

SUMMARY
Acute myeloid leukemia (AML) is a cancer of myeloid-lineage cells with limited therapeutic options. We previously combined ex vivo drug sensitivity with genomic, transcriptomic, and clinical annotations for a large cohort of AML patients, which facilitated discovery of functional genomic correlates. Here, we present a dataset, which has been harmonized with our initial report to yield a cumulative cohort of 805 patients (942 specimens). We show strong cross-cohort concordance and identify features of drug response. Further, deconvoluting transcriptomic data shows that drug sensitivity is governed broadly by AML cell differentiation state, sometimes conditionally impacting other correlates of response. Finally, modeling of clinical outcome reveals a single gene, PEAR1, to be among the strongest predictors of patient survival, especially for young patients. Collectively, this report expands a large functional genomic resource, offers avenues for mechanistic exploration and drug development, and reveals tools for predicting outcome in AML.
Keywords: hematologic malignancy, targeted therapy, LSC17, cell state, leukemia stem cell, monocyte, eigengene, JEDI, MEGF12
Graphical Abstract
eTOC
Bottomly et al. present a functional genomic resource comprised of molecular, clinical, and drug response data on acute myeloid leukemia patient specimens. Through integration of all of these data, they identify genetic and cell differentiation state features that predict drug response, and they utilize modeling to identify targetable determinants of clinical outcome.
INTRODUCTION
Acute myeloid leukemia (AML) is characterized by neoplastic proliferation of myeloid-lineage cells. Approximately 21,000 diagnoses and 10,000 AML-related deaths are reported annually in the United States (Jemal et al., 2010; SEER, 2021). Genetic features include 16 recurrent gene rearrangements and a plethora of unique, tumor-specific aberrations (Arber et al., 2016). In addition, ~60 genes exhibit recurrent point mutations with many thousand additional rarely mutated genes (Cancer Genome Atlas Research et al., 2013; Papaemmanuil et al., 2016; Tyner et al., 2018). Conventional chemotherapies combine anthracyclines with nucleoside analogs and a subsequent bone marrow transplant for ~20% of patients. This results in durable remission for only ~20% of patients (Pulte et al., 2016).
AML tumors also exhibit a diversity of cell phenotypes that can be aligned with differentiation states in healthy hematopoiesis. These phenotypes have been captured in classification schemes such as the French-American-British (FAB) system (Bennett et al., 1976) and from the World Health Organization (Arber et al., 2016). Gene expression signatures from the most primitive AML cell state, leukemic stem cells (LSC), have been shown to carry prognostic significance in AML and myelodysplastic syndromes (MDS) (Elsayed et al., 2020; Gal et al., 2006; Gentles et al., 2010; Horibata et al., 2019; Ng et al., 2016; Wang et al., 2020). Most recently, single-cell sequencing has been employed to describe gene expression signatures that define six distinct AML tumor maturation states (Van Galen et al., 2019).
Rationally targeted therapeutics for AML include all-trans retinoic acid combined with arsenic trioxide (Huang et al., 1988; Shen et al., 1997), small-molecules targeting mutated enzymes such as FLT3 (gilteritinib (Perl et al., 2019) and midostaurin (Stone et al., 2017)) or IDH1/2 (ivosidenib (DiNardo et al., 2018) and enasidenib (Stein et al., 2017)), an antibody drug conjugate targeting CD33 (gemtuzumab (Petersdorf et al., 2013)), a liposomal formulation of cytarabine and daunorubicin (CPX-351 (Lancet et al., 2018)), and the BCL2 inhibitor, venetoclax, used in combination with hypomethylating agents (azacitidine or decitabine) (DiNardo et al., 2019). Despite these tools, drug resistance and disease relapse remain a persistent problem necessitating a more complete understanding of the biological factors driving drug response.
RESULTS
To better understand the factors governing AML drug response and clinical outcome, we developed a comprehensive platform to combine clinical, cellular, and molecular features of disease. Our complete OHSU Beat AML cohort represents sample collection and characterization over a span of 10 years with integration of ex vivo drug sensitivity testing, curation of clinical annotations, and DNA- and RNA-sequencing (summarized in the Graphical Abstract). The data in Tyner et al (2018) represent the first tranche of patient sample data (denoted as “Waves 1+2”). Here we provide additional longitudinal samples for Waves 1+2, updated clinical information, as well as additional patient accrual, which represents the final two waves (“Waves 3+4”). Waves 3+4, collected over a 2.5-year period, is comprised of a total of 293 specimens from 279 patients (243 patients unique to Waves 3+4). We also provide the harmonization of these data sets together, for a cumulative cohort of 942 specimens from 805 patients, which reflects a real-world cohort of AML cases, inclusive of de novo, transformed, and therapy-related AML as well as cases at the point of initial diagnosis (70% of cases) and smaller numbers with relapsed or residual disease. A full listing of samples, available data, and clinical annotations are in Table S1. All somatic variant calls, gene expression counts, and drug response data can be explored and visualized through our interactive browser, Vizome (www.vizome.org/aml2). For all cohort-level analyses, only specimens from the first timepoint of each patient were used (defined in Table S1), with all remission samples excluded. A broad overview of clinical data showed comparable features between the two datasets with only a slightly lower percentage of cases with de novo AML in the first dataset (49% in Waves 1+2 vs. 58% in Waves 3+4). Frequencies of the most commonly mutated genes were also equivalent between cohorts (Figure 1A). Clinical outcomes, as measured by overall survival, of patients whose specimens were collected during the first cohort were indistinguishable from those collected during the second cohort (Figure 1B). The ex vivo drug sensitivity data were compared in de novo samples for each dataset, due to our prior observation that general drug sensitivity is reduced in secondary AML cases (Tyner et al., 2018), and showed high concordance (Figure 1C). We harmonized the RNA-Seq data across the waves (Figure S1A) by re-using the parameters and reference distribution of the conditional quantile normalization (CQN; (Hansen et al., 2012)) procedure applied previously. We highlight this with our prior weighted gene co-expression network analysis (WGCNA; (Zhang and Horvath, 2005)) that identified 13 gene expression modules across Waves 1+2 (Tyner et al., 2018). Part of the WGCNA methodology involves the summarization of each module by its first principal component (PC) score (termed an eigengene; (Horvath and Dong, 2008)). Using the parameters (e.g. means, standard deviations and rotations) learned from Waves 1+2, we predicted the PC scores of the Waves 3+4 samples for each module and revealed near complete overlap in every module with respect to range, distributions, and clustering (Figure S1B). Finally, our prior analysis included integration of ex vivo drug sensitivity with both mutational/cytogenetic status and gene expression to identify genomic or transcriptomic predictors of drug response. We observed strong concordance of associations between drug sensitivity and mutational status (89% concordance of Waves 1+2 and Waves 3+4), with nearly all associations showing a similar effect size in both the positive and negative direction (Figure 1D). In addition, we assessed correlation between gene expression (13 WGCNA module eigengenes) and drug response, revealing 98% concordance for these associations in Waves 1+2 vs. Waves 3+4 (Figure S1C).
Figure 1. Genomic features and outcomes for the Beat AML cohorts are concordant.
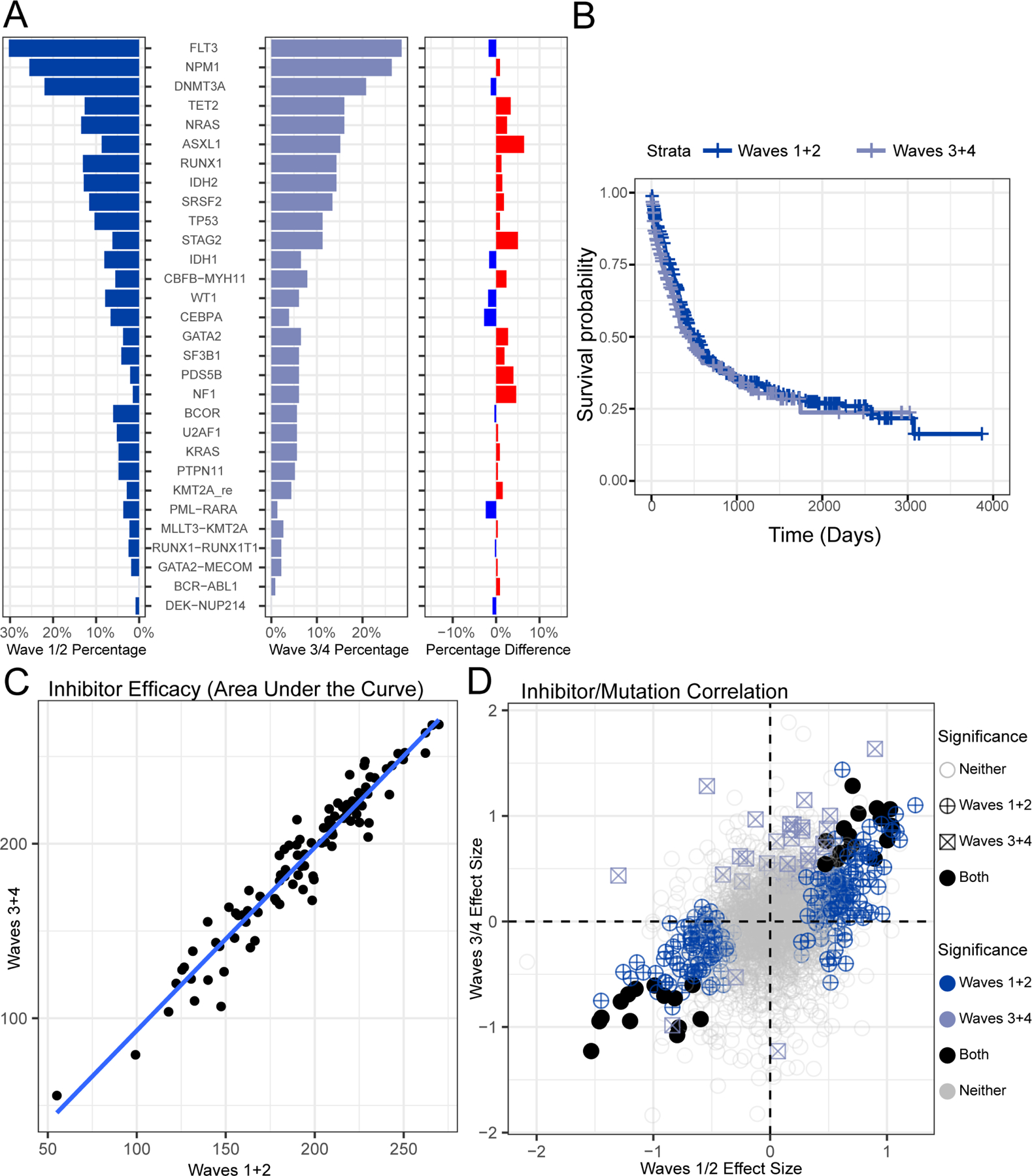
(A) The percentage of patients with a somatic mutation or gene fusion is shown for Waves 1+2 and Waves 3+4 cohorts and percentage difference is shown (average difference is 1.037%). (B) Overall survival is shown for Waves 1+2 and Waves 3+4 cohorts (p-value 0.2 log-rank test). (C). Ex vivo drug response values are shown comparing the average AUC for each drug (points) within de novo AML patients comparing Waves 1+2 to Waves 3+4 (Pearson’s correlation r=0.965). (D) Waves 3+4 can serve as validation cohort to assess prior mutation-inhibitor associations (Tyner et al., 2018). The difference in average response of specimens that are mutated versus wild type for a given gene (effect size) are plotted for Waves 1+2 on the x-axis and Waves 3+4 on the y-axis. Effect sizes were expressed as Glass’s delta with respect to the wild type group. Significance was based on the Welch’s t-test comparing mutated vs wild type and requiring a minimum of five mutations in Waves 1+2 and three mutations in Waves 3+4. Adjusted significance in Waves 1+2, Waves 3+4, or both are also annotated (qvalue < 0.05; (Storey and Tibshirani, 2003)).
Deconvoluting AML Cell Maturation State
We and others have observed that drug response patterns in AML are sometimes correlated with maturation state of AML tumor cells, with certain drugs exhibiting stronger efficacy against tumors with less differentiated cell state (e.g. BCL2i, CDK4/6i; (Kuusanmaki et al., 2020; Majumder et al., 2020; Pei et al., 2020; Romine et al., 2021; Zhang et al., 2020)) and others showing better efficacy against tumors with more differentiated state (e.g. BETi, MEKi; (Romine et al., 2021; White et al., 2021)). Analytical approaches can facilitate the deconvolution of deep sequencing data to infer proportions of distinct cell types (Avila Cobos et al., 2018), and single-cell sequencing technology has recently defined gene expression patterns for six distinct cell-types in AML (Hematopoietic Stem Cell (HSC)-like, Progenitor-like, Granulocyte-Monocyte Progenitor (GMP)-like, Promonocyte-like, Monocyte-like, conventional Dendritic Cell (cDC)-like); (Van Galen et al., 2019)). We summarized each of these six signatures by their PC1 scores (similar to the WGCNA eigengene). An example of the formation of the Monocyte-like score is shown in Figure S2A. Comparison of the HSC-like or Monocyte-like scores against FAB subtypes showed higher overall scores for HSC-like in the less differentiated M0-M2 samples and higher Monocyte-like scores in the myelomonocytic and monocytic M4-M5 groups (Figure S2B). To understand the relationship between our WGCNA modules and differences in cell-type, we compared eigengene scores of each signature. We found WGCNA modules that correlate strongly with the more differentiated cell-types (N=5), less differentiated cell-types (N=3), as well as those that exhibit correlation with early- and late-stage cell-types (N=5) (Figure 2A). Finally, we wanted to understand whether mutational or cytogenetic features showed enrichment for AML cell-type signatures. Indeed, we observed numerous significant correlations, including TP53, BCOR, and SF3B1 enriched for HSC-like state, NPM1 enriched for Promonocyte-like, RAS correlating with Monocyte-like, and numerous other associations. Interestingly, the associations were not always compartmentalized or linear, for instance RUNX1 showed strong correlation with both HSC-like (the most primitive) and cDC-like (the most differentiated), suggesting that RUNX1-mutant AML tumors may co-opt features of multiple, disparate cell maturations states (Figure 2B; for all associations see Table S2).
Figure 2. Differentiation scoring to characterize expression and mutational patterns.
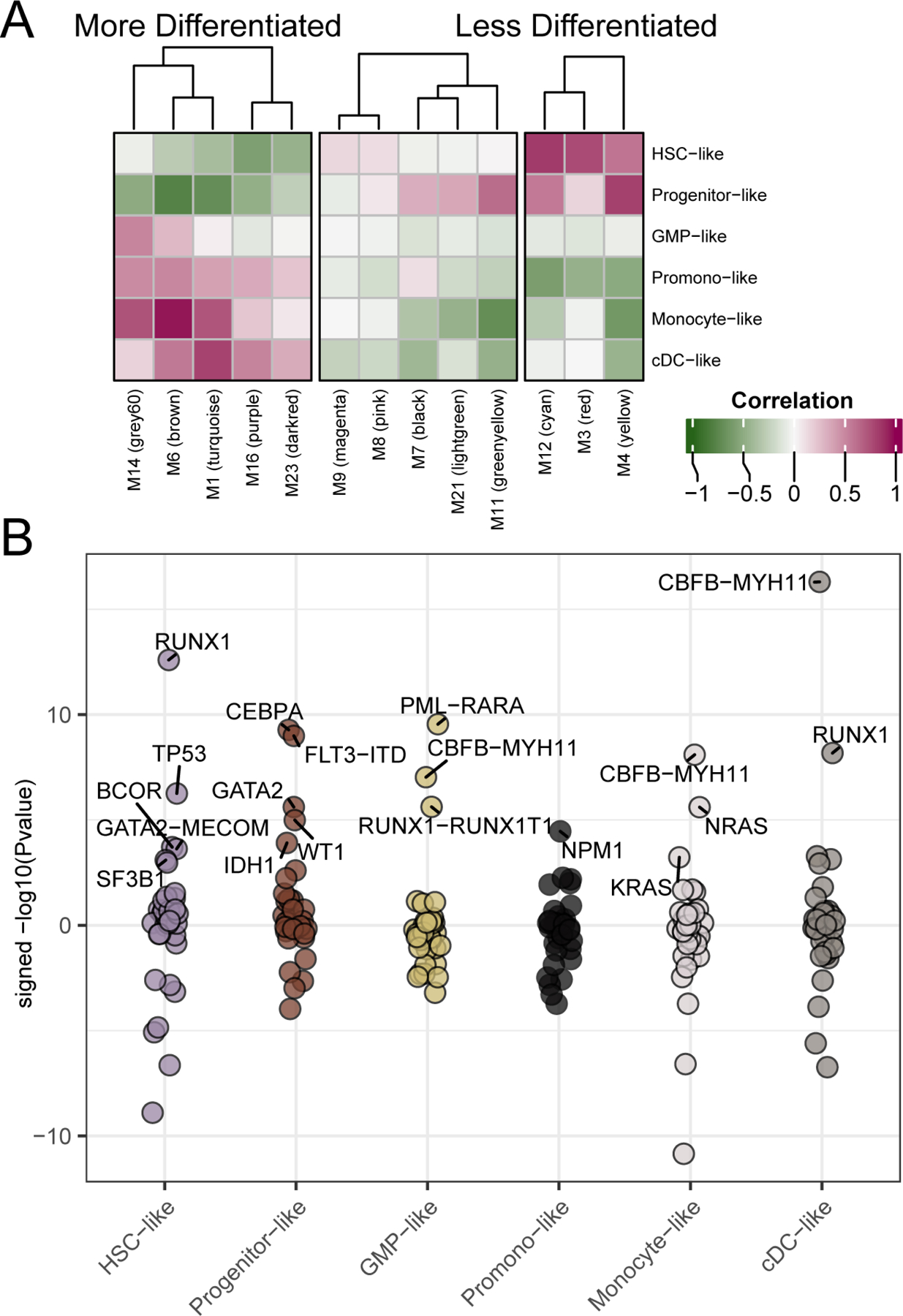
We generated malignant cell-type gene expression scores for each of our patients relative to 6 sets of 30 genes derived from expression signatures from single-cell sequencing (Van Galen et al., 2019). (A) By examining the correlation of expression module eigengenes and the cell-type scores we can see that WGCNA expression modules (X-axis) show correlation with differentiation cell-type scores (y-axis). Pearson’s correlation r-values are annotated. (B) Similarly, we can determine the mutational events significantly associated with each of the cell-type scores. Shown are the signed –log10(Welch’s t-test p-values) for the differences in cell-type score with respect to mutational status. Up to the top 5 most significant (qvalue < 0.05; (Storey and Tibshirani, 2003)) mutational events associated with enrichment of each cell-type score are highlighted. Full data are available in Table S2.
