

Abstract
Childhood-onset forms of hereditary spastic paraplegia are ultra-rare diseases and often present with complex features. Next-generation-sequencing allows for an accurate diagnosis in many cases but the interpretation of novel variants remains challenging, particularly for missense mutations. Where sufficient knowledge of the protein function and/or downstream pathways exists, functional studies in patient-derived cells can aid the interpretation of molecular findings. We here illustrate the case of a 13-year-old female who presented with global developmental delay and later mild intellectual disability, progressive spastic diplegia, spastic-ataxic gait, dysarthria, urinary urgency, and loss of deep tendon reflexes of the lower extremities. Exome sequencing showed a novel splice-site variant in trans with a novel missense variant in B4GALNT1 [NM_001478.5: c.532–1G>C / c.1556G>C (p.Arg519Pro)]. Functional studies in patient-derived fibroblasts and cell models of GM2 synthase deficiency confirmed a loss of B4GALNT1 function with no synthesis of GM2 and other downstream gangliosides. Collectively these results established the diagnosis of B4GALNT1-associated HSP (SPG26). Our approach illustrates the importance of careful phenotyping and functional characterization of novel gene variants, particularly in the setting of ultra-rare diseases, and expands the clinical and molecular spectrum of SPG26, a disorder of complex ganglioside biosynthesis.
Keywords: Hereditary spastic paraplegia, spastic paraplegia 26, spasticity, inborn error of metabolism, gangliosides, neurodegeneration
INTRODUCTION
The hereditary spastic paraplegias (HSPs) are a heterogenous group of over 80 different neurogenetic disorders (Blackstone, 2018). Early-onset complex forms of HSP pose a diagnostic challenge given the often non-specific and heterogenous clinical presentation and the fact that many are ultra-rare diseases with only a few individuals reported worldwide. Molecular testing allows for an accurate diagnosis in many cases but the interpretation of novel variants remains challenging, particularly for missense mutations. Where sufficient knowledge of the protein function and/or downstream pathways exists, functional studies in patient-derived cells can aid the interpretation of molecular findings and support a diagnosis.
Glycosphingolipids are ubiquitous constituents of cellular membranes, and their sialylated derivatives, gangliosides, play a major role in neurons and glia (Schengrund, 2015). While the disorders of ganglioside degradation are a well-known group of neurodegenerative lysosomal storage diseases, only two single gene disorders of ganglioside biosynthesis, caused by variants in the B4GALNT1 and ST3GAL5 genes, have been described thus far (Boukhris et al., 2013; Harlalka et al., 2013; Simpson et al., 2004). Reported in 13 families to date, bi-allelic loss-of-function variants in the B4GALNT1 gene, encoding beta-1,4-N-acetylgalactosaminyltransferase 1, the enzyme responsible for GM2, GA2, GD2 and GT2 synthesis, have been shown to lead to early-onset HSP (Boukhris et al., 2013; Dad et al., 2017; Harlalka et al., 2013; Wakil et al., 2014). We describe the case of a 13-year-old female who presented with a complex neurological syndrome. Exome sequencing showed a novel splice-site variant in trans with a novel missense variant in B4GALNT1. Functional studies in patient-derived fibroblasts and cell models of GM2 synthase deficiency confirmed a loss-of-function, therefore establishing the diagnosis of B4GALNT1-associated HSP (SPG26). Our approach illustrates the importance of a careful functional characterization of novel gene variants, particularly in the setting of ultra-rare diseases.
MATERIALS AND METHODS
Clinical characterization and molecular testing
This study was approved by the Institutional Review Board at Boston Children’s Hospital (IRB-P00033016). Written consent was obtained. Exome sequencing was performed at GeneDx (Gaithersburg, MD, USA). Neurofilament light chain levels in plasma were measured using the HD-X NfL kit on the SiMoA HD-X Analyzer (Quanterix).
Analysis of missense variant spectrum and modeling of B4GALNT1 protein structure
Information on the protein sequence of human B4GALNT1 was obtained from the Universal Protein Resource database (UniProt ID: Q00973). CADD scores (v1.6) (Rentzsch et al., 2019) of all possible base substitutions of the B4GALNT1 transcript were computed and mapped to the corresponding protein sequence using VarMAP (Stephenson et al., 2019). All variants were harmonized to the canonical Ensembl feature ENST00000341156.9 (NM_001478.5) of GRCh37/hg19 (Kopanos et al., 2019). Modelling of the B4GALNT1 primary protein structure and associated disease-causing variants was done using R v4.1.0 (2021-05-18) and RStudio (v1.4.1103; RStudio, Inc.) (Neuser et al., 2021). The 3D structural model of B4GALNT1 was obtained from the AlphaFold Protein Structure Database (Jumper et al., 2021) and a model of the p.Arg519Pro variant was created using Discovery Studio 2021 (Dassault Systèmes). Mutation energy was calculated with the Calculate Mutation Energy/Stability module.
Antibodies and reagents
Antibodies and reagents are listed in Supplementary Table 1.
Primary fibroblast culture
Fibroblasts from the proband and a healthy unrelated control were obtained and cultured as described previously (Behne et al., 2020; Ebrahimi-Fakhari et al., 2021).
