

Abstract
The cation channel of sperm (CatSper) is a validated target for nonhormonal male contraception, but it lacks selective blockers, hindering studies to establish its role in both motility and capacitation. Via an innovative calcium uptake assay utilizing human sperm we discovered novel inhibitors of CatSper function from a high‐throughput screening campaign of 72,000 compounds. Preliminary SAR was established for seven hit series. HTS hits or their more potent analogs blocked potassium‐induced depolarization and noncompetitively inhibited progesterone‐induced CatSper activation. CatSper channel blockade was confirmed by patch clamp electrophysiology and these compounds inhibited progesterone‐ and prostaglandin E1‐induced hyperactivated sperm motility. One of the hit compounds is a potent CatSper inhibitor with high selectivity for CatSper over hCav1.2, hNav1.5, moderate selectivity over hSlo3 and hERG, and low cytotoxicity and is therefore the most promising inhibitor identified in this study. These new CatSper blockers serve as useful starting points for chemical probe development and drug discovery efforts.
Keywords: cation channel of sperm, inhibitors, selectivity, sperm motility, electrophysiology
High‐throughput screening identified seven hit series as novel inhibitors of the cation channel of sperm (CatSper), a validated male contraceptive target. CatSper channel blockade was confirmed by patch clamp electrophysiology. The most promising analog was found to be a potent CatSper inhibitor with high selectivity for CatSper over hCav1.2, hNav1.5, moderate selectivity over hSlo3 and hERG, and low cytotoxicity.

Introduction
After the introduction of the female birth control pill in 1960, research to discover and develop a male counterpart focused on hormonal contraceptives. However, no agent has yet reached the market to date due to side effects such as decreased high‐density lipoprotein (HDL), acne, low libido, and weight gain.[ 1 , 2 ] Therefore, more recent efforts have centered on developing non‐hormonal pharmacological agents that specifically target the testis, the epididymis, or sperm function.[ 3 , 4 , 5 ] Of the increasing number of targets available to the field, the voltage‐gated calcium ion channel CatSper (cation ion of sperm)[ 6 , 7 ] is considered an exceptionally promising target for male contraception because it is expressed exclusively in sperm and it is functionally relevant only in mature sperm. Furthermore, channel malformation caused by genetic polymorphisms is sufficient for complete infertility in human men.[ 8 , 9 , 10 , 11 ] CatSper activation leads to a large influx of calcium into the tail of sperm, creating calcium oscillations that propagate towards the head of the sperm. [12] This increase in intracellular calcium ion concentration is necessary for facilitating many processes within mature sperm including the acrosome reaction, capacitation, and hyperactivated motility (HAM).[ 13 , 14 ] HAM is a mode of motility characterized by a shift in flagellar beat patterns from sinusoidal, regular motion to more dramatic, whip‐like gesticulations.[ 15 , 16 , 17 ] Sperm cells that do not achieve HAM cannot penetrate the viscous fluid of the upper reproductive tract and thus cannot fertilize the egg. [18] The endogenous steroid progesterone and several prostaglandins, particularly prostaglandin E1 (PGE1), are potent activators of CatSper.[ 19 , 20 , 21 ] In addition to these activators, alkaline environments and high external potassium concentrations activate calcium influx in sperm via CatSper.[ 22 , 23 ]
As mentioned previously, a selective CatSper blocker would affect only mature sperm. Therefore, after discontinuation of treatment and drug wash out, fertility will return upon replenishment of mature sperm population. These observations underlie the rationale for developing a novel contraceptive agent targeting CatSper. Such an agent could potentially be used as an on‐demand male contraceptive and alternatively could also be applied as a vaginal treatment to prevent sperm hypermotility and thereby pregnancy. Compounds impacting male fertility have been predominantly identified from anecdotal and off‐target effects of drugs used for other indications. Examples include anticancer agent lonidamine and related analogs adjudin [24] and gamendazole, [25] triptolide and triptonide,[ 26 , 27 ] BET bromodomain inhibitor JQ1, [28] ALDH1 A2 inhibitor WIN 18,466, [29] originally developed as an amebicide, and retinoic acid receptor antagonists originally developed for dermatological and inflammatory diseases. [30] To date, little progress has been made in developing CatSper‐specific blockers. HC‐056456 (Figure 1), a CatSper blocker identified through screening, slows the rise of intracellular calcium in sperm and prevents HAM in the low micromolar range. [31] Mibefradil (Ro 40‐5967) [31] and its close structural analog NNC 55‐0396 [32] are calcium channel blockers that were originally developed to block T‐ and L‐type channels, and later found to also block CatSper. [33] Recently, a synthetic steroidal blocker (RU 1968) of CatSper was reported, displaying micromolar potency and providing a new class of CatSper blockers. [34] We recently reported on the ability of the three steroids medroxyprogesterone acetate, levonorgestrel, and aldosterone to antagonize progesterone‐ and PGE1‐mediated calcium influx. [35] In addition, the plant triterpenoids pristimerin and lupeol were reported to inhibit progesterone‐induced CatSper currents and it was proposed that these compounds compete with progesterone at its regulatory site on ABHD2.[ 36 , 37 ] After the initial report, two subsequent studies found that these triterpenoids do not antagonize progesterone‐induced CatSper currents.[ 38 , 39 ]
Figure 1.

Structures of known CatSper inhibitors.
