

Abstract
The field of proteomics is continually improving, requiring the development of new quantitative methods. Stable isotope labeling in cell culture (SILAC) is a metabolic labeling technique originating in the early 2000s. By incorporating isotopically labeled amino acids into the media used for cell culture, unlabeled versus labeled cells can be differentiated by the mass spectrometer. Traditional SILAC labeling has been expanded to pulsed applications allowing for a new quantitative dimension of proteomics – temporal analysis. The complete introduction of Heavy SILAC labeling chased with surplus unlabeled medium mimics traditional pulse-chase experiments and allows for the loss of heavy signal to track proteomic changes over time. In a similar fashion, pulsed SILAC (pSILAC) monitors the initial incorporation of a heavy label across a period of time, which allows for the rate of protein label integration to be assessed. These innovative techniques have aided in inspiring numerous SILAC-based temporal and spatial labeling applications, including super SILAC, spike-in SILAC, spatial SILAC, and a revival in label multiplexing. This review reflects upon the evolution of SILAC and the pulsed SILAC application, introduces advances in SILAC labeling, and proposes future perspectives for this novel and exciting field.
1. Introduction
In every biological system there exists a constant fluctuation of protein synthesis and degradation.1 In the field of proteomics, we aim to study these changing protein populations. Analyzing the proteome of any species is a daunting task that requires complex instrumentation and detailed sample preparation strategies. Mass spectrometry has allowed quantitative proteomic analysis to become a common application in the bioanalytical chemistry field.
There are two major areas of quantitative proteomics: labeling and label-free quantification. Effective labeling strategies for quantitative proteomics experiments are essential to gather meaningful data.2 One of the most popular labeling strategies involves the use of isotopes.3 The use of isotopic labels has been extensively studied and has evolved through multiple avenues. Isotopic labels come in many varieties including isotope-coded infinity tags, 18O labeling, isobaric tags, and metabolic labeling by the incorporation of stable isotopes.3 The various isotopic labeling strategies can be further divided into two categories: metabolic label incorporation and postdigestive chemical labeling.3
Metabolic labeling involves the introduction of isotopic labels at the cellular level during growth. Metabolic labeling is an extremely reproducible quantification strategy as it involves labeling the proteins prior to analysis, ultimately eliminating variability that can be introduced in downstream sample preparation.4 However, while metabolic incorporation has the potential to decrease variation, this technique is limited to actively growing biological samples, as it relies on protein synthesis to incorporate these labels. An additional challenge is spectral complexity, as multiple label types and samples are combined, additional isotopic peaks are added, convoluting the mass spectrum. To address these complications, peak spacing can be increased by utilizing combined 13C- and 15N-labeled amino acid enriched mediums.3,5–7 The use of isotopically labeled amino acids was developed over twenty years ago by Matthias Mann’s research group and dubbed Stable Isotope Labeling by Amino Acids in Cell culture (SILAC).7,8
The SILAC proteomic workflow begins at the cellular level. To culture cells, medium is necessary to fulfill the nutritional requirements. Specifically, mammalian cells must be supplemented with a minimum of thirteen amino acids essential to growth: arginine, cystine, glutamine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, tyrosine, and valine.9,10 Cells grown in their preferred culture medium are transferred to a specifically formulated medium that replaces all naturally occurring isotopes in specific amino acids, typically lysine and arginine, to a solely light 12C14N lysine and 12C14N arginine or heavy 13C15N lysine and 13C15N arginine. The media is then supplemented with a dialyzed serum. The cells are maintained in this labeled medium for five doublings to ensure that the labels are fully incorporated during protein synthesis.11 The labels must achieve a high level of incorporation for this quantitative technique to be successful.
Once incorporated, the cells are lysed, and the proteins extracted. These proteins can be analyzed intact by top-down analysis, or the proteins can be digested into peptides and analyzed by bottom-up proteomic analysis. Typically, the classic SILAC experiment is associated with bottom-up proteomic analysis, but these labels can be utilized in either approach.12 Following the bottom-up workflow, the extracted proteins will then be reduced, alkylated, and digested. While protein digestion can be accomplished with a variety of proteolytic enzymes, the use of trypsin is especially complementary with the labeled lysine and arginine amino acids, as trypsin cleaves at the C-terminus of both of these residues. Essentially, this configuration allows for each peptide to be labeled and designated as either heavy or light depending on the label state of the lysine or arginine residue. These peptides can then be analyzed by a high-resolution mass spectrometer.
Analysis of SILAC labeled peptides within bottom-up proteomics has traditionally been coupled with a data dependent analysis (DDA) approach. Both heavy and light labeled peptides co-elute at the same time during the initial liquid chromatography separation. These coeluted peptides can then undergo an initial precursor ion scan. The abundance ratios of the peptides for both label states can be determined based on the signal intensities from this full precursor ion scan, making high resolution mass spectrometry necessary during this crucial step.13 These peptides must then undergo fragmentation to make accurate identifications. These data are then analyzed by complex search algorithms which utilize comparisons to specific theoretical tandem mass spectra to infer proteins by their corresponding peptides.14 The peptide abundance ratios previously established in the precursor ion scan can then be used to determine the relative protein abundance ratios, ultimately determining protein quantification for both heavy and light samples. While the SILAC DDA approach utilizes the precursor ion abundances (MS1) for quantification, this reliance has limitations due to instrumentation limits for ion accumulation and the increased spectral complexity resulting from multiplexing samples.15 However, data independent acquisition (DIA) approaches with the SILAC workflow are gaining popularity and have shown the potential for outperforming traditional DDA experiments under certain conditions.16–18
Once the MS analysis has been completed, raw files must undergo extensive searches to make protein identifications and determine abundance differences amongst samples. Data analysis for SILAC-DDA experiments can typically be addressed in a few different ways. There are multiple software packages available that can handle SILAC data including MaxQuant, MASCOT, and Proteome Discoverer.19–22 Additionally, R-based proteomics pipelines are increasingly popular for processing SILAC data.23 While these programs are suitable for handling traditional duplex SILAC experiments, additional tools can be required when dealing with more complex data, such is the case with multiplexed labels and certain spatial or temporal experiments.
The introduction of SILAC labels to bottom-up proteomics has revolutionized protein analysis and while this fundamental workflow can be useful in experiments such as label incorporation estimations, SILAC has the potential to be used in more advanced ways, specifically pulse-chase experiments. The original pulse-chase experiment was completed by Matthew Meselson and Franklin Stahl.24 In 1958, Meselson and Stahl grew bacteria Escherichia coli (E. coli) in a culture medium containing only heavy 15N nitrogen. The bacteria grew and replicated their deoxyribose nucleic acid (DNA) in the heavy labeled medium to create a parental generation. Excess light 14N nitrogen was then added to the bacterial growing medium for subsequent generations and the DNA was analyzed at set intervals using ultraviolet light photographs. Meselson and Stahl discovered that by comparing the relative band densities of the 14N DNA band and 15N DNA band to a band containing an isotopic mixture of the two types of DNA, they could define the relative abundances of both isotopes in the mixture.24 While the ultimate impacts of this study helped validate Watson and Crick’s model for DNA by demonstrating semiconservative replication, the experimental design has been celebrated as being “the most beautiful experiment in biology.”25
Pulse-chase analysis has more recently been defined as any method involving the exposure of a labeled compound to a biological system (pulse) and then switching to the same compound’s unlabeled counterpart (chase) to examine a change in cellular process over time by loss of the heavy labeled signal (Fig. 1).26–28 The combination of pulse-chase experiments with SILAC began with a curiosity to further investigate protein dynamics. While classic proteomics has focused on comparisons of protein quantities between two states, pulse chase SILAC was designed to evaluate the dynamic changes in the proteome, which can then be compared across different biological states or test conditions. Thus, the evolution of pulse-chase SILAC and, subsequently, pulsed SILAC proteome analysis was born. This review aims to examine this evolution, to highlight the many varieties of spatial and temporal SILAC experiments, and to infer possible future directions for this quickly progressing field.
Fig. 1.