B78 and L cell lines, B4GALNT1 cDNA expression plasmids and transfection
B78 and L cells were cultured as previously described (Bhuiyan et al., 2019). Mutant cDNA clones were isolated using site-directed mutagenesis from the wild-type GM2/GD2 synthase cDNA expression vector, pM2T1–1 in pMIKneo. The specific nucleotide substitution and a deletion of exon 6 were generated by inverse PCR using the KOD-Plus-Mutagenesis kit (Toyobo, #SMK-101) with primer sets for each mutation. The following primers were designed based on B4GALNT1 sequence information (Furukawa et al., 1996), and used for inverse PCR: For c.1556G>C (p.Arg519Pro): Primer F(1): 1548–1569, 5’-CAAACACCCGCTGCTCTTCTTC-3’. Primer R(1): 1529–1547, 5’-GCCATCTGGCTCTCGTCCA-3’. For c.532–1G>C (deletion of 181nt): Primer F(2): 713–732, 5’-TCCGGTTCTCCACCGAGGGA-3’. Primer R(2): 509–531, 5’-CTGGTATACCTCCTGACCAGAAG-3’. DPN-1 was used for digestion of methylated template cDNA in the PCR products. After self-ligation with T4 polynucleotide kinase and ligase, self-ligated PCR products were transformed into E. coli competent cells. Obtained clones were expanded and confirmed by DNA sequencing. B78 and L cells were transfected with the pMIKneo/GM2/GD2 synthase cDNAs using Lipofectamine 2000 (Invitrogen #52887).
Western Blotting
Western blotting was performed using cell extracts from transfected B78 cells as previously described (Bhuiyan et al., 2019).
Immunocytochemistry
Forty-eight hours after transfection, B78 cells were fixed, stained and imaged on a confocal microscope (Fluoview FV10i, Olympus) as previously described (Bhuiyan et al., 2019).
Flow cytometry
For experiments shown in Figure 2D, fibroblasts were detached with 0.25% trypsin/EDTA, and then stained with a fluorescently labeled beta subunit of cholera toxin (CTXb-FITC, 1:100) in PBS containing 1% bovine serum albumin. The cells were analyzed by Attune NxT flow cytometer (Thermo Fisher Scientific). For experiments shown in Figure 2H, cell surface expression of glycolipids was analyzed using the BDAccuri™ C6 flow cytometer (Accuri Flow Cytometers Inc.) as described previously (Ohmi et al., 2018).
Figure 2.
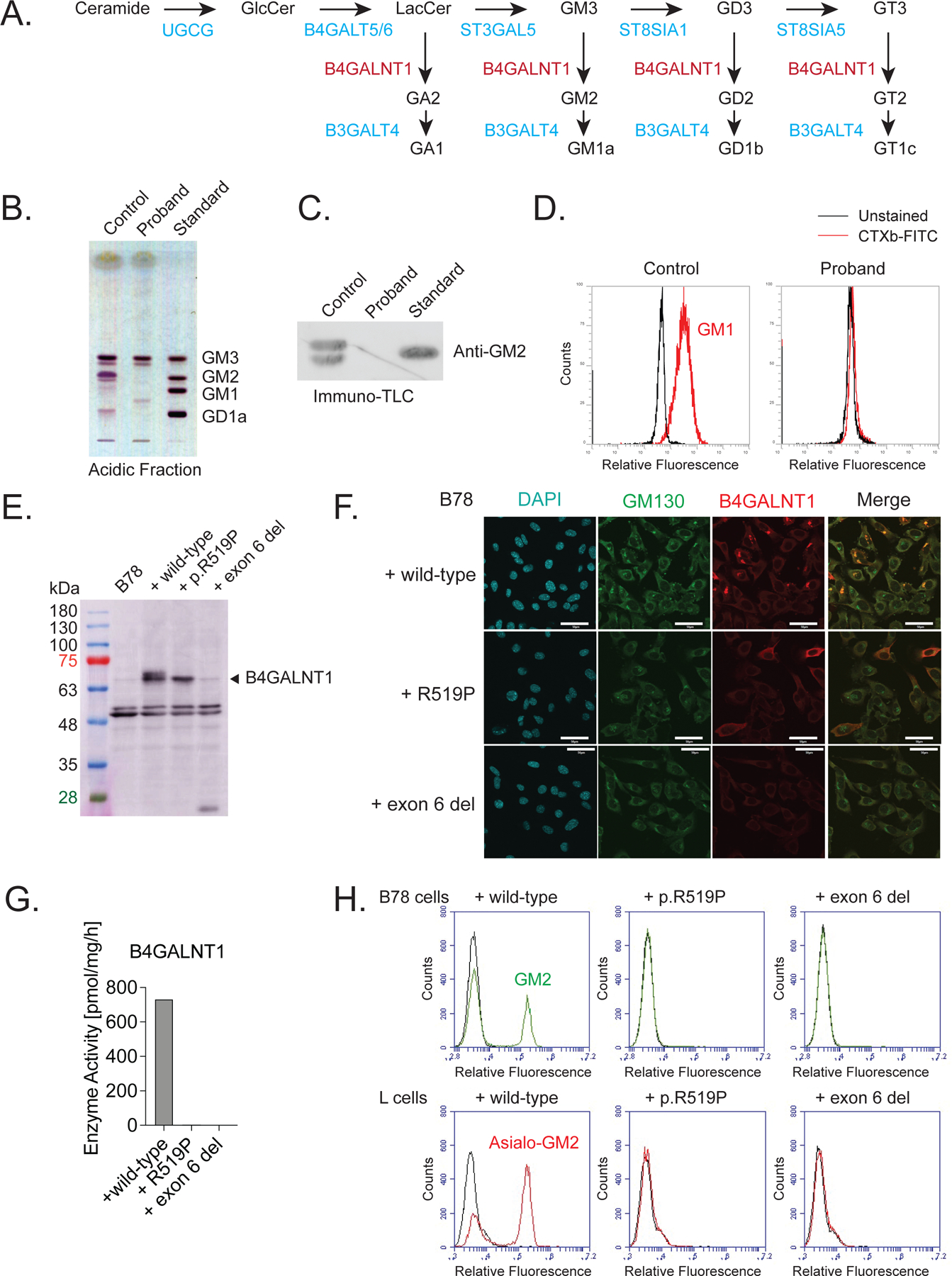
Functional assays confirm a lack of B4GALNT1 function in primary fibroblasts derived from the proband and in cell models.