To better understand the role of CatSper in reproductive biology and as a starting point for male contraceptive drug development, new CatSper channel blockers are needed. Herein, we disclose the results of a high‐throughput screening (HTS) campaign that revealed several previously unreported chemical scaffolds with the ability to block CatSper. We established the initial structure‐activity relationships (SAR) for seven screening hit series as inhibitors of high potassium/high pH‐ and progesterone‐induced calcium influx in human sperm and characterized these hit series using patch‐clamp electrophysiology in human sperm, computer‐aided sperm analysis (CASA), cytotoxicity in both sperm and somatic cells, and ion channel selectivity screening.
Results and Discussion
Assay validation, screening, and hit discovery
Heterologous expression of a functional CatSper channel has not yet been achieved and is likely due to failure in proper assembly of the CatSper complex, hampering drug discovery efforts. [40] Thus, a phenotypic screen was used as an alternative, despite challenges that can be encountered with such approaches such as hit validation or target deconvolution that require follow‐up testing in functional assays. [41] Live human sperm were used in a HTS campaign to identify compounds that inhibit CatSper function by monitoring intracellular calcium ion concentration after loading isolated cells with calcium‐binding fluorophore Fluo‐4 AM. Fluorescence was monitored in real time during test compound and channel activator addition using a FLIPR Tetra. CatSper inhibitors were identified by a reduction in fluorescence peak height produced by the activator relative to DMSO controls.
An overview of the HTS campaign utilizing high potassium/high pH to activate CatSper is shown in Figure 2. The CatSper FLIPR assay was validated by first examining the reproducibility of a series of controls (high, low, high+inhibitor, background) distributed vertically across entire assay plates to establish key metrics including signal window and Z scores. Although the signal window (S/B∼5), fluorescence peak (average 335 RFU), and Z (0.4) were relatively low, these values were considered sufficient to proceed with screening. Whereas the signal size was likely limited by the fast timescale of ion channel opening and closing, an improved peak RFU and S/B were achieved while screening by inclusion of probenecid to block dye efflux. [42] Validation of hit detection and reproducibility was then established in a pilot screen using the Library of Pharmacologically Active Compounds (LOPAC) collection tested in four independent experiments. These assay validation steps indicated that potential CatSper blockers could be identified in an HTS campaign using this FLIPR assay method.
Figure 2.
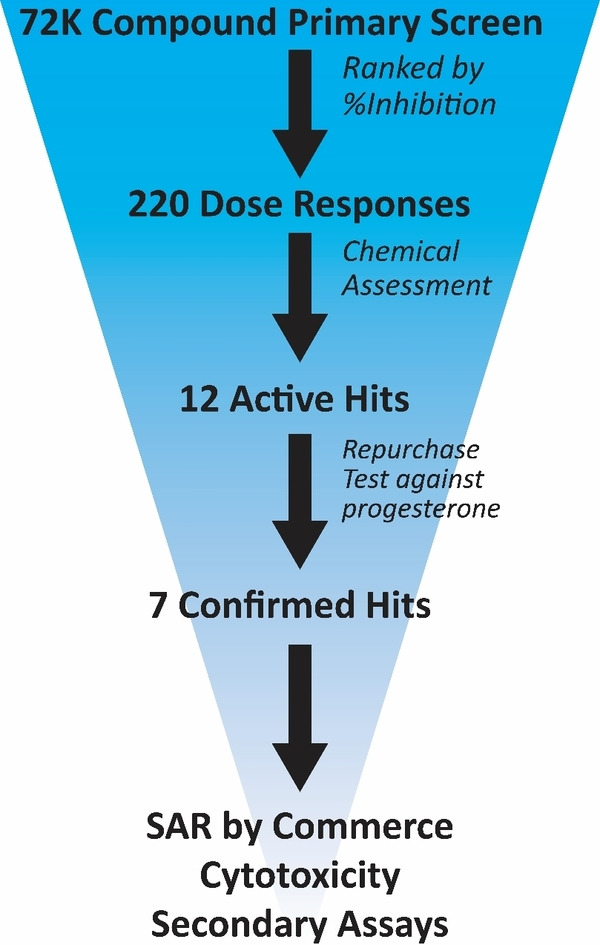
Graphical representation of HTS workflow.
Over 72,000 compounds from the following libraries were tested in the primary screen at 10 μM: the National Cancer Institute set, the ChemDiv Peptidomimetic and Kinase libraries, the TOCRIS Tocriscreen Complete and Kinase Inhibitor Library, the ChemBridge MicroFormat Library, the LOPAC library, the NIH Clinical Library, the Prestwick Chemical Library, and the MicroSource Spectrum Collection. Of the 72,000 compounds screened, 220 exhibited greater than 60 % inhibition and were selected for IC50 determinations. Approximately half of these compounds (104) showed well‐defined sigmoidal dose‐response curves with IC50 values less than 55 μM. These 104 compounds were further triaged based on fluorescence interference, unfavorable physicochemical properties, and potency, resulting in 12 hits prioritized for repurchase.
Of those 12 hits, eight compounds reconfirmed in the HTS assay using high potassium/high pH activation and in a similar assay using progesterone (1 μM) to elicit calcium influx. Inhibition of progesterone‐induced calcium influx increased confidence that the hits act by blocking CatSper, resulting in elimination of four hits from further consideration. One of the remaining hit molecules was apomorphine, which was removed from consideration because of its known properties as a non‐selective dopamine agonist, leaving seven scaffolds for further pursuit (Figure 3). [43] Hits 1 a and 2 a were identified from the LOPAC library, hits 3 a, 4 a, and 5 a came from the ChemBridge library, 6 a from the MicroSource library and compound 7 a from the ChemDiv kinase library. A representative FLIPR dose‐response trace showing calcium influx blocked by compound 4 a in human sperm is shown in Figure 4.