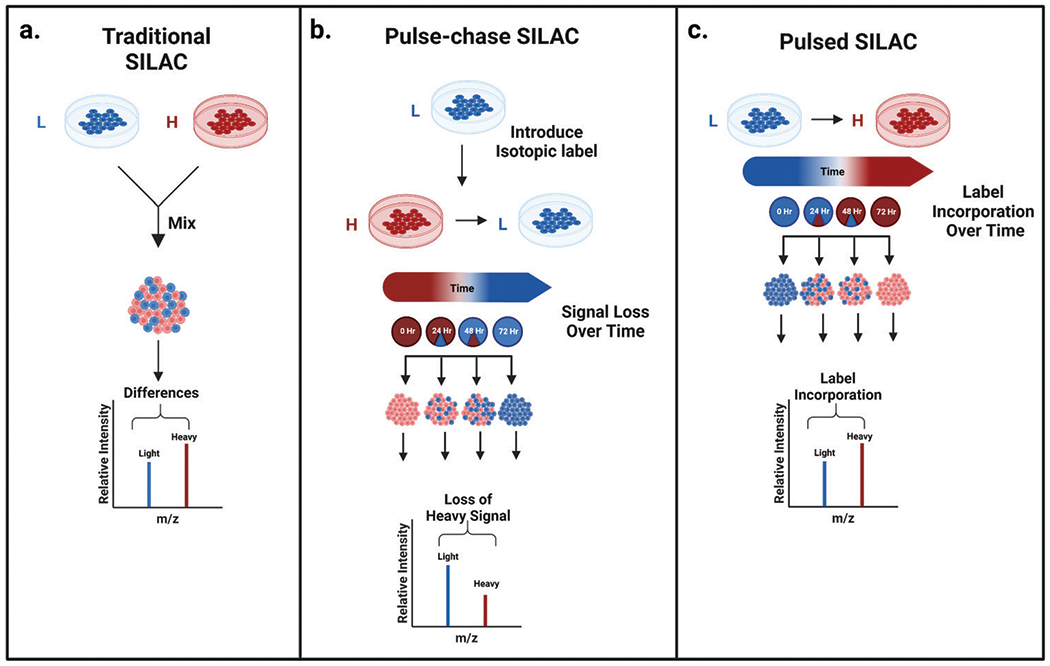
Classic SILAC experimental workflow versus pulsed and pulse-chase SILAC protein turnover experimental workflow. (a) The classic SILAC workflow combining light and heavy labeled cells prior to downstream sample processing. (b) Pulse-chase SILAC workflow demonstrating how heavy labeled cells in the presence of excess unlabeled media exhibit diminishing heavy signal over time, which can be used to monitor protein turnover. (c) Pulsed SILAC workflow showing the monitoring of heavy signal incorporation over time to determine protein turnover.28
2. Developments in SILAC applications
Elucidating the dynamic proteome: chronologically following the evolution of studying protein turnover rates with stable isotopes.
Many names have been associated with protein turnover SILAC experiments: dynamic SILAC, pulsed SILAC (pSILAC), and pulse-chase SILAC. While the jargon may be unwieldy, one constant among these experiments is that they all focus on protein dynamics. Just months after the research group of Matthias Mann published the first SILAC paper, Robert Beynon and Simon Gaskell pioneered the missing link between stable isotope labeled amino acids and protein turnover.7,29,30 This link would spur many future studies of proteome dynamics (Fig. 2).
Fig. 2.
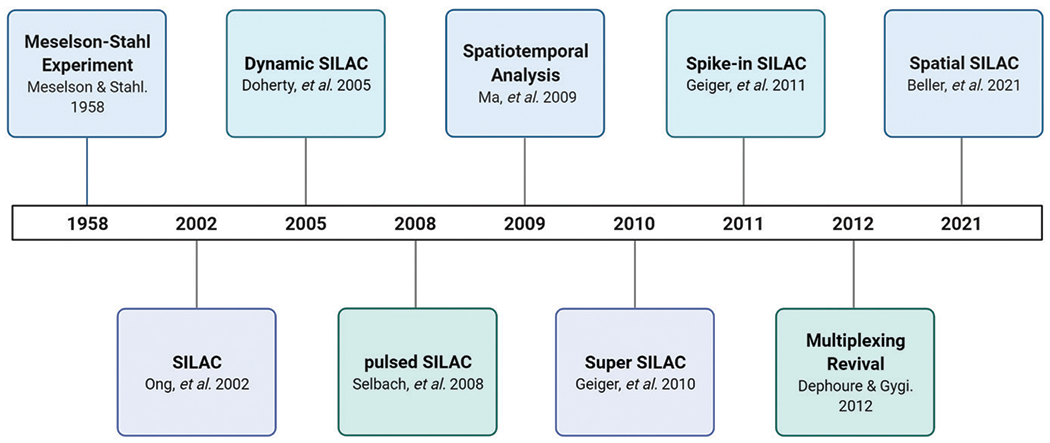
Timeline featuring the progression of pulse-chase SILAC experimentation and current innovations. From the beginning of pulse-chase with the Meselson–Stahl experiment in 1958, to the introduction of pulse-chase SILAC experiments in 2005, SILAC has expanded its applications to study the dynamic proteome. SILAC has continued to expand its applications to greater multiplexing capabilities and to studies involving spatial dimensions.7,24,27–29,44–46,54,58,73
The use of pulse-chase SILAC to study system-wide protein turnover rates using two SILAC channels was originally referred to as “Dynamic SILAC”.30,31 Robert Beynon’s research group conducted a pilot experiment to the first dynamic SILAC study. In 2002, they analyzed protein turnover within dynamically labeled amino acids and examined the degradation rates of highly abundant proteins in glucose-limited yeast cells.29 This initial study utilized culture medium containing deuterated leucine, which was then chased with excess unlabeled leucine and sampled at set time points. Peptides from each time point were analyzed with a MALDI-TOF mass spectrometer. Utilizing monoisotopic peak intensities from both the heavy and light tryptic peptides, they determined relative isotope abundances at each set time points, which were then used to calculate the rate of degradation (Fig. 3).29 This study validated the application of SILAC for protein dynamics studies, essentially unlocking a new dimension in proteome analysis.7,29
Fig. 3.
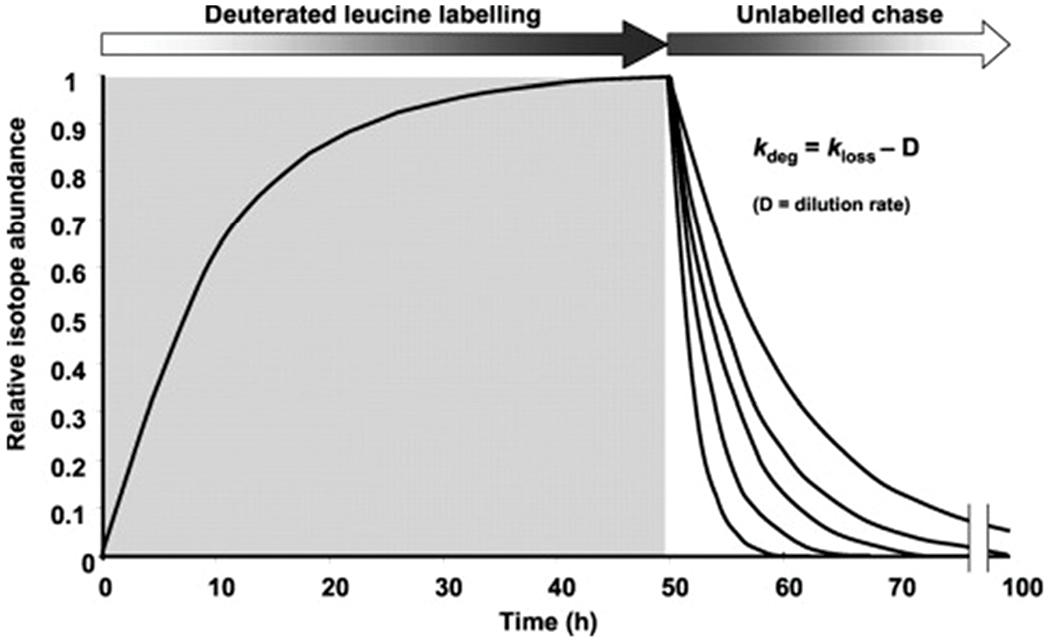
Pulse-chase SILAC protein turnover rate determination strategy. Yeast cells experienced full incorporation of a deuterated leucine label followed by an unlabeled chase. Proteins synthesized after the unlabeled chase will contain unlabeled leucine, thus allowing the rate of protein turnover to be determined by the rate of replacement (labeled to unlabeled leucine). Reprinted with permission from Pratt, J. M., et al., Dynamics of protein turnover, a missing dimension in proteomics. Mol. Cell. Proteomics, 2002, 1 (8), 579.29
As more dynamic SILAC studies were completed, attention was focused on the labels. Prior to SILAC, radioisotope labeling of proteins was used to study protein dynamics.32–39 While these radioisotopes were useful to elucidate protein dynamics on smaller scales, they are hazardous substances.39 As the push towards the usage of stable isotopes in amino acids occurred, researchers adopted less hazardous labels. Beynon and Pratt compiled a thorough review examining each amino acid’s eligibility for effective labeling.39 Deuterated amino acids were a major consideration in their review. The presence of deuterium in amino acids can result in differences in retention time when the experiment is coupled with liquid chromatography.39,40 For this reason, many recent studies have utilized 13C and or 15N heavy labeled amino acids, which do not suffer from retention time differences.39–41
While the analysis of protein dynamics within single cellular organisms furthered understanding of the dynamic nature of the proteome, the technique has been expanded to more complex systems. Just three years after their initial deep dive into protein dynamics on the diploid yeast strain BY4743, Beynon and Gaskell published on the use of stable isotopes to monitor protein turnover rates in a more complex organism: a chicken.30 The chickens were fed a strict diet containing light valine for six days, then the diet was changed to include heavy 13C valine for 120 hours, with three birds sacrificed for sampling fourteen times throughout that period. The pectoralis muscle from the chicken was extracted and analyzed with a MALDI-TOF mass spectrometer. This study determined individual protein turnover rates in these complex organisms, ultimately paving the way for future vertebrate dynamic SILAC studies.30
From single cell organisms to multicellular complex organisms, dynamic SILAC has also been utilized for studies in human cell lines. Analysis of human cells using variations of pulsed SILAC was accomplished in 2005 by Matthias Mann and colleagues as they studied nucleolar proteome dynamics.42 Subsequent studies included major histocompatibility complex peptides from human ovarian cancer cells, human atrial trabeculae, and human A549 adenocarcinoma cells.41,43,44 As applications for the SILAC approach broadened, so did the variations of this technique. Multiple research groups have made this approach their own by coining individual names for the many variations, including pulsed SILAC, super SILAC, and spiked-in SILAC.45–48
Matthias Selbach and Björn Schwanhäusser et al. coined the phrase “pulsed SILAC” (pSILAC); they defined a platform involving light labeled cell populations pulsed with two different forms of heavy isotopic labels to quantify relative differences in de novo protein synthesis of two samples.31,47,49 Schwanhäusser’s initial study tracked cellular proteome changes at the translation level.50 They began by growing HeLa cells in a light cell culture medium. These cells were then split into two populations with differential treatment and grown in either a heavy or medium labeled culture medium. These differing pulse treatments allowed them to evaluate the respective “heavy/medium” SILAC abundance ratios to determine the differences in translation of the corresponding proteins.49 This groundbreaking work introduced an exciting new perspective into how protein dynamics can be studied.