A) Overview of the ganglioside biosynthesis pathway. B4GALNT1 is responsible for the synthesis of GA2, GM2, GD2 and GT2 and downstream complex gangliosides. B) Thin-layer chromatography of acidic glycosphingolipids from fibroblasts of a healthy control and the proband. Glycosphingolipids were detected using orcinol-sulfuric acid reagent. The proband’s fibroblasts lack GM2 and downstream gangliosides. C) Immuno-thin-layer chromatography of acidic glycosphingolipids from control and proband fibroblasts, detected using anti-GM2 antibody. This confirms the lack of GM2 in the proband’s fibroblasts. D) Flow cytometry analysis of cell-surface staining of GM1 using CTXb-FITC (red line). The black line indicates an unstained control sample. The proband’s fibroblasts show no detectable levels of GM1. E) Western blotting for B4GALNT1 in whole cell lysates of B78 cells transfected with full length, wild-type B4GALNT1 or plasmids carrying the p.R519P mutation or a deletion of exon 6. Untransfected B78 cells do not express detectable levels of endogenous B4GALNT1. B78 cells transfected with wild-type cDNA show robust levels of B4GALNT1, whereas the p.R519P mutant protein is expressed at a lower level and the plasmid carrying an exon 6 deletion shows no expression above background. F) Immunocytochemistry for B4GALNT1 in B78 cells transfected with full length, wild-type B4GALNT1 or plasmids carrying the p.R519P mutation or a deletion of exon 6. Wild-type B4GALNT1 localizes predominantly to the Golgi network, marked by GM130. p.R519P mutant B4GALNT1 shows a more diffuse, cytoplasmic distribution and exon 6 deleted B4GALNT1 shows no expression. Scale bar = 50μm. G) In vitro B4GALNT1 enzyme activity is diminished below the detection level in B78 cells expressing either B4GALNT1 variant. H) Flow cytometry analysis of GM2 and GA2 (also known as asialo-GM2) in B78 cells and L cells, respectively, transfected with full length, wild-type B4GALNT1 or plasmids carrying the p.R519P mutation or a deletion of exon 6. Expression of either B4GALNT1 variant does not lead to any detectable levels of GM2 and GA2, confirming loss of B4GALNT1 enzyme activity.
B4GALNT1 enzyme assay
Forty-eight hours after transfection, B78 cells were harvested and washed. Membrane fractions were collected from cell pellets in ice-cold PBS containing 1mM PMSF using a nitrogen cavitation apparatus (Parr Instrument Co.) at 300p.s.i. for 30min. After removing nuclei by centrifugation, the supernatants were centrifuged at 40,000rpm for 1h at 4°C, and membrane fractions were dissolved in cold 0.1M sodium cacodylate buffe (pH 7.2) and used to measure B4GALNT1 enzyme activity (Bhuiyan et al., 2019).
Immuno-thin-layer chromatogram (TLC)
Glycosphingolipid analysis by TLC and immuno-TLC was performed as described previously (Go et al., 2017). Briefly, total cellular lipids were extracted using chloroform/methanol, and acidic glycosphingolipids were separated using a DEAE-Sephadex A-25 column, followed by mild alkaline hydrolysis. After desalting, the acidic glycosphingolipids were spotted on a TLC plate and developed with chloroform/methanol/0.2%CaCl2 (55:45:10, v/v/v). Glycosphingolipids were visualized by orcinol/sulfuric acid staining. For immuno-TLC, a developed TLC plate was dried, dipped in cyclohexane containing 0.1% (w/v) poly-(isobutyl methacrylate) for 1min, blocked with PBS containing 1% BSA, and stained with anti-GM2 antibody followed by horseradish peroxidase-conjugated anti-mouse IgM.