Figure 3.

Hit compounds resulting from HTS campaign.
Figure 4.
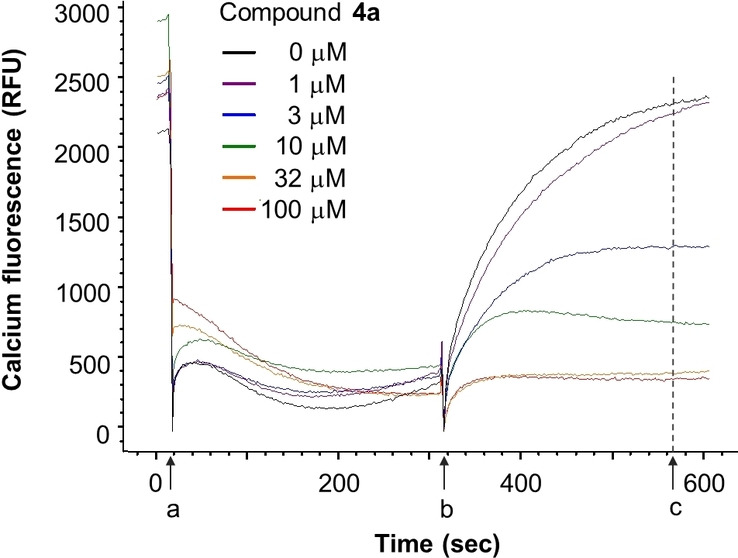
Representative FLIPR traces showing blockade of K+ depolarization‐induced increase in intracellular calcium by compound 4 a in human sperm. a=addition of test compound to sperm pre‐loaded with Fluo‐4‐AM dye; b=addition of activation buffer in the absence (control) or presence of test compound (zeroed here); c=endpoint reading taken. The 1st phase (a to b interval) is used to assess nonspecific compound effects, whereas CatSper channel blockade is determined during the 2nd phase (b to c interval).
Several rounds of SAR studies were carried out with the seven confirmed CatSper hits using commercially available molecules. First, because only a fraction of the HTS library was tested in the primary screen, a Tanimoto similarity search was performed for each confirmed hit against the entire HTS compound collection containing ∼250,000 compounds. This generated a list of analogs from which 50 compounds were selected, cherry‐picked from DMSO stocks, and tested in concentration‐response in both the K+‐ and progesterone‐induced activation assays. Additionally, a commercial chemical database (eMolecules) was queried using each hit compound. Again, a Tanimoto similarity or substructure search guided compound selection. A total of 67 analogs were selected for purchase based on structural diversity, lack of undesirable chemical functionalities, absence of known biological liabilities, and compound cost and availability.
Preliminary SAR of seven hit compounds
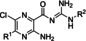
Hit series 1 (Table 1) is characterized by a guanidine group linked via an amide bond to a substituted pyrazine core. The most potent compound in this series, the original hit compound 1 a (5‐hexamethylene amiloride), carries an azepane group at C5 (R1) and is unsubstituted at the guanidinium moiety (R2=H). Placing less bulky amine substituents at the C5 position decreased potency (1 b–1 d). The epithelial sodium channel blocker amiloride (1 d) itself is inactive at CatSper. However, adding a hydrophobic benzyl group to the guanidinium moiety restored some activity, even when the azepane group was absent (1 e vs. 1 d). Since analog 1 f, carrying a 2,4‐dichloro group (R2), showed potency like the original hit compound 1 a, we concluded from this result that lipophilicity may play an important role in defining the activity of this series. The dichloro substitution present on the phenyl moiety of 1 f appears to be important, since a 3‐fold loss in potency is observed when the 2,4‐dichloro motif is replaced with a simple benzyl (1 e) or phenyl (1 h) group.
Table 1.
Guanidinium hit series 1 SAR determined in the human sperm calcium influx assay.[a]
|
| |||
|---|---|---|---|
|
Compd |
R1 |
R2 |
IC50 [μM] |
|
1 a |
|
H |
5.4±0.6 |
|
1 b |
|
H |
9.1±0.4 |
|
1 c |
|
H |
67±12 |
|
1 d |
|
H |
>100 |
|
1 e |
|
|
23±6 |
|
1 f |
|
|
7.8* |
|
1 g |
|
>100 |
|
|
1 h |
|
|
26* |
|
1 i |
|
8.1* |
|
|
1 j |
|
>100* |
|
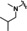
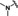
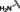
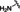
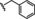
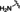
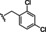
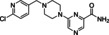
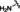
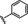
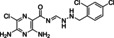
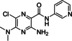
[a] CatSper activated by 3 μM progesterone. IC50 values are means±SEM of three independent experiments. *IC50 values are means of two independent experiments.
The guanidine group of compound 1 f can be converted to a hydrazinylmethylenamide group found in analog 1 i without negatively affecting potency. However, replacement of the guanidinium group with pyridine abolishes activity (1 c vs. 1 j). Pyrazine amide analog 1 g was also inactive. The C5 azepane group and an alkyl group appended to the guanidinium group appear to confer increased potency in this series, likely through increasing the lipophilicity of the molecule. Hit series 1 blockers were not pursued further because these compounds contain a guanidinium group present in sodium channel pore blockers, such as tetrodotoxin and saxitoxin; these compounds are also analogs of the epithelial sodium channel blocker amiloride, suggesting that selective CatSper blockers would be difficult to achieve in this series.
