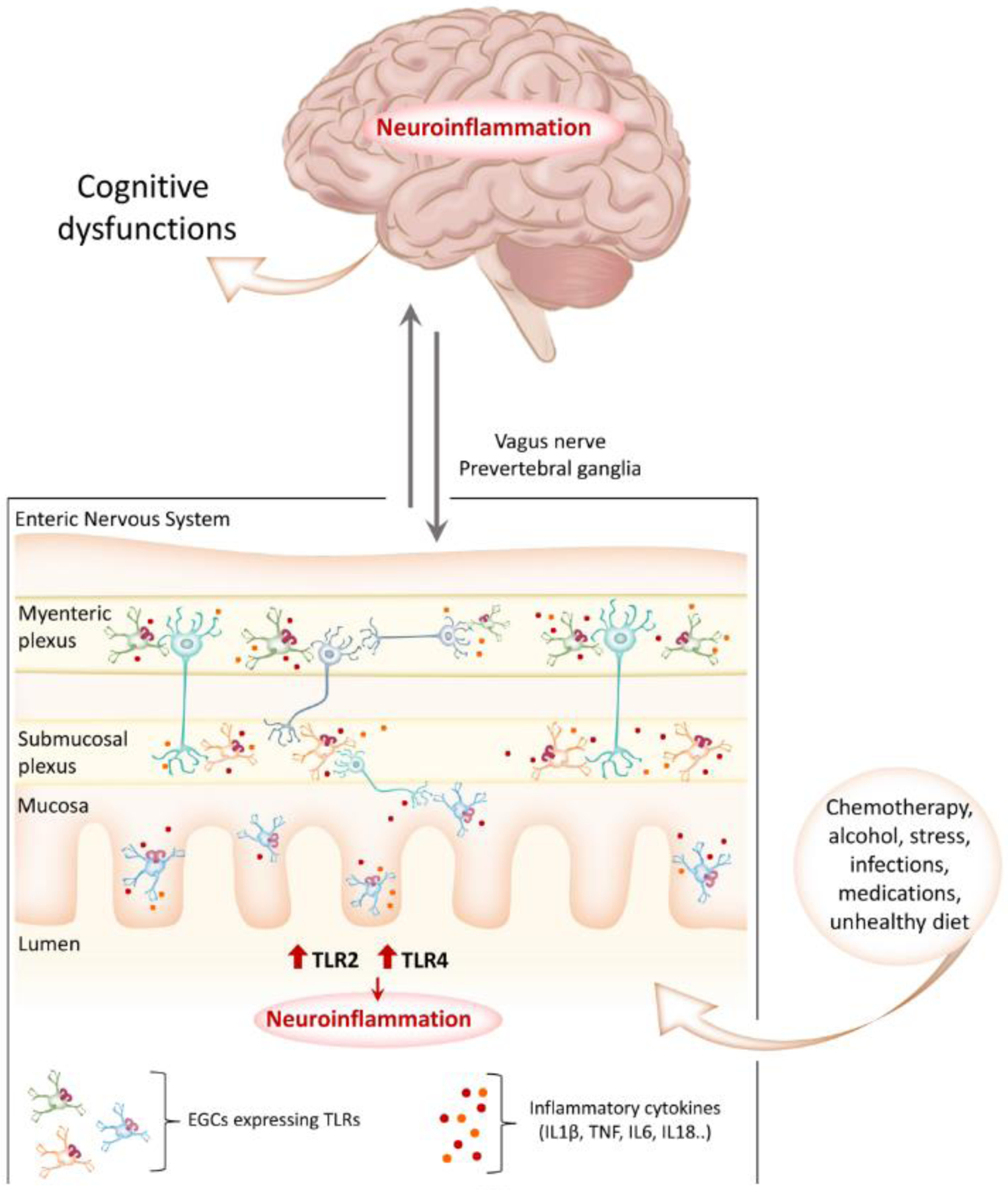
There is mounting evidence that signs of glial activation and neuronal dysfunction in the enteric nervous system (ENS) can be detected long before neurodegeneration in the CNS and the appearance of dementia. Enteric glial cells (EGCs) share several structural and functional characteristics with the central astrocytes and express higher levels of TLRs than neurons. It has become evident that external insults such as chemotherapy, alcohol consumption, medications (e.g., antibiotics, morphine) as well as an unbalanced diet can cause the dysregulation of TLR signaling in the ENS, leading to the overproduction of inflammatory cytokines and the generation of a persistent inflammatory state. Neuroinflammation and pathological aggregates of misfolded proteins then spread to the brain through the vagus nerve, ultimately leading to the development of cognitive dysfunctions.
TLRs are essential mediators in ENS physiological functions and in the neuroimmune communication along the microbiota-gut-brain axis [86]. Different temporal and spatial expression patterns of TLR4 and TLR7 expression in the ENS were reported during embryonic development, suggesting a role for these receptors during morphogenesis of the ENS [87]. TLR2 activation promotes neurogenesis and maintains ENS structure and function after insult [88]. However, TLR up-regulation and chronic activation in the ENS have been also linked with cognitive decline associated with normal aging or pathological conditions [89–91]. It has been proposed that microbial dysbiosis-induced TLR2 and TLR4 signaling alteration can promote α-synuclein aggregation in ENS neurons: α-synuclein aggregates can then spread to the peripheral nerves and the brain, contributing to PD pathogenesis and progression [82]. In support, a recent study in a mouse model of PD showed that 1) pre-symptomatic animals had a dysregulation of TLR2 in colonic tissues and 2) that enteric α-synuclein induced caspase-1, IL1β and TNF increase in the gut, with compromised neuroimmune crosstalk before the emergence of the central pathology and PD symptoms [92]. To investigate the extra-CNS origin of AD, amyloid-β, which is a ligand for TLR2/TLR4, was injected into the gastrointestinal tract of 2-months old mice. One year later, Aβ deposits were found in the vagus nerve and mice displayed both cerebral amyloidosis and cognitive dysfunctions [93]. Crohn’s disease (CD) is a type of inflammatory bowel disease whose symptoms often include cognitive impairment in patients [94]. CD patients show a dysregulation of serotonin levels in the serum, and it was recently shown that inhibition of TLR2 in the gut with the novel small-molecule antagonist MMG-11 restores serotoninergic signaling and attenuated p38MAPK-mediated neuroinflammation [95].
