

Abstract
Coffin-Siris syndrome (CSS) is an autosomal dominant neurodevelopmental syndrome that can present with a variety of structural birth defects. Pathogenic variants in 12 genes have been shown to cause CSS. Most of these genes encode proteins that are a part of the mammalian switch/sucrose non-fermentable (mSWI/SNF; BAF) complex. An association between genes that cause CSS and congenital diaphragmatic hernia (CDH) has been suggested based on case reports and the analysis of CSS and CDH cohorts. Here, we describe an unpublished individual with CSS and CDH, and we report additional clinical information on four published cases. Data from these individuals, and a review of the literature, provide evidence that deleterious variants in ARID1B, ARID1A, SMARCB1, SMARCA4, SMARCE1, ARID2, DPF2, and SMARCC2, which are associated with CSS types 1-8, respectively, are associated with the development of CDH. This suggests that additional genetic testing to identify a separate cause of CDH in an individual with CSS may be unwarranted, and that comprehensive genetic testing for individuals with non-isolated CDH should include an evaluation of CSS-related genes. These data also suggest that the mSWI/SNF (BAF) complex may play an important role in diaphragm development.
Keywords: congenital diaphragmatic hernia, CDH, Coffin-Siris syndrome, exome sequencing
INTRODUCTION
Coffin-Siris syndrome (CSS) is an autosomal dominant neurodevelopmental syndrome that was first described over 50 years ago (Coffin & Siris, 1970). Features of CSS include intellectual disability and developmental delay, hypotonia, distinct dysmorphic features that include coarse features and ectodermal abnormalities (hirsutism, sparse scalp hair, and abnormal dentition), sensorineural hearing loss, vision abnormalities, gastrointestinal anomalies, and cardiac defects (Mannino, Miyawaki, Santen, & Schrier Vergano, 2018; Vasko, Drivas, & Schrier Vergano, 2021). This syndrome is sometimes termed “fifth digit syndrome”, due to a short fifth digit and underdeveloped nails, seen in about 65% and 76% of patients, respectively, in one cohort (Mannino et al., 2018) and in 88% and 97% in another (Kosho et al., 2014).
There are currently 12 different types of CSS, each associated with a different gene. By order, these are ARID1B (MIM# 135900), ARID1A (MIM# 614607), SMARCB1 (MIM# 614608), SMARCA4 (MIM# 614609), SMARCE1 (MIM# 616938), ARID2 (MIM# 617808), DPF2 (MIM# 618027), SMARCC2 (MIM# 618362), SOX11 (MIM# 615866), SOX4 (618506), SMARCD1 (MIM# 618779) and BICRA (MIM# 619325). With the exception of SOX11 and SOX4, these genes encode proteins related to the mammalian switch/sucrose non-fermentable (mSWI/SNF; BAF) complex (Bögershausen & Wollnik, 2018). This complex has a role in chromatin remodeling and is known to mediate cell differentiation and lineage specification (Mittal & Roberts, 2020; Pagliaroli et al., 2021).
Congenital diaphragmatic hernia (CDH) is a severe, life-threatening malformation (McGivern et al., 2015), usually associated with pulmonary hypoplasia. Its prevalence has been estimated between 2.3 to 15.9 in 10,000 births (Munim et al., 2013; Paoletti et al., 2020). CDH can be isolated or appear as part of a broader syndrome. Although it is not considered a common feature of CSS, an association between CSS and CDH has been suggested based on case reports and the analysis of CSS and CDH cohorts (Bartin et al., 2018; Delvaux et al., 1998; Knapp et al., 2019; Kosho et al., 2014; Mannino et al., 2018; Scott et al., 2022; Shang et al., 2015; Slavotinek et al., 2022; Sweeney et al., 2018; Tsurusaki et al., 2012; van der Sluijs et al., 2022; Vasko et al., 2021).
In this paper, we present a previously unpublished individual with CSS and CDH and provide additional clinical information on four previously reported cases. We also summarize all previously reported cases, to provide a comprehensive review of the association between these two disorders.