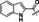
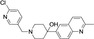
The most active compound in hit series 2, compound 2 a (selective dopamine D2 receptor antagonist L741,626 [44] ) containing a 4‐phenylpiperidin‐4‐ol core was identified from the LOPAC library and its CatSper SAR is shown in Table 2. Replacing the indole of 2 a with a phenyl group (2 b) decreases potency significantly, although the introduction of a 2‐fluorobenzyl group recovered some of the lost potency (2 c). The 4‐chlorophenyl group of 2 a was replaced with piperidine‐4‐azepane in compound 2 d, which was only ∼2‐fold less potent, suggesting that a hydrogen bond acceptor/donor is needed at this position rather than strictly the tertiary hydroxyl group. Addition of a ketone to the methylene linker in 2 a completely abolished activity as shown for analogs 2 e–2 j. Replacing the indole of 2 e with a 2‐methylquinoline (2 f) also led to an inactive compound. Adding a 4‐(2‐methylquinolin‐6‐yl) group to the scaffold resulted in inactive 2 k and compound 2 l, 3‐(1‐benzylpiperidin‐4‐yl)‐1H‐indole, is inactive as well. These two compounds indicate that this scaffold is not amenable to changes in connectivity or composition. To summarize, a basic piperidine nitrogen appears to be necessary for activity in this scaffold series and subtle changes are needed if this compound is to be developed further. However, it is promising that the tertiary alcohol may not be needed for activity since the 4‐phenylpiperidin‐4‐ol substructure has the potential to be converted to MPTP‐like compounds, which are known to be neurotoxic, causing Parkinson's disease‐like symptoms. [45]
Table 2.
Piperidine hit series 2 SAR determined in the human sperm calcium influx assay.[a]
|
| |||
|---|---|---|---|
|
Compd |
R1 |
R2 |
IC50 [μM] |
|
2 a |
|
Cl |
7.5±1.3 |
|
2 b |
|
H |
92±14 |
|
2 c |
|
Cl |
18±2 |
|
2 d |
|
18±7 |
|
|
2 e |
|
Cl |
>100 |
|
2 f |
|
Cl |
>100 |
|
2 g |
|
Cl |
>100 |
|
2 h |
|
Cl |
>100* |
|
2 i |
|
Cl |
>100* |
|
2 j |
|
Cl |
87* |
|
2 k |
|
>100* |
|
|
2 l |
|
>100* |
|
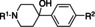
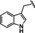
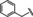
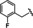
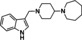
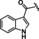
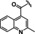
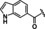
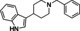
[a] CatSper activated by 3 μM progesterone. IC50 values are means±SEM of three independent experiments. *IC50 values are means of two independent experiments.
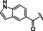
Hit compound 3 a (Table 3) was identified from the ChemBridge library and is characterized by a 2‐methylquinolin‐4‐amine core structure. As very few commercially available analogs of the quinoline core were available, only a limited SAR survey for this scaffold was performed and will need to be explored further in future studies. Compared to the initial hit molecule 3 a, the loss of the 6,8‐dimethyl substitutions in the quinoline core (3 b) decreased potency 4‐fold and removing the ethylene linker between the amine and the benzene ring reduced potency 3‐fold (3 a vs. 3 c). The 4‐amino analog (3 d) displayed little activity, and amide groups in place of the amine were not tolerated (3 e and 3 f). Changing the attachment point of the 2‐phenylacetamide group from the 4‐position to the 5‐position on quinoline 3 g also abolished activity. Although a relatively limited number of analogs were explored, the current data suggests that only subtle modifications are tolerated in this series.
Table 3.
Quinolone hit series 3 SAR determined in the human sperm calcium influx assay.[a]
|
| |||
|---|---|---|---|
|
Compd |
R1 |
R2 |
IC50 [μM] |
|
3 a |
Me |
|
3.6±0.6 |
|
3 b |
H |
|
15±2 |
|
3 c |
Me |
|
10.7±0.3 |
|
3 d |
Me |
H |
96±9 |
|
3 e |
Me |
|
>100 |
|
3 f |
H |
|
>100 |
|
3 g |
|
>100 |
|
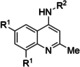
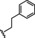
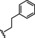
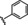
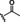
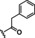
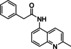
[a] CatSper activated by 3 μM progesterone. IC50 values are means±SEM of three independent experiments.
Hit series 4 (Table 4) features a N,4‐diphenylpiperazine‐1‐carboxamide core. Concentration‐dependent decreases in progesterone‐induced calcium uptake for compound 4 a (Figure 4) and selected analogs in this series with a range of potencies are shown in Figure 5. The most potent compounds, including initial hit molecule 4 a and analogs 4 b and 4 c have halogenated, electron‐withdrawing substituents on both benzene rings. Removing either of the 3‐trifluoromethyl groups on the aromatic rings in compound 4 a abolished potency, as seen for compounds 4 d and 4 e, and changing R2 to the 2‐trifluoromethyl group (4 f) also abolished potency. Given the vast commercial availability of this scaffold, a general Topliss scheme [46] for aromatic substituents was followed in which analogs with varying substitutions on the two benzene rings were explored. Replacement of the 3‐trifluromethyl groups in the R2 position with 4‐methyl (4 g), 4‐chloro (4 h), and 4‐methoxy (4 i) groups resulted in inactive compounds. Other combinations of aromatic substitution, like those observed for compounds 4 j, 4 k, and 4 l also led to weak or inactive analogs. Furthermore, replacing the phenylurea in 4 a with a N,N‐diethylurea group seen for 4 m reduced activity 8‐fold. Changing to a pyridine (4 n) resulted in a complete loss of activity. The data suggest that electron withdrawing groups in the 3‐position of both aromatic rings are favored, but that substitution with electron withdrawing groups in the 4‐position, as seen with 4 b, should be explored further. Notably, derivatives of the N,4‐diarylpiperazine‐1‐carboxamide core, have been described as agonists and antagonists for the transient receptor potential vanilloid receptor 1 (TRPV1) calcium ion channel, [47] indicating that this scaffold could interact with multiple ion channels.