It has become evident that neuroinflammation in the ENS is an early event in the development of cognitive impairment during normal aging and in pathological states (Figure 2). Considering their prominent role in cell communication and in the propagation of inflammatory signaling, studying TLRs could provide new knowledge in our understanding on the pre-symptomatic evolution of neurodegenerative diseases.
Concluding remarks and future perspective
Besides their role in innate immunity, TLRs have emerged as major contributors to the progression of glia-mediated peripheral and central inflammation underpinning cognitive decline. TLR2 and TLR4 have been so far the most extensively studied, and their downstream signaling pathways, in both neurons and glia, overlap in the pathogenesis of several diseases associated with dementia. This suggests the possibility that the same molecular mechanisms also underlie the development of cognitive impairment in those clinical conditions whose etiologies remain mostly unknown. An example is the recent findings on Chemotherapy-Induced Cognitive Impairment (CICI), a major neurotoxicity that affects memory, attention, and executive functions in >50% of patients during and after the completion of chemotherapy [96]. It was reported that TLR4 activation in the hippocampus and prefrontal cortex is the linchpin in cisplatin-induced NLRP3 inflammasome activation and increased IL1β formation leading to cognitive impairment in mice (S. Squillace et al., unpublished). Similar results were obtain with doxorubicin, another widely used chemotherapeutic [97]. Since compelling evidence suggests an involvement of the microbiota-gut-brain axis also in CICI [98–100], it is important to investigate if dysregulation of TLRs signaling in enteric glia anticipates CNS neurodegeneration (see Outstanding Questions).
Outstanding Questions.
Emerging evidence suggests that microglia, astrocytes and EGCs are not a homogenous cell type but are rather constituted by several functionally and morphologically distinct subpopulations. Are TLRs differentially express among glia subtypes and how do they contribute to the different reactive glial subtypes’ responses during neuroinflammation/disease progression?
Dysregulation of the gut microbiota is gaining increasing attention in the investigation of the extra CNS origin of dementia, however the underlying molecular mechanisms occurring in glia and neurons are still mostly unknown. What is the role of TLRs in the neuroimmune communication along the gut-brain axis in physiological and pathological conditions?
Several microRNAs were found dysregulated in blood, serum, and cerebrospinal fluid of patients with mild cognitive impairment, and it has been hypothesized they may contribute to the transition between normal aging and the development of severe forms of dementia. As microRNAs are TLRs ligands, more in depth investigations should be addressed to elucidate the role of their interaction not only in major neurodegenerative disorders, but also in other pathological settings characterized by cognitive decline.
Several TLRs agonists and antagonists are currently advancing in clinical trials for cancer, inflammatory and autoimmune diseases, and others are being tested in preclinical studies. However, successful in vitro and/or in vivo evaluation of TLRs agonists and antagonists often fail during clinical trials, due to the different expression pattern of TLRs among tissues or the extent to which TLRs respond to their ligands. How is it possible to improve the clinical translatability of drug targeting TLRs signaling?
Misfolded proteins, DAMPs and microRNA can activate TLRs during early pathological stages, establishing a positive loop that increases their own expression as well as the production of inflammatory cytokines, promoting disease progression. Even if selective TLR agonists and antagonists have been developed and are being tested in clinical trials for cancer, inflammatory and autoimmune diseases [101, 102], they have not been intensively studied in the context of cognitive functions. Molecular strategies such as antibody binding and decoy receptors, as well as global TLRs knockouts, have helped consolidate and expand our knowledge on the role of TLRs in cognition.
Current treatments for neurocognitive disorders attenuate the symptoms but cannot restore lost neurons and neuronal networks. For this reason, future studies aimed at further elucidating TLR-mediated neuroinflammation in the CNS and in the ENS are of pivotal importance for the development of TLRs-based therapeutics and could also overturn the concept of cognitive dysfunction as a central disease, driving development of more efficient drugs to prevent neurodegeneration in the brain and cognitive decline.
Figure I (Box 1). Aging-related structural and functional changes in the brain can increase the risk of developing dementia.