Many pulsed SILAC applications have encountered the limitations of requiring complete metabolic labeling of cell cultures, ultimately eliminating the study of human tissue.46 A few different techniques have tried to address some of these constraints such as super SILAC and spike-in SILAC.45,46,50 Super SILAC refers to the combination of multiple SILAC labeled cell cultures from differing cell lines.46 In the technique’s debut by the Matthias Mann research group, four different breast cancer cell lines from various stages and origins were grown with heavy SILAC labels. The resulting proteins from each of these cell lines were normalized and combined with unlabeled tumor tissue. The samples were digested with filter-aided sample preparation (FASP) method, and fractionated with strong anion exchange in a StageTip format.46,51 The resulting analyses showed increased protein identification and quantification accuracy.46 The success of these experiments performed with the super SILAC technique inspired the spike-in SILAC approaches.45
Spike-in SILAC labeling is another innovative SILAC variation. Spike-in SILAC is different in application from its counterparts and uses heavy isotopically labeled cells as a spiked-in standard, introduced to non-labeled samples after any perturbation experiments have been performed.45 This technique eliminates many of the inherent barriers that the use of SILAC can present, such as the need for complete labeling and strict dietary limitations. Additionally, this technique differs from the previous super SILAC approach as it is not limited to digestion and fractionation techniques, further increasing applicability. While this approach does not fit the traditional structure of a pulsed experiment, this method deserves mention here as it has still helped to further studies of protein dynamics.
Many successful experiments have utilized this method or variations of it. Spike-in SILAC has been used to identify a new member of a protein family in newt hearts, recording real-time protein-mediated cell–cell interactions, and large-scale phosphosite quantification of tissues.52–54 The many variations of SILAC (Fig. 4) have allowed for groundbreaking discoveries. As SILAC and its many variations have evolved, so has the nature of our experiments, leading us next to examine dynamic studies in other dimensions.
Fig. 4.
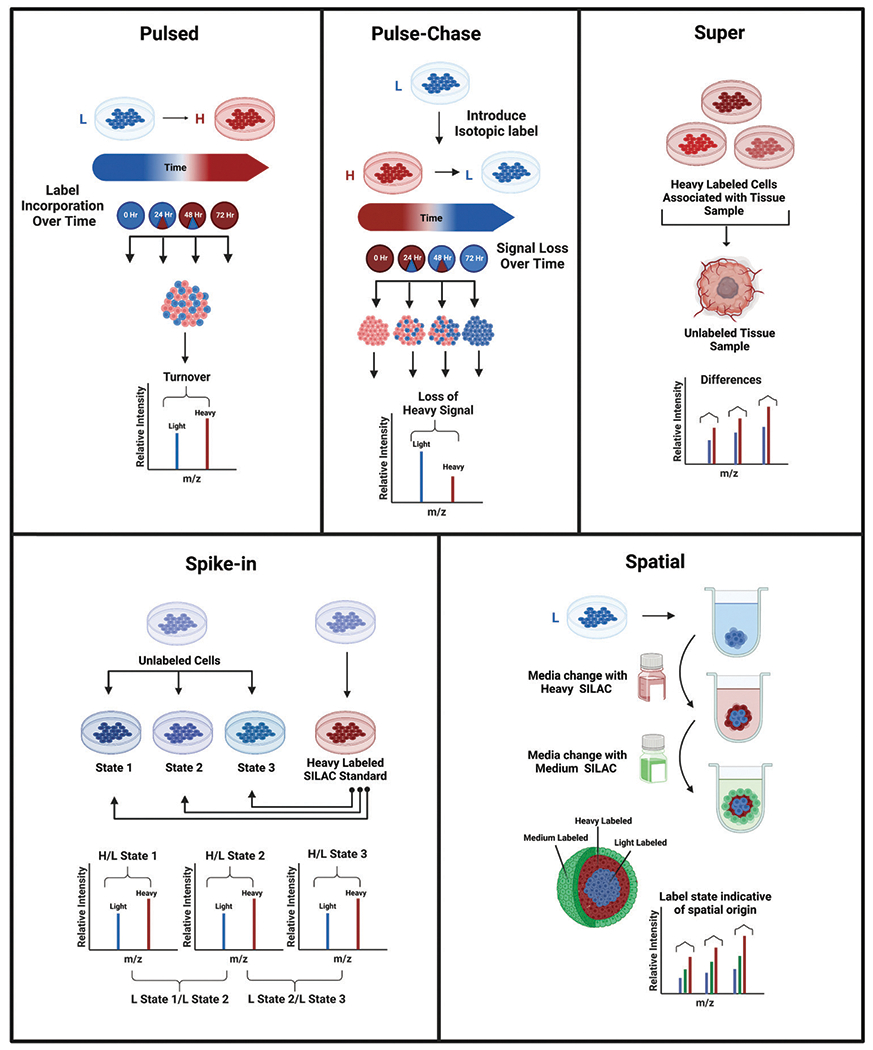
Workflows demonstrating and comparing a few of the many variations of SILAC including pulsed, pulse-chase, super, spike-in, and spatial SILAC. (a) Pulsed SILAC monitors the rate of incorporation of a heavy label over time. (b) Pulse-chase SILAC begins with a labeled pulse to fully incorporate a heavy label, followed by a chase of excess unlabeled media to monitor the subsequent signal change over time. (c) Super SILAC introduces isotopically labeled cells associated with a tissue type respective to the unlabeled tissue sample. (d) Spike-in SILAC involves spiking a heavy labeled SILAC cell population representative of the corresponding cells into a second cell population of various treatment conditions to act as an internal standard. (e) Spatial SILAC involves pulsing labeled media into a 3D cell culture at strategic points during its growth to isotopically label specific spheroid regions for proteomic analysis.28,58
Another dimension of pulse-chase SILAC: spatial demonstrations
Stable isotope labeling and its many variations have allowed for the expansion of proteomic analysis to both the spatial and temporal dimensions. The ability to examine proteome changes with respect to a localized area versus globally presents opportunities to both further our understanding of biological systems and to explore targeted therapeutics in greater detail. From cells grown in a monolayer to animal models, the spatial applications of SILAC can be used to elucidate another dimension of the dynamic proteome.
The study of spatial SILAC protein dynamics in vitro emerged shortly after the rise of both the spike-in and Super SILAC variations. One of the first mentions of spatial analysis with SILAC involved both a heavy and light SILAC labeled HEK-293 human cell population that was stimulated with a tumor necrosis factor at set time points as performed by the Rong Zeng research group.55 These samples then underwent nuclear component enrichment with subcellular fractionation before being combined for digestion and analysis. This analysis allowed for measured levels of signaling-associated proteins to be correlated with their respective spatial delineations.55 This study inspired additional methods for resolving spatial proteome dynamics including enzyme targeting, spatial SILAC, and DIPPER, a spatiotemporal proteomics atlas of human intervertebral discs.56–59
Additional studies involving cell culture models have utilized a variety of techniques to achieve spatial information. While the use of subcellular fractionation has proven to be valuable, spatial information can also be achieved by combining SILAC with enzymes genetically targeted to cellular regions of interest.55–57,60 Alice Ting’s research group has utilized peroxidases for their ability to biotinylate nearby proteins. These proteins can then be enriched and combined with different SILAC label states to determine spatial orientation.56,57 These studies have helped pave the way for spatial dynamics to be studied in larger and more complex systems.