We next wanted to determine the breadth of drugs with response that is correlated with AML cell differentiation state and, accordingly, compared the six cell-type scores with drug response across our full panel. Overall, 73 drugs showed response that significantly correlated with at least one cell-type. Hierarchical clustering of these 73 drugs revealed three main groups. One cluster contained a series of inhibitors with greater activity against HSC- and Progenitor-like states and with less efficacy against the more differentiated cell-types. Another cluster displayed the inverse pattern with greatest activity against the cDC- and Monocyte-like states. The third cluster showed some, albeit muted, activity against both HSC-/Progenitor-like and cDC-/Monocyte-like states with the most resistance conferred by GMP- and Promonocyte-like states (Figure 3A; for all associations see Table S3). Finally, there were two drugs, venetoclax and the HDAC inhibitor, panobinostat, that were extreme outliers and exhibited inverse patterns of response (Figure S3A). We next wanted to understand whether any of the associations between drug response and cell-type score might be conditional on mutational or cytogenetic events. Numerous instances of significant interactions between cell-type score and mutational/cytogenetic events were identified, mostly involving the Monocyte-like score (Figure 3B). For example, sorafenib is a potent inhibitor of the FLT3 receptor tyrosine kinase and exhibits significantly greater efficacy in FLT3-ITD mutant samples. However, inclusion of cell-type scores reveals that sorafenib shows stronger efficacy in cases with both FLT3-ITD and a high Progenitor-like score, while the existence of a prominent Monocyte-like signal confers resistance of FLT3-ITD-positive AML cases to sorafenib (Figure S3B).
Figure 3. Influence of cell-type on inhibitor response.
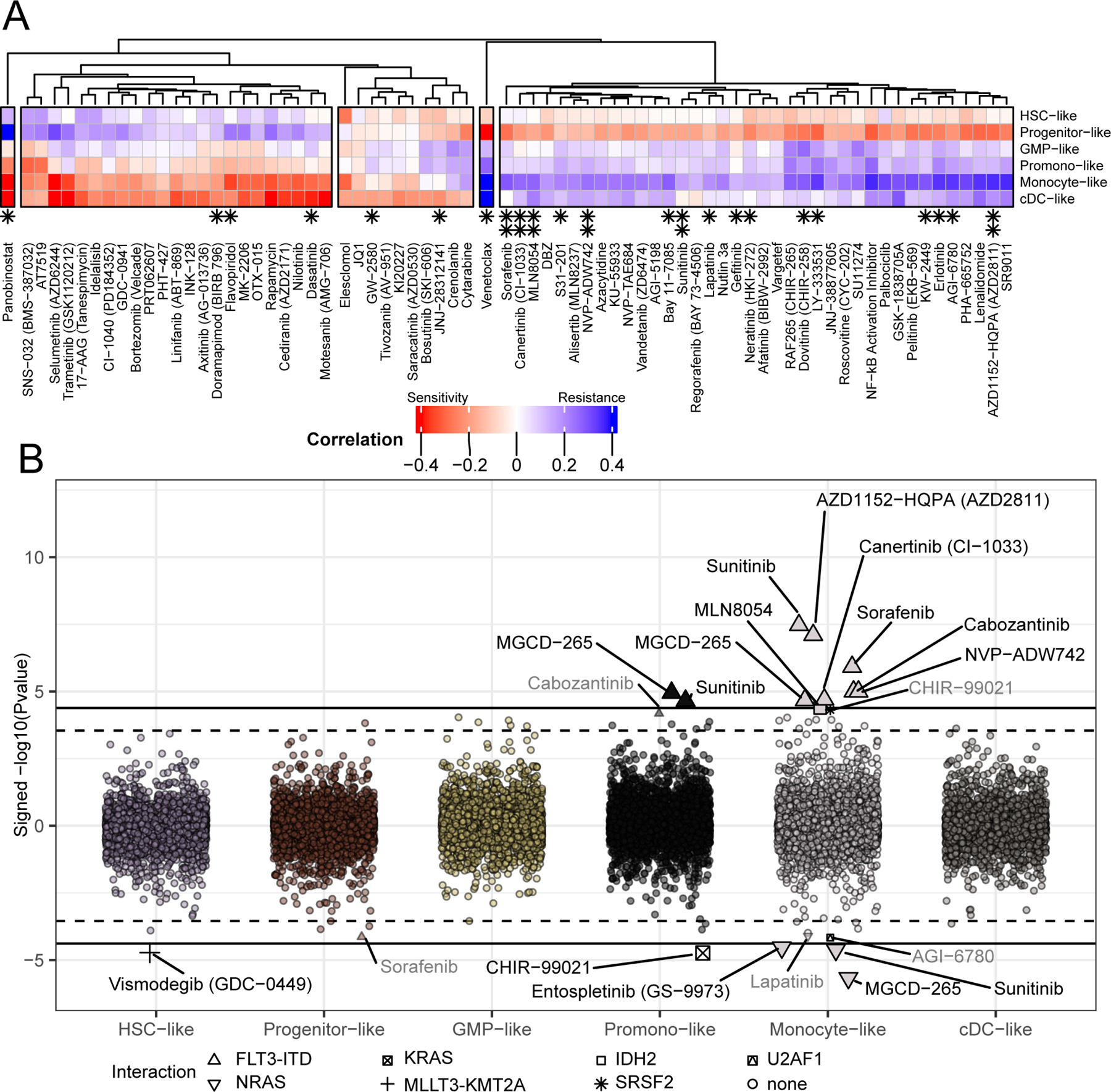
(A) We compared the cell-type gene expression scores, developed in Figure 2, with ex vivo drug response. Pearson’s correlation of ex vivo inhibitor response (measured by AUC) with each of the six cell-type scores reveals relationship between differentiation and resistance (blue) or sensitivity (red). Note, all drugs displayed in the heatmap showed a significant correlation with at least one cell-type (BY FDR < 0.05; (Benjamini and Yekutieli, 2001)). Panobinostat and Venetoclax are shown correlated with Monocyte-like score in Figure S3A. Asterisks indicate those inhibitors in which there is an interaction between mutation and cell-type, shown below in panel B (single asterisk, qvalue < 0.1; double asterisk, qvalue < 0.05; (Storey and Tibshirani, 2003)). Full data are available in Table S3. (B) We examined whether correlations between drug response and cell-type score changed due to mutational status. Drug response modification was quantified by examining the significance (Y-axis) of the interaction between each mutational event and cell-type (x-axis) for each inhibitor, requiring a minimum of 10 mutations. Interactions with qvalue at two thresholds (q < 0.1; q < 0.05) are called out by text and shape and distinguished by a dashed and solid line, respectively. As an example, the conditional relationships between sorafenib, FLT3-ITD, and cell-type scores are shown in Figure S3B, where sorafenib activity is robust in FLT3-ITD cases with a high progenitor-like score, but activity is lost in FLT3-ITD cases with a high monocyte-like score.
Drug Family Responses
The increased sample size of the harmonized dataset yields numerous correlations of drug response with mutational status that achieve statistical significance compared with Waves 1+2 alone (65 added associations, 27% increase). However, we next wanted to extract additional signal by organizing drug response data into groups of inhibitors based on shared targets or pathways. Accordingly, we aggregated drug-target relationship data from the Cancer Targetome (Blucher et al., 2017; Blucher et al., 2019; Choonoo et al., 2019) and other sources (Davis et al., 2011) to yield 25 drug families with some overlap of drugs between families based on known polypharmacology (Table S4). We observed variable overall response of drug families across all samples tested, with some families (e.g. Phosphatidyl inositol 3’ kinase-related kinases (PIKK)) showing greater overall activity and others (e.g. Aurora Kinase (AurK)) showing generally less sensitivity (Figure 4A). By examining response of each drug family in specimens with wild type versus mutated status, we were able to identify numerous instances where mutational events exhibit significantly greater sensitivity or resistance to drug family classes (Figure 4B; for all associations see Table S5). In addition to expected associations such as FLT3-ITD with Type III receptor tyrosine kinase (RTK) inhibitors and RAS mutations with STE7 (MEK) inhibitors, we identified numerous unexpected associations, including several treatment-refractory genetic subtypes (mutation of TP53, STAG2, RUNX1) showing greater sensitivity to phosphatidylinositol kinase family (PIK) inhibitors and N/KRAS mutation conferring greater sensitivity to cyclin-dependent kinase inhibitors. We also assessed correlations between drug family activity and tumor cell-type scores and found strong correlations with one or more tumor cell-type signature for 18 out of the 25 drug families (Figure 5A; for all associations see Table S6). Finally, we also observed a number of drug family correlations with cell-type scores that were conditional on mutational/cytogenetic events, such as for PIK family inhibitors showing strong activity against NPM1-mutated cases that also exhibit a high cDC-like score, but PIK inhibitor resistance for NPM1-mutated cases with high HSC-like scores (Figure 5B).
Figure 4. Genomic associations of drug family response.
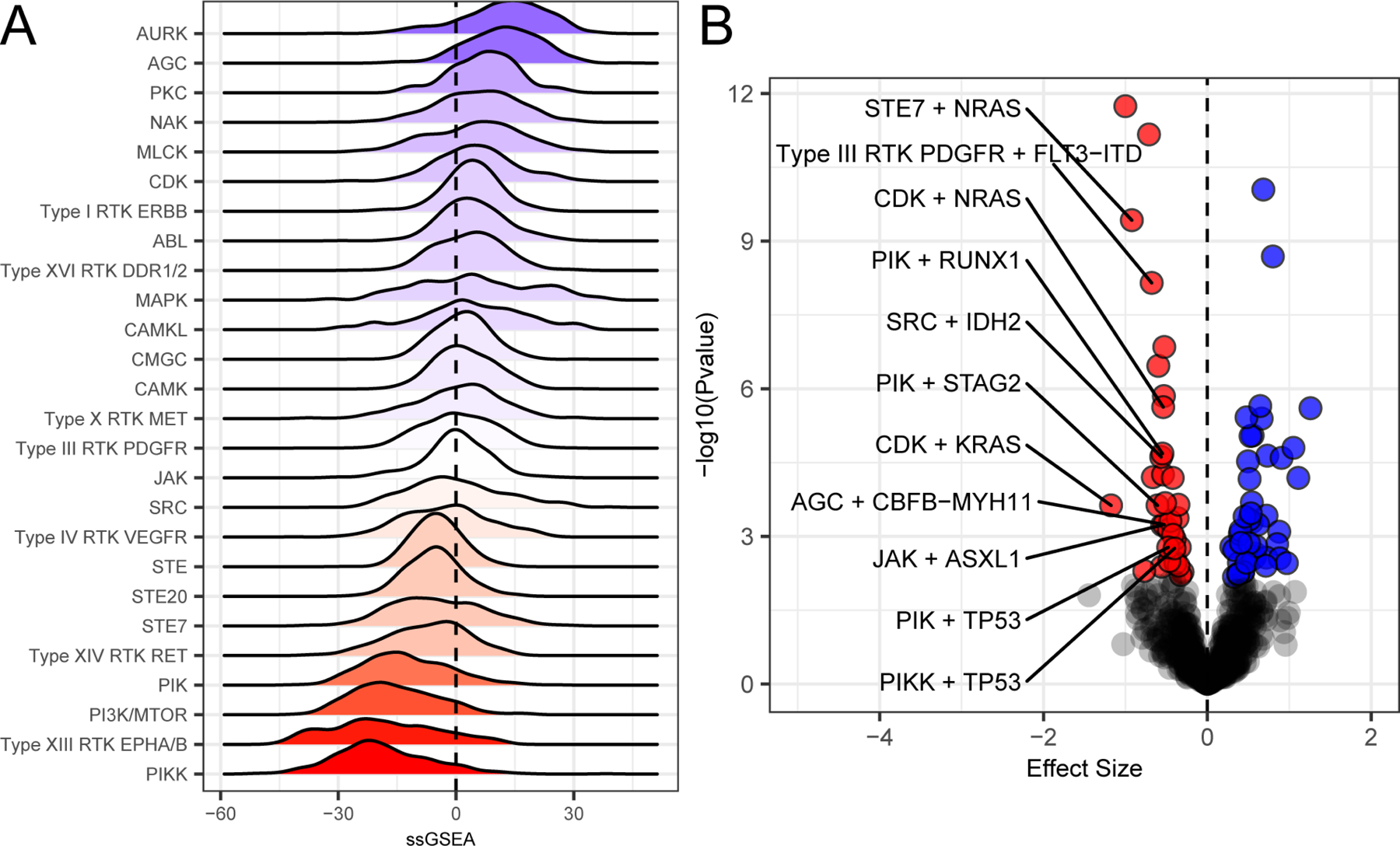
As in Figure 3A, we define sensitivity by red and resistant with blue and with white indicating intermediate. (A) Drug target information, where available, was annotated for all drugs on the panel, and these targets were mapped onto pathways to create drug families that share common targets and pathways. Full mapping of drug families is available in Table S4. Family-level drug response summaries allow us to identify cohort-level responses. Some like the Aurora Kinase family (Aurk) show overall resistance and others like Type XIII RTKs (Eph) or PIKK are overall sensitive. (B). We examined correlations between mutational status and drug family response. Mutational status associates with drug family response either in terms of significant (qvalue < 0.05; (Storey and Tibshirani, 2003)) sensitivity (red) or resistance (blue). Association tests were performed using Welch’s T-test comparing mutated vs wild type and requiring at least 5 mutations for a given mutational event. Effect size was based off of Glass’s delta using wild type as the reference. Full data are available in Table S5.
Figure 5. Drug family response is influenced by and conditional on cell-type.