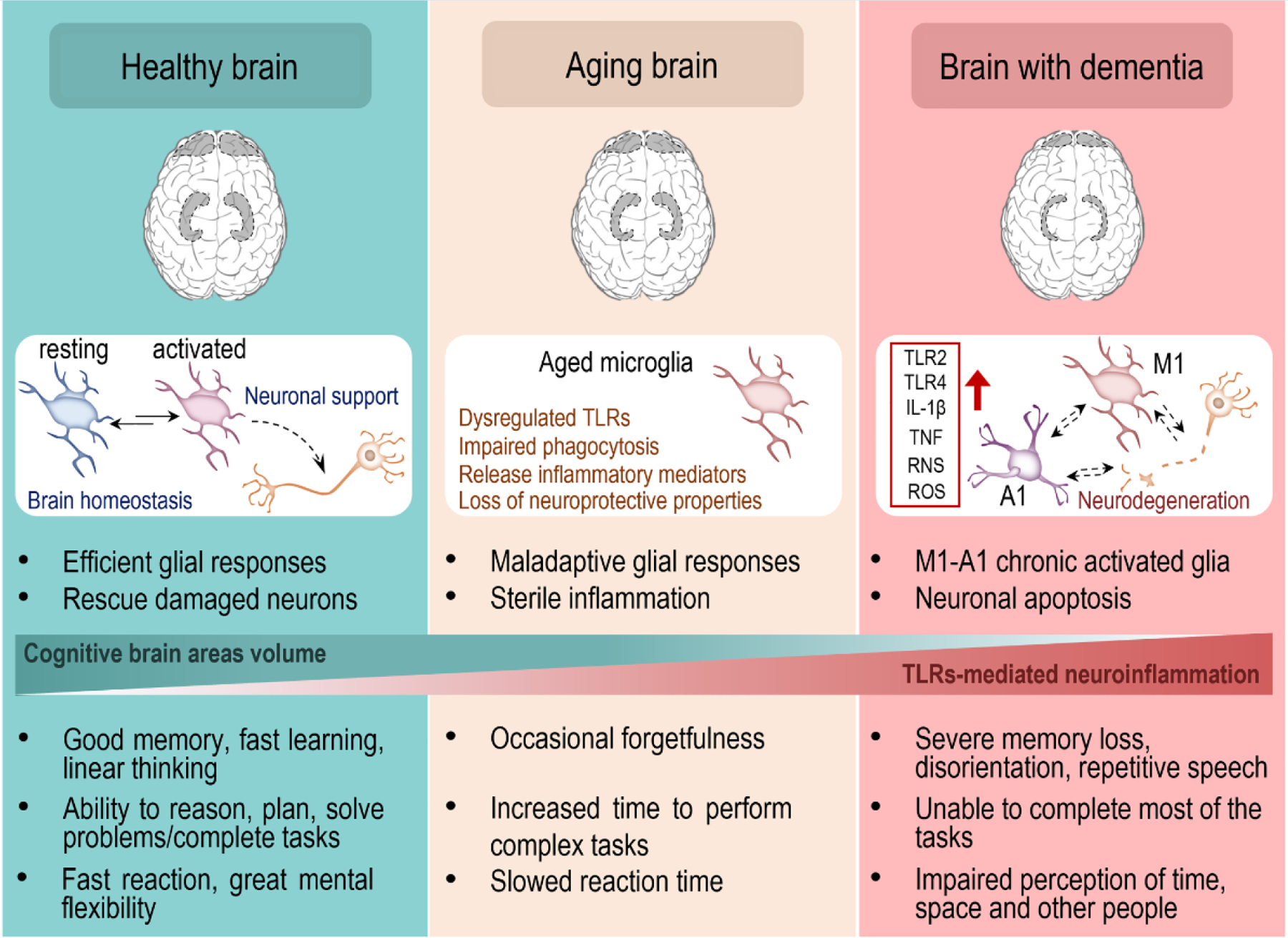
It has been reported that prefrontal cortex and hippocampus, both critically involved in cognitive functions, progressively shrink throughout the lifetime due to neuronal loss. This phenomenon correlates with an increased expression/activation of TLR2, TLR4 and their cofactors on glial cells. In particular, aged microglia show maladaptive cell responses, with overproduction of inflammatory mediators following stimulation. This generates a persistent low state of inflammation that eventually leads to neuronal degeneration and can increase the risk of developing dementia. Dysregulation of TLR2 and TLR4 cascades in glial cells underlies cognitive impairment in both normal aging and in multiple neurodegenerative disorders.
Table 1 -.
TLRs involvement in the onset and progression of cognitive impairment in major neurodegenerative diseases
| Pathology | TLRs involved | Animal and Tissue/Cell type | Functions | Ref |
|---|---|---|---|---|
| Alzheimer’s disease | TLR2 TLR4 |
Mouse microglia | Early stage – Increased microglia recruitment and phagocytosis of Aβ deposits; attenuation of AD-related tauopathy; improvement of cognitive performance | [20–22] |
| Late stage - Sustained TLR2/TLR4 activation induces M1 neurotoxic phenotype, extensive neuroinflammation and neuronal death | [23, 25–28] | |||
|
| ||||
| TLR3 | Human microglial cells | Upregulation of TLR3 on microglia correlates with the progression of the pathology | [32] | |
|
| ||||
| TLR5 | Human frontal cortex | TLR5 is upregulated in the frontal cortex of AD patients | [33] | |
| Mouse brain | TLR5 decoy receptor-based strategy counteracts Aβ aggregation | [34] | ||
|
| ||||
| TLR7 | Human cerebrospinal fluid | The miRNA let-7 is increased in AD patients and triggers | [35, 36] | |
| Mouse neurons | TLR7/Caspase3-mediated neurodegeneration in mice | |||
|
| ||||
| TLR9 | Mouse cortex and hippocampus | TLR9 agonism (CpG ODNs, i.p.) ameliorates cognitive performance; reduces all the major pathological hallmarks of AD; increases plasma levels of neuroprotective IL10 | [37, 38] | |
|
| ||||
| Vascular dementia | TLR4 | Rat hippocampus | TLR4/MyD88/NF-κB pathway drives neural death and cognitive impairment | [39] |
| Rat prefrontal cortex | Inhibition of the TLR4/NF-κB pathway improves working memory | [40] | ||
| Rat hippocampal microglia | The microRNA-93 activates TLR4/MyD88/NF-κB pathway and contributes to memory deficits | [41] | ||
|
| ||||
| Parkinson’s disease | TLR2 | PD patients’ brain – neurons and microglia | Microglial TLR2 increases neuroinflammation; neuronal TLR2 impairs α-synuclein clearance through AKT/mTOR leading to neurodegeneration | [49, 50] |
| Mouse striatum, substantia nigra and motor cortex Mouse microglia |
TLR2 antagonism (wtTIDM and wtNBD) reduces microglia-derived inflammatory mediators, decreases α-synuclein accumulation and spreading, and protects dopaminergic neurons | [51] | ||
|
| ||||
| TLR4 | Mouse microglia | Neuroprotective role - Microglia-mediated α-synuclein clearance; prevents neuronal degeneration and improves behavioral responses | [43, 44] | |
| Rodent and human neurons, microglia, and astrocytes | Neurotoxic effects – Sustained activation induces neurotoxic glial phenotype; TLR4 antagonists (TAK242 or RSLA) protect neurons from α-synuclein-induced toxicity | [45] | ||
| Mouse midbrain Mouse microglia |
Genetic TLR4 ablation or indirect antagonism (Salidroside, p.o.) prevents striatal dopaminergic neurons loss, inhibits glial activation, oxidative stress and NLRP3 inflammasome pathway | [46–48] | ||
|
| ||||
| TLR5 | Mouse microglia | α-synuclein-induces microglial NLRP3/caspase1/IL1β signaling activation through TLR5; Anti-TLR5 antibody attenuates α-synuclein-mediated IL1β and CXCL2 release from microglia | [52] | |
|
| ||||
| TLR7 TLR8 |
Mouse midbrain | TLR7/8 deletion reduces α-synuclein aggregation, astrogliosis, microgliosis and T-cell activation, preventing the loss of dopaminergic neurons and the onset of motor and non-motor PD symptoms | [53] | |
|
| ||||
| TLR9 | Mouse midbrain Microglia in the substantia nigra of human PD brains |
Downregulation of microglia glucocorticoid receptors, which leads to dopaminergic neurodegeneration in parkinsonism, involves TLR9 activation | [55] | |