Pulsed SILAC has more recently been used to provide spatial information for three-dimensional cell culture model systems. Our research group recently described a method of utilizing pulsed SILAC to label the different cellular subpopulations within a multicellular tumor aggregate, or spheroid (Fig. 5).58 By switching the isotopic labels in the cell culture medium at specific times during the spheroid’s development, we created a triplex labeled spheroid in which the core, middle, and outer layers all contained a distinct regional isotopic label (Fig. 5). This preliminary study in spatial labeling utilized a technique called serial trypsinization to dissociate layers of cells from the spheroids allowing for individual layer proteome analysis to confirm the discrete presence of the pulsed-in isotopic labels. This technique can be utilized as a model system for performing both pharmacokinetic and pharmacodynamic drug evaluation studies.58
Fig. 5.
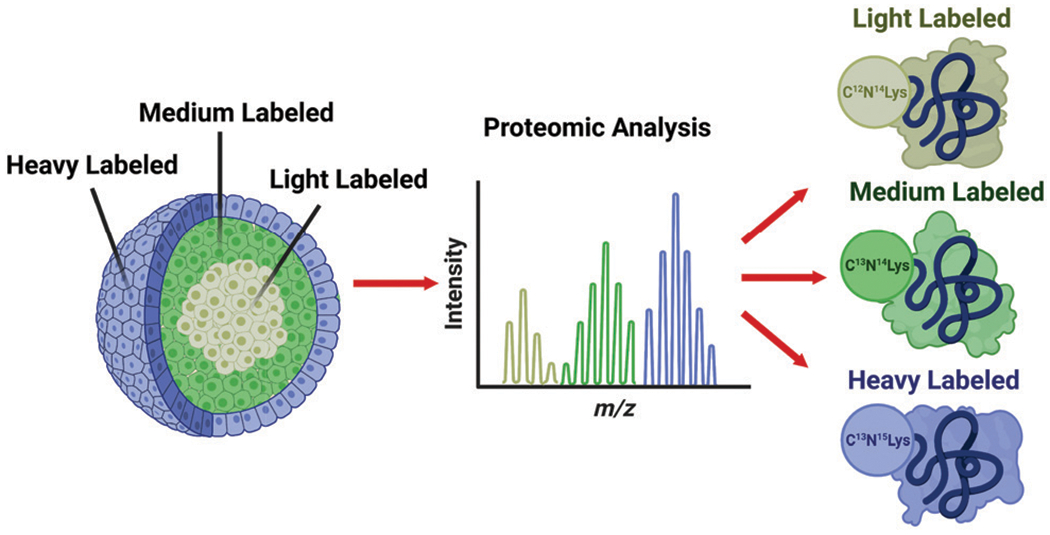
Spatial SILAC labeling of three-dimensional multicellular tumor aggregate (spheroid). Isotopically labeled cell culture media is introduced at strategic time points during the spheroid’s growth. The pulsed labels effectively create an “isotopic zip-code” allowing labeled proteins to be correlated with a layer of origin within the spheroid. Reprinted with permission from Beller, N. C., et al., Spatial stable isotopic labeling by amino acids in cell culture: pulse-chase labeling of three-dimensional multicellular spheroids for global proteome analysis. Anal. Chem. 2021, 93 (48), 15990–15999.58
Similar techniques have also been performed in tissue via ex vivo SILAC labeling. Danny Chan and colleagues have utilized a technique they coined “DIPPER” to connect high-resolution point-reference maps of static spatial proteomes to dynamic SILAC proteomic data within intervertebral discs.59 Utilizing an established high-resolution point-reference map of static spatial proteomes along the lateral and anteroposterior directions of intervertebral discs, they demonstrated how point-reference proteomes can be utilized via integration with dynamic proteome datasets.59 These spatial connections are not limited to cells and tissue cultures; they have also been observed in whole animal models.
SILAC has been expanded from use solely as a cell culture labeling technique to whole animal models, an approach referred to as SILAM-Stable Isotope Labeling of Mammals.61–63 Matthias Mann and colleagues are credited with generating the first fully labeled SILAC mouse population and their group was able to successfully compare proteomes between knock-out and wild-type mice, ultimately determining critical protein activity and function within a more dynamic model system.61
The dynamic SILAM model system has enabled research on targeted tissue types. One such example focused on in vivo pulsed SILAC labeling of mice to elucidate turnover rates of collagens within articular cartilage and bone and age-dependent changes in protein incorporation into collagen-rich tissues.64,65 Many additional studies have been performed utilizing pulse-chase SILAM to elucidate development, aging, and disease states within mouse brains.66–69 Jeffrey Savas’s research group has recently utilized this model system to study Alzheimer’s disease by evaluating amyloid beta peptide accumulation through presynaptic protein turnover (Fig. 6).69 The ability to measure proteome turnover in animal models, and specifically trace these changes back to their origins, is a valuable tool to further understanding of disease progression and therapeutic treatments.
Fig. 6.
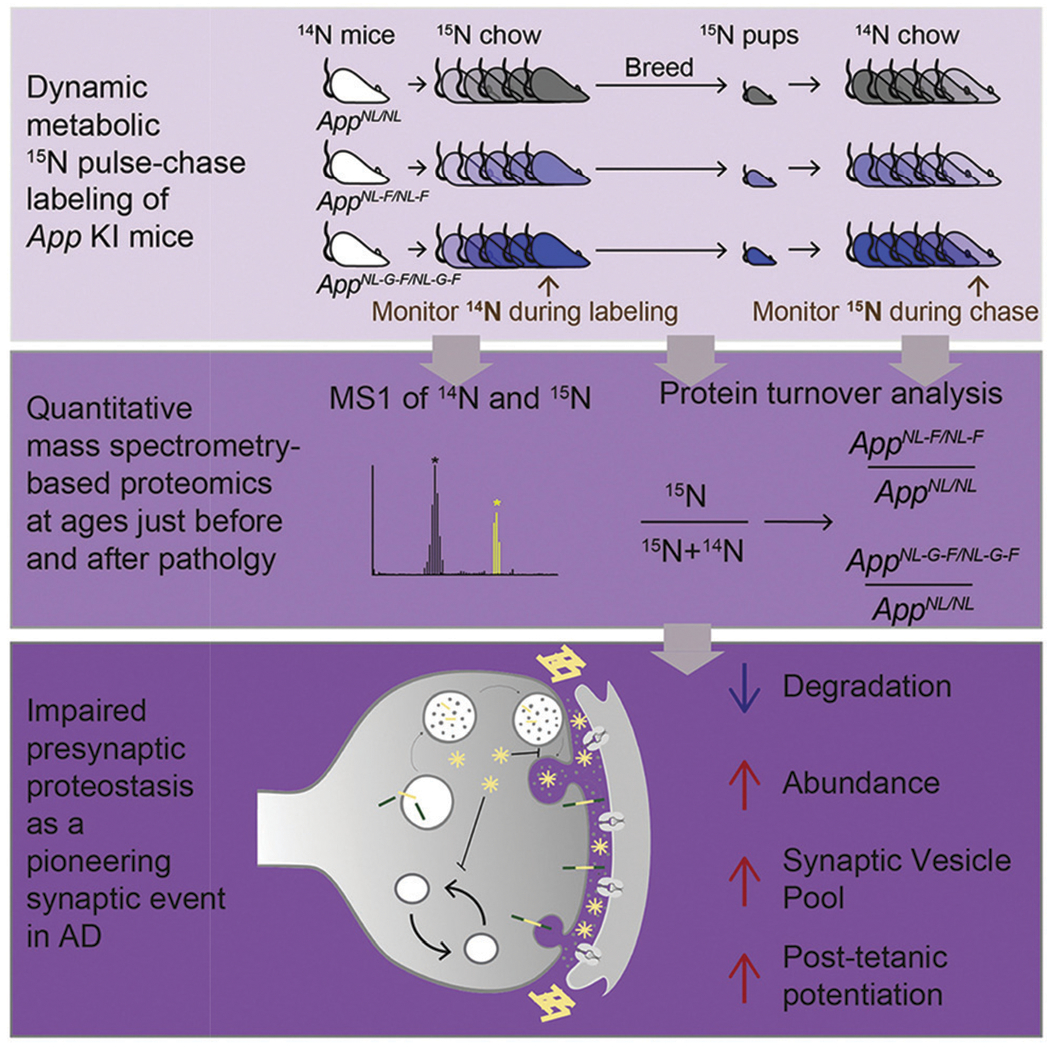
Metabolic pulse-chase labeling of App KI mice to study synaptic proteomic changes. An example of the pulse-chase SILAM model, mice were monitored throughout two phases: the heavy 15N pulse label incorporation period, followed by an unlabeled chase. During this time, protein turnover was monitored and ultimately revealed an association between the presynaptic terminal and initial Alzheimer’s disease etiology. Reprinted with permission from Hark, T.J., et al., Pulse-chase proteomics of the app knockin mouse models of Alzheimer’s disease reveals that synaptic dysfunction originates in presynaptic terminals. Cell Systems 2021, 12 (2), 141–158.e9.69
Higher order multiplexing: do you believe in a higher power?