MATERIALS AND METHODS
Editorial Policies and Ethical Considerations
Subjects were enrolled in research studies in accordance with protocols approved by local institutional review boards, or anonymized data is being reported as allowed by a research protocol approved by the institutional review board of Baylor College of Medicine (H-47546). The procedures followed were in accordance with the ethical standards of Baylor College of Medicine’s committee on human research and were in keeping with international standards.
Molecular Testing
Trio exome sequencing for Subject 1 was performed on a clinical basis at Baylor Genetics (Meng et al., 2017).
In silico variant analyses
In silico analyses of sequence variants were performed using MutationTaster (http://www.mutationtaster.org/), and Combined Annotation Dependent Depletion (CADD; https://cadd.gs.washington.edu/) (Rentzsch et al., 2021; Schwarz et al., 2014). All variant descriptions were checked using Mutalyzer (https://mutalyzer.nl/).
CLINICAL REPORTS
Subject 1
Subject 1 is a 20-month-old female and the only child of her parents, after a single miscarriage. At 16 weeks of gestational age, an ultrasound exam revealed a left-sided CDH. She also had an atrial septal defect. She was born at 36 weeks and 5 days of gestational age and had a birth weight of 2.18 kg (8th centile). Due to respiratory difficulties, she was placed on extracorporeal membrane oxygenation (ECMO). At the age of 20 days, 3 days after coming off ECMO, her CDH was repaired. During surgery, a rim of diaphragm was observed from the anterior clavicular to the posterior clavicular line with some primary closure medially. However, the near absence of the left-hemidiaphragm required placement of a 6 mm Gore-Tex patch. No sequestration or hernia sac was appreciated. Later in life, she underwent a second hernia repair, due to re-herniation. A tracheostomy was placed at 5 months of age due to a chronic need for ventilator support and unsuccessful extubation attempts. At 1 year of age, she had cardiac arrest, requiring 3 minutes of successful cardiopulmonary resuscitation.
Over time, she was diagnosed with developmental delay. She was able to hold her head up at 10 months, was able to sit at 14 months, and rolled over at 18 months. At 20 months she was able to stand with support but could not walk. She said her first words at 14 months, and she only used 2 to 3 words at 20 months. A brain MRI without contrast performed at 15 months showed mild-to-moderate cerebral volume loss with prominent ventricles, borderline caliber of a dysplastic corpus callosum, signs of a chronic subdural hematoma, a pars intermedia/Rathke cleft cyst in the sella turcica, numerous tortuous small vessels in the peripheral CSF space, and diffuse calvarial and skull base thickening.
Her physical examination at 20 months of age showed dysmorphic features which included sparse hair, bitemporal narrowing, coarse facies, broad eyebrows, ptosis, a flat nasal bridge, a broad nose, anteverted nares, a thin upper vermilion, and a thick lower vermilion (Figure 1A–C). An umbilical hernia was also observed. Her fifth fingers were mildly short, but she had normal nails. Her height was 77.5cm (3rd centile), weight was 11 kg (56th centile) and head circumference at 18 months was 49.5cm (98th centile).
Figure 1. Photographs of Subject 1 and her father who carry a pathogenic, frameshift variant in ARID1A.