|
| ||||
| Multiple sclerosis | TLR3 | Mouse spinal cord | TLR3 agonism (Poly I:C, i.p.) is neuroprotective and suppresses relapsing demyelination | [59] |
|
| ||||
| TLR4 | Rat oligodendrocytes | Blocking TLR4 (in vitro, L48H37; C20) increases myelination capacity, and reduced inflammation and nitroxidative stress | [57] | |
Highlights.
Toll-like receptors (TLRs), known for their role in innate and adaptive immune responses, have gained attention for their involvement in brain physiology as well as in peripheral and central inflammation underlying normal aging and the onset of clinical conditions characterized by cognitive decline.
TLR2 and TLR4 are the most investigated in the context of cognitive impairment, and their downstream pathways, in both neurons and glia, overlap in the pathogenesis of several diseases associated with dementia.
Increasing evidence support TLRs-mediated neuroinflammation in the enteric nervous system as an early event in cognitive decline, suggesting an extra central nervous system origin for neurocognitive diseases.
Drugs targeting TLRs-driven neuroinflammation allow an early intervention, preventing neuronal loss in the brain with a more efficient treatment of cognitive dysfunctions.
Acknowledgements
We would like to acknowledge support from NIH grant RO1CA230512 and RO1NS111120 (DS). We thank Dr J. Eissenberg (Saint Louis University) for editorial help.
Glossary
- Adaptor
Protein containing multiple binding domains whose main function is to promote the formation of short-lived protein complexes to allow a molecular signal to be transmitted. Adaptor proteins have a crucial role in signal transduction, as different adaptors can facilitate the creation of multiple signaling pathways from a single stimulus.
- Amyloid-β (Aβ)
Peptides (38–42 amino acids) produced through the proteolytic cleavage of a large transmembrane protein, the amyloid precursor protein (APP), by specific secretases. Extracellular Aβ accumulation generates insoluble and neurotoxic aggregates, called plaques, which are one of the main hallmarks of Alzheimer’s disease.
- Decoy receptor
Receptor able to recognize and bind specific ligands like a functional receptor. However, it is not structurally able to activate the downstream signaling cascade, so it acts as an inhibitor, preventing the ligand from activating its natural receptor.
- Human endogenous retroviruses (HERVs)
Endogenous retroviral elements are present in all vertebrates as vestiges of retroviral infections that became part of the host genome. HERVs represents up to 8% of the human genome and, even if unable to self-replicate, they can be expressed and have been linked to the pathogenesis of different diseases.
- Envelope protein
Small surface or transmembrane protein involved in multiple aspects of the virus life cycle, including the fusion of the viral membrane with the host cellular membrane. Envelope proteins can stimulate the secretion of inflammatory cytokines and chemokines as part of the innate immune response.
- Microbiota
The gut microbiota is the community of microorganisms (e.g., bacteria, viruses, fungi and protozoa) normally present in the gastrointestinal tract. The gut microbiota is composed of about 100 trillion microorganisms, producing thousands of metabolites that participate in several of the functions of the host, consequently influencing the host’s fitness, phenotype and health.
- Neuroimmune
Term that indicates all the biochemical and electrophysiological processes occurring in the bi-directional communication between the nervous system and the immune system.
- Nitroxidative stress
Following injuries to the nervous system, a rapid increase in reactive oxygen and nitrogen species formation occurs in neuronal, glial and immune cells. Once generated, these nitroxidative species initiate a cascade of redox reactions resulting in a long-lasting inflammatory condition known as nitroxidative stress.
- Plexus
A bundle of intersecting nerves, blood vessels, or lymphatic vessels in the organism. The myenteric and the submucosal plexuses in the gut are formed by a network of different neuronal and glial subpopulations that participate in digestive functions as well as in the crosstalk between the ENS and the CNS.