RESULTS
Clinical presentation and neuroimaging
The patient was the first child of non-consanguineous parents of Puerto-Rican descent. Pregnancy and the peri- and neonatal period were uncomplicated. First concerns arose in infancy when the patient presented with axial hypotonia, poor head control and feeding difficulties. Motor milestones were delayed: Unsupported sitting was achieved at 18 months, standing at 24 months and independent walking at 30 months. Gait remained unsteady, leading to frequent falls. Over time, the patient developed a spastic diplegia. Other domains of development were delayed as well. She remained nonverbal until about the age of 4 years. Now at age 13 years, she can ambulate short distances independently and longer distances with a posterior walker. She is unable to climb stairs without assistance. Longer walking distances, i.e. more than 2–3 blocks, are associated with fatigue and muscle cramps. She uses about 20 words and can form 2- or 3-word sentences. Her speech has become progressively dysarthric over the years. She has urinary urgency and occasional episodes of urinary incontinence. Autonomic symptoms include episodic discoloration of her distal limbs usually in the setting of cold temperatures. She attends a school for children with disabilities and requires help with all activities of daily living. There is no history of swallowing dysfunction or seizures.
Upon last evaluation, at the age of 13-years, anthropometric data were within normal range (height: 29%ile, weight: 28%ile, head circumference: 73%ile). There were no distinctive facial features. Neurological examination at age 13 years was notable for mild dysarthria, slow horizontal saccades, pseudoathetosis of the hands, weakness of the proximal legs, a spastic diplegia with flexion-inversion contractures of the ankles, and a broad-based spastic-ataxic gait. Muscle bulk in the lower extremities was reduced and reflexes in the lower extremities were attenuated. Babinski sign was positive. The Spastic Paraplegia Rating Scale score was 20/52.
Overall, this presentation seemed consistent with a syndrome in the category of early-onset complex HSP, characterized by global developmental delay with prominently delayed motor milestones and later mild intellectual disability, followed by progressive distal spastic paraplegia with weakness and ankle contractures.
Routine laboratory investigations were unremarkable. Brain and lumbar spine MR imaging showed no abnormalities. Plasma neurofilament light chain levels were elevated (203.98 ± 4.80 (CV%) vs. 25.19 ± 7.78 (CV%) in the patient’s mother). Fundoscopic exam and optical coherence tomography of the retinal nerve fiber layer were normal. EKG and a trans-thoracic echocardiogram showed no abnormalities.
Molecular testing
Chromosomal microarray analysis was normal. Testing for repeat expansion disorders (ATXN1, ATXN2, ATXN3, CACNA1A, ATXN7, ATXN8OS, ATXN10, PPP2R2B, TBP, ATN1, FXN, FMR1, RFC1) was negative. Exome sequencing showed compound heterozygous variants in B4GALNT1 (NM_001478.5): c.532–1G>C / c.1556G>C (p.Arg519Pro). The paternally-inherited c.532–1G>C variant is predicted to disrupt a splice site leading to a null variant and was thus classified as likely pathogenic based on ACMG criteria (PVS1, PM2). The variant is absent from gnomAD exomes and genomes (coverage = 32.2) and received pathogenic computational verdict based on 6 pathogenic predictions from BayesDel_addAF, DANN, EIGEN, FATHMM-MKL, MutationTaster and scSNV-Splicing versus no benign predictions. The maternally-inherited c.1556G>C (p.Arg519Pro) missense variant is present in gnomAD genomes with an allele count of 9 and no homozygous cases (coverage = 31.2). It was previously classified as a variant of unclear significance (ClinVar: VCV000848217.2, based on PM2 and BP4) with the majority of in silico prediction tools favoring a benign verdict. Figure 1A shows all reported variants (also see Supplementary Table 2) and an additive model of CADD PHRED v1.6 values for all possible missense variants in B4GALNT1 across the linear protein structure. This analysis revealed high CADD scores for most possible variants in the catalytic domain with lower scores for those variants found closer to the N-terminal region. Most reported disease-associated variants are located within the catalytic domain, emphasizing the functional importance of this region. The 3D structural model of B4GALNT1 from AlphaFold database shows that arginine 519 is located at the center of the helix (Figure 1B). Since proline is established as a helix breaker, the arginine-to-proline mutation identified in this patient likely destabilizes the helix. Energy calculation using Discovery Studio indicates that the variant is less stable than wild-type by 5.6 kcal/mol. This suggests that the p.Arg519Pro variant will likely destabilize the protein and consequently abolish its catalytic activity. Additional gene variants, all classified as variants of uncertain significance, discovered through exome sequencing in the proband are listed in Supplementary Table 3.
Figure 1.