Table 4.
Biphenylpiperazine hit series 4 SAR determined in the human sperm calcium influx assay.[a]
|
| |||
|---|---|---|---|
|
Compd |
R1 |
R2 |
IC50 [μM] |
|
4 a |
m‐CF3 |
m‐CF3 |
4.9±0.6 |
|
4 b |
p‐F |
m‐CF3 |
8.9±0.4 |
|
4 c |
m‐CF3 |
m‐Cl |
6.0±0.4 |
|
4 d |
m‐CF3 |
H |
>100 |
|
4 e |
H |
m‐CF3 |
>100 |
|
4 f |
m‐CF3 |
o‐CF3 |
>100 |
|
4 g |
m‐CF3 |
p‐Me |
>100 |
|
4 h |
m‐CF3 |
p‐Cl |
>100 |
|
4 i |
m‐CF3 |
p‐OMe |
>100 |
|
4 j |
m‐Me |
o‐CF3 |
>100 |
|
4 k |
p‐Me |
o‐CF3 |
>100 |
|
4 l |
o‐Me |
o‐CF3 |
64±18 |
|
4 m |
|
39* |
|
|
4 n |
|
>100 |
|
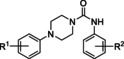
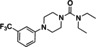
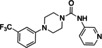
[a] CatSper activated by 3 μM progesterone. IC50 values are means±SEM of three independent experiments. *IC50 value is the mean of two independent experiments.
Figure 5.
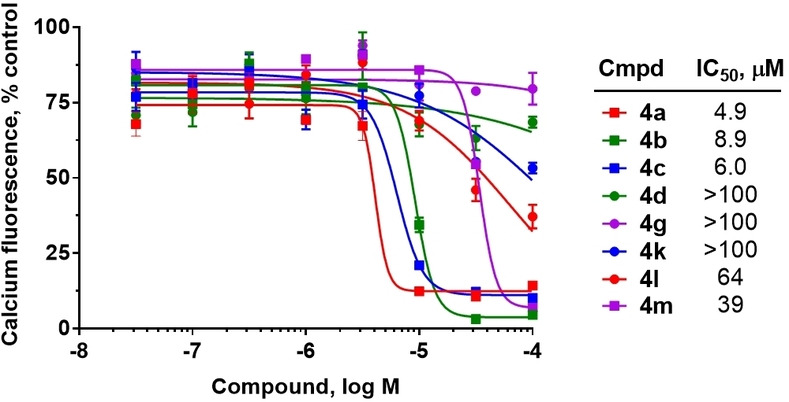
Inhibition of progesterone‐induced calcium uptake in human sperm by selected analogs in hit series 4. Data presented as the mean of 3 independent experiments (except 4 m, n=2) with error bars representing the SEM.
Hit series 5 (Table 5) is based on an alkylated benzimidazolidin‐2‐iminium core and most analogs tested in this series had similar potencies in the single or double digit micromolar range. Similar benzimidazoles are part of the so‐called Malaria Box that has been used to find antimalarial agents. [48] Replacement of the dimethylaminomethyl substituent of initial hit compound 5 a with a methyl group at R2 (5 b) increased potency slightly, whereas shortening to an ethyl group from the propyl chain reversed this trend (5 c vs. 5 b). The western phenyl moiety displayed an obvious SAR trend favoring electron withdrawing substitutions, since changing from a weakly electron‐withdrawing 4‐bromo group to 4‐unsubstituted and further to the strongly donating 4‐methoxy group (5 a to 5 d to 5 e) resulted in increasing losses of potency. A similar but less pronounced trend is noticed with analogs 5 f through 5 h, each carrying a piperidine moiety instead of a dimethylaminomethyl group. A piperidine group is tolerated as an R2 substituent, as seen in 5 h, even while retaining the strong electron‐donating p‐methoxy group at R1. Compound 5 f, 5 i, and 5 j show improved potencies over initial hit compound 5 a. Further, cyclization of the side chain observed in 5 k is tolerated. In general, this series contains several analogs with up to 3.5‐fold improvement in potency over initial hit 5 a. Hit series 5 contains a dialkylarylguanidinium group, a known pore blocker of cationic channels,[ 49 , 50 , 51 ] suggesting that identifying selective CatSper blockers in this series may be challenging.
Table 5.
Benzimidazolidin‐2‐iminium hit series 5 SAR determined in the human sperm calcium influx assay.[a]
|
| |||
|---|---|---|---|
|
Compd |
R1 |
R2 |
IC50 [μM] |
|
5 a |
p‐Br |
|
12±2 |
|
5 b |
p‐Br |
Me |
7.7±0.1 |
|
5 c |
p‐Br |
H |
19±3 |
|
5 d |
H |
|
71±1 |
|
5 e |
p‐OMe |
|
>100 |
|
5 f |
m‐Br |
|
3.4±0.4 |
|
5 g |
H |
|
9.8±1.4 |
|
5 h |
p‐OMe |
|
12* |
|
5 i |
m,p‐(Cl)2 |
|
5.0±1.1 |
|
5 j |
m,p‐(Cl)2 |
|
4.1* |
|
5 k |
|
14±2 |
|
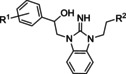
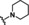
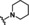
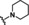
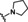
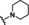
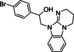
[a] CatSper activated by 3 μM progesterone. IC50 values are means±SEM of three independent experiments. *IC50 value is the mean of two independent experiments.