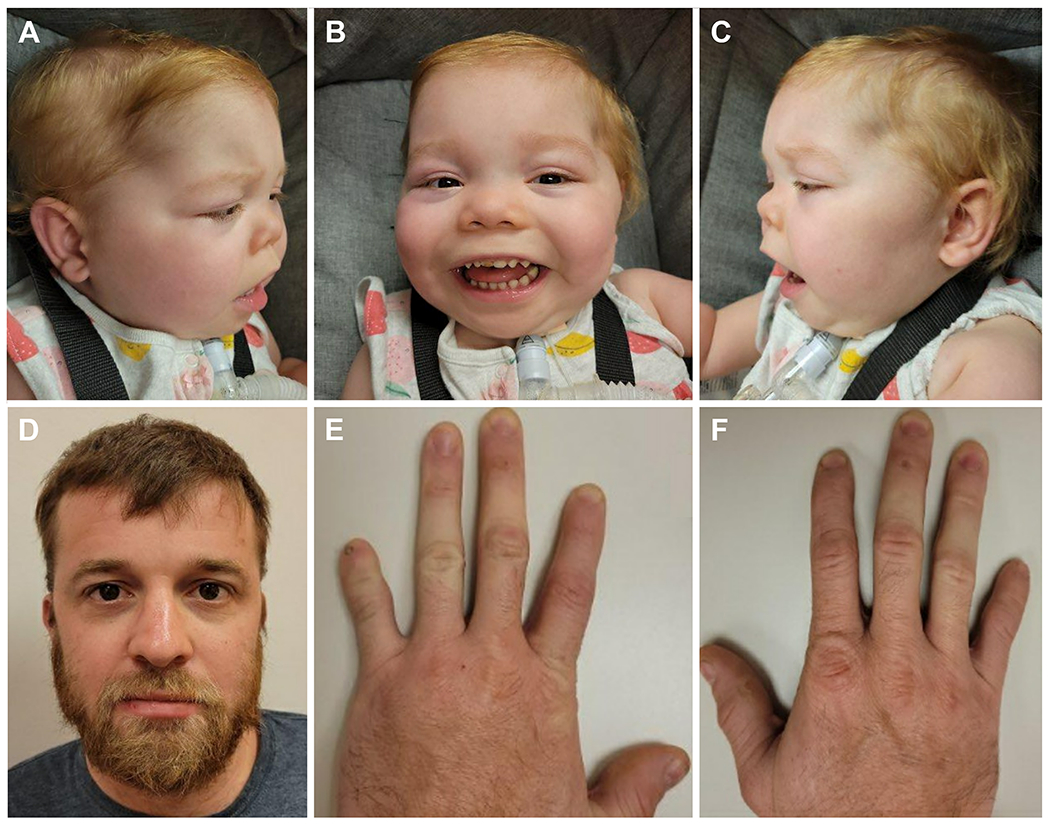
A-C) Subject 1 was born with an atrial septal defect and a large, left-sided CDH that was repaired with a Gore-Tex patch. A tracheostomy was placed due to a chronic need for ventilator support. She has developmental delay, sparse hair, bitemporal narrowing, coarse facies, broad eyebrows, ptosis, a flat nasal bridge, a broad nose, anteverted nares, a thin upper vermilion, and a thick lower vermilion. D-F) Subjects 1’s father was born with a ventricular septal defect that spontaneously resolved. His speech development was slow, and he required additional help throughout his school years. He has fifth finger hypoplasia bilaterally and hypoplasia of the first and second fingers on the left.
Subject 1’s father was 23 years old at the birth of his daughter. He had a history of a ventricular septal defect (VSD) that resolved without intervention by 12 years of age. While no delays in motor skills were noticed, his speech development was described as slower than expected. He was diagnosed with a learning disability in the 2nd grade and required additional help throughout his school years. He had urinary retention until 12 years of age and a history of enuresis. He did not pursue higher education and currently works in a foundry. He has a hearing impairment, thought to be caused by exposure to excessive noise generated by heavy machinery. His physical examination was positive for hypoplasia of the fifth fingers bilaterally and hypoplastic first and second fingers on the left (Figure 1E–F).
A chromosomal microarray analysis and rapid fluorescence in situ hybridization testing (FISH) for trisomy 13, 18 and 21 were performed for Subject 1 and were negative. Trio exome sequencing was performed, and revealed a paternally inherited pathogenic c.98_107del, p.(Glu33Glyfs*15) [NM_006015.6] frameshift variant in ARID1A. This variant has not been previously reported and is not found in the gnomAD v2.1.1 dataset (https://gnomad.broadinstitute.org/) or in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/).
A paternally inherited c.1880A>G, p.(Lys627Arg) [NM_016604.4] missense variant of unknown significance (ACMG criteria PM2 and PP3) was also identified in KDM3B. Autosomal dominant pathogenic variants in KDM3B are associated with Diets-Jongmans syndrome (DIJOS; MIM# 618846). Individuals with this syndrome have pointed chins, long ears, and wide mouths, findings that were not seen in Subject 1 or in her father. However, short stature, a broad nasal tip and ptosis are also seen in DIJOS and were seen in Subject 1 but not in her father. The variant appears once in gnomAD (allele frequency of 0.00000401), has a CADD score of 25.7 and classified as “disease causing” by MutationTaster. It is possible that this variant contributed to the CDH in Subject 1 since CDH is sometimes seen in individuals with DIJOS (Diets et al., 2019).