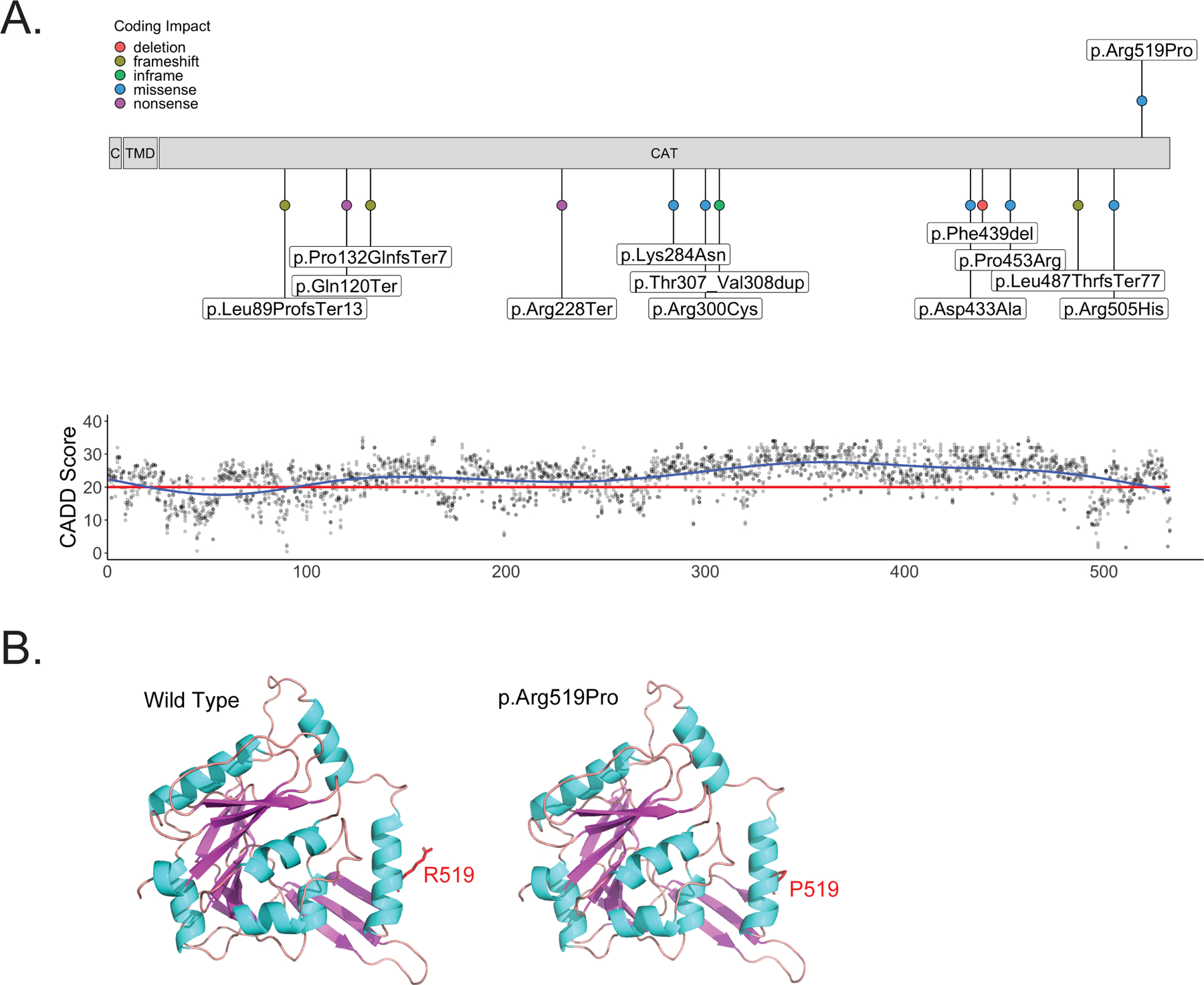
B4GALNT1 protein structure with variant distribution.
A) Schematic of B4GALNT1 protein structure. The p.Arg519Pro missense variant discovered in the proband is shown above the schematic. All previously published exonic variants are shown below the schematic. The lower panel shows CADD PHRED v1.6 values for all possible missense variants in B4GALNT1 across the linear protein structure. The red line marks the recommended cut off of 20. B) 3D structural model of the B4GALNT1 catalytic domain (273–533) derived from the AlphaFold database (left) and a model of the p.Arg519Pro variant generated using Discovery Studio (right). The 3D structures are shown in a ribbon model and Arg519 (left) and Pro519 (right) are shown in stick model. The figure was prepared using PyMol. Since proline is established as a helix breaker, the arginine-to-proline mutation is predicted to destabilize the helix structure.
Functional characterization of novel B4GALNT1 variants
To explore the impact of the two novel variants on B4GALNT1 (GM2/GD2/GA2 synthase) function, experiments were carried out in cell models and primary fibroblasts from the proband. A general overview of ganglioside biosynthesis is shown in Figure 2A. In fibroblasts of the proband, we found a lack of GM2 and downstream gangliosides GM1 and GD1a, measured by thin layer chromatography (Figure 2B). This was confirmed by immuno-TLC (Figure 2C). Flowcytometry for cholera toxin-labeled GM1 confirmed an absence of this ganglioside in fibroblasts of the proband (Figure 2D). To further investigate the impact of each mutation separately, we turned to functional assays in a cell model. B78 cells, which lack endogenous GM2 expression (Bhuiyan et al., 2019), were transiently transfected with cDNA plasmid encoding full-length wild-type B4GALNT1, a plasmid carrying the c.1556G>C (p.Arg519Pro) missense variant or a plasmid carrying a deletion of exon 6 since the c.532–1G>C variant is predicted to lead to a loss of exon 6. Western blot from whole cell lysates showed strong expression of B4GALNT1 in B78 cells transfected with wild-type cDNA, whereas the c.1556G>C (p.Arg519Pro) mutant protein was expressed at a lower level and the plasmid carrying an exon 6 deletion showed no expression above background (Figure 2E). Immunocytochemistry revealed that wild-type B4GALNT1 co-localized predominately with the Golgi marker GM130, consistent with prior reports (Bhuiyan et al., 2019). The c.1556G>C (p.Arg519Pro) variant showed a more dispersed localization throughout the cytoplasm and no enrichment in the Golgi region. Exon 6 deleted B4GALNT1 showed no staining, confirming lack of expression of a detectable protein (Figure 2F). A cell-free in vitro enzyme assay using membrane fractions from transfected B78 cells, showed no detectable GM2/GD2/GA2 synthase activity for both B4GALNT1 variants (Figure 2G), contrasting with the robust enzyme activity in membranes from cells transfected with the wild-type construct. This was confirmed by flow cytometry for GM2 and asialo-GM2 (also known as GA2), in which cells transfected with cDNA of either of the B4GALNT1 variants revealed no evidence of GM2 or GA2 synthesis (Figure 2H).