While many of the previously mentioned techniques utilize either duplex or triplex SILAC labeling, recent experiments have turned to the coupling of pulse-chase SILAC with alternative labeling techniques. As SILAC labels are incorporated into a cell’s proteome during growth, SILAC is a metabolic labeling technique with MS1 level quantification. As previously discussed, there are many advantages to using metabolic labeling over conventional chemical labeling techniques. However, chemical labeling has the potential to label a wider variety of samples, as labels are incorporated after cell lysis (Fig. 7).3,15,70 For this reason, many chemical labels can be used in tandem with SILAC labeling to provide additional quantitative information across a variety of samples.
Fig. 7.
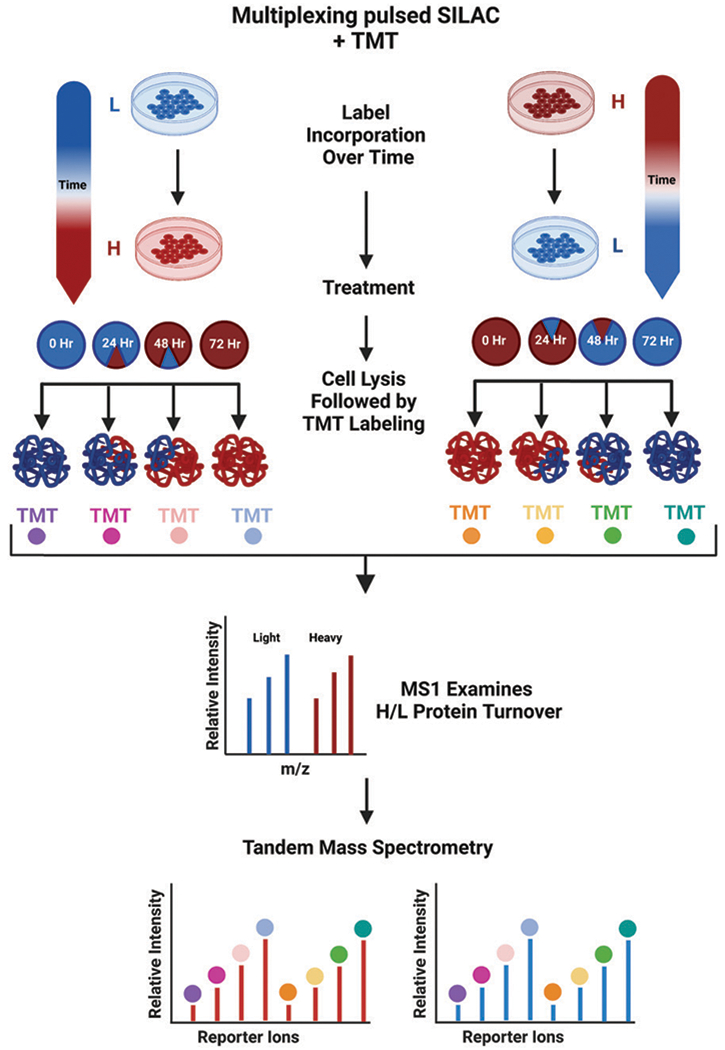
General workflow for multiplexing pulsed SILAC with TMT labeling. Cells are pulsed with a heavy label for set periods of time, after which they are lysed and chemically labeled. The resulting peptides will contain both the metabolically incorporated SILAC label and the chemically added TMT labels, which can both be detected by a high-resolution mass spectrometer.28
The concept of multiplexing for shotgun proteomics has existed for over twenty years as chemical labeling strategies pushed the sampling limitations further and further.71 However, the process of “higher order multiplexing” or “hyperplexing” has been a more recent endeavor in the last fifteen years, especially in the past four years.15,71,72 Higher order multiplexing refers to the process of combining MS1 and MS2 level quantification techniques. Typically, higher order multiplexing references the combination of a chemical labeling technique with metabolic labeling.
Chemical labeling has many variations, and the most common techniques involve the use of isobaric stable isotope labeling tags. Two popular techniques include isobaric tags for relative and absolute quantification (iTRAQ) and tandem mass tags (TMT). These techniques allow for multiplexing of up to 11 samples.71,73 Unlike SILAC, iTRAQ and TMT labels are introduced after digestion at the peptide level, and relative quantification of these peptides is performed during tandem mass spectrometry scans.73 These differing aspects of isobaric tag labeling make them an ideal partner for higher order multiplexing with metabolic labels.
The first study to combine MS1 and MS2 quantification for higher order multiplexing was performed in 2010 by Jayapal et al.74 They expanded upon the concept of pulse-chase SILAC by following each pulsed sample with an iTRAQ reagent in order to decouple peptide identification and quantification of degradation dynamics at both MS1 and tandem MS scans.74 The term hyperplexing can be traced back to Noah Dephoure and Steven Gygi in 2012 as they combined triplex metabolic labeling with six-plex isobaric tags to investigate temporal abundance profiles for yeast proteins.72 This approach allowed for 18 different samples to be monitored simultaneously, identifying thousands of proteins in just a fraction of the time required for immunoblotting.72 Since this initial approach, many others have had success with higher order multiplexing ultimately leading to a recent revival in the field.
The recent revival in higher order multiplexing has led to many exciting discoveries and a focus on combining pulsed SILAC with this technique. Starting in 2016, Ajay Kumar et al. combined biorthogonal noncanonical amino acid tagging (BONCAT) with pSILAC and iTRAQ labeling, which was then elaborated upon by Thomas Neubert’s group, creating a technique they coined (BONLAC) (Fig. 8).75–77 This technique was later expanded upon by Daniel Rothenberg et al. to combine TMT labeling with pSILAC in what they called MITNCAT: Multiplex Isobaric Tagging/Noncanonical Amino acid Tagging.78 Additional studies utilizing pSILAC studies with higher order multiplexing have elucidated the secretome, complexome, and translation proteome dynamics.75,79–82 These recent advances provide a promising future for explicating spatiotemporal proteome dynamics.
Fig. 8.
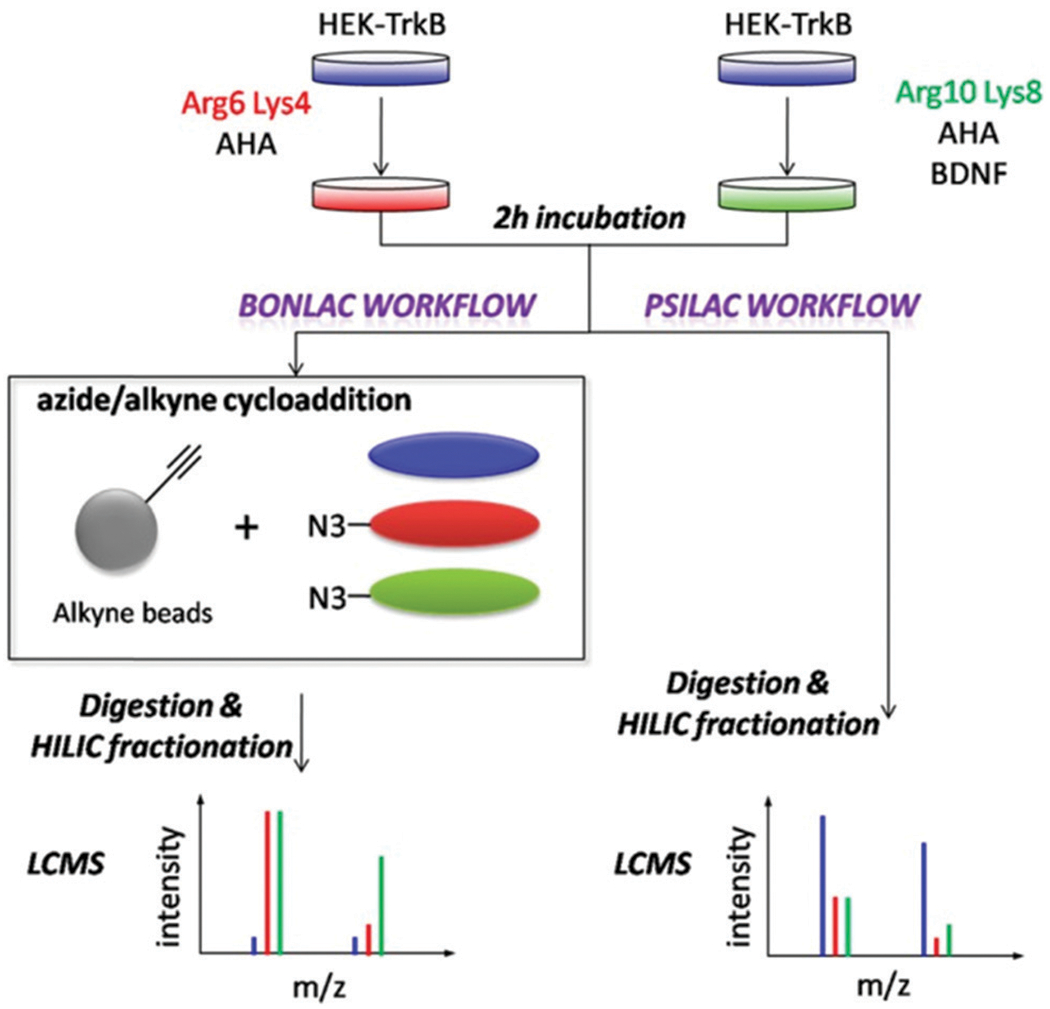
BONLAC pSILAC workflow showing proteomic changes induced by BDNF signaling. Cells undergo pulsed SILAC labeling followed by bioorthogonal noncanonical amino acid tagging (BONCAT) to study and compare de novo protein synthesis. Reprinted with permission from Zhang, G., et al., In-depth quantitative proteomic analysis of de novo protein synthesis induced by brain-derived neurotrophic factor. J. Proteome Res. 2014, 13 (12), 5707–5714.77
Current perspectives and the future of pulse-chase SILAC
Pulse-chase SILAC is a versatile technique that is almost as dynamic as the proteome itself. With so many new innovations in the field, pulse-chase SILAC has a bright future. From the introduction of SILAC labels twenty years ago to current spatial and higher order multiplexing studies, progress is actively accelerating. However, as with all innovations, no technique is without its flaws.