- Tauopathy
Neurodegenerative disorder characterized by the aggregation of misfolded tau protein in the brain. Predominantly expressed in the neurons, the main function of tau is to maintain the stability of microtubules in axons. Examples of tauopathies are Alzheimer’s disease, some forms of parkinsonism and certain frontotemporal dementias.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Resources
References
- 1.Chen CY, et al. (2019) Beyond defense: regulation of neuronal morphogenesis and brain functions via Toll-like receptors. J Biomed Sci 26, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mielcarska MB, et al. (2021) Cell Surface Expression of Endosomal Toll-Like Receptors—A Necessity or a Superfluous Duplication? Frontiers in Immunology 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bezhaeva T, et al. (2022) The Intriguing Role of TLR Accessory Molecules in Cardiovascular Health and Disease. Front Cardiovasc Med 9, 820962–820962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang J, et al. (2020) TLR4 Cross-Talk With NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer’s Disease. Frontiers in Immunology 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fiebich BL, et al. (2018) Role of Microglia TLRs in Neurodegeneration. Frontiers in Cellular Neuroscience 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Negi N and Das BK (2018) CNS: Not an immunoprivilaged site anymore but a virtual secondary lymphoid organ. Int Rev Immunol 37, 57–68 [DOI] [PubMed] [Google Scholar]
- 7.Woodburn SC, et al. (2021) The semantics of microglia activation: neuroinflammation, homeostasis, and stress. Journal of Neuroinflammation 18, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu Y, et al. (2021) Toll-like receptor-2 gene knockout results in neurobehavioral dysfunctions and multiple brain structural and functional abnormalities in mice. Brain, Behavior, and Immunity 91, 257–266 [DOI] [PubMed] [Google Scholar]
- 9.Connolly MG, et al. (2020) Toll-like receptor 4 differentially regulates adult hippocampal neurogenesis in an age- and sex-dependent manner. Hippocampus 30, 958–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Connolly MG, et al. (2021) Effects of Toll-like receptor 4 inhibition on spatial memory and cell proliferation in male and female adult and aged mice. Brain, Behavior, and Immunity 97, 383–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hung YF, et al. (2018) Endosomal TLR3, TLR7, and TLR8 control neuronal morphology through different transcriptional programs. The Journal of cell biology 217, 2727–2742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seong K-J, et al. (2022) Toll-Like Receptor 5 Promotes the Neurogenesis From Embryonic Stem Cells and Adult Hippocampal Neural Stem Cells in Mice. Stem Cells 40, 303–317 [DOI] [PubMed] [Google Scholar]
- 13.Ifuku M, et al. (2020) Activation of Toll-like receptor 5 in microglia modulates their function and triggers neuronal injury. Acta Neuropathologica Communications 8, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schilling S, et al. (2021) TLR2- and TLR3-activated microglia induce different levels of neuronal network dysfunction in a context-dependent manner. Brain Behav Immun [DOI] [PubMed] [Google Scholar]
- 15.(2021) 2021 Alzheimer’s disease facts and figures. Alzheimers Dement 17, 327–406 [DOI] [PubMed] [Google Scholar]
- 16.Lotz M, et al. (2005) Amyloid beta peptide 1–40 enhances the action of Toll-like receptor-2 and −4 agonists but antagonizes Toll-like receptor-9-induced inflammation in primary mouse microglial cell cultures. J Neurochem 94, 289–298 [DOI] [PubMed] [Google Scholar]
- 17.Minoretti P, et al. (2006) Effect of the functional toll-like receptor 4 Asp299Gly polymorphism on susceptibility to late-onset Alzheimer’s disease. Neuroscience letters 391, 147–149 [DOI] [PubMed] [Google Scholar]
- 18.Jana M, et al. (2008) Fibrillar amyloid-beta peptides activate microglia via TLR2: implications for Alzheimer’s disease. J Immunol 181, 7254–7262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walter S, et al. (2007) Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell Physiol Biochem 20, 947–956 [DOI] [PubMed] [Google Scholar]
- 20.Song M, et al. (2011) TLR4 mutation reduces microglial activation, increases Aβ deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. J Neuroinflammation 8, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen K, et al. (2006) Activation of Toll-like receptor 2 on microglia promotes cell uptake of Alzheimer disease-associated amyloid beta peptide. The Journal of biological chemistry 281, 3651–3659 [DOI] [PubMed] [Google Scholar]
- 22.Qin Y, et al. (2016) Stimulation of TLR4 Attenuates Alzheimer’s Disease–Related Symptoms and Pathology in Tau-Transgenic Mice. The Journal of Immunology 197, 3281–3292 [DOI] [PubMed] [Google Scholar]
- 23.Zhou Y, et al. (2020) TLR4 Targeting as a Promising Therapeutic Strategy for Alzheimer Disease Treatment. Frontiers in neuroscience 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adhikarla SV, et al. (2021) TLR-Mediated Signal Transduction and Neurodegenerative Disorders. Brain sciences 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu S, et al. (2012) TLR2 is a primary receptor for Alzheimer’s amyloid β peptide to trigger neuroinflammatory activation. J Immunol 188, 1098–1107 [DOI] [PubMed] [Google Scholar]
- 26.Cui W, et al. (2020) Inhibition of TLR4 Induces M2 Microglial Polarization and Provides Neuroprotection via the NLRP3 Inflammasome in Alzheimer’s Disease. Frontiers in neuroscience 14, 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruganzu JB, et al. (2021) Downregulation of TREM2 expression exacerbates neuroinflammatory responses through TLR4-mediated MAPK signaling pathway in a transgenic mouse model of Alzheimer’s disease. Molecular immunology 142, 22–36 [DOI] [PubMed] [Google Scholar]