Taken together this set of experiments confirms a loss of B4GALNT1 function with lack of GM2 biosynthesis because of the novel B4GALNT1 variants discovered in the proband. We therefore re-classify the c.1556G>C (p.Arg519Pro) missense variant as likely pathogenic.
DISCUSSION
This case of a young female with a complex form of HSP highlights the concept that functional studies in patient-derived cells can aid the interpretation of molecular findings and help establish a diagnosis. This is particularly important for ultra-rare diseases and requires sufficient understanding of the protein function and/or downstream pathways that are potentially affected by variants in a particular gene. B4GALNT1 is a key enzyme in the synthesis of complex gangliosides (Furukawa et al., 2002) and knowledge of the substrate and product of this enzyme allowed us to explore its function through various established assays. We show that the c.532–1G>C splicing variant leads to lack of expression of a functional protein whereas the c.1556G>C (p.Arg519Pro) missense variant leads to reduced expression of B4GALNT1, a loss of enzyme function and abnormal intracellular localization. Consistent with a loss of function, fibroblasts of the proband show a lack of expression of GM2 and other downstream gangliosides.
The spectrum of disease-associated variants in B4GALNT1 consists of 15 variants with most exonic variants localizing to the catalytic domain (Supplementary Table 1). The spectrum consists of frameshift, non-sense, in frame deletions and insertions, and missense variants. Prior work established that all these variants lead to a loss of enzyme function (Bhuiyan et al., 2019). Gangliosides play complex roles in CNS development, myelination, and synaptic signaling. The motor phenotypes of B4galnt1 knockout mice resemble that of SPG26 with progressive hindlimb motor impairment (Chiavegatto et al., 2000) and are thought to be the result of progressive dysmyelination and axonal degeneration (Sheikh et al., 1999). There seems to be at least some redundancy in the function of gangliosides in the CNS and elevated levels of GM3 might partially compensate for the lack of GM2 in B4galnt1 knockout mice (Takamiya et al., 1996). This hypothesis is supported by the observation that patients with ST3GAL5 (GM3 synthase) deficiency have a more severe clinical phenotype (Simpson et al., 2004).
The clinical phenotype of our proband consisted of early-onset developmental delay and mild intellectual disability, spastic paraplegia, gait impairment, dysarthria, and urinary symptoms. Interestingly, there were pseudoathetoid movements of the hands and deep tendon reflexes in the lower extremity were absent, potentially indicating a peripheral neuropathy, which is supported by nerve conduction studies performed in a small number of patients (Boukhris et al., 2013), and similar testing in knockout mice (Takamiya et al., 1996). The core clinical features of B4GALNT1-associated HSP are summarized in Table 1. Disease onset is typically in early childhood with developmental delay. Progressive spasticity of the legs is a universal feature leading to gait impairment. Distal muscle atrophy and contractures are common. Speech is characterized by mild to moderate dysarthria. There is intellectual impairment in most patients, typically in the mild to moderate range. Brain MR imaging is normal or shows a pattern of non-specific white matter changes or atrophy in a subset of patients (Boukhris et al., 2013). Current treatment for B4GALNT1-associated HSP is directed at ameliorating symptoms but an advanced understanding of the underlying molecular biology and the availability of viral vectors for gene delivery for CNS disorders carries the potential for targeted therapies, i.e. gene replacement (Yang et al., 2021).
Table 1.
Summary of core clinical features of all patients reported with B4GALNT1-associated HSP.