Subject 2
Subject 2 is a 3-year-old Caucasian male, who was previously reported by Scott et al. without detailed clinical information (Scott et al., 2022). Pregnancy was complicated by polyhydramnios requiring amnioreduction at 34 weeks of gestation. He was born with a left-sided CDH with gastrointestinal malrotation and underwent surgical repair at 9 weeks age. He also had partial corpus callosum agenesis, a hard palate cleft, hypospadias, a non-palpable left testis, a right-sided hydrocele, and a slightly low-lying conus identified on spinal ultrasound. He was later diagnosed with hypermetropia and conductive hearing loss due to small auditory canals. Developmental delay was noticed with sitting documented at 17 months of age. He has a history of seizures and is currently fed through a gastrostomy tube.
At 6 months of age, his head circumference was 42 cm (11th centile). At 13 months, he was 77 cm tall (50th centile) and weighed 9.03 kg (16th centile). His physical examination was positive for hypotonia, but no hirsutism or sparse scalp hair was seen. Fifth digit anomalies were not documented.
Trio exome sequencing revealed a de novo, pathogenic c.2936G>A, p.(Arg979Gln) [NM_001128849.3] variant in SMARCA4. The CSS registry (Mannino et al., 2018) contains three other individuals with the same genetic variant. However, these individuals do not have CDH.
Subject 3
Subject 3 is a 6-year-old female, who was previously reported by Wild et al. without detailed clinical information (Wild et al., 2022). She was prenatally diagnosed with a left-sided CDH containing virtually the entire left lobe of the liver, stomach, spleen and bowel. The hernia was repaired at 3 weeks of life. Around 1 year of age, she was noted to have prolonged sinopulmonary and other infections. Testing revealed low IgG levels, and she was ultimately diagnosed with specific antibody deficiency. At 5 years of age, she was diagnosed with very-early-onset inflammatory bowel disease (VEO-IBD). Her parents also reported some sleeping difficulties. A brain MRI performed at 2 months of age showed no structural abnormalities. A second MRI performed at 3 years of age revealed a short corpus callosum posteriorly, prominent ventricles, and thin optic nerves.
A prenatal chromosomal microarray showed a maternally-inherited 1.3 Mb duplication-arr[hg19] 16p13.11(15,058,820-16,328,840)x3, including 21 genes that was classified as a variant of unknown significance. Subsequently, clinically-based exome sequencing showed a de novo, likely pathogenic c.3595G>A, p.(Val1199Met) [NM_001128849.3] variant in SMARCA4.
Subject 4
Subject 4 is a 6-year-old female, who was previously reported by Mannino et al. without clinical data (Mannino et al., 2018). Prenatal testing revealed an atrioventricular canal defect. She was born small for her gestational age at 37 weeks. Her physical examination revealed dysmorphic features and hypoplasia of fifth digits/nails. She was later diagnosed with a Morgagni-type CDH, pyloric stenosis, and sensorineural hearing loss. She has developmental delay and intellectual disability with documentation of sitting at 11 months of age. Genetic testing revealed a de novo, likely pathogenic c.218A>G, p.(Tyr73Cys) [NM_003079.5] missense variant in SMARCE1.
Subject 5
Subject 5 was a female who died in the perinatal period. She was previously reported by Scott et al. without detailed clinical information (Scott et al., 2022). She had a left-sided CDH with nearly all the bowel and a large part of the liver within the left chest. In addition, she had a double-outlet right ventricle with D-malposed great arteries, a large VSD, pulmonary valve hypoplasia, peripheral cystic dysplasia of the right kidney, a two-vessel cord, and lumbosacral segmentation anomalies. Dysmorphic features included low-set, mildly posteriorly rotated ears, mildly down-slanting palpebral fissures, a saddle nose, and mild micrognathia. Her family history was notable for a mother with unilateral renal agenesis. Duo exome sequencing revealed a c.1651-2A>G, p.(?) variant of unknown significance in SMARCC2 that was not maternally inherited. A presumed diagnosis of CSS was made by the treating geneticist who believed the SMARCC2 variant explained the patient’s phenotypes. However, given the renal agenesis in Subject 5’s mother, it is possible that unidentified, maternally-inherited genetic factors may have also contributed to these phenotypes.