- 28.Chen R, et al. (2021) Targeting the TLR4/NF-κB pathway in β-amyloid-stimulated microglial cells: A possible mechanism that oxysophoridine exerts anti-oxidative and anti-inflammatory effects in an in vitro model of Alzheimer’s disease. Brain Res Bull 175, 150–157 [DOI] [PubMed] [Google Scholar]
- 29.Zhang B, et al. (2021) DL0410 Alleviates Memory Impairment in D-Galactose-Induced Aging Rats by Suppressing Neuroinflammation via the TLR4/MyD88/NF-κB Pathway. Oxid Med Cell Longev 2021, 6521146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McDonald CL, et al. (2016) Inhibiting TLR2 activation attenuates amyloid accumulation and glial activation in a mouse model of Alzheimer’s disease. Brain Behav Immun 58, 191–200 [DOI] [PubMed] [Google Scholar]
- 31.Huang NQ, et al. (2017) TLR4 is a link between diabetes and Alzheimer’s disease. Behavioural brain research 316, 234–244 [DOI] [PubMed] [Google Scholar]
- 32.Walker DG, et al. (2018) Increased expression of toll-like receptor 3, an anti-viral signaling molecule, and related genes in Alzheimer’s disease brains. Experimental neurology 309, 91–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herrera-Rivero M, et al. (2019) Dysregulation of TLR5 and TAM Ligands in the Alzheimer’s Brain as Contributors to Disease Progression. Molecular Neurobiology 56, 6539–6550 [DOI] [PubMed] [Google Scholar]
- 34.Chakrabarty P, et al. (2018) TLR5 decoy receptor as a novel anti-amyloid therapeutic for Alzheimer’s disease. Journal of Experimental Medicine 215, 2247–2264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lehmann SM, et al. (2012) An unconventional role for miRNA: let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat Neurosci 15, 827–835 [DOI] [PubMed] [Google Scholar]
- 36.Derkow K, et al. (2018) Distinct expression of the neurotoxic microRNA family let-7 in the cerebrospinal fluid of patients with Alzheimer’s disease. PloS one 13, e0200602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scholtzova H, et al. (2014) Amyloid β and Tau Alzheimer’s disease related pathology is reduced by Toll-like receptor 9 stimulation. Acta Neuropathol Commun 2, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scholtzova H, et al. (2017) Innate Immunity Stimulation via Toll-Like Receptor 9 Ameliorates Vascular Amyloid Pathology in Tg-SwDI Mice with Associated Cognitive Benefits. The Journal of neuroscience : the official journal of the Society for Neuroscience 37, 936–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang L, et al. (2021) Ginkgolide B Alleviates Learning and Memory Impairment in Rats With Vascular Dementia by Reducing Neuroinflammation via Regulating NF-κB Pathway. Front Pharmacol 12, 676392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang F, et al. (2022) Low-Intensity Focused Ultrasound Stimulation Ameliorates Working Memory Dysfunctions in Vascular Dementia Rats via Improving Neuronal Environment. Frontiers in aging neuroscience 14, 814560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L, et al. (2020) Acupuncture Attenuates Inflammation in Microglia of Vascular Dementia Rats by Inhibiting miR-93-Mediated TLR4/MyD88/NF-κB Signaling Pathway. Oxid Med Cell Longev 2020, 8253904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Su R and Zhou T (2021) Alpha-Synuclein Induced Immune Cells Activation and Associated Therapy in Parkinson’s Disease. Frontiers in aging neuroscience 13, 769506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Choi I, et al. (2020) Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nature Communications 11, 1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Venezia S, et al. (2021) Toll-like receptor 4 deficiency facilitates α-synuclein propagation and neurodegeneration in a mouse model of prodromal Parkinson’s disease. Parkinsonism & Related Disorders 91, 59–65 [DOI] [PubMed] [Google Scholar]
- 45.Hughes CD, et al. (2019) Picomolar concentrations of oligomeric alpha-synuclein sensitizes TLR4 to play an initiating role in Parkinson’s disease pathogenesis. Acta Neuropathol 137, 103–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Campolo M, et al. (2019) TLR4 absence reduces neuroinflammation and inflammasome activation in Parkinson’s diseases in vivo model. Brain Behav Immun 76, 236–247 [DOI] [PubMed] [Google Scholar]
- 47.Shao Q. h., et al. (2019) TLR4 deficiency has a protective effect in the MPTP/probenecid mouse model of Parkinson’s disease. Acta Pharmacologica Sinica 40, 1503–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang X, et al. (2020) Salidroside ameliorates Parkinson’s disease by inhibiting NLRP3-dependent pyroptosis. Aging (Albany NY) 12, 9405–9426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dzamko N, et al. (2017) Toll-like receptor 2 is increased in neurons in Parkinson’s disease brain and may contribute to alpha-synuclein pathology. Acta Neuropathol 133, 303–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kwon S, et al. (2019) Targeting Microglial and Neuronal Toll-like Receptor 2 in Synucleinopathies. Exp Neurobiol 28, 547–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dutta D, et al. (2021) Selective targeting of the TLR2/MyD88/NF-κB pathway reduces α-synuclein spreading in vitro and in vivo. Nature Communications 12, 5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scheiblich H, et al. (2021) Microglial NLRP3 Inflammasome Activation upon TLR2 and TLR5 Ligation by Distinct α-Synuclein Assemblies. J Immunol 207, 2143–2154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Campolo M, et al. (2020) TLR7/8 in the Pathogenesis of Parkinson’s Disease. Int J Mol Sci 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ros-Bernal F, et al. (2011) Microglial glucocorticoid receptors play a pivotal role in regulating dopaminergic neurodegeneration in parkinsonism. Proceedings of the National Academy of Sciences of the United States of America 108, 6632–6637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maatouk L, et al. (2018) TLR9 activation via microglial glucocorticoid receptors contributes to degeneration of midbrain dopamine neurons. Nat Commun 9, 2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zheng C, et al. (2020) Inflammatory Role of TLR-MyD88 Signaling in Multiple Sclerosis. Frontiers in Molecular Neuroscience 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Göttle P, et al. (2021) TLR4 Associated Signaling Disrupters as a New Means to Overcome HERV-W Envelope-Mediated Myelination Deficits. Frontiers in Cellular Neuroscience 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Göttle P, et al. (2019) Rescuing the negative impact of human endogenous retrovirus envelope protein on oligodendroglial differentiation and myelination. Glia 67, 160–170 [DOI] [PubMed] [Google Scholar]