While the numerous advantages of using pulse-chase SILAC have been discussed, it is critical to also highlight limitations. First, consideration must be taken when examining the high costs of using metabolic or chemical labels. Higher order multiplexing ultimately combines the cost of both techniques, significantly limiting its application.83 SILAC labeling also is generally limited to organisms that can fully incorporate the labels metabolically, but, as previously mentioned, techniques like super SILAC and spike-in SILAC have aided in addressing this issue.8,50,54 Additionally, hyperplexing with TMT or iTRAQ chemical labels comes with the inherent risk of systemic quantitative ratio compression due to peptide cofragmentation.84
With advances in data independent analysis (DIA), many labs are turning to SILAC DIA experiments.16–18,85 Notably, Yansheng Liu’s group has recently developed a technique Boxcarmax to enable multiplexed MS1 and MS2 acquisition.85 Alternative approaches have also considered neutron encoded labeling strategies to increase the multiplexing capabilities of DIA.86–88 These advances offer an alternative approach to conventional multiplexing with DDA.
Finally, considerations must also be taken when examining the complexity of the data produced. As the field moves towards more complicated spatiotemporal analysis and higher order multiplexing, the data subsequently becomes more convoluted, often beyond the capabilities of current data processing algorithms.71 This conundrum will ultimately require advances in the data analytics field to develop new software to handle these evolving approaches. In the next ten years, we predict that higher order multiplexing will see increasing popularity and will hopefully be expanded to even greater orders of hyperplex magnitudes. We also expect to see this technique used to gain a more complete spatiotemporal understanding within the dynamic proteomes of additional complex model systems and organisms. We hope to see this technique address a plethora of disease states and hopefully begin its journey into clinical applications to gain insights into therapeutic testing.
3. Conclusions
In this review, we have thoroughly examined the evolution of pulse-chase SILAC and the future direction of SILAC labeling. From its origins in 1958 with the Meselson–Stahl experiment, the pulse-chase technique has significantly evolved with SILAC labeling to become a commonplace experiment for the evaluation of protein turnover. As this platform progressed, its applications were expanded to not only temporal studies, but also to spatial studies in complex multicellular organisms. These advances brought forth many variations of SILAC, many of which overcame previous limitations of the base SILAC labeling. This growth led to new combinations and permutations of both chemical and metabolic labeling to increase sample pools to larger multiplexes.
The rapid growth of SILAC approaches is a testament to the scientific community’s desire for fuller comprehension of the dynamic proteome. From unicellular organisms to animal models, the intricate dynamics of the elusive proteome remain a mystery waiting to be unraveled. SILAC has shown great potential for finally unlocking these mysteries, and future advances in the field provide hope for ultimately illuminating the rates and motivations for change of the dynamic proteome. With the versatility of this technique, its future applications hold limitless potential.
Biographies

Nicole C. Beller
Nicole Beller received her BS in Biochemistry from Wayne State University in 2018. That same year she joined the Ohio State University where she earned her MS in Chemistry. Nicole is currently a fourth-year graduate student working under the guidance of Dr Amanda Hummon. Nicole’s current research focuses on bottom-up proteomic analysis of pulse-chase SILAC labeled threedimensional multicellular spheroids.

Amanda B. Hummon
Amanda Hummon earned her AB in chemistry at Cornell University and her PhD in analytical chemistry at the University of Illinois, in the laboratory of Prof. Jonathan Sweedler. She completed her postdoctoral work at the National Cancer Institute. She is currently a Professor in the Department of Chemistry and Biochemistry and the Comprehensive Cancer Center at The Ohio State University. Her laboratory develops mass spectrometric methods to explore cancer tissues, cell cultures, and organoids. She has been recognized with an NSF CAREER award, the Presidential Early Career Award for Scientists and Engineers (PECASE) and a Fulbright Scholar Award.
Footnotes
Conflicts of interest
There are no conflicts to declare.
References
- 1.Laskowska E, Kuczyńska-Wiśnik D and Lipińska B, Proteomic analysis of protein homeostasis and aggregation, J. Proteomics, 2019, 198, 98–112. [DOI] [PubMed] [Google Scholar]
- 2.Karen AS and Jeroen AAD, Labeling Methods in Mass Spectrometry Based Quantitative Proteomics, in Integrative Proteomics, ed. Hon-Chiu Leung TKM and Ricardo F, Intech Open, 2012. [Google Scholar]
- 3.Sap K and Demmers J, Labeling methods in mass spectrometry based quantitative proteomics, in Integrative Proteomics, ed. Leung HE, Man T and Flores RJ, IntechOpen, London, 2012, pp. 111–132. [Google Scholar]
- 4.Gouw JW, Krijgsveld J and Heck AJR, Quantitative proteomics by metabolic labeling of model organisms, Mol. Cell. Proteomics, 2010, 9(1), 11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oda Y, Huang K, Cross FR, Cowburn D and Chait BT, Accurate quantitation of protein expression and site-specific phosphorylation, Proc. Natl. Acad. Sci. U. S. A, 1999, 96(12), 6591–6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krijgsveld J, Ketting RF, Mahmoudi T, Johansen J, Artal-Sanz M, Verrijzer CP, Plasterk RH and Heck AJ, Metabolic labeling of C. elegans and D. melanogaster for quantitative proteomics, Nat. Biotechnol, 2003, 21(8), 927–931. [DOI] [PubMed] [Google Scholar]
- 7.Ong S-E, Blagoev B, Kratchmarova I, Kristensen D, Steen H, Pandey A and Mann M, Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics, Mol. Cell. Proteomics, 2002, 1, 376–386. [DOI] [PubMed] [Google Scholar]
- 8.Mann M, Fifteen Years of Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC), in Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC): Methods and Protocols, ed. Warscheid B, Springer, New York, 2014, pp. 1–7. [DOI] [PubMed] [Google Scholar]
- 9.Eagle H, Amino Acid Metabolism in Mammalian Cell Cultures, Science, 1959, 130(3373), 432–437. [DOI] [PubMed] [Google Scholar]
- 10.Yamamoto K and Niwa A, Amino acid and vitamin requirements in mammalian cultured cells, Amino Acids, 1993, 5(1), 1–16. [DOI] [PubMed] [Google Scholar]
- 11.Ong S-E and Mann M, A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC), Nat. Protoc, 2006, 1(6), 2650–2660. [DOI] [PubMed] [Google Scholar]
- 12.Waanders LF, Hanke S and Mann M, Top-down quantitation and characterization of SILAC-labeled proteins, J. Am. Soc. Mass Spectrom, 2007, 18(11), 2058–2064. [DOI] [PubMed] [Google Scholar]
- 13.Li Z, Adams RM, Chourey K, Hurst GB, Hettich RL and Pan C, Systematic comparison of label-free, metabolic labeling, and isobaric chemical labeling for quantitative proteomics on LTQ orbitrap velos, J. Proteome Res, 2012, 11(3), 1582–1590. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, Fonslow BR, Shan B, Baek M-C and Yates JR 3rd, Protein analysis by shotgun/bottom-up proteomics, Chem. Rev, 2013, 113(4), 2343–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pappireddi N, Martin L and Wühr M, A review on quantitative multiplexed proteomics, ChemBioChem, 2019, 20(10), 1210–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pino LK, Baeza J, Lauman R, Schilling B and Garcia BA, Improved SILAC quantification with data-independent acquisition to investigate bortezomib-induced protein degradation, J. Proteome Res, 2021, 20(4), 1918–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y, Borel C, Li L, Müller T, Williams EG, Germain PL, Buljan M, Sajic T, Boersema PJ, Shao W, Faini M, Testa G, Beyer A, Antonarakis SE and Aebersold R, Systematic proteome and proteostasis profiling in human Trisomy 21 fibroblast cells, Nat. Commun, 2017, 8(1), 1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salovska B, Zhu H, Gandhi T, Frank M, Li W, Rosenberger G, Wu C, Germain PL, Zhou H, Hodny Z, Reiter L and Liu Y, Isoform-resolved correlation analysis between mRNA abundance regulation and protein level degradation, Mol. Syst. Biol, 2020, 16(3), e9170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tyanova S, Temu T and Cox J, The MaxQuant computational platform for mass spectrometry-based shotgun proteomics, Nat. Protoc, 2016, 11, 2301–2319. [DOI] [PubMed] [Google Scholar]
- 20.Cox J, Matic I, Hilger M, Nagaraj N, Selbach M, Olsen JV and Mann M, A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics, Nat. Protoc, 2009, 4, 698–705. [DOI] [PubMed] [Google Scholar]
- 21.Koenig T, Menze BH, Kirchner M, Monigatti F, Parker KC, Patterson T, Steen JJ, Hamprecht FA and Steen H, Robust prediction of the MASCOT score for an improved quality assessment in mass spectrometric proteomics, J. Proteome Res, 2008, 7(9), 3708–3717. [DOI] [PubMed] [Google Scholar]
- 22.Orsburn BC, Proteome discoverer-a community enhanced data processing suite for protein informatics, Proteomes, 2021, 9(1), 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gatto L and Christoforou A, Using R and Bioconductor for proteomics data analysis, Biochimica et Biophysica Acta, 2014, 1844(1 Pt A), 42–51. [DOI] [PubMed] [Google Scholar]
- 24.Meselson M and Stahl FW, The replication of DNA in Escherichia coli, Proc. Natl. Acad. Sci. U. S. A, 1958, 44(7), 671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holmes FL, Meselson, Stahl, and the Replication of DNA: A History of “the Most Beautiful Experiment in Biology”, Yale University Press, 2001. [Google Scholar]
- 26.Takahashi M and Ono Y, Pulse-Chase Analysis of Protein Kinase C, in Protein Kinase C Protocols, ed. Newton AC, Humana Press, Totowa, NJ, 2003, pp. 163–170. [DOI] [PubMed] [Google Scholar]
- 27.Pulse-chase. In Merriam-Webster, Merriam-Webster.com Medical Dictionary, 2022. [Google Scholar]
- 28.Created with BioRender.com. (accessed Jan 10, 2022).