DISCUSSION
CSS is a neurodevelopmental syndrome associated with several congenital anomalies and a specific finding of 5th finger hypoplasia. CDH is not considered a common finding of CSS based on reviews of large cohorts of individuals with this disorder (Mannino et al., 2018; Vasko et al., 2021), with only two of the 13 patients reviewed here (Subjects 2 and 4) appearing in the Cofin-Siris Registry maintained by Cofin-Siris Syndrome Foundation (https://www.coffinsiris.org/). However, an association between CSS and CDH has been suggested based on individual case reports and reviews of CSS and CDH cohorts (Bartin et al., 2018; Delvaux et al., 1998; Knapp et al., 2019; Kosho et al., 2014; Mannino et al., 2018; Scott et al., 2022; Shang et al., 2015; Slavotinek et al., 2022; Sweeney et al., 2018; Tsurusaki et al., 2012; Vasko et al., 2021).
Here, we describe one previously unreported individual with CSS (Subject 1), and four individuals (Subject 2–5) that were published without complete clinical descriptions, who have CDH. Including these subjects, 13 individuals with molecularly confirmed CSS and CDH have now been reported (Table 1). While Subject 1’s KDM3B variant may have also contributed to her CDH and some of the physical features seen in her and her father, we believe there is sufficient evidence to suggest that an association between ARID1A and CDH.
Table 1.
Molecular and clinical data from individuals with CSS and CHD reported here and in the literature.
| Reference; Subject | CSS Type # | Gene | cDNA Change | Protein Change | Inheritance | ACMG Variant Call and Criteria; MutationTaster Call; CADD Score | CDH Type |
|---|---|---|---|---|---|---|---|
| (Delvaux et al., 1998) | ? | ND | ND | ND | ND | N/A | ND |
| (Sweeney et al., 2018) | 1 | ARID1B | c.3096_3100delCAAAG [NM_020732.3] | p.(Lys1033Argfs*32) | De Novo | Likely Pathogenic; PVS1, PS2, PM2; Disease causing; N/A | Left-sided |
| (Bartin, Corizzi, Melle, & Mechler, 2018) | 2 | ARID1A | ND | ND | De Novo | N/A | Left-sided |
| (Slavotinek et al., 2021) | 2 | ARID1A | Deletion of exon 1; (chr1: 26,797,508-27,052,080; hg19) | p.? | ND | VUS; BP2; N/A; N/A | Right-sided |
| Subject 1 | 2 | ARID1A | c.98_107del [NM_006015.6] | p.(Glu33Glyfs*15) | Paternal (affected) | Pathogenic; PVS1, PM2, PP4; Disease causing; N/A | Left-sided |
| (Tsurusaki et al., 2012) | 3 | SMARCB1 | ND | ND | ND | N/A | ND |
| (Kosho et al., 2014); Patient Y11 | 3 | SMARCB1 | c.1130G>A [NM_003073.5} | p.(Arg377His) | ND | VUS; PM2, PP3: Disease causing; 27 | ND |
| Subject 2; (Scott et al., 2021) Patient 31 | 4 | SMARCA4 | c.2936G>A [NM_001128849.3] | p.(Arg979Gln) | De Novo | Pathogenic; PS2,PS4,PM2,PP3; Disease causing; 32 | Left-sided |
| Subject 3; (Wild et al., 2022) | 4 | SMARCA4 | c.3595G>A NM_001128849.3] | p.(Val1199Met) | De Novo | Likely Pathogenic; PS2, PM2, PP3; Disease causing; 32 | Left-sided |
| (Wild et al., 2022) | 4 | SMARCA4 | c.3728G>A; [NM_001128844.3] | p.(Arg1243Gln) | De Novo | Likely Pathogenic; PS2, PM2, PP3; Disease causing; 29.7 | Left-sided |
| Subject 4; (Mannino et al., 2018) | 5 | SMARCE1 | c.218A>G [NM_003079.5] | p.(Tyr73Cys) | De Novo | Likely Pathogenic; PS2, PM2, PP3; Disease causing; 29.7 | Morgagni |