- 59.Touil T, et al. (2006) Cutting Edge: TLR3 stimulation suppresses experimental autoimmune encephalomyelitis by inducing endogenous IFN-beta. J Immunol 177, 7505–7509 [DOI] [PubMed] [Google Scholar]
- 60.McInnes K, et al. (2017) Mild Traumatic Brain Injury (mTBI) and chronic cognitive impairment: A scoping review. PloS one 12, e0174847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yao X, et al. (2021) Sodium houttuyfonate attenuates neurological defects after traumatic brain injury in mice via inhibiting NLRP3 inflammasomes. J Biochem Mol Toxicol 35, e22850. [DOI] [PubMed] [Google Scholar]
- 62.Rosa JM, et al. (2021) TLR4 pathway impairs synaptic number and cerebrovascular functions through astrocyte activation following traumatic brain injury. British journal of pharmacology 178, 3395–3413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ved R, et al. (2021) Disulfide HMGB1 acts via TLR2/4 receptors to reduce the numbers of oligodendrocyte progenitor cells after traumatic injury in vitro. Sci Rep 11, 6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu N, et al. (2021) Recombinant annexin A2 inhibits peripheral leukocyte activation and brain infiltration after traumatic brain injury. J Neuroinflammation 18, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Farré-Alins V, et al. (2021) Serum Amyloid A1/Toll-Like Receptor-4 Axis, an Important Link between Inflammation and Outcome of TBI Patients. Biomedicines 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.He G-Y, et al. (2021) S100A8 Promotes Inflammation via Toll-Like Receptor 4 After Experimental Traumatic Brain Injury. Frontiers in neuroscience 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang X, et al. (2020) Toll-Like Receptor 2 Attenuates Traumatic Brain Injury-Induced Neural Stem Cell Proliferation in Dentate Gyrus of Rats. Neural plasticity 2020, 9814978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen C-Y, et al. (2019) Beyond defense: regulation of neuronal morphogenesis and brain functions via Toll-like receptors. Journal of Biomedical Science 26, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pervin Z and Stephen JM (2021) Effect of alcohol on the central nervous system to develop neurological disorder: pathophysiological and lifestyle modulation can be potential therapeutic options for alcohol-induced neurotoxication. AIMS Neurosci 8, 390–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McCarthy GM, et al. (2018) Chronic ethanol consumption: role of TLR3/TRIF-dependent signaling. Addict Biol 23, 889–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang X, et al. (2020) Chronic ethanol exposure induces neuroinflammation in H4 cells through TLR3 / NF-κB pathway and anxiety-like behavior in male C57BL/6 mice. Toxicology 446, 152625. [DOI] [PubMed] [Google Scholar]
- 72.Blednov YA, et al. (2021) Deletion of Tlr3 reduces acute tolerance to alcohol and alcohol consumption in the intermittent access procedure in male mice. Addict Biol 26, e12932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Warden AS, et al. (2019) Toll-like receptor 3 activation increases voluntary alcohol intake in C57BL/6J male mice. Brain Behav Immun 77, 55–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pascual M, et al. (2018) Role of the innate immune system in the neuropathological consequences induced by adolescent binge drinking. Journal of neuroscience research 96, 765–780 [DOI] [PubMed] [Google Scholar]
- 75.Montesinos J, et al. (2015) TLR4 elimination prevents synaptic and myelin alterations and long-term cognitive dysfunctions in adolescent mice with intermittent ethanol treatment. Brain Behav Immun 45, 233–244 [DOI] [PubMed] [Google Scholar]
- 76.Pascual M, et al. (2017) TLR4 response mediates ethanol-induced neurodevelopment alterations in a model of fetal alcohol spectrum disorders. J Neuroinflammation 14, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Coleman LG, et al. (2017) Microglial-derived miRNA let-7 and HMGB1 contribute to ethanol-induced neurotoxicity via TLR7. Journal of Neuroinflammation 14, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang C, et al. (2020) Exosomes of Antler Mesenchymal Stem Cells Improve Postoperative Cognitive Dysfunction in Cardiopulmonary Bypass Rats through Inhibiting the TLR2/TLR4 Signaling Pathway. Stem Cells International 2020, 2134565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lin F, et al. (2021) Toll-like receptor 2 activation and up-regulation by high mobility group box-1 contribute to post-operative neuroinflammation and cognitive dysfunction in mice. J Neurochem [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Muscat SM, et al. (2021) Postoperative cognitive dysfunction is made persistent with morphine treatment in aged rats. Neurobiol Aging 98, 214–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen C, et al. (2019) Extracellular RNAs-TLR3 signaling contributes to cognitive decline in a mouse model of postoperative cognitive dysfunction. Brain Behav Immun 80, 439–451 [DOI] [PubMed] [Google Scholar]