- 29.Pratt JM, Petty J, Riba-Garcia I, Robertson DHL, Gaskell SJ, Oliver SG and Beynon RJ, Dynamics of protein turnover, a missing dimension in proteomics, Mol. Cell. Proteomics, 2002, 1(8), 579. [DOI] [PubMed] [Google Scholar]
- 30.Doherty MK, Whitehead C, McCormack H, Gaskell SJ and Beynon RJ, Proteome dynamics in complex organisms: Using stable isotopes to monitor individual protein turnover rates, Proteomics, 2005, 5(2), 522–533. [DOI] [PubMed] [Google Scholar]
- 31.Ross AB, Langer JD and Jovanovic M, Proteome turnover in the spotlight: Approaches, applications, and perspectives, Mol. Cell. Proteomics, 2021, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marshall S, Okuyama R and Rumberger JM, Turnover and characterization of UDP-N-acetylglucosaminyl transferase in a stably transfected HeLa cell line, Biochem. Biophys. Res. Commun, 2005, 332(1), 263–270. [DOI] [PubMed] [Google Scholar]
- 33.Afify EA, Turnover of μ-opioid receptors in neuroblastoma cells, Mol. Brain Res, 2002, 106(1), 83–87. [DOI] [PubMed] [Google Scholar]
- 34.Rechinger KB, Siegumfeldt H, Svendsen I and Jakobsen M, “Early” protein synthesis of Lactobacillus delbrueckii ssp. bulgaricus in milk revealed by [35S] methionine labeling and two-dimensional gel electrophoresis, Electrophoresis, 2000, 21(13), 2660–2669. [DOI] [PubMed] [Google Scholar]
- 35.Vasseur C, Labadie J and Hébraud M, Differential protein expression by Pseudomonas fragi submitted to various stresses, Electrophoresis, 1999, 20(11), 2204–2213. [DOI] [PubMed] [Google Scholar]
- 36.Sangerman J and Goodman SR, Measurement of the synthesis, turnover, and assembly of α- and β-erythroid and nonerythroid spectrins in cultured rat hippocampal neurons, Brain Res. Protoc, 2001, 6(3), 141–147. [DOI] [PubMed] [Google Scholar]
- 37.Jones BR, Li W, Cao J, Hoffman TA, Gerk PM and Vore M, The role of protein synthesis and degradation in the post-transcriptional regulation of rat multidrug resistance-associated protein 2 (Mrp2, Abcc2), Mol. Pharmacol, 2005, 68(3), 701–710. [DOI] [PubMed] [Google Scholar]
- 38.Choi KL, Wang Y, Tse CA, Lam KS, Cooper GJ and Xu AJP, Proteomic analysis of adipocyte differentiation: Evidence that α2 macroglobulin is involved in the adipose conversion of 3T3 L1 preadipocytes, Proteomics, 2004, 4(6), 1840–1848. [DOI] [PubMed] [Google Scholar]
- 39.Doherty MK and Beynon RJ, Protein turnover on the scale of the proteome, Expert Rev. Proteomics, 2006, 3(1), 97–110. [DOI] [PubMed] [Google Scholar]
- 40.Beynon RJ and Pratt JM, Metabolic labeling of proteins for proteomics, Mol. Cell. Proteomics, 2005, 4(7), 857–872. [DOI] [PubMed] [Google Scholar]
- 41.Doherty MK, Hammond DE, Clague MJ, Gaskell SJ and Beynon RJ, Turnover of the human proteome: Determination of protein intracellular stability by dynamic SILAC, J. Proteome Res, 2009, 8(1), 104–112. [DOI] [PubMed] [Google Scholar]
- 42.Andersen JS, Lam YW, Leung AKL, Ong S-E, Lyon CE, Lamond AI and Mann M, Nucleolar proteome dynamics, Nature, 2005, 433(7021), 77–83. [DOI] [PubMed] [Google Scholar]
- 43.Milner E, Barnea E, Beer I and Admon A, The turnover kinetics of major histocompatibility complex peptides of human cancer cells, Mol. Cell. Proteomics, 2006, 5(2), 357–365. [DOI] [PubMed] [Google Scholar]
- 44.Lampert FM, Matt P, Grapow M, Lefkovits I, Zerkowski H-R and Grussenmeyer T, “Turnover Proteome” of Human Atrial Trabeculae, J. Proteome Res, 2007, 6(11), 4458–4468. [DOI] [PubMed] [Google Scholar]
- 45.Geiger T, Wisniewski JR, Cox J, Zanivan S, Kruger M, Ishihama Y and Mann M, Use of stable isotope labeling by amino acids in cell culture as a spike-in standard in quantitative proteomics, Nat. Protoc, 2011, 6(2), 147–157. [DOI] [PubMed] [Google Scholar]
- 46.Geiger T, Cox J, Ostasiewicz P, Wisniewski JR and Mann M, Super-SILAC mix for quantitative proteomics of human tumor tissue, Nat. Methods, 2010, 7(5), 383–385. [DOI] [PubMed] [Google Scholar]
- 47.Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R and Rajewsky N, Widespread changes in protein synthesis induced by microRNAs, Nature, 2008, 455(7209), 58–63. [DOI] [PubMed] [Google Scholar]
- 48.Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W and Selbach M, Global quantification of mammalian gene expression control, Nature, 2011, 473(7347), 337–342. [DOI] [PubMed] [Google Scholar]
- 49.Schwanhäusser B, Gossen M, Dittmar G and Selbach M, Global analysis of cellular protein translation by pulsed SILAC, Proteomics, 2009, 9(1), 205–209. [DOI] [PubMed] [Google Scholar]
- 50.Shenoy A and Geiger T, Super-SILAC: Current trends and future perspectives, Expert Rev. Proteomics, 2015, 12(1), 13–19. [DOI] [PubMed] [Google Scholar]
- 51.Wiśniewski JR, Zougman A and Mann M, Combination of FASP and stagetip-based fractionation allows in-depth analysis of the hippocampal membrane proteome, J. Proteome Res, 2009, 8(12), 5674–5678. [DOI] [PubMed] [Google Scholar]
- 52.Looso M, Michel CS, Konzer A, Bruckskotten M, Borchardt T, Krüger M and Braun T, Spiked-in pulsed in vivo labeling identifies a new member of the CCN family in regenerating newt hearts, J. Proteome Res, 2012, 11(9), 4693–4704. [DOI] [PubMed] [Google Scholar]
- 53.Wang X, He Y, Ye Y, Zhao X, Deng S, He G, Zhu H, Xu N and Liang S, SILAC-based quantitative MS approach for real-time recording protein-mediated cell-cell interactions, Sci. Rep, 2018, 8(1), 8441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Monetti M, Nagaraj N, Sharma K and Mann M, Large-scale phosphosite quantification in tissues by a spike-in SILAC method, Nat. Methods, 2011, 8(8), 655–658. [DOI] [PubMed] [Google Scholar]
- 55.Ma D.-j, Li S-J, Wang L-S, Dai J, Zhao S.-l and Zeng R, Temporal and spatial profiling of nuclei-associated proteins upon TNF-α/NF-κB signaling, Cell Res, 2009,19(5), 651–664. [DOI] [PubMed] [Google Scholar]
- 56.Hung V, Udeshi ND, Lam SS, Loh KH, Cox KJ, Pedram K, Carr SA and Ting AY, Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2, Nat. Protoc, 2016, 11(3), 456–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rhee H-W, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA and Ting AY, Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging, Science, 2013, 339(6125), 1328–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beller NC, Lukowski JK, Ludwig KR and Hummon AB, Spatial stable isotopic labeling by amino acids in cell culture: Pulse-chase labeling of three-dimensional multicellular spheroids for global proteome analysis, Anal. Chem, 2021, 93(48), 15990–15999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tam V, Chen P, Yee A, Solis N, Klein T, Kudelko M, Sharma R, Chan WC, Overall CM, Haglund L, Sham PC, Cheah KSE and Chan D, DIPPER, a spatiotemporal proteomics atlas of human intervertebral discs for exploring ageing and degeneration dynamics, eLife, 2020, 9, e64940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boisvert F-M, Ahmad Y, Gierliński M, Charrière F, Lamont D, Scott M, Barton G and Lamond AI, A quantitative spatial proteomics analysis of proteome turnover in human cells, Mol. Cell. Proteomics, 2012, 11, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krüger M, Moser M, Ussar S, Thievessen I, Luber CA, Forner F, Schmidt S, Zanivan S, Fässler R and Mann M, SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function, Cell, 2008, 134(2), 353–364. [DOI] [PubMed] [Google Scholar]