| (Shang et al., 2015) | 6 | ARID2 | c.1028T>A [NM_152641.4] | p.(Leu343*) | ND | Likely Pathogenic; PVS1, PM2; Disease causing; 35 | ND |
| (Knapp et al., 2019) | 7 | DPF2 | c.1066T>G [NM_006268.4] | p.(Cys356Gly) | De Novo | Likely Pathogenic; PS2, PM2, PP3; Disease causing; 28 | Right-sided |
| Subject 5; (Scott et al., 2021) Patient 32 | 8 | SMARCC2 | c.1651-2A>G [NM_003075.3] | p.? | Not Maternal | VUS; PVS1, PM2; N/A, 33 | Left-sided |
N/A = not applicable, ND = not described, VUS = variant of unknown significance
Currently, CSS is divided into 12 types based on their associated genes. CDH has now been reported in association with genes that cause CSS types 1-8: ARID1B, ARID1A, SMARCB1, SMARCA4, SMARCE1, ARID2, DPF2, SMARCC2. All of these genes encode proteins that are part of the mSWI/SNF (BAF) complex that plays a role in chromatin remodeling and is known to mediate cell differentiation and lineage specification (Mittal & Roberts, 2020). As expected, the mouse homologs of these genes, and the mouse homologs of the genes associated with CSS types 9-12, are expressed in the developing mouse diaphragm at embryonic day (E)11.5, E12.5 and E16.5 based on the whole-transcriptome expression profiles reported by Russell et al (Russell et al., 2012).
Although no individuals with CSS type 10, which is caused by pathogenic variants in SOX4, have been reported to have CDH, a subset of mice that are homozygous for a Sox4 hypomorphic allele develop CDH (Foronda et al., 2014). CDH has not been described in association with transgenic mouse models involving the homologs of genes associated with CSS types 1-8. We note, however, that dedicated studies may be needed to identify low penetrance CDH in mouse models (Beck et al., 2013; Jordan et al., 2018). Mouse models involving the homologs of genes associated with CSS types 9, 11 and 12 have not been generated. Hence, it is possible that future studies in mice may provide additional evidence in support of an association between CSS genes and CDH.
Based on these data, we conclude that CDH is a low penetrance phenotype associated with CSS, especially types 1-8 which are caused by variants in ARID1B, ARID1A, SMARCB1, SMARCA4, SMARCE1, ARID2, DPF2, SMARCC2 (Table 1). Hence, in individuals with a molecular diagnosis of CSS, additional testing to identify a separate cause of CDH may not be warranted. These data also suggest that sequencing and deletion/duplication analysis of CSS-related genes should be included in the comprehensive genetic evaluation of individuals with non-isolated CDH. The association between CSS genes and CDH also suggests the mSWI/SNF (BAF) complex may play an important role in the development of the diaphragm.
ACKNOWLEDGEMENTS
The authors thank family members for participating in this research study. This work was supported by National Institutes of Health/Eunice Kennedy Shriver National Institute of Child Health and Human Development grant R01HD098458 to D.A.S.
Footnotes
CONFLICTS OF INTEREST
The Department of Molecular & Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing completed at Baylor Genetics.
DATA AVAILABILITY
The ARID1A variant seen in Subject 1 and her father has been submitted to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/).