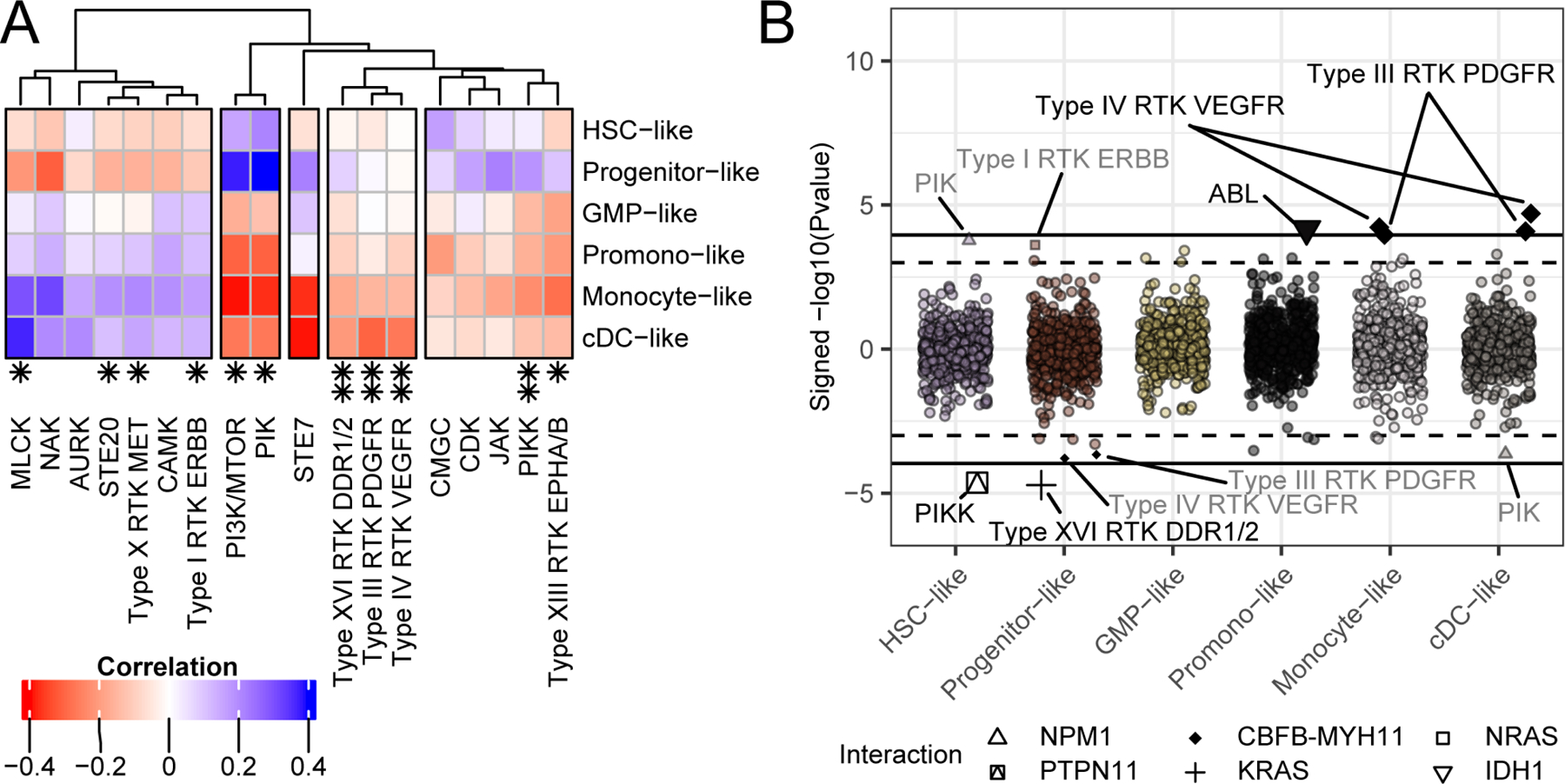
(A) Similar to the analysis of single-inhibitor responses and their correlation with cell-type score, shown in Figure 3A, we can group the drug families from Figure 4 by their association with cell-type differentiation scores based on their Pearson’s correlation (BY FDR < 0.05; (Benjamini and Yekutieli, 2001)). (B) We can also determine instances where drug family correlation with mutational state is conditional on cell-type (as we did for individual drugs in Figure 3B). The mutations that play a significant drug response modification role based on the statistical interaction between cell-type score and mutation, requiring at least 10 mutations per event is shown. The Y-axis indicates the signed -log10 (Pvalue), the X-axis indicates cell-type. The interactions are listed by text (qvalue < 0.1 and qvalue < 0.05; (Storey and Tibshirani, 2003)) and distinguished by a dashed and solid line respectively. Full data are available in Table S6.
Determinants of Clinical Outcome
Finally, we wanted to explore correlations between the WGCNA expression modules and cell-type scores with clinical variables in our dataset, including overall survival, tumor burden and cell composition, age, transformed or therapy-related status, and prognostic categories. To ensure robust associations with overall survival, we limited this analysis to samples collected at initial acute leukemia diagnosis and with RNASeq data (n=435). Clustering of these features revealed one branch that contained three WGCNA modules (3, 9, and 12) as well the HSC-like cell-type score that associated with shorter overall survival (Figure 6A). Of these, modules 3 and 9 showed the strongest associations with survival. We prioritized the genes in module 3 (n=203) and module 9 (n=119) by computing the correlation with the eigengene (termed kME; (Fuller et al., 2007)) to quantify each gene’s degree of hub status. This revealed a single gene, platelet endothelial aggregation receptor 1 (PEAR1), that correlated far more strongly with the module 3 eigengene than did any other module gene member (Figure 6B). No similar singular driver gene was identified for module 9. Additionally, both module 3 and PEAR1 expression were positively correlated with HSC-like score (Figure 6B; a full list of module component genes is in Table S7). We next applied conditional inference forest methodology (cforest; (Hothorn et al., 2006)) to understand the relative importance of patient age, WGCNA module eigengenes, cell-type scores, and mutational/cytogenetic status on overall survival. For this analysis, we placed a further restriction to only evaluate cases of de novo AML (n=298) because of the observed association between AML type and other variables. Validating known risk factors, this methodology identified age and mutated TP53 (Papaemmanuil et al., 2016) being among the strongest predictors of survival. However, the WGCNA module 3 was also included as one of the strongest predictors of poor outcome. For young (<45 years) and TP53-wild type middle (45–60 years) age group patients, our analysis revealed module 3 expression to be the strongest determinant of outcome (Figure 6C).
Figure 6. Comprehensive analysis of clinical variables identifies PEAR1 expression as a prognostic factor in AML.
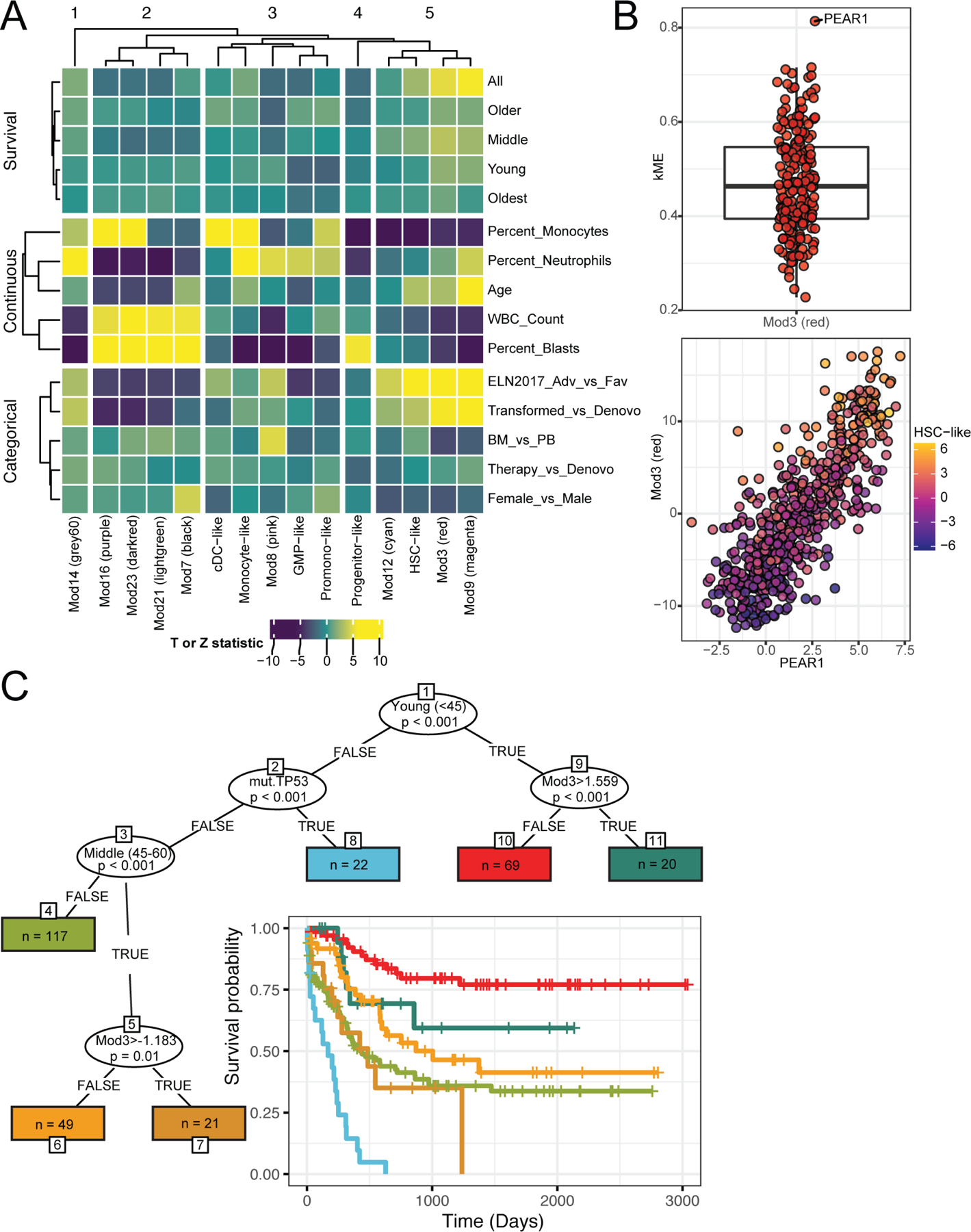
(A) We analyzed univariate associations between the 6 cell-type scores and the set of 9 non-redundant WGCNA eigengenes with categorical and continuous clinical variables as well as overall survival across and within age subsets. Each variable type was grouped on the Y-axis and the displayed association values were derived from the corresponding test statistics. These include the Z statistic for categorical (logistic regression) and survival outcomes (Cox Proportional Hazards) and T-statistic for the continuous outcomes (general linear model). Average linkage hierarchical clustering was applied to rows (within groups) and columns (B; top) To understand which gene(s) among the module 3 WGCNA signature are the strongest drivers of the overall signature, we calculated the correlation of each gene with the module eigengene (kME). Of all the 203 genes in module 3, PEAR1 has the highest correlation. (B; bottom) The patient expression values (dots) of PEAR1 (x-axis) correlated highly with the Mod3 module eigengene (y-axis) and both are also correlated with the ‘HSC-like’ malignant cell-type as indicated by the cell-type purple(low)-yellow(high) gradient as defined in Figure S2A. Full module gene components are available in Table S7. (C) A strategy based on a forest of conditional inference trees (cforest) was used to determine variables that differentiate overall survival. Age groups, expression modules (denoted with Mod*), gene mutation, AML gene fusions, and cell-type scores from in the harmonized dataset were all included in the model. The age groups are young (<45), middle (45–60), older (60–75), oldest (>75). Depiction of the resulting tree where oval splits indicate variables that most significantly split overall survival. Lines indicate values that were split on, for instance, “TRUE” for the ‘young’ variable, which indicates that the subgroup is <45 years old. The resulting subgroups of patients, rectangles that denote ‘terminal nodes’, are listed with the subgroup size (denoted as n) and are colored to match corresponding survival curves.
We next explored association of individual PEAR1 gene expression with clinical parameters, since PEAR1 was so strongly associated with the overall module 3 score. Assessment of PEAR1 expression as it relates to European LeukemiaNet (ELN) prognostic categories indicated a significant difference in PEAR1 expression between Adverse and Favorable risk categories in patients 60 and older (3.765 mean increase in Adverse; Pvalue=3.276e-25; N=172). PEAR1 expression, however, shows a smaller but significant difference in the middle age group (1.497 mean increase; Pvalue=0.006; N=72) and is not significantly associated with Adverse vs. Favorable ELN risk in young patients (Pvalue=0.170; N=79) despite its strong association with outcome in this group (Figure 7A). We compared PEAR1 expression to the leukemic stem cell 17-gene signature (LSC17) (Ng et al., 2016) in this young subset of patients and found that expression of the single PEAR1 gene could distinguish outcomes in young patients to a similar degree as the 17-gene LSC17 signature (Figure 7B). To further compare PEAR1 with LSC17, we assessed the hazard ratio for PEAR1 and LSC17 in all age groups and in the young subset. Additionally, we further stratified cases where the specimen obtained was a bone marrow aspirate, due to improved splitting of outcomes observed with bone marrow compared with peripheral blood specimens (Figure S4). The hazard ratios reveal similar performance of PEAR1 with LSC17 (Figure 7C), noting predictive range is influenced by subset and split type. Since PEAR1 shows strong upregulation in transformed AML, which is known to exhibit inferior outcome (Figure 6A, S5A,B), and since a similar association was observed with LSC17 and transformed AML (Pvalue=2.18e-07), we wanted to compare PEAR1 and LSC17 in all cases with specimens obtained at initial acute leukemia diagnosis, inclusive of transformed and therapy-related AML cases. This analysis revealed PEAR1 to perform equivalently to LSC17 both in the young subset and across all age groups (Figure S6). Finally, to validate the prognostic significance of PEAR1 using independent patient cohorts, we examined the capacity of PEAR1 expression to stratify overall survival using a dataset from Malani and colleagues as well as from The Cancer Genome Atlas (Cancer Genome Atlas Research et al., 2013; Malani et al., 2022). We found that elevated PEAR1 expression conferred shortened overall survival in a strongly significant manner in both datasets (Figure S7A, B).
Figure 7. Integrative modeling shows PEAR1 expression is a single, independent predictor of poor prognosis.
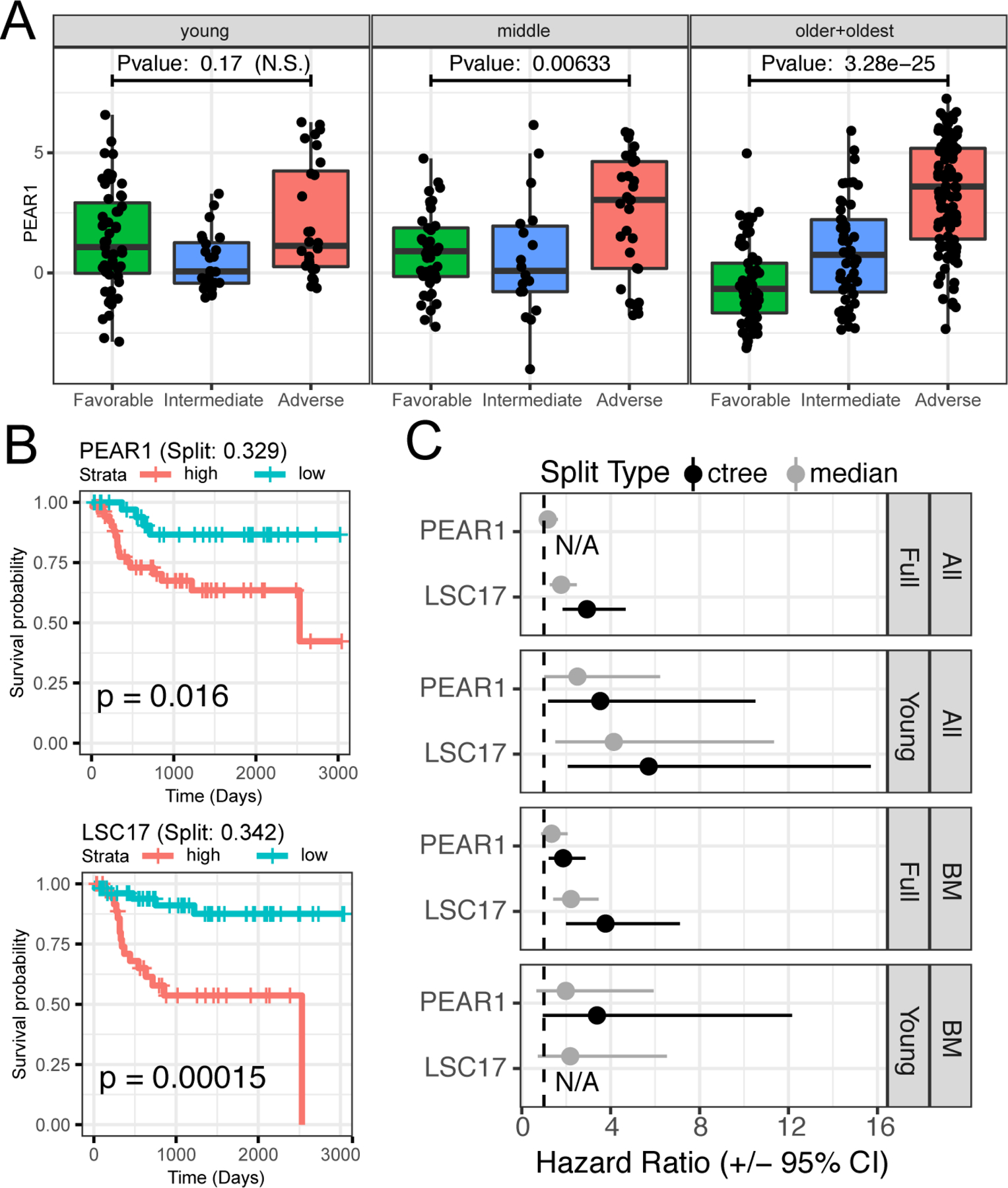
(A) PEAR1 expression (y-axis) is significantly higher in Adverse compared to Favorable ELN 2017 risk categorization (x-axis) for patients 60 and over (dots; older + oldest group), but the pattern is less pronounced in patients < 60. Significance determined using a using a Welch’s T-test. (B) PEAR1 expression differentiates survival for young patients equivalently to the LSC17 signature. Categorization of samples into high and low expression groups for PEAR1 and LSC17 was determined using the ctree methodology to facilitate comparison with PEAR1. (C) We compared performance of PEAR1 and LSC17 using both ctree and the median expression values for each. Shown are the hazard ratios (HRs) and 95% confidence intervals (CIs) for PEAR1 and LSC17 in the entire cohort or in the younger patients as well as in all sample types or only in bone marrow samples. The categories were high vs low expression as determined by either median threshold or significant splits using ctree (i.e., split type). In two instances, the ctree method failed to identify a split, and these are denoted with “N/A”.