- 82.Gorecki AM, et al. (2021) TLR2 and TLR4 in Parkinson’s disease pathogenesis: the environment takes a toll on the gut. Translational Neurodegeneration 10, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jacobson A, et al. (2021) The intestinal neuro-immune axis: crosstalk between neurons, immune cells, and microbes. Mucosal Immunology 14, 555–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rao M and Gershon MD (2016) The bowel and beyond: the enteric nervous system in neurological disorders. Nat Rev Gastroenterol Hepatol 13, 517–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Seguella L and Gulbransen BD (2021) Enteric glial biology, intercellular signalling and roles in gastrointestinal disease. Nature Reviews Gastroenterology & Hepatology 18, 571–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Layunta E, et al. (2021) Crosstalk Between Intestinal Serotonergic System and Pattern Recognition Receptors on the Microbiota-Gut-Brain Axis. Front Endocrinol (Lausanne) 12, 748254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Arnaboldi F, et al. (2020) Expression of Toll-like receptors 4 and 7 in murine peripheral nervous system development. Ann Anat 231, 151526. [DOI] [PubMed] [Google Scholar]
- 88.Yarandi SS, et al. (2020) Intestinal Bacteria Maintain Adult Enteric Nervous System and Nitrergic Neurons via Toll-like Receptor 2-induced Neurogenesis in Mice. Gastroenterology 159, 200–213.e208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lin C, et al. (2019) Microbiota-gut-brain axis and toll-like receptors in Alzheimer’s disease. Comput Struct Biotechnol J 17, 1309–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Perez-Pardo P, et al. (2019) Role of TLR4 in the gut-brain axis in Parkinson’s disease: a translational study from men to mice. Gut 68, 829–843 [DOI] [PubMed] [Google Scholar]
- 91.Wu ML, et al. (2021) Age-related cognitive decline is associated with microbiota-gut-brain axis disorders and neuroinflammation in mice. Behavioural brain research 402, 113125. [DOI] [PubMed] [Google Scholar]
- 92.Pellegrini C, et al. (2022) Enteric α-synuclein impairs intestinal epithelial barrier through caspase-1-inflammasome signaling in Parkinson’s disease before brain pathology. npj Parkinson’s Disease 8, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sun Y, et al. (2020) Intra-gastrointestinal amyloid-β1–42 oligomers perturb enteric function and induce Alzheimer’s disease pathology. J Physiol 598, 4209–4223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.van Langenberg DR, et al. (2017) Cognitive impairment in Crohn’s disease is associated with systemic inflammation, symptom burden and sleep disturbance. United European Gastroenterol J 5, 579–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Qasem A, et al. (2021) Enteropathogenic infections modulate intestinal serotonin transporter (SERT) function by activating Toll-like receptor 2 (TLR-2) in Crohn’s disease. Scientific Reports 11, 22624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ongnok B, et al. (2020) Doxorubicin and cisplatin induced cognitive impairment: The possible mechanisms and interventions. Experimental neurology 324, 113118. [DOI] [PubMed] [Google Scholar]
- 97.Ibrahim SS, et al. (2021) Nose-to-brain delivery of chrysin transfersomal and composite vesicles in doxorubicin-induced cognitive impairment in rats: Insights on formulation, oxidative stress and TLR4/NF-kB/NLRP3 pathways. Neuropharmacology 197, 108738. [DOI] [PubMed] [Google Scholar]
- 98.Juan Z, et al. (2022) Probiotic supplement attenuates chemotherapy-related cognitive impairment in patients with breast cancer: a randomised, double-blind, and placebo-controlled trial. Eur J Cancer 161, 10–22 [DOI] [PubMed] [Google Scholar]
- 99.Brown T, et al. (2021) Implications of Breast Cancer Chemotherapy-Induced Inflammation on the Gut, Liver, and Central Nervous System. Biomedicines 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Subramaniam CB, et al. (2020) The microbiota-gut-brain axis: An emerging therapeutic target in chemotherapy-induced cognitive impairment. Neurosci Biobehav Rev 116, 470–479 [DOI] [PubMed] [Google Scholar]
- 101.Farooq M, et al. (2021) Toll-Like Receptors as a Therapeutic Target in the Era of Immunotherapies. Frontiers in Cell and Developmental Biology 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Anwar MA, et al. (2019) Recent clinical trends in Toll-like receptor targeting therapeutics. Medicinal research reviews 39, 1053–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huang J, et al. (2015) DAMPs, ageing, and cancer: The ‘DAMP Hypothesis’. Ageing Res Rev 24, 3–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Feldman N, et al. (2015) DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res Rev 24, 29–39 [DOI] [PubMed] [Google Scholar]
- 105.Hardcastle C, et al. (2020) Contributions of Hippocampal Volume to Cognition in Healthy Older Adults. Frontiers in aging neuroscience 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fonken LK, et al. (2016) The Alarmin HMGB1 Mediates Age-Induced Neuroinflammatory Priming. The Journal of neuroscience : the official journal of the Society for Neuroscience 36, 7946–7956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fei X, et al. (2022) TLR4 Deletion Improves Cognitive Brain Function and Structure in Aged Mice. Neuroscience [DOI] [PubMed] [Google Scholar]
- 108.Zhong Q, et al. (2020) Toll-like receptor 4 deficiency ameliorates β2-microglobulin induced age-related cognition decline due to neuroinflammation in mice. Mol Brain 13, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fang X, et al. (2021) Evaluation of the Anti-Aging Effects of a Probiotic Combination Isolated From Centenarians in a SAMP8 Mouse Model. Front Immunol 12, 792746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zuo W, et al. (2021) MD2 contributes to the pathogenesis of perioperative neurocognitive disorder via the regulation of α5GABA(A) receptors in aged mice. J Neuroinflammation 18, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]