| Clinical Feature | Present Case | Wilkinson et al. 2005 (1 family, n=5) | Boukhris et al. 2013 (7 families, n=18) | Harlalka et al. 2013 (2 families, n=6) | Wakil et al. 2013 (2 families, n=9) | Dad et al. 2017 (1 family, n=3) | Total (14 families, n=42) |
|---|---|---|---|---|---|---|---|
| Consan-guinity | No | Yes | Yes: 4/7 | Yes: 2/2 | Yes: 2/2 | Yes | 71% |
| Age at onset | Infancy | ~ 7 years | 2–19 years | Childhood | Childhood | 5–8 years | Childhood |
| Progressive spastic diplegia | Yes | 5/5 | 18/18 | 6/6 | 9/9 | 3/3 | 100% |
| Gait | Spastic-ataxic | Spastic: 4/5 Spastic-ataxic: 1/5 |
Impaired in 18/18 | Spastic: 6/6 | Impaired in 9/9 | Impaired in 3/3 | Impaired gait: 100% |
| Lower extremity reflexes | Absent | Hyperreflexia: 4/5 Absent: 1/5 |
Hyperreflexia: 9/18 Absent or reduced: 9/18 |
Absent: 2/4 Hyperreflexia: 1/4 Normal: 1/4 |
n.a. | Normal: 3/3 | Hyperreflexia: 45% Absent or reduced: 42% Normal: 13% |
| Distal muscle atrophy | Yes | 5/5 | 9/18 | 2/2 | 9/9 | 0/3 | 68% |
| Dysarthria | Mild to moderate | Severe: 4/5 | 7/18 | n.a. | 5/9 | 1/3 | 50% |
| Intellectual impairment | Mild | Mild: 5/5 | Mild: 13/18 Moderate: 5/18 |
Mild: 3/6 Moderate: 2/6 Severe: 1/6 |
Mild: 5/9 Moderate: 4/9 |
None: 3/3 | Mild: 64% Moderate: 26% Severe: 3% None: 7% |
| Urinary symptoms | Yes | n.a. | 6/18 | 0/2 | n.a. | n.a. | 33% |
| Brain MRI | Normal | Normal: 5/5 | Normal: 4/10 Abnormal: 6/10 |
Normal: 2/2 | Normal: 9/9 | Normal: 3/3 | Normal: 80% |
In conclusion, we report a patient with B4GALNT1-associated HSP and expand the molecular spectrum associated with this ultra-rare form of childhood-onset HSP. We highlight the importance of a translational approach to evaluating novel variants using functional assays in cell-based disease models.
Supplementary Material
ACKNOWLEDGEMENT / FUNDING
The authors thank the patient and her family for supporting this study. The authors also thank J. Fushimi for technical assistance. J.E.A. is supported by the Deutsche Forschungsgemeinschaft (German Research Foundation, 270949263/GRK2162), the German National Academic Foundation, the Max Weber-Program of the State of Bavaria and the German Academic Exchange Service (DAAD). K.I. is supported by JSPS KAKENHI Grant Number JP21K06531. A.S. is supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – SA 4171/1-1. M.Z. is support by the German National Academic Foundation. J.I. is supported by a Grant-in-Aid for Scientific Research (B) from MEXT (20H03452), Project for Rare/Intractable Diseases from AMED, and the Takeda Foundation. This study was supported by Grants-in-Aids from the Ministry of Education, Culture, Sports and Technology of Japan (MEXT) (19K22518 (Ko.F.), 19K07393 (Y.O.), 21K06828 (Ke.F.), 21H02699 (Ko.F.)) and by JST-CREST (JPMJCR17H2 (Ko.F.). D.E.F. received support from the CureAP4 Foundation, the CureSPG50 Foundation, the Spastic Paraplegia Foundation (U.S.A.), the Manton Center for Orphan Disease Research and the National Institute of Health / National Institute of Neurological Disorders and Stroke (1K08NS123552–01).
Footnotes
CONFLICT OF INTEREST
There are no relevant conflicts of interest.
DATA AVAILABILITY STATEMENT
All data are available from the corresponding authors upon reasonable request.
REFERENCES
- Behne R, Teinert J, Wimmer M, D’Amore A, Davies AK, Scarrott JM, … Ebrahimi-Fakhari D (2020). Adaptor protein complex 4 deficiency: a paradigm of childhood-onset hereditary spastic paraplegia caused by defective protein trafficking. Human Molecular Genetics, 29(2), 320–334. 10.1093/hmg/ddz310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhuiyan RH, Ohmi Y, Ohkawa Y, Zhang P, Takano M, Hashimoto N, … Furukawa K (2019). Loss of Enzyme Activity in Mutated B4GALNT1 Gene Products in Patients with Hereditary Spastic Paraplegia Results in Relatively Mild Neurological Disorders: Similarity with Phenotypes of B4galnt1 Knockout Mice. Neuroscience, 397, 94–106. 10.1016/j.neuroscience.2018.11.034 [DOI] [PubMed] [Google Scholar]
- Blackstone C (2018). Hereditary spastic paraplegia. Handbook of Clinical Neurology, 148, 633–652. 10.1016/b978-0-444-64076-5.00041-7 [DOI] [PubMed] [Google Scholar]
- Boukhris A, Schule R, Loureiro JL, Lourenco CM, Mundwiller E, Gonzalez MA, … Stevanin G (2013). Alteration of ganglioside biosynthesis responsible for complex hereditary spastic paraplegia. American Journal of Human Genetics, 93(1), 118–123. 10.1016/j.ajhg.2013.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiavegatto S, Sun J, Nelson RJ, & Schnaar RL (2000). A functional role for complex gangliosides: motor deficits in GM2/GD2 synthase knockout mice. Experimental Neurology, 166(2), 227–234. 10.1006/exnr.2000.7504 [DOI] [PubMed] [Google Scholar]