DISCUSSION
The impact of AML LSCs on tumor biology and therapeutics has been explored and suggested to prime disease pathogenesis and seed relapse. However, a broader and more nuanced understanding of the full range of AML tumor cell maturation states, and the way in which these different cell states are tied to other disease features such as genetics and drug response, has been lacking. While some of these maturation states were roughly captured in the historical FAB classification system for AML, this system has been all but abandoned in current classification schemes that utilize primarily (or only) genetics and prior clinical history (Arber et al., 2016; Dohner et al., 2017). In addition, while targeting of LSCs has been a major goal for AML research with the notion that eliminating LSCs would induce tumor collapse, this result has proved elusive with the observation that relapse can be seeded by more mature cells, depending on the administered therapy. Through broad mapping of distinct AML cell maturation states with both genetic features and response to broad families of drugs, we find that tumors can display features of multiple, disparate cell maturation states and that a majority of drugs and drug families exhibit cell maturation state-biased response. Our expansive modeling of clinical outcome has also led to a single, targetable gene that is a strong determinant of overall survival in AML, PEAR1.
PEAR1, also known as JEDI or MEGF12, was originally identified to signal during platelet aggregation (Nanda et al., 2005) and is a homolog of Draper (D. melanogaster) and ced-1 (C. elegans). It is a type I transmembrane protein with 15 extracellular epidermal growth factor-like repeats, a domain sharing homology with NOTCH ligands, and it contains tyrosine and serine residues that can become phosphorylated (Krivtsov et al., 2007; Nanda et al., 2005). Genetic polymorphisms in the PEAR1 locus associate with with cardiovascular events, response to anti-platelet aggregation therapies (Eicher et al., 2016; Faraday et al., 2011; Herrera-Galeano et al., 2008; Johnson et al., 2010; Jones et al., 2009; Lewis et al., 2013), and endothelial cell biology (Fisch et al., 2015; Vandenbriele et al., 2015; Zhan et al., 2020). Methylation of the PEAR1 locus correlates with megakaryopoiesis and platelet function (Izzi et al., 2019; Izzi et al., 2018; Izzi et al., 2016). On glial precursor cells, PEAR1 triggers clathrin-dependent engulfment of apoptotic neurons generated during development of peripheral ganglia (Sullivan et al., 2014; Wu et al., 2009). This phagocytic process signaling involves SYK, SRC-family kinases, and MAPK8 (JNK; JUN N-terminal kinase) and/or their D. melanogaster homologs (Hilu-Dadia et al., 2018; Scheib et al., 2012). PEAR1 ligands include Fc Epsilon Receptor Ia (Sun et al., 2015), dextran sulfate (Vandenbriele et al., 2016), and sulfated fucose-based polysaccharides (fucoidans) (Kardeby et al., 2019), and signaling in platelets involves SRC-family kinases and PIK3C/AKT (Kauskot et al., 2012; Kauskot et al., 2013). In healthy hematopoietic cells, PEAR1 expression is highest in HSC and in megakaryocyte-erythroid progenitor cells. Forced expression of PEAR1 in bone marrow cells or in fibroblast stromal cells was shown to reduce clonogenic myeloid colony formation (Krivtsov et al., 2007). This effect seems counterintuitive given the clear association of elevated PEAR1 expression with worse outcome in AML and warrants further investigation.
A recent study examined AML TCGA (Cancer Genome Atlas Research et al., 2013) to identify an immune prognostic signature for AML, which included PEAR1 as well as another gene, PYCARD (Dao et al., 2021). This model showed correlation with T-cell enrichment and expression of immune checkpoints. It was also shown to correlate with poor clinical outcome in several AML datasets; however, a role for PEAR1 in shaping leukemic cell biology or cell-type was not considered.
Here, we show that PEAR1 expression predicts outcome in young AML patients independently of ELN category, performs equivalently to the LSC17 gene signature, and correlates strongly with an HSC-like signature. Increased PEAR1 was confirmed in cases with mutation of TP53, RUNX1, and ASXL1 (as shown in (Dao et al., 2021)), and we also observe elevation in other genetic subsets with poor prognostic markers (SRSF2 mutation, GATA2-MECOM) and lower expression with certain good prognostic features (NPM1, CEBPA) (Figure S5C). Finally, while there was an increasing trend in expression of initial diagnosis PEAR1 across ethnicities (Non-Hispanic white: mean 1.395, N=359; African Heritage: mean 2.205, N=13), the smaller sample size in the non-Caucasian groups warrants further investigation in a validation cohort. This is potentially important due to recent findings of outcome disparities among Black AML patients (Bhatnagar et al., 2021) as well as several studies reporting racially-skewed representation of PEAR1 polymorphisms that impact on PEAR1 expression/function (Herrera-Galeano et al., 2008; Keramati et al., 2019; Qayyum et al., 2015).
Elevation of PEAR1 expression in ASXL1-mutated AML is notable, since ASXL1 is commonly mutated in clonal hematopoiesis, which is also associated with elevated cardiovascular events (Genovese et al., 2014; Jaiswal et al., 2014). As noted above, PEAR1 genetics and function have been tied to the biology of cardiovascular events, raising the intriguing possibility that these findings could be connected. Platelet levels have been shown to decline with age (Biino et al., 2013), and a leukocyte pool with enhanced platelet aggregation potential – possibly facilitated by increased PEAR1 downstream of somatic mutational events such as ASXL1 – could represent a selective advantage within an aging hematopoietic microenvironment.
All of these data raise the possibility of PEAR1 as a therapeutic target. Since PEAR1 may function as an active signaling protein, small-molecules could mitigate PEAR1 signaling. Antibodies to PEAR1 have been developed, so a large-molecule approach may also be feasible. Indeed, we have performed preliminary validation of this concept by showing PEAR1 surface expression on tumor cells from AML patient samples (Figure S7C) with the degree of surface expression strongly correlated with PEAR1 transcript levels (Figure S7D). Interruption of normal PEAR1 function must be considered for any targeting strategy; however, platelet function was not compromised in a PEAR1-null mouse (Criel et al., 2016).
There are other important limitations to consider. First, the prognostic significance of PEAR1 has, thus far, been demonstrated in datasets where therapies employed were almost entirely anthracycline-based. As drug regimens are now more diversified, it will be important to study the prognostic significance of PEAR1 in patients receiving alternative treatments. In this sense, and in general, the clinical utility of measuring PEAR1 expression for prognostication and/or of therapeutically targeting PEAR1 must be confirmed in future clinical studies.
Collectively, this study exemplifies the utility of integrative analysis that incorporates functional testing of large cohorts of patient specimens. This includes broad association of drug sensitivity with tumor cell differentiation state and drug/mutation correlations that are sometimes conditional on cell-type. Given the strong associations that can be seen between cell-type score and drug response, the distribution of these cell phenotypes should be assessed when evaluating clinical differences in drug sensitivity. Testing interactions between cell-type and other disease or patient features can help identify potential confounders or modifiers of drug response. For key comparisons, cell-type consideration should be done as routine as population substructure adjustment. Finally, our analysis of clinical outcome has yielded a prognostic and potentially targetable feature of AML that merits further testing and exploration.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contacts, Dr. Shannon McWeeney (mcweeney@ohsu.edu) and Dr. Jeffrey Tyner (tynerj@ohsu.edu).
Materials Availability
This study did not generate unique reagents.
Data and Code Availability
All raw and processed sequencing data, along with relevant clinical annotations have been submitted to dbGaP and Genomic Data Commons and are publicly available. The dbGaP study ID is 30641 and accession ID is phs001657.v2.p1 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001657.v2.p1). The clinical annotations and data availability are found in Table S1. In addition, all data can be accessed and queried through our online, interactive user interface, Vizome, at www.vizome.org/aml2. Original code for replicating the paper analyses based on publicly available data has been deposited at https://github.com/biodev/beataml2_manuscript. The DOI is listed in the key resources table. A frequently asked questions page for the dataset is found at https://biodev.github.io/BeatAML2/.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BV650 Mouse Anti-Human PEAR1, clone 492621 | BD Biosciences | Catalog # 748063 |
| Human TruStain FcX (Fc Receptor Blocking Solution) | Biolegend | Catalog # 422301 |
| Bacterial and virus strains | ||
| n/a | ||
| Biological samples | ||
| AML Patient Samples | Oregon Health & Science University (OHSU), University of Utah, University of Texas Medical Center (UT Southwestern), Stanford University, University of Miami, University of Colorado, University of Florida, National Institutes of Health (NIH), Fox Chase Cancer Center and University of Kansas (KUMC) | OHSU; IRB#9570; #4422; NCT01728402 |
| Healthy Bone Marrow CD34+ Cells | AllCells | Custom Order |
| Chemicals, peptides, and recombinant proteins | ||
| Drug | Source | Catalog # | CAS # |
| 17-AAG (Tanespimycin) | LC Labs | A-6880 | 75747–14-7 |
| A-674563 | Selleck | S2670 | 552325–73-2 |
| ABT-737 | Selleck | S1002 | 852808–04-9 |
| Afatinib (BIBW-2992) | LC Labs | A-8644 | 850140–72-6 |
| AGI-5198 | Selleck | S7185 | 1355326–35-0 |
| AGI-6780 | Selleck | S7241 | 1432660–47-3 |
| AKT Inhibitor IV | Millipore Sigma (EMD Biosciences; Calbiochem) | 124011 | 681281–88-9 |
| AKT Inhibitor X | Millipore Sigma (EMD Biosciences; Calbiochem) | 124020 | 925681–41-0 |
| Alisertib (MLN8237) | Selleck | S1133 | 1028486–01-2 |
| AMPK Inhibitor | Millipore Sigma (EMD Biosciences; Calbiochem) | 171260 | 866405–64-3 |
| Arsenic trioxide | Fisher | NC0244675 | 1327–53-3 |
| Artemisinin | Selleck | S1282 | 63968–64-9 |
| AST487 | Selleck | S6662 | 630124–46-8 |
| AT-101 | Selleck | S2812 | 866541–93-7 |
| AT7519 | Selleck | S1524 | 844442–38-2 |
| Axitinib (AG-013736) | LC Labs | A-1107 | 319460–85-0 |
| Azacytidine | Selleck | S1782 | 320–67-2 |
| AZD1480 | Selleck | S2162 | 935666–88-9 |
| Baicalein | Selleck | S2268 | 491–67-8 |
| Barasertib (AZD1152-HQPA) | Selleck | S1147 | 722544–51-6 |
| Bay 11–7085 | Selleck | S7352 | 196309–76-9 |
| BEZ235 | LC Labs | N-4288 | 915019–65-7 |
| BI-2536 | Selleck | S1109 | 755038–02-9 |
| Birinapant | Selleck | S7015 | 1260251–31-7 |
| BLZ945 | Selleck | S7725 | 953769–46-5 |
| BMS-345541 | Selleck | S8044 | 445430–58-0 |
| BMS-754807 | Selleck | S1124 | 1001350–96-4 |
| Bortezomib (Velcade) | LC Labs | B-1408 | 179324–69-7 |
| Bosutinib (SKI-606) | LC Labs | B-1788 | 380843–75-4 |
| Cabozantinib (XL184) | Selleck | S1119 | 849217–68-1 |
| Canertinib (CI-1033) | LC Labs | C-1201 | 289499–45-2 |
| Cediranib (AZD2171) | LC Labs | C-4300 | 288383–20-0 |
| CHIR-99021 | LC Labs | C-6556 | 252917–06-9 |
| CI-1040 (PD184352) | LC Labs | P-8499 | 212631–79-3 |
| Crenolanib | Selleck | S2730 | 670220–88-9 |
| Crizotinib (PF-2341066) | LC Labs | C-7900 | 877399–52-5 |
| CYT387 | Selleck | S2219 | 1056634–68-4 |
| Cytarabine | Selleck | S1648 | 147–94-4 |
| Dasatinib | LC Labs | D-3307 | 302962–49-8 |
| DBZ | Tocris | 4489 | 209984–56-5 |
| Doramapimod (BIRB 796) | LC Labs | D-2744 | 285983–48-4 |
| Dovitinib (CHIR-258) | LC Labs | D-3608 | 405169–16-6 |
| Elesclomol | Selleck | S1052 | 488832–69-5 |
| Entospletinib (GS-9973) | Selleck | S7523 | 1229208–44-9 |
| Entrectinib | Selleck | S7998 | 1108743–60-7 |
| Erlotinib | LC Labs | E-4007 | 183319–69-9 |
| Etomoxir | Selleck | S8244 | 828934–41-4 |
| Everolimus | Selleck | S1120 | 159351–69-6 |
| Flavopiridol | Selleck | S1230 | 131740–09-5 |
| Foretinib (XL880) | LC Labs | F-4185 | 849217–64-7 |
| GDC-0879 | Selleck | S1104 | 905281–76-7 |
| GDC-0941 | LC Labs | G-9252 | 957054–30-7 |
| Gefitinib | LC Labs | G-4408 | 84475–35-2 |
| Gilteritinib | Selleck | S7754 | 1254053–43-4 |
| Go6976 | LC Labs | G-6203 | 136194–77-9 |
| GSK-2879552 | Selleck | S7796 | 1902123–72-1 |
| GSK690693 | Selleck | S1113 | 937174–76-0 |
| GW-2580 | LC Labs | G-5903 | 870483–87-7 |
| H-89 | LC Labs | H5239 | 127243–85-0 |
| Ibrutinib (PCI-32765) | LC Labs | I-3311 | 936563–96-1] |
| Idelalisib (GS-1101) | Selleck | S2226 | 870281–82-6 |
| Imatinib | LC Labs | I5577 | 152459–95-5 |
| Indisulam | Selleck | S9742 | 165668–41-7 |
| INK-128 | Selleck | S2811 | 1224844–38-5 |
| JAK Inhibitor I | Millipore Sigma (EMD Biosciences; Calbiochem) | 420099 | 457081–03-7 |
| JNJ-28312141 | SYN Kinase | SYN-1154 | 885692–52-4 |
| JNJ-38877605 | Selleck | S1114 | 943540–75-8 |
| JNJ-7706621 | Selleck | S1249 | 443797–96-4 |
| JNK Inhibitor II (SP600126) | LC Labs | S-7979 | 129–56-6 |
| JQ1 | Selleck | S7110 | 1268524–70-4 |
| KI20227 | Tocris | 4481 | 623142–96-1 |
| KN92 | Tocris | 4130 | 1135280–28-2 |
| KN93 | Tocris | 1278 | 139298–40-1 |
| KU-55933 | Selleck | S1092 | 587871–26-9 |
| KW-2449 | Selleck | S2158 | 1000669–72-6 |
| Lapatinib | Selleck | S2111 | 231277–92-2 |
| Lenalidomide | Selleck | S1029 | 191732–72-6 |
| Lenvatinib | Selleck | S1164 | 857890–39-2 |
| Lestaurtinib (CEP-701) | LC Labs | L-6307 | 111358–88-4 |
| Linifanib (ABT-869) | Selleck | S1003 | 796967–16-3 |
| Lovastatin | Selleck | S2061 | 75330–75-5 |
| LY-333531 | Tocris | 4738 | 169939–93-9 |
| LY294002 | LC Labs | L-7962 | 154447–36-6 |
| Masitinib (AB-1010) | LC Labs | M-7007 | 790299–79-5 |
| MEK Inhibitor VII | Millipore Sigma (EMD Biosciences; Calbiochem) | 444939 | 305350–87-2 |
| Metformin | Selleck | S5958 | 657–24-9 |
| MGCD-265 | Selleck | S1361 | 875337–44-3 |
| Midostaurin | LC Labs | P-7600 | 120685–11-2 |
| MK-2206 | Selleck | S1078 | 1032350–13-2 |
| MLN120B | MedChem Express | HY-15473 | 783348–36-7 |
| MLN8054 | Selleck | S1100 | 869363–13-3 |
| Motesanib (AMG-706) | LC Labs | M-2900 | 857876–30-3 |
| Neratinib (HKI-272) | Selleck | S2150 | 698387–09-6 |
| NF-kB Activation Inhibitor | Selleck | S4902 | 545380–34-5 |
| Nilotinib | Selleck | S1033 | 641571–10-0 |
| Nutlin 3a | Selleck | S8059 | 675576–98-4 |
| NVP-ADW742 | Selleck | S1088 | 475488–23-4 |
| NVP-AEW541 | Selleck | S1034 | 475489–16-8 |
| NVP-TAE684 | Selleck | S1108 | 761439–42-3 |
| Olaparib | Selleck | S1060 | 763113–22-0 |
| OTX-015 | Selleck | S7360 | 202590–98-5 |
| p38 MAP Kinase Inhibitor | Millipore Sigma (EMD Biosciences; Calbiochem) | 506126 | 219138–24-6 |
| Palbociclib | Selleck | S4482 | 571190–30-2 |
| Panobinostat | Selleck | S1030 | 404950–80-7 |
| Pazopanib (GW786034) | LC Labs | P6706 | 444731–52-6 |
| PD153035 | Millipore Sigma (EMD Biosciences; Calbiochem) | SML0564 | 183322–45-4 |
| PD173955 | Symansis | SY-PD173955 | 260415–63-2 |
| PD98059 | LC Labs | P-4313 | 167869–21-8 |
| Pelitinib (EKB-569) | Selleck | S1392 | 257933–82-7 |
| Perhexiline maleate | Selleck | S6959 | 6724–53-4 |
| PH-797804 | Selleck | S2726 | 586379–66-0 |
| PHA-665752 | Selleck | S1070 | 477575–56-7 |
| PHT-427 | Selleck | S1556 | 1191951–57-1 |
| PI-103 | Selleck | S1038 | 371935–74-9 |
| PLX-4720 | Selleck | S1152 | 918505–84-7 |
| Ponatinib (AP24534) | LC Labs | P-7022 | 943319–70-8 |
| PP2 | Millipore Sigma (EMD Biosciences; Calbiochem) | 529573 | 172889–27-9 |
| PP242 | LC Labs | P-6666 | 1092351–67-1 |
| PRT062070 HCL | Selleck | S7634 | 1369761–01-2 |
| PRT062607 | Selleck | S8032 | 1370261–97-4 |
| Quizartinib (AC220) | LC Labs | Q-4747 | 950769–58-1 |
| R406 | Selleck | S2194 | 841290–81-1 |
| R547 | Selleck | S2688 | 741713–40-6 |
| RAF265 (CHIR-265) | Selleck | S2161 | 927880–90-8 |
| Ralimetinib (LY2228820) | Selleck | S1494 | 862507–23-1 |
| Ranolazine (free base) | Selleck | S1799 | 95635–55-5 |
| Rapamycin | LC Labs | R-5000 | 53123–88-9 |
| Regorafenib (BAY 73–4506) | Selleck | S1178 | 755037–03-7 |
| Roscovitine (CYC-202) | LC Labs | R-1234 | 186692–46-6 |