- 62.McClatchy DB, Liao L, Park SK, Xu T, Lu B and Yates JR III, Differential proteomic analysis of mammalian tissues using SILAM, PLoS One, 2011, 6(1), e16039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu CC, MacCoss MJ, Howell KE, Matthews DE and Yates JR, Metabolic labeling of mammalian organisms with stable isotopes for quantitative proteomic analysis, Anal. Chem, 2004, 76(17), 4951–4959. [DOI] [PubMed] [Google Scholar]
- 64.Ariosa-Morejon Y, Charles P, Davis S, Fischer R and Vincent T, In vivo pulsed SILAC labeling reveals distinctive, age-dependent, turnover rates of collagens of articular cartilage and bone, Osteoarthr. Cartil, 2018, 26, S30–S31. [Google Scholar]
- 65.Ariosa-Morejon Y, Santos A, Fischer R, Davis S, Charles P, Thakker R, Wann AKT and Vincent TL, Age-dependent changes in protein incorporation into collagen-rich tissues of mice by in vivo pulsed SILAC labelling, eLife, 2021, 10, e66635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rauniyar N, McClatchy DB and Yates JR, Stable isotope labeling of mammals (SILAM) for in vivo quantitative proteomic analysis, Methods, 2013, 61(3), 260–268. [DOI] [PubMed] [Google Scholar]
- 67.Ng SS, Park JE, Meng W, Chen CP, Kalaria RN, McCarthy NE and Sze SK, Pulsed SILAM reveals in vivo dynamics of murine brain protein translation, ACS Omega, 2020, 5(23), 13528–13540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Heo S, Diering Graham H, Na Chan H, Nirujogi Raja S, Bachman Julia L, Pandey A and Huganir Richard L, Identification of long-lived synaptic proteins by proteomic analysis of synaptosome protein turnover, Proc. Natl. Acad. Sci. U. S. A, 2018, 115(16), E3827–E3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hark TJ, Rao NR, Castillon C, Basta T, Smukowski S, Bao H, Upadhyay A, Bomba-Warczak E, Nomura T, O’Toole ET, Morgan GP, Ali L, Saito T, Guillermier C, Saido TC, Steinhauser ML, Stowell MHB, Chapman ER, Contractor A and Savas JN, Pulse-chase proteomics of the app knockin mouse models of Alzheimer’s disease reveals that synaptic dysfunction originates in presynaptic terminals, Cell Syst., 2021, 12(2), 141–158.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Becker GW, Stable isotopic labeling of proteins for quantitative proteomic applications, Briefings Funct. Genomics, 2008, 7(5), 371–382. [DOI] [PubMed] [Google Scholar]
- 71.Aggarwal S, Talukdar NC and Yadav AK, Advances in higher order multiplexing techniques in proteomics, J. Proteome Res, 2019, 18(6), 2360–2369. [DOI] [PubMed] [Google Scholar]
- 72.Dephoure N and Gygi SP, Hyperplexing: A method for higher-order multiplexed quantitative proteomics provides a map of the dynamic response to rapamycin in yeast, Sci. Signaling, 2012, 5(217), rs2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zecha J, Satpathy S, Kanashova T, Avanessian SC, Kane MH, Clauser KR, Mertins P, Carr SA and Kuster A, TMT labeling for the masses: A robust and cost-efficient, in-solution labeling approach, Mol. Cell. Proteomics, 2019, 18(7), 1468–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jayapal KP, Sui S, Philp RJ, Kok Y-J, Yap MGS, Griffin TJ and Hu W-S, Multitagging proteomic strategy to estimate protein turnover rates in dynamic systems, J. Proteome Res, 2010, 9(5), 2087–2097. [DOI] [PubMed] [Google Scholar]
- 75.Kumar A, Jamwal S, Midha MK, Hamza B, Aggarwal S, Yadav AK and Rao KVS, Dataset generated using hyperplexing and click chemistry to monitor temporal dynamics of newly synthesized macrophage secretome post infection by mycobacterial strains, Data Brief, 2016, 9, 349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dieterich DC, Link AJ, Graumann J, Tirrell DA and Schuman D, Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT), Proc. Natl. Acad. Sci. U. S. A, 2006, 103(25), 9482–9487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang G, Bowling H, Hom N, Kirshenbaum K, Klann E, Chao MV and Neubert TA, In-depth quantitative proteomic analysis of de novo protein synthesis induced by brain-derived neurotrophic factor, J. Proteome Res, 2014, 13(12), 5707–5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rothenberg DA, Taliaferro JM, Huber SM, Begley TJ, Dedon PC, White FM and Proteomics A, Approach to profiling the temporal translational response to stress and growth, iScience, 2018, 9, 367–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Klann K and Münch C, Unbiased translation proteomics upon cell stress, Mol. Cell. Oncol, 2020, 7(4), 1763150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wittig I and Malacarne PF, Complexome profiling: Assembly and remodeling of protein complexes, Int. J. Mol. Sci, 2021, 22, 7809–7820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zecha J, Gabriel W, Spallek R, Chang Y-C, Mergner J, Wilhelm M, Bassermann F and Kuster B, Linking post-translational modifications and protein turnover by site-resolved protein turnover profiling, Nat. Commun, 2022, 13(1), 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Eichelbaum K, Winter M, Diaz MB, Herzig S and Krijgsveld J. J. N. b, Selective enrichment of newly synthesized proteins for quantitative secretome analysis, Nat. Biotechnol, 2012, 30(10), 984–990. [DOI] [PubMed] [Google Scholar]
- 83.Lau H-T, Suh HW, Golkowski M and Ong S-E, Comparing SILAC- and stable isotope dimethyl-labeling approaches for quantitative proteomics, J. Proteome Res, 2014, 13(9), 4164–4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Savitski MM, Mathieson T, Zinn N, Sweetman G, Doce A, Becher I, Pachl F, Kuster B and Bantscheff M, Measuring and managing ratio compression for accurate iTRAQ/TMT quantification, J. Proteome Res, 2013, 12(8), 3586–3598. [DOI] [PubMed] [Google Scholar]
- 85.Salovska B, Li W, Di Y and Liu Y, BoxCarmax: A high-selectivity data-independent acquisition mass spectrometry method for the analysis of protein turnover and complex samples, Anal. Chem, 2021, 93(6), 3103–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Minogue CE, Hebert AS, Rensvold JW, Westphall MS, Pagliarini DJ and Coon JJ, Multiplexed Quantification for Data-Independent Acquisition, Anal. Chem, 2015, 87(5), 2570–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Di Y, Zhang Y, Zhang L, Tao T and Lu H, MdFDIA: A mass defect based four-plex data-independent acquisition strategy for proteome quantification, Anal. Chem, 2017, 89(19), 10248–10255. [DOI] [PubMed] [Google Scholar]
- 88.Zhong X, Frost DC, Yu Q, Li M, Gu T and Li L, Mass defect-based DiLeu tagging for multiplexed data-independent acquisition, Anal. Chem, 2020, 92(16), 11119–11126. [DOI] [PMC free article] [PubMed] [Google Scholar]