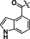
Hit series 6 contains a phthalazinone core, exemplified by HTS hit 6 a or azelastine, a histamine H1 receptor antagonist (Table 6) that also inhibits histamine release from mast cells and is used in a nasal spray. [52] The only compound in this series that retained activity was 6 b, the N‐desmethyl analog of the hit compound, which is an active metabolite of azelastine. The multiple changes in compounds 6 c–6 e resulted in complete loss of potency. Limited commercial availability of analogs of these compounds precluded a thorough study and description of the SAR of this scaffold. Further synthetic endeavors would be needed to explore this scaffold, taking into consideration that azelastine is a potent blocker of the hERG channel. [53]
Table 6.
Phthalazinone core hit series 6 SAR determined in the human sperm calcium influx assay.[a]
|
| |||
|---|---|---|---|
|
Compd |
R1 |
R2 |
IC50 [μM] |
|
6 a |
|
|
5.4±0.4 |
|
6 b |
|
|
8.3±1.1 |
|
6 c |
|
|
>100 |
|
6 d |
|
|
>100 |
|
6 e |
|
|
>100 |
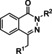
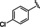
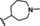
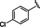
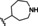
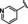
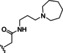
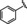
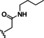
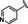
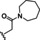
[a] CatSper activated by 3 μM progesterone. IC50 values are means±SEM of three independent experiments.
Hit series 7 (Table 7) is characterized by a triazolopyridazine core with flanking benzene and piperidine rings. Preliminary SAR studies explored the position of the methyl group on the phenyl moiety (R1), while also testing a range of amines at the piperidine amide. The majority of compounds tested within this series displayed potencies similar to initial hit molecule 7 a, except aliphatic amine 7 b; this compound was the only R2 substitution not bearing an amine on its side chain. As seen in compounds 7 c–7 e, little effect on potency was observed for cyclization of the terminal amine, although dimethyl amine 7 c was 3‐fold less potent, suggesting hydrophobic interactions are beneficial. Extending the terminal nitrogen by one carbon in 7 f reduced potency threefold relative to 7 d, suggesting a reduction in hydrogen bonding. Somewhat surprisingly, a piperidine sidechain (7 g) retained potency despite lacking a terminal nitrogen, perhaps by providing a hydrogen bond acceptor. No improvement in potency over initial hit 7 a was observed for any analogs tested, but the consistent activity of several analogs indicates this scaffold is amenable to changes and may present opportunities for future efforts.
Table 7.
Triazolopyridazine hit series 7 SAR determined in the human sperm calcium influx assay.[a]
|
| |||
|---|---|---|---|
|
Compd |
R1 |
R2 |
IC50 [μM] |
|
7 a |
m‐Me |
|
20±1 |
|
7 b |
m‐Me |
|
>100 |
|
7 c |
m‐Me |
|
58±5 |
|
7 d |
p‐Me |
|
25±3 |
|
7 e |
p‐Me |
|
21.7±0.4 |
|
7 f |
p‐Me |
|
68±6 |
|
7 g |
m‐Me |
|
35±10 |
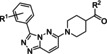
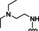
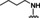
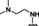
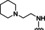
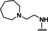
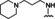
[a] CatSper activated by 3 μM progesterone. IC50 values are means±SEM of three independent experiments.
Mode of inhibition studies
Progesterone activates CatSper indirectly by stimulating activity of the lipid hydrolase ABDH2, which removes tonic inhibition of CatSper by 2‐achrachidonoylglycerol (2‐AG). [54] This finding indicates that the screening hits could inhibit progesterone‐induced activation of ABHD2 rather than acting directly at the CatSper channel. To distinguish between these two possibilities, the effect of increasing concentrations of inhibitors on the dose‐dependent activation of CatSper by progesterone was determined (Figure 6). Inhibitors acting directly at CatSper are expected to reduce the maximum level of progesterone‐induced activation, whereas reduced progesterone potency would indicate competitive inhibition of progesterone activation of ABDH2. All hit scaffolds produced insurmountable inhibition consistent with direct blockade of the channel, rather than indirect inhibition of ABHD2 activation, consistent with their structural dissimilarity to the steroid hormone. Moreover, representative members of all seven hit classes blocked CatSper activated by high potassium/high pH (Table S1) with similar potencies to those determined for progesterone‐induced CatSper inhibition (Tables 1–7), suggesting that their mechanism of inhibition is unrelated to antagonism of progesterone activation of ABHD2.
Figure 6.

Hit compounds from seven series, NNC 55‐0396, and mibefradil produce an insurmountable inhibition of progesterone‐induced calcium increases in human sperm. Concentration‐response curves for progesterone‐induced calcium fluorescence in sperm in the presence of fixed concentrations of compounds. The data are presented as the mean of three independent experiments with error bars representing the SEM.