| Ruxolitinib, free base | LC Labs | R-6600 | 120685–11-2 |
| S31–201 | Santa Cruz Biotech | sc-204304 | 501919–59-1 |
| Saracatinib (AZD0530) | LC Labs | S-8906 | 379231–04-6 |
| SB-202190 | LC Labs | S-1700 | 152121–30-7 |
| SB-203580 | LC Labs | S-3400 | 152121–47-6 |
| SB-431542 | Selleck | S1067 | 301836–41-9 |
| Selinexor | Selleck | S7252 | 1393477–72-9 |
| Selumetinib (AZD6244) | LC Labs | S-4490 | 606143–52-6 |
| SGX-523 | Selleck | S1112 | 1022150–57-7 |
| SNS-032 (BMS-387032) | Selleck | S1145 | 345627–80-7 |
| Sorafenib tosylate | LC Labs | S-8502 | 475207–59-1 |
| SR9011-HCL | MedChem Express | HY-16988A | 2070014–94-5 |
| Staurosporine, free base | LC Labs | S-9300 | 62996–74-1] |
| STO609 | Millipore Sigma (EMD Biosciences; Calbiochem) | 570250 | 52029–86-4 |
| SU11274 | Selleck | S1080 | 658084–23-2 |
| SU14813 | Selleck | S0504 | 627908–92-3 |
| Sunitinib malate | LC Labs | S-8803 | 341031–54-7 |
| SYK Inhibitor | Millipore Sigma (EMD Biosciences; Calbiochem) | 574711 | 622387–85-3 |
| Tandutinib (MLN518) | LC Labs | T-7802 | 387867–13-2 |
| TG100–115 | Selleck | S1358 | 677297–51-7 |
| TG101348 | Active Biochem | A-1140 | 936091–26-8 |
| Tivozanib (AV-951) | LC Labs | T-6466 | 475108–18-0 |
| Tofacitinib (CP-690550) | LC Labs | T-1377 | 477600–75-2 |
| Tozasertib (VX-680) | LC Labs | T-2304 | 639089–54-6 |
| Trametinib (GSK1120212) | LC Labs | T-8132 | 871700–17-3 |
| Vandetanib (ZD6474) | LC Labs | V-9402 | 443913–73-3 |
| Vargetef (Nintedanib) | LC Labs | N-9077 | 656247–17-5 |
| Vatalanib (PTK787) | LC Labs | V-8303 | 212141–51-0 |
| Vemurafenib (PLX4032) | LC Labs | V-2800 | 918504–65-1 |
| Venetoclax | LC Labs | V-3579 | 1257044–40-8 |
| Vismodegib (GDC-0449) | LC Labs | V-4050 | 879085–55-9 |
| Volasertib (BI-6727) | Selleck | S2235 | 755038–65-4 |
| VX-745 | Tocris | 3915 | 209410–46-8 |
| XAV-939 | Selleck | S1180 | 284028–89-3 |
| XMD 8–87 | Selleck | S8272 | 1234480–46-6 |
| YM-155 | Selleck | S1130 | 781661–94-7 |
| Other Chemical | Source | Catalog # |
| Zombie Aqua Fixable Viability Dye | Biolegend | Catalog # 423101 |
| Critical commercial assays | ||
| Nextera RapidCapture Exome | Illumina | |
| SureSelect Strand-Specific RNA Library Preparation Kit | Agilent | |
| DNeasy Blood & Tissue Kit | Qiagen | |
| RNeasy Mini Kit | Qiagen | |
| Deposited data | ||
| DNA Sequencing Data | This paper | http://vizome.org/aml2/, dbGaP study ID is 30641 and accession ID is phs001657.v2.p1 |
| RNA Sequencing Data | This paper | http://vizome.org/aml2/, dbGaP study ID is 30641 and accession ID is phs001657.v2.p1 |
| Experimental models: Cell lines | ||
| n/a | ||
| Experimental models: Organisms/strains | ||
| n/a | ||
| Oligonucleotides | ||
| Custom capture probes for DNA sequencing (11.9 MB) | Roche-Nimblegen | Sequencing coordinates listed Table S14 of (Tyner et al., 2018) |
| Forward Primer FLT3: 5′- AGCA ATT TAG GTA TGA AAG CCA GCTA - 3′ | Eurofins | (Kottaridis et al., 2001) |
| Reverse Primer FLT3: 5′ - CTT TCA GCA TTT TGA CGG CAA CC - 3′ | Eurofins | (Kottaridis et al., 2001) |
| Forward Primer NPM1: 5′ - GTT TCT TTT TTT TTT TTT CCA GGC TAT TCA AG - 3′ | Eurofins | (Falini et al., 2007) |
| Reverse Primer NPM1: 5′ - CAC GGT AGG GAA AGT TCT CAC TCT GC - 3′ | Eurofins | (Falini et al., 2007) |
| Recombinant DNA | ||
| n/a | ||
| Software and algorithms | ||
| R version 4.03 | R Core Team | https://www.r-project.org |
| R package GSVA v1.38 | Bioconductor | DOI: 10.18129/B9.bioc.GSVA |
| R package cqn v1.36 | Bioconductor | DOI: 10.18129/B9.bioc.cqn |
| R package qvalue v2.22.0 | Bioconductor | DOI: 10.18129/B9.bioc.qvalue |
| R package partykit v1.2–15 | CRAN | https://CRAN.R-project.org/package=partykit |
| R package survival v3.2–7 | CRAN | https://CRAN.R-project.org/package=survival |
| R package WGCNA v1.69 | CRAN | https://CRAN.R-project.org/package=WGCNA |
| R package data.table v1.13.6 | CRAN | https://CRAN.R-project.org/package=data.table |
| Manuscript analysis workflow |
This paper | DOI: 10.5281/zenodo.6773715 |
| Other | ||
| Ex Vivo Drug Sensitivity Data | This paper | http://vizome.org/aml2/ and https://biodev.github.io/BeatAML2/ |
| Clinical Annotations | This paper | Table S1 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patient Sample Collection and Cohort Organization
The complete OHSU Beat AML cohort represents sample collection and characterization over a span of 10 years. The initial cohort, first reported in (Tyner et al., 2018) represent the first two waves of patient accrual and sample data (denoted “Waves 1+2”). Additional longitudinal samples for Waves 1+2, in addition to patient accrual represents the final two waves (“Waves 3+4”). Harmonization of these datasets together for specific analyses is denoted as the “harmonized data”. For this manuscript we only utilized specimens from the first available timepoint (defined by a 5-day interval) for each patient such that each patient was represented by a single sample for a given datatype. Additionally, we removed samples taken while a patient was in remission from consideration.
All patients gave informed consent to participate in this study, which had the approval and guidance of the institutional review boards at Oregon Health & Science University (OHSU), University of Utah, University of Texas Medical Center (UT Southwestern), University of Miami, University of Colorado, University of Florida, National Institutes of Health (NIH), and University of Kansas (KUMC). Samples were sent to the coordinating center (OHSU; IRB#9570; #4422; NCT01728402) where they were coded and processed. Specific names of centers associated with each specimen were coded and centers providing less than 5 samples were aggregated together and given one center identifier. Clinical, prognostic, genetic, cytogenetic, and pathologic lab values as well as treatment and outcome data were manually curated from patient electronic medical records. Patients were assigned a specific diagnosis based on the prioritization of genetic and clinical factors as determined by WHO guidelines (Arber et al., 2016). To prevent re-identification, any patient over the age of 90 was placed into a >90 aggregated age bracket. Genetic characterization of the leukemia samples included results of clinical deep-sequencing panels of genes commonly mutated in hematologic malignancies (Sequenom and GeneTrails (OHSU); Foundation Medicine (UTSW); Genoptix; and Illumina).
METHOD DETAILS
Patient Specimen Processing
Processing of patient specimens, isolation of nucleic acids, DNA- and RNA-sequencing, detection of FLT3-ITD and 4-base pair NPM1 insertion, performance of ex vivo drug sensitivity assays, and initial analytical workflows were performed in identical fashion as in our prior study (Tyner et al., 2018). Any changes that were made to analytical approaches and techniques employed in this study are described below.
DNA Sequencing
For DNA sequencing, we used the 11.9 megabase custom capture library that was developed to provide coverage of all variants previously reported in AML (including all variants that were detected from exome sequencing in our prior study). The genes, variants, and capture regions for this custom library were reported in the Supplementary Information of our prior study (Table S14 in (Tyner et al., 2018)), as well as detailed methods for pre-processing and analysis (Supplemental Methods).
European Leukemia Network Prognostic Categorization
Some updates were implemented to our pipeline for calling ELN categories. First, we confined calls in this study to specimens taken at initial acute leukemia diagnosis, and did not place specimens that were taken in remission, at relapse, or from cases with a non-AML diagnosis (e.g. MDS) into ELN categories. Second, for consideration of mutation of ASXL1, RUNX1, or TP53, we used our internal deep sequencing data as the primary source of information, rather than clinical sequencing results curated from the electronic medical record. This was due to a higher percentage of samples with available sequencing data for these three genes from our internal versus curated clinical data. We compared our internal results with available clinical sequencing results for these genes, and we manually adjudicated the minority of instances where discrepancies existed. We also manually adjudicated calls for bi-allelic CEBPA, using both our internal sequencing and results from clinical sequencing. Relevant cytogenetic events were parsed from clinical karyotype and cytogenetic results, and we used our consensus FLT3-ITD and NPM1 calls as described in our prior study (Tyner et al., 2018), including data from internal PCR amplicon size-based evaluation using published primers (Falini et al., 2007; Kottaridis et al., 2001). All of these results were used to call Favorable, Intermediate, or Adverse categories according to the ELN 2017 criteria established in (Dohner et al., 2017) with a minority of cases requiring indication of uncertainty due to lack of knowledge regarding FLT3-ITD allelic ratio. Finally, we discontinued usage of calls based on the 2008 ELN guidelines.
Ex vivo Functional Drug Screen Data Processing
Dose response curves for all the drugs were fit using probit models (Kurtz et al., 2017) similar to the flagship Beat AML manuscript (Tyner et al., 2018). However, the QC and summarization approach was modified for the situations where multiple replicates were available. In the description below, a profile indicates a sample - inhibitor pair. Profiles with no replicates were subject to probit curve fit thresholds. Specifically, profiles with both Deviance > 2 and AIC > 12 were removed. Profiles with replicates were first assessed based on the variability of the readouts (normalized viability capped at 100%). For each plate, the standard deviation (SD) across replicates for each of the 7 concentration points was assessed and the plate was removed if 4 or more of the concentrations were considered highly variable (SD > 25). If replicates were available across multiple plates, the viability at each concentration point was first averaged per plate. The SD across plates was computed per concentration point similar to above and an AUC was computed from probit fits to each plate. Similar to the initial replicate filter, profiles were removed if both 4 or more concentrations had highly variable SD and the SD(AUC) > 50. Finally, a single probit curve was used to determine the AUC and IC values for each profile. As with (Tyner et al., 2018), drug response is indicated by color coding scheme (i.e., Red – sensitivity, white - intermediate, Blue - resistance).
Organization of Drugs into Families Based on Targets and Pathways
We utilized two primary data sources for information regarding drug/target relationships, Targetome (Blucher et al., 2017; Blucher et al., 2019; Choonoo et al., 2019) and KINOMEscan (Davis et al., 2011). For determination of top tier targets of each drug from KINOMEscan and Targetome V2 data, we used a threshold that was 10-fold higher than the second lowest Kd for each drug (termed Tier1 hits). We defined a set of high confidence drug/target relationships requiring either a Tier1 with at most a Kd of 25 in KINOMEscan and/or Targetome. Additionally, we required Targetome to have at least two references with at least two unique supporting assay values. This high confidence set was combined with a small number of additional interactions annotated manually (see Table S4 for full annotations of drug family targets). We utilized the IUPHAR family classifications for targets (Armstrong et al., 2020). Targets were assigned to the lowest level of the family hierarchy as well as up to two higher levels. The resulting inhibitor/family relationships were additionally manually curated.
Inhibitor family response
To generate gene family scores, we first rescaled the inhibitor AUC responses to be between 0 and 1 in order to ensure comparability between inhibitors with different concentration ranges. We then used the single sample GSEA (ssGSEA; (Barbie et al., 2009)) approach as implemented in GSVA package (Hanzelmann et al., 2013) to generate a score per family. We required at least 5 inhibitors per family and did not implement the cohort range normalization as only a single patient was provided at a time. Note that the use of ssGSEA as opposed to a principal component-based scoring was driven by the need to account for differences in drug coverage amongst patients.
RNA Sequencing
Gene-level RNASeq counts were generated as in the previous Beat AML manuscript (Tyner et al., 2018). Again, we used conditional quantile normalization (CQN; (Hansen et al., 2012)). In order to facilitate comparison with previous results we used the CQN reference distribution parameters learned from the Waves 1+2 samples to apply the normalization to the Waves 3+4 samples.
Weighted Gene Co-expression Network Analysis
Module Formation and Summarization
We started with the original set of 14 gene expression modules (13 + Mod0 (grey) ‘outlier’ module) from the Beat AML manuscript derived using the weighted gene co-expression network analysis (WGCNA) (Zhang and Horvath, 2005) methodology. With the exception of Mod0 (grey), these gene sets are typically summarized by their ‘eigengene’ which is their first principal component (PC) score (Horvath and Dong, 2008). As the mod0 (grey) gene-set was more heterogenous, we kept the first 5 PC scores for the eigengene. Since the CQN normalization approach above did not change the original data, we were able to directly ‘predict’ the PC scores of the Waves 3+4 cohort for each module. First, we centered and scaled (C/S) the Waves 3+4 cohort expression matrix using the mean and standard deviations estimated from the original cohort. We then formed scores per Waves 3+4 cohort patient as the linear combination of their C/S expression values and the corresponding column of the original matrix of eigenvectors/rotations.
Module Membership
A standard WGCNA methodology is the formation of kME values which are defined as the correlation of gene expression with the module eigengene (PC1 score as described above) (Langfelder et al., 2008). Genes with high kME are considered to have higher (fuzzy) membership in each module or alternatively can be seen as more ‘hub-like’ in a network context (Horvath and Dong, 2008). In this instance the correlation used was the robust biweight midcorrelation (Langfelder and Horvath, 2012).
Module Associations
To relate WGCNA modules to external continuous variables, we assessed direct correlation of a variable with a module eigengene which indicates whether the pattern of a module covaries with the variable (Horvath et al., 2006).
Cell-type Scores
We first divided the Waves 1+2 cohort with RNASeq samples by whether they were bone marrow derived or from peripheral blood/leukapheresis and centered and scaled them separately. Using the top 30 genes for each of six single-cell AML tumor-derived signatures (Van Galen et al., 2019), we computed gene set scores. Similar to the WGCNA eigengenes, our scores were based on the first principal component and aligned with the average expression. This is similar to the approach used in the context of pathway-based geneset analysis (PLAGE; (Tomfohr et al., 2005)). Waves 3+4 cell-type scores were ‘predicted’ from ‘Waves 1+2’ using the same approach described for the WGCNA eigengenes but this time using separate centering and scaling for the specimen types.
Formation of Genomic Features
We first combined the set of expression module and cell-type scores from the RNASeq samples. The two continuous datatypes were correlated, often with modules having opposing correlations with cell-types associated with low and high differentiation. To address this, we removed the expression modules seen as being both positively and negatively correlated (abs ( r )>=.6) with a cell-type. The consensus AML fusions and mutation calls were then added in as binary features.
Random Survival Forest
Using the combined set of genomic features, we fit a conditional random forest (cforest) model using the `partykit` package with post-diagnosis survival, measured in days, as the outcome (Hothorn et al., 2006). Our model utilized 1,000 trees and 63.2% subsampling as opposed to the bootstrap as previously suggested (Strobl et al., 2007). We used the predictions (i.e. log10 median survival) from this ‘model to fit a single surrogate conditional inference tree model to facilitate interpretation (Pearson’s correlation between model predictions: .877).