REFERENCES
- Bartin R, Corizzi F, Melle L, Mechler C, Gavard L, Boutaud de la Combe l., … Picone O (2018). OP23.09: Prenatal diagnosis of Coffin Siris syndrome. Ultrasound in Obstetrics & Gynecology, 52, 136–137. doi: 10.1002/uog.19612 [DOI] [Google Scholar]
- Beck TF, Veenma D, Shchelochkov OA, Yu Z, Kim BJ, Zaveri HP, … Scott DA (2013). Deficiency of FRAS1-related extracellular matrix 1 (FREM1) causes congenital diaphragmatic hernia in humans and mice. Human Molecular Genetics, 22(5), 1026–1038. doi: 10.1093/hmg/dds507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bögershausen N, & Wollnik B (2018). Mutational Landscapes and Phenotypic Spectrum of SWI/SNF-Related Intellectual Disability Disorders. Frontiers in Molecular Neuroscience, 11, 252. doi: 10.3389/fnmol.2018.00252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffin GS, & Siris E (1970). Mental retardation with absent fifth fingernail and terminal phalanx. American journal of diseases of children (1960), 119(5), 433–439. doi: 10.1001/archpedi.1970.02100050435009 [DOI] [PubMed] [Google Scholar]
- Delvaux V, Moerman P, & Fryns JP (1998). Diaphragmatic hernia in the Coffin-Siris syndrome. Genetic counseling (Geneva, Switzerland), 9(1), 45–50. [PubMed] [Google Scholar]
- Diets IJ, van der Donk R, Baltrunaite K, Waanders E, Reijnders MRF, Dingemans AJM, … Jongmans MCJ (2019). De novo and inherited pathogenic variants in KDM3B cause intellectual disability, short stature, and facial dysmorphism. American Journal of Human Genetics, 104(4), 758–766. doi: 10.1016/j.ajhg.2019.02.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foronda M, Martínez P, Schoeftner S, Gómez-López G, Schneider R, Flores JM, … Blasco MA (2014). Sox4 links tumor suppression to accelerated aging in mice by modulating stem cell activation. Cell reports, 8(2), 487–500. doi: 10.1016/j.celrep.2014.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan VK, Beck TF, Hernandez-Garcia A, Kundert PN, Kim B-J, Jhangiani SN, … Scott DA (2018). The role of FREM2 and FRAS1 in the development of congenital diaphragmatic hernia. Human Molecular Genetics, 27(12), 2064–2075. doi: 10.1093/hmg/ddy110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp KM, Poke G, Jenkins D, Truter W, & Bicknell LS (2019). Expanding the phenotypic spectrum associated with DPF2: A new case report. American Journal of Medical Genetics. Part A, 179(8), 1637–1641. doi: 10.1002/ajmg.a.61262 [DOI] [PubMed] [Google Scholar]
- Kosho T, Okamoto N, & Coffin-Siris Syndrome International Collaborators. (2014). Genotype-phenotype correlation of Coffin-Siris syndrome caused by mutations in SMARCB1, SMARCA4, SMARCE1, and ARID1A. American Journal of Medical Genetics. Part C, Seminars in Medical Genetics, 166C(3), 262–275. doi: 10.1002/ajmg.c.31407 [DOI] [PubMed] [Google Scholar]
- Mannino EA, Miyawaki H, Santen G, & Schrier Vergano SA (2018). First data from a parent-reported registry of 81 individuals with Coffin-Siris syndrome: Natural history and management recommendations. American Journal of Medical Genetics. Part A, 176(11), 2250–2258. doi: 10.1002/ajmg.a.40471 [DOI] [PubMed] [Google Scholar]
- McGivern MR, Best KE, Rankin J, Wellesley D, Greenlees R, Addor M-C, … Martos C (2015). Epidemiology of congenital diaphragmatic hernia in Europe: a register-based study. Archives of Disease in Childhood. Fetal and Neonatal Edition, 100(2), F137–44. doi: 10.1136/archdischild-2014-306174 [DOI] [PubMed] [Google Scholar]
- Mittal P, & Roberts CWM (2020). The SWI/SNF complex in cancer - biology, biomarkers and therapy. Nature Reviews. Clinical Oncology, 17(7), 435–448. doi: 10.1038/s41571-020-0357-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munim S, Maheen H, Zainab G, & Fatima S (2013). Fetal outcome of cases with diaphragmatic hernia. The Journal of Maternal-Fetal & Neonatal Medicine, 26(14), 1439–1442. doi: 10.3109/14767058.2013.783814 [DOI] [PubMed] [Google Scholar]