Electrophysiological confirmation
To confirm that the compound‐induced reduction in intracellular calcium is due to CatSper blockade, we turned to whole cell patch clamp electrophysiology to measure CatSper‐dependent currents in human sperm. Baseline ICatSper was monitored in cesium divalent free media (CsDVF) followed by addition of 10 μM test compound. After the voltage ramp to elicit current, compound washout was initiated by flowing CsDVF media through the chamber to observe if any effects on ICatSper were reversible. As shown in Figure 7 and Table S2, the most potent inhibitors of calcium influx in the FLIPR assay from hit series 2 through 7 all partially or completely block ICatSper at 10 μM. Representative ICatSper traces for each hit scaffold indicate that all blockers reduce both inward and outward ICatSper (Figures 7 and S1‐S5). As seen in the current density plot in Figure 7, compounds 3 a and 6 b show near complete inhibition of the inward ICatSper at 10 μM. Compounds 4 a and 7 a display 84 % and 89 % block, respectively, whereas 2 a and 5 f are the least effective blockers (65 % and 47 % inhibition, respectively). The block by all tested compounds was reversible when washed out with CsDVF, except 5 f, which produced an apparent irreversible loss of channel function (data not shown), similar to mibefradil and NNC 55–0396 reported previously.[ 20 , 21 ] In all cases, each tested compound appeared more potent in the electrophysiology experiments than the FLIPR assay.
Figure 7.
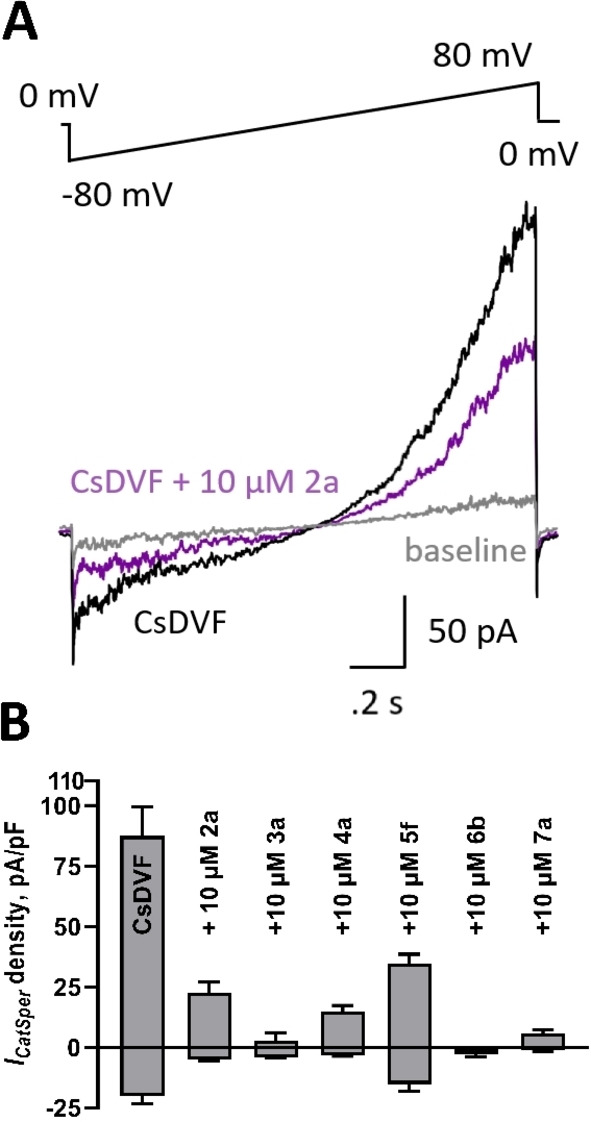
Inhibition of human ICatSper by compounds from each hit series. A. Representative ICatSper recording from human sperm in cesium divalent free media (CsDVF) elicited by the voltage ramp in the absence (black) and presence (purple) of test compound 2 a. Baseline signal in high saline (HS) buffer shown in grey. B. Current density plots of tested compounds showing inhibition of ICatSper at +80 mV (positive scale) or −80 mV (negative scale). Data are displayed as means±SEM of at least 3 independent experiments.
Computer‐aided sperm analysis
Computer‐aided sperm analysis (CASA) utilizes recordings of sperm cells to determine several kinematic parameters, including total, progressive and HAM; HAM is of particular interest, as it results directly from CatSper activation. While several scaffolds had minimal effects on total and progressive motility, compounds 4 a, 5 f, and 7 a substantially reduced both total and progressive motility at 10 μM (Figure 8). As described below, these compounds were more potent inhibitors of HAM, suggesting that they selectively block CatSper, although they may interfere with other molecular targets at higher concentrations. Similar attenuation of progressive motility has been observed with NNC 55–0396, [55] suggesting that this CatSper blocker has nonspecific effects on sperm.
Figure 8.

Effect of compounds from each hit series on total and progressive motility determined by CASA. Increasing concentrations of compounds were incubated for 4 h with human sperm after which time total (solid fill) and progressive (dotted fill) were measured. Data are plotted as a percent of total sperm population and are the mean±SEM of at least 3 individual experiments. **p <0.005, ***p <0.0005 and ****p <0.0001.
To determine the effect of compounds on HAM, we established reliable conditions to generate dose‐dependent increases in populations of sperm displaying HAM based on previous reports.[ 13 , 36 , 37 , 56 ] The ability of each scaffold to diminish HAM elicited by 100 nM progesterone is shown in Figure 9A and 100 nM PGE1 in Figure 9B. Each tested compound generally had a more pronounced HAM inhibition induced by PGE1 than progesterone, suggesting specific differences in CatSper‐mediated HAM. With respect to progesterone‐induced HAM, all scaffolds showed a prominent decrease at 10 μM, though only the reduction by compounds 4 a, 6 b, and 7 a was statistically significant. In the presence of 100 nM PGE1, all compounds produced robust inhibition of HAM at 1 μM, and scaffold 2 a greatly reduced HAM at 100 nM, representing one of the most potent inhibitors of HAM reported to date.