Where indicated, high vs low group categorizations were also performed using ctree but limiting depth to a single split and requiring minimum group size of 20.
LSC17
For comparisons with the LSC17 signature (Ng et al 2017), we replicated the signature using the coefficients in the manuscript, which was reported as the optimized signature by the authors (which was also patented WO2017132749A1). Categorization of expression for Hazard Ratio and other analyses was done using two different “split types”: median as described in (Ng et al., 2016) or ctree to facilitate comparison with PEAR1.
Clinical curation via Natural Language Processing (NLP)
An NLP workflow was developed to automatically extract key clinical data elements that were only available in the Electronic Health Records (EHR) in unstructured clinical notes. The NLP pipeline was written in Python which incorporated a commercially available text miner - Linguamatics’ Interactive Information Extraction (I2E) platform, as a major component, along with Python-based document preprocessing, results output logic, and evaluation components. Data from the manually curated Gold Standard Data Set (GSDS) from Waves 1+2 were used to evaluate the performance of the NLP system. An NLP Data Set (NLPDS) was assembled that contained as far as possible the source pathology documents set from which the GSDS was originally obtained by manual review supervised by the data manager. This was subdivided into 5 partitions, and used in an iterative training-development-validation cycle to optimize the NLP and Python code. For evaluation purposes, we treated partitions 1 and 2 as a single training set and partitions 3, 4, and 5 as a single test set. Overall, the training set consisted of 108 patients with 134 specimens and 241 documents, and the test set consisted of 252 patients with 289 specimens and 532 documents. Evaluation of NLP results led in some cases to discovery of missing or incorrect data in the GSDS (which was estimated to have an overall error rate of 9%), further improving data quality. For the NLP data by data element, Accuracy ranged from 79% to 93%, Precision 85% to 96%, Recall from 76% to 93% and F1-score (the harmonic mean of the precision and recall) 81% to 94%.
PEAR1 Flow Cytometry Analysis
AML patient samples were thawed, washed in pre-warmed media, and resuspended in phosphate-buffered saline (PBS). Cells were then stained for viability via zombie aqua dye (Biolegend, #423101) for 15 minutes. Cells were washed in FACS buffer (PBS, 2% fetal bovine serum, sodium azide, EDTA) and then Fc receptors were blocked for 5 minutes (Biolegend, #422301) prior to staining with anti-PEAR1 antibody conjugated to brilliant violet 650 (clone 492621; BD Biosciences Catalog #748063) at a dilution of 1:40 for 30 minutes covered on ice. Cells were then fixed with 2% paraformaldehyde for 20 minutes and analyzed with a Cytek Aurora flow cytometer. Analysis and figure generation was performed with FlowJo Software. PEAR1 positive cells were identified by first gating single cells using forward and side scatter, then gating only live cells (zombie aqua dye negative), then gating for PEAR1+ cells as compared to a fluorescence minus one (FMO) control.
QUANTIFICATION AND STATISTICAL ANALYSIS
In all cases, statistical analyses of data, including tests used, exact value of n (where n indicates the number of patient specimens that were available for a given analysis), definition of center, dispersion, and precision measures, and correction for false discovery rate (FDR) are reported in the descriptive sections of the results, Method Details section of STAR Methods, in corresponding figure legends, and in figures, themselves, where possible. Measures of significance or correlation are reported as p- or r-values, with p-values corrected for FDR, where appropriate. All patient specimens with material sufficient for analysis and with data passing quality control thresholds (described above in Method Details section of STAR Methods and in (Tyner et al., 2018)) were included in the cohort and dataset.
ADDITIONAL RESOURCES
All data can be accessed and queried through our online, interactive user interface, Vizome, at www.vizome.org/aml2. Original code for replicating the paper analyses based on publicly available data has been deposited at https://github.com/biodev/beataml2_manuscript. The DOI is listed in the key resources table. A frequently asked questions page for the dataset is found at https://biodev.github.io/BeatAML2/.
Supplementary Material
Table S1. Clinical Summary, Related to Figure 1.
Table S2. Mutation-cell type associations, Related to Figure 2. Results from a Welch’s t-test (t and pval columns) comparing the average cell-type scores between mutated vs wild type patients. The sample size for each comparison is provided in terms of the number of mutated and wildtype (num_muts and num_neg). The effect size is given in terms of both raw average difference (estimate) and Glass’s delta (glass_d) relative to the wildtype group. Multiple-testing adjustments were performed using qvalue.
Table S3. Drug-cell type correlations with top mutation interactions, Related to Figure 3. Pearson’s correlation (cor) and resulting BY FDR (cor_fdr) are provided for each drug and cell_type along with results from the top gene (inter_gene) ranked by interaction pvalue. Reported for the interaction terms are the interaction coefficient (inter_est), the number of mutations (inter_muts) and the qvalue.
Table S4. Drug Family annotations, Related to Figure 4. Provided are a series of mapping files supporting the assignment of drugs to families. Specifically, we provided separate sheets describing (1) the family to synonym mapping, (2) drug to family mapping as well as (3) the inhibitor to gene/family mapping. In this latter tab, in addition to drug and gene family information we provide a summary of the evidence linking drugs to genes. This includes the minimum assay value reported (db_min_kd and targetome_min_assay), the amount of literature support and number of distinct values reported in Targetome (Blucher et al., 2017; Blucher et al., 2019; Choonoo et al., 2019) for assay values < 25 as well as our tier assignments. For each tab, manual adjustments are indicated by TRUE/FALSE.
Table S5. Associations between drug families and mutations, Related to Figure 4B. Provided are results from a Welch’s t-test (t and pval columns) comparing the difference in average drug enrichment score between patient mutation groups for a given gene. Each family is represented by both the IUPHAR name (family) as well as our synonym. Sample size is provided in terms of the number of patients with and without mutations (num_muts and num_neg respectively). The effect size is given in terms of both raw average difference (estimate) and Glass’s delta (glass_d) relative to the wildtype group. Multiple-testing adjustments were performed using qvalue.
Table S6. Drug family-cell type score correlations with top interactions, Related to Figure 5. The Pearson’s correlation between each drug family enrichment score and cell-type score is provided (cor) along with BY FDR adjustments (cor_fdr). Results for the corresponding top mutation ranked by interaction pvalue are provided. Reported for the interaction terms are the interaction coefficient (inter_est), the number of mutations (inter_muts) and the qvalue.
Table S7. WGCNA module membership, Related to Figure 6B. Gene assignments are provided for each WGCNA expression module including module 3 which is highlighted in Figure 6B. Modules are indicated by both label and corresponding color. Genes are described by Ensembl unique identifier and related annotation. The kME for the combined expression set (comb_kme) indicates the correlation of each gene relative to its module’s eigengene. As Mod0 (grey) is not a cohesive module but more of a container for ‘outlier’ genes, we do not report corresponding kME’s.
Highlights.
Acute myeloid leukemia patient cohort with clinical, molecular, drug response data
Validation and discovery of diverse biological features of drug response
Broad mapping of tumor cell differentiation state impacting response to drugs
Modeling reveals a strong and targetable determinant of clinical outcome
ACKNOWLEDGEMENTS
We thank all of our patients at all sites for donating precious time and tissue. DNA and RNA quality assessments, library creation, and short read sequencing assays were performed by the OHSU Massively Parallel Sequencing Shared Resource. We thank Oscar Brück, Disha Malani, Olli Kallioniemi, and Kimmo Porkka for rapidly facilitating analysis of PEAR1 expression correlation with overall survival in their AML patient dataset (Malani et al., 2022) and Oscar Brück for rapidly executing our analysis scripts from this study on those data.
Funding
Funding for this project was provided in part by The Leukemia & Lymphoma Society (for the Waves 1+2 dataset) and the Knight Cancer Research Institute (Oregon Health & Science University, OHSU). Supported by grants from the National Cancer Institute (U01CA217862, U54CA224019, U01CA214116) and NIH/NCATS CTSA UL1TR002369 (SKM, BW). CET receives grant support from the National Cancer Institute (R01CA214428). AA is supported by grants from the National Cancer Institute (R01CA229875), National Heart, Lung, and Blood Institute (R01HL155426), American Cancer Society (RSG-17–187-01-LIB), Alex Lemonade/Babich RUNX1 Foundation, EvansMDS Foundation, and a V foundation Scholar award. Funding was provided to TPB by an American Society of Hematology Research Restart Award, an American Society of Hematology Scholar Award, and 1 K08 CA245224 from NCI. SKJ is supported by the ARCS Scholar Foundation, The Paul & Daisy Soros Fellowship, and the National Cancer Institute (F30CA239335). JEM receives funding from the American Cancer Society (RSG-19–184-01 – LIB) and NIH/NCI (R01 CA247943). HZ received grants from the National Cancer Institute (R00 5K99CA237630) and the Oregon Medical Research Foundation New Investigator Award. Some patient samples used in this work were provided by the Division of Hematology Biorepository at Huntsman Cancer Institute, University of Utah, which is supported by the National Cancer Institute of the National Institutes of Health under Award Number P30CA042014. Additional funding came from the Huntsman Center of Excellence in Hematologic Malignancies and Hematology at Huntsman Cancer Institute, University of Utah. CRC received a Scholar in Clinical Research Award from The Leukemia & Lymphoma Society (2400–13), was distinguished with a Pierre Chagnon Professorship in Stem Cell Biology and Blood & Marrow Transplant and a UF Research Foundation Professorship. This work was supported in part by the Intramural Research Program of the National Heart, Lung, and Blood Institute of the National Institutes of Health. BJD received funding from the Howard Hughes Medical Institute. JWT received grants from the V Foundation for Cancer Research, the Gabrielle’s Angel Foundation for Cancer Research, the Mark Foundation for Cancer Research, the Silver Family Foundation, and the National Cancer Institute (R01CA245002, R01CA262758).
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
Competing Interests CET receives research support from Notable Labs and serves as a scientific liaison for AstraZeneca. JEM receives research funding from Gilead Pharmaceutical and serves on a scientific advisory board for Ionis Pharmaceuticals. MWD serves on the advisory boards and/or as a consultant for Novartis, Incyte, and BMS and receives research funding from BMS and Gilead. CSH receives research funding from Sellas. TLL consults for Jazz Pharmaceuticals and receives research funding from Tolero, Gilead, Prescient, Ono, Bio-Path, Mateon, Genentech/Roche, Trovagene, AbbVie, Pfizer, Celgene, Imago, Astellas, Karyopharm, Seattle Genetics, and Incyte. DAP receives research funding from Pfizer and Agios and served on advisory boards for Pfizer, Celyad, Agios, Celgene, AbbVie, Argenx, Takeda and Servier. BJD serves on the advisory boards for Aileron Therapeutics, Aptose, Blueprint Medicines, Cepheid, EnLiven Therapeutics, Gilead, GRAIL, Iterion Therapeutics, Nemucore Medical Innovations, the Novartis CML Molecular Monitoring Steering Committee, Recludix Pharma, the RUNX1 Research Program, ALLCRON Pharma, VB Therapeutics, and Vincerx Pharma, the Board of Directors for Amgen, and receives research funding from EnLiven and Recludix. BJD is principal investigator or coinvestigator on Novartis, BMS, and Pfizer clinical trials. His institution, Oregon Health & Science University, has contracts with these companies to pay for patient costs, nurse and data manager salaries, and institutional overhead. He does not derive salary, nor does his laboratory receive funds from these contracts. JWT has received research support from Acerta, Agios, Aptose, Array, AstraZeneca, Constellation, Genentech, Gilead, Incyte, Janssen, Kronos, Meryx, Petra, Schrodinger, Seattle Genetics, Syros, Takeda, and Tolero and serves on the advisory board for Recludix Pharma. The authors certify that all compounds tested in this study were chosen without input from any of our industry partners. A subset of findings from this manuscript have been included in a pending patent application.
REFERENCES
- Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, and Vardiman JW (2016). The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127, 2391–2405. [DOI] [PubMed] [Google Scholar]
- Armstrong JF, Faccenda E, Harding SD, Pawson AJ, Southan C, Sharman JL, Campo B, Cavanagh DR, Alexander SPH, Davenport AP, et al. (2020). The IUPHAR/BPS Guide to PHARMACOLOGY in 2020: extending immunopharmacology content and introducing the IUPHAR/MMV Guide to MALARIA PHARMACOLOGY. Nucleic Acids Res 48, D1006–D1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila Cobos F, Vandesompele J, Mestdagh P, and De Preter K (2018). Computational deconvolution of transcriptomics data from mixed cell populations. Bioinformatics 34, 1969–1979. [DOI] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. (2009). Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Yekutieli D (2001). The Control of the False Discovery Rate in Multiple Testing Under Dependency. The Annals of Statistics 29, 1165–1188. [Google Scholar]
- Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, and Sultan C (1976). Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol 33, 451–458. [DOI] [PubMed] [Google Scholar]
- Bhatnagar B, Kohlschmidt J, Mrozek K, Zhao Q, Fisher JL, Nicolet D, Walker CJ, Mims AS, Oakes C, Giacopelli B, et al. (2021). Poor Survival and Differential Impact of Genetic Features of Black Patients with Acute Myeloid Leukemia. Cancer Discov 11, 626–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biino G, Santimone I, Minelli C, Sorice R, Frongia B, Traglia M, Ulivi S, Di Castelnuovo A, Gogele M, Nutile T, et al. (2013). Age- and sex-related variations in platelet count in Italy: a proposal of reference ranges based on 40987 subjects’ data. PLoS One 8, e54289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blucher AS, Choonoo G, Kulesz-Martin M, Wu G, and McWeeney SK (2017). Evidence-Based Precision Oncology with the Cancer Targetome. Trends in pharmacological sciences 38, 1085–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blucher AS, McWeeney SK, Stein L, and Wu G (2019). Visualization of drug target interactions in the contexts of pathways and networks with ReactomeFIViz. F1000Research 8, 908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N., Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ Jr., Laird PW, et al. (2013). Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368, 2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choonoo G, Blucher AS, Higgins S, Boardman M, Jeng S, Zheng C, Jacobs J, Anderson A, Chamberlin S, Evans N, et al. (2019). Illuminating biological pathways for drug targeting in head and neck squamous cell carcinoma. PLoS One 14, e0223639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criel M, Izzi B, Vandenbriele C, Liesenborghs L, Van Kerckhoven S, Lox M, Cludts K, Jones EA, Vanassche T, Verhamme P, and Hoylaerts M (2016). Absence of Pear1 does not affect murine platelet function in vivo. Thromb Res 146, 76–83. [DOI] [PubMed] [Google Scholar]
- Dao FT, Wang J, Yang L, and Qin YZ (2021). Development of a poor-prognostic-mutations derived immune prognostic model for acute myeloid leukemia. Scientific reports 11, 4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, and Zarrinkar PP (2011). Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, Frankfurt O, Konopleva M, Wei AH, Kantarjian HM, et al. (2019). Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 133, 7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, Swords R, Collins RH, Mannis GN, Pollyea DA, et al. (2018). Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N Engl J Med 378, 2386–2398. [DOI] [PubMed] [Google Scholar]
- Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, Dombret H, Ebert BL, Fenaux P, Larson RA, et al. (2017). Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129, 424–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eicher JD, Chami N, Kacprowski T, Nomura A, Chen MH, Yanek LR, Tajuddin SM, Schick UM, Slater AJ, Pankratz N, et al. (2016). Platelet-Related Variants Identified by Exomechip Meta-analysis in 157,293 Individuals. Am J Hum Genet 99, 40–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsayed AH, Rafiee R, Cao X, Raimondi S, Downing JR, Ribeiro R, Fan Y, Gruber TA, Baker S, Klco J, et al. (2020). A six-gene leukemic stem cell score identifies high risk pediatric acute myeloid leukemia. Leukemia 34, 735–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falini B, Nicoletti I, Martelli MF, and Mecucci C (2007). Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc+ AML): biologic and clinical features. Blood 109, 874–885. [DOI] [PubMed] [Google Scholar]
- Faraday N, Yanek LR, Yang XP, Mathias R, Herrera-Galeano JE, Suktitipat B, Qayyum R, Johnson AD, Chen MH, Tofler GH, et al. (2011). Identification of a specific intronic PEAR1 gene variant associated with greater platelet aggregability and protein expression. Blood 118, 3367–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisch AS, Yerges-Armstrong LM, Backman JD, Wang H, Donnelly P, Ryan KA, Parihar A, Pavlovich MA, Mitchell BD, O’Connell JR, et al. (2015). Genetic Variation in the Platelet Endothelial Aggregation Receptor 1 Gene Results in Endothelial Dysfunction. PLoS One 10, e0138795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller TF, Ghazalpour A, Aten JE, Drake TA, Lusis AJ, and Horvath S (2007). Weighted gene coexpression network analysis strategies applied to mouse weight. Mamm Genome 18, 463–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal H, Amariglio N, Trakhtenbrot L, Jacob-Hirsh J, Margalit O, Avigdor A, Nagler A, Tavor S, Ein-Dor L, Lapidot T, et al. (2006). Gene expression profiles of AML derived stem cells; similarity to hematopoietic stem cells. Leukemia 20, 2147–2154. [DOI] [PubMed] [Google Scholar]
- Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, et al. (2014). Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 371, 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentles AJ, Plevritis SK, Majeti R, and Alizadeh AA (2010). Association of a leukemic stem cell gene expression signature with clinical outcomes in acute myeloid leukemia. JAMA 304, 2706–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, Irizarry RA, and Wu Z (2012). Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics 13, 204–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzelmann S, Castelo R, and Guinney J (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC bioinformatics 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera-Galeano JE, Becker DM, Wilson AF, Yanek LR, Bray P, Vaidya D, Faraday N, and Becker LC (2008). A novel variant in the platelet endothelial aggregation receptor-1 gene is associated with increased platelet aggregability. Arterioscler Thromb Vasc Biol 28, 1484–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilu-Dadia R, Hakim-Mishnaevski K, Levy-Adam F, and Kurant E (2018). Draper-mediated JNK signaling is required for glial phagocytosis of apoptotic neurons during Drosophila metamorphosis. Glia 66, 1520–1532. [DOI] [PubMed] [Google Scholar]
- Horibata S, Gui G, Lack J, DeStefano CB, Gottesman MM, and Hourigan CS (2019). Heterogeneity in refractory acute myeloid leukemia. Proc Natl Acad Sci U S A 116, 10494–10503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, and Dong J (2008). Geometric interpretation of gene coexpression network analysis. PLoS computational biology 4, e1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Zhang B, Carlson M, Lu KV, Zhu S, Felciano RM, Laurance MF, Zhao W, Qi S, Chen Z, et al. (2006). Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc Natl Acad Sci U S A 103, 17402–17407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hothorn T, Hornik K, and Zeileis A (2006). Unbiased Recursive Partitioning: A Conditional Inference Framework. Journal of Computational and Graphical Statistics 15, 651–674. [Google Scholar]
- Huang ME, Ye YC, Chen SR, Chai JR, Lu JX, Zhoa L, Gu LJ, and Wang ZY (1988). Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 72, 567–572. [PubMed] [Google Scholar]
- Izzi B, Gianfagna F, Yang WY, Cludts K, De Curtis A, Verhamme P, Di Castelnuovo A, Cerletti C, Donati MB, de Gaetano G, et al. (2019). Variation of PEAR1 DNA methylation influences platelet and leukocyte function. Clin Epigenetics 11, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzi B, Noro F, Cludts K, Freson K, and Hoylaerts MF (2018). Cell-Specific PEAR1 Methylation Studies Reveal a Locus that Coordinates Expression of Multiple Genes. Int J Mol Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzi B, Pistoni M, Cludts K, Akkor P, Lambrechts D, Verfaillie C, Verhamme P, Freson K, and Hoylaerts MF (2016). Allele-specific DNA methylation reinforces PEAR1 enhancer activity. Blood 128, 1003–1012. [DOI] [PubMed] [Google Scholar]
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. (2014). Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371, 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Xu J, and Ward E (2010). Cancer statistics, 2010. CA: a cancer journal for clinicians 60, 277–300. [DOI] [PubMed] [Google Scholar]
- Johnson AD, Yanek LR, Chen MH, Faraday N, Larson MG, Tofler G, Lin SJ, Kraja AT, Province MA, Yang Q, et al. (2010). Genome-wide meta-analyses identifies seven loci associated with platelet aggregation in response to agonists. Nat Genet 42, 608–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CI, Bray S, Garner SF, Stephens J, de Bono B, Angenent WG, Bentley D, Burns P, Coffey A, Deloukas P, et al. (2009). A functional genomics approach reveals novel quantitative trait loci associated with platelet signaling pathways. Blood 114, 1405–1416. [DOI] [PubMed] [Google Scholar]
- Kardeby C, Falker K, Haining EJ, Criel M, Lindkvist M, Barroso R, Pahlsson P, Ljungberg LU, Tengdelius M, Rainger GE, et al. (2019). Synthetic glycopolymers and natural fucoidans cause human platelet aggregation via PEAR1 and GPIbalpha. Blood advances 3, 275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauskot A, Di Michele M, Loyen S, Freson K, Verhamme P, and Hoylaerts MF (2012). A novel mechanism of sustained platelet alphaIIbbeta3 activation via PEAR1. Blood 119, 4056–4065. [DOI] [PubMed] [Google Scholar]
- Kauskot A, Vandenbriele C, Louwette S, Gijsbers R, Tousseyn T, Freson K, Verhamme P, and Hoylaerts MF (2013). PEAR1 attenuates megakaryopoiesis via control of the PI3K/PTEN pathway. Blood 121, 5208–5217. [DOI] [PubMed] [Google Scholar]
- Keramati AR, Yanek LR, Iyer K, Taub MA, Ruczinski I, Becker DM, Becker LC, Faraday N, and Mathias RA (2019). Targeted deep sequencing of the PEAR1 locus for platelet aggregation in European and African American families. Platelets 30, 380–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, Walker H, Wheatley K, Bowen DT, Burnett AK, et al. (2001). The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood 98, 1752–1759. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Rozov FN, Zinovyeva MV, Hendrikx PJ, Jiang Y, Visser JW, and Belyavsky AV (2007). Jedi--a novel transmembrane protein expressed in early hematopoietic cells. J Cell Biochem 101, 767–784. [DOI] [PubMed] [Google Scholar]
- Kurtz SE, Eide CA, Kaempf A, Khanna V, Savage SL, Rofelty A, English I, Ho H, Pandya R, Bolosky WJ, et al. (2017). Molecularly targeted drug combinations demonstrate selective effectiveness for myeloid- and lymphoid-derived hematologic malignancies. Proc Natl Acad Sci U S A 114, E7554–E7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuusanmaki H, Leppa AM, Polonen P, Kontro M, Dufva O, Deb D, Yadav B, Bruck O, Kumar A, Everaus H, et al. (2020). Phenotype-based drug screening reveals association between venetoclax response and differentiation stage in acute myeloid leukemia. Haematologica 105, 708–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancet JE, Uy GL, Cortes JE, Newell LF, Lin TL, Ritchie EK, Stuart RK, Strickland SA, Hogge D, Solomon SR, et al. (2018). CPX-351 (cytarabine and daunorubicin) Liposome for Injection Versus Conventional Cytarabine Plus Daunorubicin in Older Patients With Newly Diagnosed Secondary Acute Myeloid Leukemia. J Clin Oncol 36, 2684–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, and Horvath S (2012). Fast R Functions for Robust Correlations and Hierarchical Clustering. Journal of Statistical Software 46. [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Zhang B, and Horvath S (2008). Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics 24, 719–720. [DOI] [PubMed] [Google Scholar]
- Lewis JP, Ryan K, O’Connell JR, Horenstein RB, Damcott CM, Gibson Q, Pollin TI, Mitchell BD, Beitelshees AL, Pakzy R, et al. (2013). Genetic variation in PEAR1 is associated with platelet aggregation and cardiovascular outcomes. Circ Cardiovasc Genet 6, 184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder MM, Leppa AM, Hellesoy M, Dowling P, Malyutina A, Kopperud R, Bazou D, Andersson E, Parsons A, Tang J, et al. (2020). Multi-parametric single cell evaluation defines distinct drug responses in healthy hematological cells that are retained in corresponding malignant cell types. Haematologica 105, 1527–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malani D, Kumar A, Bruck O, Kontro M, Yadav B, Hellesoy M, Kuusanmaki H, Dufva O, Kankainen M, Eldfors S, et al. (2022). Implementing a Functional Precision Medicine Tumor Board for Acute Myeloid Leukemia. Cancer Discov 12, 388–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda N, Bao M, Lin H, Clauser K, Komuves L, Quertermous T, Conley PB, Phillips DR, and Hart MJ (2005). Platelet endothelial aggregation receptor 1 (PEAR1), a novel epidermal growth factor repeat-containing transmembrane receptor, participates in platelet contact-induced activation. J Biol Chem 280, 24680–24689. [DOI] [PubMed] [Google Scholar]
- Ng SW, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, Arruda A, Popescu A, Gupta V, Schimmer AD, et al. (2016). A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 540, 433–437. [DOI] [PubMed] [Google Scholar]
- Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F, Bolli N, et al. (2016). Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med 374, 2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei S, Pollyea DA, Gustafson A, Stevens BM, Minhajuddin M, Fu R, Riemondy KA, Gillen AE, Sheridan RM, Kim J, et al. (2020). Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Patients with Acute Myeloid Leukemia. Cancer Discov 10, 536–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S, Montesinos P, Baer MR, Larson RA, Ustun C, et al. (2019). Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N Engl J Med 381, 1728–1740. [DOI] [PubMed] [Google Scholar]
- Petersdorf SH, Kopecky KJ, Slovak M, Willman C, Nevill T, Brandwein J, Larson RA, Erba HP, Stiff PJ, Stuart RK, et al. (2013). A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 121, 4854–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulte D, Jansen L, Castro FA, Krilaviciute A, Katalinic A, Barnes B, Ressing M, Holleczek B, Luttmann S, Brenner H, and Group GCSW (2016). Survival in patients with acute myeloblastic leukemia in Germany and the United States: Major differences in survival in young adults. International journal of cancer 139, 1289–1296. [DOI] [PubMed] [Google Scholar]
- Qayyum R, Becker LC, Becker DM, Faraday N, Yanek LR, Leal SM, Shaw C, Mathias R, Suktitipat B, and Bray PF (2015). Genome-wide association study of platelet aggregation in African Americans. BMC Genet 16, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romine KA, Nechiporuk T, Bottomly D, Jeng S, McWeeney SK, Kaempf A, Corces MR, Majeti R, and Tyner JW (2021). Monocytic differentiation and AHR signaling as Primary Nodes of BET Inhibitor Response in Acute Myeloid Leukemia. Blood Cancer Discov 2, 518–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheib JL, Sullivan CS, and Carter BD (2012). Jedi-1 and MEGF10 signal engulfment of apoptotic neurons through the tyrosine kinase Syk. J Neurosci 32, 13022–13031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEER (2021). National Cancer Institute: Surveillance, Epidemiology, and End Results Program In https://seer.cancer.gov/statfacts/html/amyl.html.
- Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, Zhu J, Tang W, Sun GL, Yang KQ, et al. (1997). Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood 89, 3354–3360. [PubMed] [Google Scholar]
- Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, Stone RM, DeAngelo DJ, Levine RL, Flinn IW, et al. (2017). Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 130, 722–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, Thiede C, Prior TW, Dohner K, Marcucci G, et al. (2017). Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med 377, 454–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, and Tibshirani R (2003). Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100, 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobl C, Boulesteix AL, Zeileis A, and Hothorn T (2007). Bias in random forest variable importance measures: illustrations, sources and a solution. BMC bioinformatics 8, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan CS, Scheib JL, Ma Z, Dang RP, Schafer JM, Hickman FE, Brodsky FM, Ravichandran KS, and Carter BD (2014). The adaptor protein GULP promotes Jedi-1-mediated phagocytosis through a clathrin-dependent mechanism. Mol Biol Cell 25, 1925–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Vandenbriele C, Kauskot A, Verhamme P, Hoylaerts MF, and Wright GJ (2015). A Human Platelet Receptor Protein Microarray Identifies the High Affinity Immunoglobulin E Receptor Subunit alpha (FcepsilonR1alpha) as an Activating Platelet Endothelium Aggregation Receptor 1 (PEAR1) Ligand. Mol Cell Proteomics 14, 1265–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomfohr J, Lu J, and Kepler TB (2005). Pathway level analysis of gene expression using singular value decomposition. BMC bioinformatics 6, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, Long N, Schultz AR, Traer E, Abel M, et al. (2018). Functional genomic landscape of acute myeloid leukaemia. Nature 562, 526–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Galen P, Hovestadt V, Wadsworth Ii MH, Hughes TK, Griffin GK, Battaglia S, Verga JA, Stephansky J, Pastika TJ, Lombardi Story J, et al. (2019). Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell 176, 1265–1281 e1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbriele C, Kauskot A, Vandersmissen I, Criel M, Geenens R, Craps S, Luttun A, Janssens S, Hoylaerts MF, and Verhamme P (2015). Platelet endothelial aggregation receptor-1: a novel modifier of neoangiogenesis. Cardiovasc Res 108, 124–138. [DOI] [PubMed] [Google Scholar]
- Vandenbriele C, Sun Y, Criel M, Cludts K, Van Kerckhoven S, Izzi B, Vanassche T, Verhamme P, and Hoylaerts MF (2016). Dextran sulfate triggers platelet aggregation via direct activation of PEAR1. Platelets 27, 365–372. [DOI] [PubMed] [Google Scholar]
- Wang YH, Lin CC, Yao CY, Hsu CL, Hou HA, Tsai CH, Chou WC, and Tien HF (2020). A 4-gene leukemic stem cell score can independently predict the prognosis of myelodysplastic syndrome patients. Blood advances 4, 644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White BS, Khan SA, Mason MJ, Ammad-Ud-Din M, Potdar S, Malani D, Kuusanmäki H, Druker BJ, Heckman C, Kallioniemi O, et al. (2021). Bayesian multi-source regression and monocyte-associated gene expression predict BCL-2 inhibitor resistance in acute myeloid leukemia. NPJ Precis Oncol 5, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HH, Bellmunt E, Scheib JL, Venegas V, Burkert C, Reichardt LF, Zhou Z, Farinas I, and Carter BD (2009). Glial precursors clear sensory neuron corpses during development via Jedi-1, an engulfment receptor. Nat Neurosci 12, 1534–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Q, Ma X, and He Z (2020). PEAR1 suppresses the proliferation of pulmonary microvascular endothelial cells via PI3K/AKT pathway in ALI model. Microvasc Res 128, 103941. [DOI] [PubMed] [Google Scholar]
- Zhang B, and Horvath S (2005). A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4, Article 17. [DOI] [PubMed] [Google Scholar]
- Zhang H, Nakauchi Y, Köhnke T, Stafford M, Bottomly D, Thomas R, Wilmot B, McWeeney SK, Majeti R, and Tyner JW (2020). Integrated analysis of patient samples identifies biomarkers for venetoclax efficacy and combination strategies in acute myeloid leukemia. Nat Cancer 1, 826–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Clinical Summary, Related to Figure 1.
Table S2. Mutation-cell type associations, Related to Figure 2. Results from a Welch’s t-test (t and pval columns) comparing the average cell-type scores between mutated vs wild type patients. The sample size for each comparison is provided in terms of the number of mutated and wildtype (num_muts and num_neg). The effect size is given in terms of both raw average difference (estimate) and Glass’s delta (glass_d) relative to the wildtype group. Multiple-testing adjustments were performed using qvalue.
Table S3. Drug-cell type correlations with top mutation interactions, Related to Figure 3. Pearson’s correlation (cor) and resulting BY FDR (cor_fdr) are provided for each drug and cell_type along with results from the top gene (inter_gene) ranked by interaction pvalue. Reported for the interaction terms are the interaction coefficient (inter_est), the number of mutations (inter_muts) and the qvalue.
Table S4. Drug Family annotations, Related to Figure 4. Provided are a series of mapping files supporting the assignment of drugs to families. Specifically, we provided separate sheets describing (1) the family to synonym mapping, (2) drug to family mapping as well as (3) the inhibitor to gene/family mapping. In this latter tab, in addition to drug and gene family information we provide a summary of the evidence linking drugs to genes. This includes the minimum assay value reported (db_min_kd and targetome_min_assay), the amount of literature support and number of distinct values reported in Targetome (Blucher et al., 2017; Blucher et al., 2019; Choonoo et al., 2019) for assay values < 25 as well as our tier assignments. For each tab, manual adjustments are indicated by TRUE/FALSE.
Table S5. Associations between drug families and mutations, Related to Figure 4B. Provided are results from a Welch’s t-test (t and pval columns) comparing the difference in average drug enrichment score between patient mutation groups for a given gene. Each family is represented by both the IUPHAR name (family) as well as our synonym. Sample size is provided in terms of the number of patients with and without mutations (num_muts and num_neg respectively). The effect size is given in terms of both raw average difference (estimate) and Glass’s delta (glass_d) relative to the wildtype group. Multiple-testing adjustments were performed using qvalue.
Table S6. Drug family-cell type score correlations with top interactions, Related to Figure 5. The Pearson’s correlation between each drug family enrichment score and cell-type score is provided (cor) along with BY FDR adjustments (cor_fdr). Results for the corresponding top mutation ranked by interaction pvalue are provided. Reported for the interaction terms are the interaction coefficient (inter_est), the number of mutations (inter_muts) and the qvalue.
Table S7. WGCNA module membership, Related to Figure 6B. Gene assignments are provided for each WGCNA expression module including module 3 which is highlighted in Figure 6B. Modules are indicated by both label and corresponding color. Genes are described by Ensembl unique identifier and related annotation. The kME for the combined expression set (comb_kme) indicates the correlation of each gene relative to its module’s eigengene. As Mod0 (grey) is not a cohesive module but more of a container for ‘outlier’ genes, we do not report corresponding kME’s.