- Pagliaroli L, Porazzi P, Curtis AT, Scopa C, Mikkers HMM, Freund C, … Trizzino M (2021). Inability to switch from ARID1A-BAF to ARID1B-BAF impairs exit from pluripotency and commitment towards neural crest formation in ARID1B-related neurodevelopmental disorders. Nature Communications, 12(1), 6469. doi: 10.1038/s41467-021-26810-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti M, Raffler G, Gaffi MS, Antounians L, Lauriti G, & Zani A (2020). Prevalence and risk factors for congenital diaphragmatic hernia: A global view. Journal of Pediatric Surgery. doi: 10.1016/j.jpedsurg.2020.06.022 [DOI] [PubMed] [Google Scholar]
- Rentzsch P, Schubach M, Shendure J, & Kircher M (2021). CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Medicine, 13, 31. 10.1186/s13073-021-00835-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell MK, Longoni M, Wells J, Maalouf FI, Tracy AA, Loscertales, …Donahoe PK. (2012). Congenital diaphragmatic hernia candidate genes derived from embryonic transcriptomes. Proceeding of the National Academy of Science U S A 109:2978–83 doi: 10.1073/pnas.1121621109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Cooper DN, Schuelke M, & Seelow D (2014). MutationTaster2: mutation prediction for the deep-sequencing age. Nature Methods, 11, 361–362. 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- Scott TM, Campbell IM, Hernandez-Garcia A, Lalani SR, Liu P, Shaw CA, … Scott DA (2022). Clinical exome sequencing data reveal high diagnostic yields for congenital diaphragmatic hernia plus (CDH+) and new phenotypic expansions involving CDH. Journal of Medical Genetics, 59(3), 270–278. doi: 10.1136/jmedgenet-2020-107317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang L, Cho MT, Retterer K, Folk L, Humberson J, Rohena L, … Chung WK (2015). Mutations in ARID2 are associated with intellectual disabilities. Neurogenetics, 16(4), 307–314. doi: 10.1007/s10048-015-0454-0 [DOI] [PubMed] [Google Scholar]
- Slavotinek A, Lefebvre M, Brehin A-C, Thauvin C, Patrier S, Sparks TN, … Huang E (2022). Prenatal presentation of multiple anomalies associated with haploinsufficiency for ARID1A. European Journal of Medical Genetics, 65(2), 104407. doi: 10.1016/j.ejmg.2021.104407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney NM, Nahas SA, Chowdhury S, Campo MD, Jones MC, Dimmock DP, … RCIGM Investigators. (2018). The case for early use of rapid whole-genome sequencing in management of critically ill infants: late diagnosis of Coffin-Siris syndrome in an infant with left congenital diaphragmatic hernia, congenital heart disease, and recurrent infections. Molecular Case Studies, 4(3). doi: 10.1101/mcs.a002469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsurusaki Y, Okamoto N, Ohashi H, Kosho T, Imai Y, Hibi-Ko Y, … Matsumoto N (2012). Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nature Genetics, 44(4), 376–378. doi: 10.1038/ng.2219 [DOI] [PubMed] [Google Scholar]
- van der Sluijs PJ, Joosten M, Alby C, Attié-Bitach T, Gilmore K, Dubourg C, … Ahlers KP (2022). Discovering a new part of the phenotypic spectrum of Coffin-Siris syndrome in a fetal cohort. Genetics in Medicine. doi: 10.1016/j.gim.2022.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasko A, Drivas TG, & Schrier Vergano SA (2021). Genotype-Phenotype Correlations in 208 Individuals with Coffin-Siris Syndrome. Genes, 12(6). doi: 10.3390/genes12060937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild KT, Schindewolf E, Hedrick HL, Rintoul NE, Hartman T, Gebb J, … Krantz ID (2022). The genomics of congenital diaphragmatic hernia: A 10 year retrospective review. The Journal of Pediatrics. doi: 10.1016/j.jpeds.2022.04.012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The ARID1A variant seen in Subject 1 and her father has been submitted to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/).