- Dad R, Walker S, Scherer SW, Hassan MJ, Alghamdi MD, Minassian BA, & Alkhater RA (2017). Febrile ataxia and myokymia broaden the SPG26 hereditary spastic paraplegia phenotype. Neurol Genet, 3(3), e156. 10.1212/NXG.0000000000000156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi-Fakhari D, Alecu JE, Brechmann B, Ziegler M, Eberhardt K, Jumo H, … Sahin M (2021). High-throughput imaging of ATG9A distribution as a diagnostic functional assay for adaptor protein complex 4-associated hereditary spastic paraplegia. Brain Commun, 3(4), fcab221. 10.1093/braincomms/fcab221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa K, Soejima H, Niikawa N, & Shiku H (1996). Genomic organization and chromosomal assignment of the human beta1, 4-N-acetylgalactosaminyltransferase gene. Identification of multiple transcription units. Journal of Biological Chemistry, 271(34), 20836–20844. 10.1074/jbc.271.34.20836 [DOI] [PubMed] [Google Scholar]
- Furukawa K, Takamiya K, & Furukawa K (2002). Beta1,4-N-acetylgalactosaminyltransferase--GM2/GD2 synthase: a key enzyme to control the synthesis of brain-enriched complex gangliosides. Biochimica et Biophysica Acta, 1573(3), 356–362. 10.1016/s0304-4165(02)00403-8 [DOI] [PubMed] [Google Scholar]
- Go S, Go S, Veillon L, Ciampa MG, Mauri L, Sato C, … Inokuchi JI (2017). Altered expression of ganglioside GM3 molecular species and a potential regulatory role during myoblast differentiation. Journal of Biological Chemistry, 292(17), 7040–7051. 10.1074/jbc.M116.771253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlalka GV, Lehman A, Chioza B, Baple EL, Maroofian R, Cross H, … Crosby AH (2013). Mutations in B4GALNT1 (GM2 synthase) underlie a new disorder of ganglioside biosynthesis. Brain, 136(Pt 12), 3618–3624. 10.1093/brain/awt270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, … Hassabis D (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583–589. 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, & Massouras A (2019). VarSome: the human genomic variant search engine. Bioinformatics, 35(11), 1978–1980. 10.1093/bioinformatics/bty897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuser S, Brechmann B, Heimer G, Brosse I, Schubert S, O’Grady L, … Ebrahimi-Fakhari D (2021). Clinical, neuroimaging, and molecular spectrum of TECPR2-associated hereditary sensory and autonomic neuropathy with intellectual disability. Human Mutation. 10.1002/humu.24206 [DOI] [PubMed] [Google Scholar]
- Ohmi Y, Kambe M, Ohkawa Y, Hamamura K, Tajima O, Takeuchi R, … Furukawa K (2018). Differential roles of gangliosides in malignant properties of melanomas. PloS One, 13(11), e0206881. 10.1371/journal.pone.0206881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentzsch P, Witten D, Cooper GM, Shendure J, & Kircher M (2019). CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Research, 47(D1), D886–D894. 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schengrund CL (2015). Gangliosides: glycosphingolipids essential for normal neural development and function. Trends in Biochemical Sciences, 40(7), 397–406. 10.1016/j.tibs.2015.03.007 [DOI] [PubMed] [Google Scholar]
- Sheikh KA, Sun J, Liu Y, Kawai H, Crawford TO, Proia RL, … Schnaar RL (1999). Mice lacking complex gangliosides develop Wallerian degeneration and myelination defects. Proceedings of the National Academy of Sciences of the United States of America, 96(13), 7532–7537. 10.1073/pnas.96.13.7532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson MA, Cross H, Proukakis C, Priestman DA, Neville DC, Reinkensmeier G, … Crosby AH (2004). Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nature Genetics, 36(11), 1225–1229. 10.1038/ng1460 [DOI] [PubMed] [Google Scholar]
- Stephenson JD, Laskowski RA, Nightingale A, Hurles ME, & Thornton JM (2019). VarMap: a web tool for mapping genomic coordinates to protein sequence and structure and retrieving protein structural annotations. Bioinformatics, 35(22), 4854–4856. 10.1093/bioinformatics/btz482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamiya K, Yamamoto A, Furukawa K, Yamashiro S, Shin M, Okada M, … Aizawa S (1996). Mice with disrupted GM2/GD2 synthase gene lack complex gangliosides but exhibit only subtle defects in their nervous system. Proceedings of the National Academy of Sciences of the United States of America, 93(20), 10662–10667. 10.1073/pnas.93.20.10662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakil SM, Monies DM, Ramzan K, Hagos S, Bastaki L, Meyer BF, & Bohlega S (2014). Novel B4GALNT1 mutations in a complicated form of hereditary spastic paraplegia. Clinical Genetics, 86(5), 500–501. 10.1111/cge.12312 [DOI] [PubMed] [Google Scholar]
- Yang H, Brown RH Jr., Wang D, Strauss KA, & Gao G (2021). AAV-Mediated Gene Therapy for Glycosphingolipid Biosynthesis Deficiencies. Trends in Molecular Medicine, 27(6), 520–523. 10.1016/j.molmed.2021.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available from the corresponding authors upon reasonable request.