Data Availability Statement
All raw and processed sequencing data, along with relevant clinical annotations have been submitted to dbGaP and Genomic Data Commons and are publicly available. The dbGaP study ID is 30641 and accession ID is phs001657.v2.p1 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001657.v2.p1). The clinical annotations and data availability are found in Table S1. In addition, all data can be accessed and queried through our online, interactive user interface, Vizome, at www.vizome.org/aml2. Original code for replicating the paper analyses based on publicly available data has been deposited at https://github.com/biodev/beataml2_manuscript. The DOI is listed in the key resources table. A frequently asked questions page for the dataset is found at https://biodev.github.io/BeatAML2/.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BV650 Mouse Anti-Human PEAR1, clone 492621 | BD Biosciences | Catalog # 748063 |
| Human TruStain FcX (Fc Receptor Blocking Solution) | Biolegend | Catalog # 422301 |
| Bacterial and virus strains | ||
| n/a | ||
| Biological samples | ||
| AML Patient Samples | Oregon Health & Science University (OHSU), University of Utah, University of Texas Medical Center (UT Southwestern), Stanford University, University of Miami, University of Colorado, University of Florida, National Institutes of Health (NIH), Fox Chase Cancer Center and University of Kansas (KUMC) | OHSU; IRB#9570; #4422; NCT01728402 |
| Healthy Bone Marrow CD34+ Cells | AllCells | Custom Order |
| Chemicals, peptides, and recombinant proteins | ||
| Drug | Source | Catalog # | CAS # |
| 17-AAG (Tanespimycin) | LC Labs | A-6880 | 75747–14-7 |
| A-674563 | Selleck | S2670 | 552325–73-2 |
| ABT-737 | Selleck | S1002 | 852808–04-9 |
| Afatinib (BIBW-2992) | LC Labs | A-8644 | 850140–72-6 |
| AGI-5198 | Selleck | S7185 | 1355326–35-0 |
| AGI-6780 | Selleck | S7241 | 1432660–47-3 |
| AKT Inhibitor IV | Millipore Sigma (EMD Biosciences; Calbiochem) | 124011 | 681281–88-9 |
| AKT Inhibitor X | Millipore Sigma (EMD Biosciences; Calbiochem) | 124020 | 925681–41-0 |
| Alisertib (MLN8237) | Selleck | S1133 | 1028486–01-2 |
| AMPK Inhibitor | Millipore Sigma (EMD Biosciences; Calbiochem) | 171260 | 866405–64-3 |
| Arsenic trioxide | Fisher | NC0244675 | 1327–53-3 |
| Artemisinin | Selleck | S1282 | 63968–64-9 |
| AST487 | Selleck | S6662 | 630124–46-8 |
| AT-101 | Selleck | S2812 | 866541–93-7 |
| AT7519 | Selleck | S1524 | 844442–38-2 |
| Axitinib (AG-013736) | LC Labs | A-1107 | 319460–85-0 |
| Azacytidine | Selleck | S1782 | 320–67-2 |
| AZD1480 | Selleck | S2162 | 935666–88-9 |
| Baicalein | Selleck | S2268 | 491–67-8 |
| Barasertib (AZD1152-HQPA) | Selleck | S1147 | 722544–51-6 |
| Bay 11–7085 | Selleck | S7352 | 196309–76-9 |
| BEZ235 | LC Labs | N-4288 | 915019–65-7 |
| BI-2536 | Selleck | S1109 | 755038–02-9 |
| Birinapant | Selleck | S7015 | 1260251–31-7 |
| BLZ945 | Selleck | S7725 | 953769–46-5 |
| BMS-345541 | Selleck | S8044 | 445430–58-0 |
| BMS-754807 | Selleck | S1124 | 1001350–96-4 |
| Bortezomib (Velcade) | LC Labs | B-1408 | 179324–69-7 |
| Bosutinib (SKI-606) | LC Labs | B-1788 | 380843–75-4 |
| Cabozantinib (XL184) | Selleck | S1119 | 849217–68-1 |
| Canertinib (CI-1033) | LC Labs | C-1201 | 289499–45-2 |
| Cediranib (AZD2171) | LC Labs | C-4300 | 288383–20-0 |
| CHIR-99021 | LC Labs | C-6556 | 252917–06-9 |
| CI-1040 (PD184352) | LC Labs | P-8499 | 212631–79-3 |
| Crenolanib | Selleck | S2730 | 670220–88-9 |
| Crizotinib (PF-2341066) | LC Labs | C-7900 | 877399–52-5 |
| CYT387 | Selleck | S2219 | 1056634–68-4 |
| Cytarabine | Selleck | S1648 | 147–94-4 |
| Dasatinib | LC Labs | D-3307 | 302962–49-8 |
| DBZ | Tocris | 4489 | 209984–56-5 |
| Doramapimod (BIRB 796) | LC Labs | D-2744 | 285983–48-4 |
| Dovitinib (CHIR-258) | LC Labs | D-3608 | 405169–16-6 |
| Elesclomol | Selleck | S1052 | 488832–69-5 |
| Entospletinib (GS-9973) | Selleck | S7523 | 1229208–44-9 |
| Entrectinib | Selleck | S7998 | 1108743–60-7 |
| Erlotinib | LC Labs | E-4007 | 183319–69-9 |
| Etomoxir | Selleck | S8244 | 828934–41-4 |
| Everolimus | Selleck | S1120 | 159351–69-6 |
| Flavopiridol | Selleck | S1230 | 131740–09-5 |
| Foretinib (XL880) | LC Labs | F-4185 | 849217–64-7 |
| GDC-0879 | Selleck | S1104 | 905281–76-7 |
| GDC-0941 | LC Labs | G-9252 | 957054–30-7 |
| Gefitinib | LC Labs | G-4408 | 84475–35-2 |
| Gilteritinib | Selleck | S7754 | 1254053–43-4 |
| Go6976 | LC Labs | G-6203 | 136194–77-9 |
| GSK-2879552 | Selleck | S7796 | 1902123–72-1 |
| GSK690693 | Selleck | S1113 | 937174–76-0 |
| GW-2580 | LC Labs | G-5903 | 870483–87-7 |
| H-89 | LC Labs | H5239 | 127243–85-0 |
| Ibrutinib (PCI-32765) | LC Labs | I-3311 | 936563–96-1] |
| Idelalisib (GS-1101) | Selleck | S2226 | 870281–82-6 |
| Imatinib | LC Labs | I5577 | 152459–95-5 |
| Indisulam | Selleck | S9742 | 165668–41-7 |
| INK-128 | Selleck | S2811 | 1224844–38-5 |
| JAK Inhibitor I | Millipore Sigma (EMD Biosciences; Calbiochem) | 420099 | 457081–03-7 |
| JNJ-28312141 | SYN Kinase | SYN-1154 | 885692–52-4 |
| JNJ-38877605 | Selleck | S1114 | 943540–75-8 |
| JNJ-7706621 | Selleck | S1249 | 443797–96-4 |
| JNK Inhibitor II (SP600126) | LC Labs | S-7979 | 129–56-6 |
| JQ1 | Selleck | S7110 | 1268524–70-4 |
| KI20227 | Tocris | 4481 | 623142–96-1 |
| KN92 | Tocris | 4130 | 1135280–28-2 |
| KN93 | Tocris | 1278 | 139298–40-1 |
| KU-55933 | Selleck | S1092 | 587871–26-9 |
| KW-2449 | Selleck | S2158 | 1000669–72-6 |
| Lapatinib | Selleck | S2111 | 231277–92-2 |
| Lenalidomide | Selleck | S1029 | 191732–72-6 |
| Lenvatinib | Selleck | S1164 | 857890–39-2 |
| Lestaurtinib (CEP-701) | LC Labs | L-6307 | 111358–88-4 |
| Linifanib (ABT-869) | Selleck | S1003 | 796967–16-3 |
| Lovastatin | Selleck | S2061 | 75330–75-5 |
| LY-333531 | Tocris | 4738 | 169939–93-9 |
| LY294002 | LC Labs | L-7962 | 154447–36-6 |
| Masitinib (AB-1010) | LC Labs | M-7007 | 790299–79-5 |
| MEK Inhibitor VII | Millipore Sigma (EMD Biosciences; Calbiochem) | 444939 | 305350–87-2 |
| Metformin | Selleck | S5958 | 657–24-9 |
| MGCD-265 | Selleck | S1361 | 875337–44-3 |
| Midostaurin | LC Labs | P-7600 | 120685–11-2 |
| MK-2206 | Selleck | S1078 | 1032350–13-2 |
| MLN120B | MedChem Express | HY-15473 | 783348–36-7 |
| MLN8054 | Selleck | S1100 | 869363–13-3 |
| Motesanib (AMG-706) | LC Labs | M-2900 | 857876–30-3 |
| Neratinib (HKI-272) | Selleck | S2150 | 698387–09-6 |
| NF-kB Activation Inhibitor | Selleck | S4902 | 545380–34-5 |
| Nilotinib | Selleck | S1033 | 641571–10-0 |
| Nutlin 3a | Selleck | S8059 | 675576–98-4 |
| NVP-ADW742 | Selleck | S1088 | 475488–23-4 |
| NVP-AEW541 | Selleck | S1034 | 475489–16-8 |
| NVP-TAE684 | Selleck | S1108 | 761439–42-3 |
| Olaparib | Selleck | S1060 | 763113–22-0 |
| OTX-015 | Selleck | S7360 | 202590–98-5 |
| p38 MAP Kinase Inhibitor | Millipore Sigma (EMD Biosciences; Calbiochem) | 506126 | 219138–24-6 |
| Palbociclib | Selleck | S4482 | 571190–30-2 |
| Panobinostat | Selleck | S1030 | 404950–80-7 |
| Pazopanib (GW786034) | LC Labs | P6706 | 444731–52-6 |
| PD153035 | Millipore Sigma (EMD Biosciences; Calbiochem) | SML0564 | 183322–45-4 |
| PD173955 | Symansis | SY-PD173955 | 260415–63-2 |
| PD98059 | LC Labs | P-4313 | 167869–21-8 |
| Pelitinib (EKB-569) | Selleck | S1392 | 257933–82-7 |
| Perhexiline maleate | Selleck | S6959 | 6724–53-4 |
| PH-797804 | Selleck | S2726 | 586379–66-0 |
| PHA-665752 | Selleck | S1070 | 477575–56-7 |
| PHT-427 | Selleck | S1556 | 1191951–57-1 |
| PI-103 | Selleck | S1038 | 371935–74-9 |
| PLX-4720 | Selleck | S1152 | 918505–84-7 |
| Ponatinib (AP24534) | LC Labs | P-7022 | 943319–70-8 |
| PP2 | Millipore Sigma (EMD Biosciences; Calbiochem) | 529573 | 172889–27-9 |
| PP242 | LC Labs | P-6666 | 1092351–67-1 |
| PRT062070 HCL | Selleck | S7634 | 1369761–01-2 |
| PRT062607 | Selleck | S8032 | 1370261–97-4 |
| Quizartinib (AC220) | LC Labs | Q-4747 | 950769–58-1 |
| R406 | Selleck | S2194 | 841290–81-1 |
| R547 | Selleck | S2688 | 741713–40-6 |
| RAF265 (CHIR-265) | Selleck | S2161 | 927880–90-8 |
| Ralimetinib (LY2228820) | Selleck | S1494 | 862507–23-1 |
| Ranolazine (free base) | Selleck | S1799 | 95635–55-5 |
| Rapamycin | LC Labs | R-5000 | 53123–88-9 |
| Regorafenib (BAY 73–4506) | Selleck | S1178 | 755037–03-7 |
| Roscovitine (CYC-202) | LC Labs | R-1234 | 186692–46-6 |
| Ruxolitinib, free base | LC Labs | R-6600 | 120685–11-2 |
| S31–201 | Santa Cruz Biotech | sc-204304 | 501919–59-1 |
| Saracatinib (AZD0530) | LC Labs | S-8906 | 379231–04-6 |
| SB-202190 | LC Labs | S-1700 | 152121–30-7 |
| SB-203580 | LC Labs | S-3400 | 152121–47-6 |
| SB-431542 | Selleck | S1067 | 301836–41-9 |
| Selinexor | Selleck | S7252 | 1393477–72-9 |
| Selumetinib (AZD6244) | LC Labs | S-4490 | 606143–52-6 |
| SGX-523 | Selleck | S1112 | 1022150–57-7 |
| SNS-032 (BMS-387032) | Selleck | S1145 | 345627–80-7 |
| Sorafenib tosylate | LC Labs | S-8502 | 475207–59-1 |
| SR9011-HCL | MedChem Express | HY-16988A | 2070014–94-5 |
| Staurosporine, free base | LC Labs | S-9300 | 62996–74-1] |
| STO609 | Millipore Sigma (EMD Biosciences; Calbiochem) | 570250 | 52029–86-4 |
| SU11274 | Selleck | S1080 | 658084–23-2 |
| SU14813 | Selleck | S0504 | 627908–92-3 |
| Sunitinib malate | LC Labs | S-8803 | 341031–54-7 |
| SYK Inhibitor | Millipore Sigma (EMD Biosciences; Calbiochem) | 574711 | 622387–85-3 |
| Tandutinib (MLN518) | LC Labs | T-7802 | 387867–13-2 |
| TG100–115 | Selleck | S1358 | 677297–51-7 |
| TG101348 | Active Biochem | A-1140 | 936091–26-8 |
| Tivozanib (AV-951) | LC Labs | T-6466 | 475108–18-0 |
| Tofacitinib (CP-690550) | LC Labs | T-1377 | 477600–75-2 |
| Tozasertib (VX-680) | LC Labs | T-2304 | 639089–54-6 |
| Trametinib (GSK1120212) | LC Labs | T-8132 | 871700–17-3 |
| Vandetanib (ZD6474) | LC Labs | V-9402 | 443913–73-3 |
| Vargetef (Nintedanib) | LC Labs | N-9077 | 656247–17-5 |
| Vatalanib (PTK787) | LC Labs | V-8303 | 212141–51-0 |
| Vemurafenib (PLX4032) | LC Labs | V-2800 | 918504–65-1 |
| Venetoclax | LC Labs | V-3579 | 1257044–40-8 |
| Vismodegib (GDC-0449) | LC Labs | V-4050 | 879085–55-9 |
| Volasertib (BI-6727) | Selleck | S2235 | 755038–65-4 |
| VX-745 | Tocris | 3915 | 209410–46-8 |
| XAV-939 | Selleck | S1180 | 284028–89-3 |
| XMD 8–87 | Selleck | S8272 | 1234480–46-6 |
| YM-155 | Selleck | S1130 | 781661–94-7 |
| Other Chemical | Source | Catalog # |
| Zombie Aqua Fixable Viability Dye | Biolegend | Catalog # 423101 |
| Critical commercial assays | ||
| Nextera RapidCapture Exome | Illumina | |
| SureSelect Strand-Specific RNA Library Preparation Kit | Agilent | |
| DNeasy Blood & Tissue Kit | Qiagen | |
| RNeasy Mini Kit | Qiagen | |
| Deposited data | ||
| DNA Sequencing Data | This paper | http://vizome.org/aml2/, dbGaP study ID is 30641 and accession ID is phs001657.v2.p1 |
| RNA Sequencing Data | This paper | http://vizome.org/aml2/, dbGaP study ID is 30641 and accession ID is phs001657.v2.p1 |
| Experimental models: Cell lines | ||
| n/a | ||
| Experimental models: Organisms/strains | ||
| n/a | ||
| Oligonucleotides | ||
| Custom capture probes for DNA sequencing (11.9 MB) | Roche-Nimblegen | Sequencing coordinates listed Table S14 of (Tyner et al., 2018) |
| Forward Primer FLT3: 5′- AGCA ATT TAG GTA TGA AAG CCA GCTA - 3′ | Eurofins | (Kottaridis et al., 2001) |
| Reverse Primer FLT3: 5′ - CTT TCA GCA TTT TGA CGG CAA CC - 3′ | Eurofins | (Kottaridis et al., 2001) |
| Forward Primer NPM1: 5′ - GTT TCT TTT TTT TTT TTT CCA GGC TAT TCA AG - 3′ | Eurofins | (Falini et al., 2007) |
| Reverse Primer NPM1: 5′ - CAC GGT AGG GAA AGT TCT CAC TCT GC - 3′ | Eurofins | (Falini et al., 2007) |
| Recombinant DNA | ||
| n/a | ||
| Software and algorithms | ||
| R version 4.03 | R Core Team | https://www.r-project.org |
| R package GSVA v1.38 | Bioconductor | DOI: 10.18129/B9.bioc.GSVA |
| R package cqn v1.36 | Bioconductor | DOI: 10.18129/B9.bioc.cqn |
| R package qvalue v2.22.0 | Bioconductor | DOI: 10.18129/B9.bioc.qvalue |
| R package partykit v1.2–15 | CRAN | https://CRAN.R-project.org/package=partykit |
| R package survival v3.2–7 | CRAN | https://CRAN.R-project.org/package=survival |
| R package WGCNA v1.69 | CRAN | https://CRAN.R-project.org/package=WGCNA |
| R package data.table v1.13.6 | CRAN | https://CRAN.R-project.org/package=data.table |
| Manuscript analysis workflow |
This paper | DOI: 10.5281/zenodo.6773715 |
| Other | ||
| Ex Vivo Drug Sensitivity Data | This paper | http://vizome.org/aml2/ and https://biodev.github.io/BeatAML2/ |
| Clinical Annotations | This paper | Table S1 |