Figure 9.

Compounds from each hit series inhibit hyperactivated motility measured by CASA. A. Progesterone‐induced HAM. B. PGE1‐induced HAM. Compounds at the indicated concentration were incubated with human sperm in the presence of progesterone or PGE1 and HAM was measured after 4 h. Data are plotted as a percent of total sperm population and are the mean±SEM of at least 3 individual experiments. *p <0.05, **p <0.005.
Cytotoxicity studies
To exclude the possibility that the effects observed from these compounds are the result of cytotoxicity, cell viability experiments were performed on the most potent compound from each series as well as reference compounds mibefradil and NNC 55–0396 (Figure 10, Table 8). The assays were performed using human sperm and IMR‐90 fibroblasts, the latter serving as a non‐transformed, non‐germline control. All compounds had little or no effect on sperm viability at concentrations at which full CatSper inhibition was observed, indicating that the observed CatSper inhibition is not due to a cytotoxic effect in sperm. IMR‐90 cells also remained viable up to 100 μM, although compounds 2 a and 6 b appear to reduce viability slightly at concentrations fully inhibiting CatSper, suggesting a lower therapeutic window for these scaffolds. These AlamarBlue viability assays were performed at 2 h to allow for adequate fluorescent signal generation. As such, it is unlikely that a compound exhibiting minimal cytotoxicity at this time point would show any cytotoxicity after 10 min in the FLIPR assay. In contrast, reference compounds mibefradil and NNC 55–0396 both display a low separation between their potencies for blocking calcium influx and cytotoxicity, indicating that caution should be used when interpreting data generated with these commonly used CatSper blockers.
Figure 10.

Activity and cytotoxicity of seven potent compounds from each hit series and two reference compounds. Activity is expressed as the percent of control calcium‐dependent fluorescence elicited by progesterone and high potassium‐induced activation of CatSper in human sperm. Cytotoxicity indicated by the reduction in viability of in both human sperm and IMR‐90 fibroblasts using the AlamarBlue assay.
Table 8.
Cytotoxicity profiling of most potent hits and reference compounds.[a]
|
Compound |
Cytotoxicity: IC50 [μM] |
|
|---|---|---|
|
Sperm |
IMR‐90 |
|
|
Mibefradil |
25±2 |
22±1 |
|
NNC 55–0396 |
62±21 |
13±2 |
|
1 a |
>100 |
>100 |
|
2 a |
>100 |
>100 |
|
3 a |
>100 |
96±5 |
|
4 a |
>100 |
>100 |
|
5 f |
>100 |
83±14 |
|
6 b |
84±10 |
68±5 |
|
7 a |
>100 |
>100 |
[a] Cytotoxicity was measured using AlamarBlue fluorescence. IC50 values are the mean±SEM of three independent experiments. *Compounds increased fluorescence at higher concentrations.
Selectivity studies
Assessing the selectivity of hit compounds is an important step in probe and drug discovery to identify the most promising series of compounds for further analog design and synthesis. We have previously shown that sperm of mice lacking SLO3 have reduced motility and are infertile. [57] SLO3 is expressed only in spermatozoa and therefore has been identified as an important potential target for non‐hormonal male contraception. SLO3 is the main voltage‐gated K+ channel in mouse and human sperm cells[ 58 , 59 ] and is activated by membrane depolarization, increase in pHi and, in human sperm, by intracellular calcium. [60] SLO3‐dependent K+ efflux, which initiates cell membrane hyperpolarization, is involved in the regulation of voltage‐sensitive ion channels like CatSper. [60]
The seven compounds were tested on HEK293 cells co‐expressed with human SLO3 and LRRC52 at a concentration of 10 μM (Figure 11). LRRC52 is an auxiliary subunit of SLO3, co‐expressing with SLO3 channels. It is required for physiological expression and voltage activation of SLO3. Compound 1 a has no significant effect on hSLO3 currents at +60 mV, the current in the presence of 1 a was 101.5±6.4 % of control (n=5, p=0.737); however, the other 6 compounds all significantly inhibited hSLO3 currents at +60 mV. In the presence of 2 a, the current was 72.2±12.9 % of control (n=5, p=0.013); in the presence of 3 a, the current was 58.5±12.5 % of control (n=5, p=0.001); in the presence of 4 a, the current was 24.6±9.1 % of control (n=5, p=0.002); in the presence of 5 f, the current was 83.2±7.8 % of control (n=5, p=0.006); in the presence of 6 b, the current was 66.3±8.5 % of control (n=5, p=0.008); in the presence of 7 a, the current was 37.5±5.6 % of control (n=5, p=0.001).
Figure 11.

Effects of test compounds on human Slo3 currents. Whole‐cell recordings were carried from HEK cells stably transfected with human SLO3 and its auxiliary subunit LRRC52. Currents were evoked from a holding potential of −60 mV by steps from −80 to +150 mV in 10 mV increments. (A to G ) Representative whole‐cell currents in the absence (control) or presence of 10 μM test compounds. Current trace at +60 mV is depicted in red. Each compound was tested against 5 cells. (H) Averaged fractional unblock recorded at +60 mV in the presence of indicated test compounds. Fractional unblock was determined by dividing current amplitudes in the presence of test compounds by the amplitude in the absence of the corresponding compound from the same cell. Data are represented as mean±SEM from 5 cells for each compound. *p<0.05.
In addition, we tested the ability of each compound to inhibit the ion channels hCav1.2, hNav1.5, and hERG (Table 9). Not only are hCav1.2, hNav1.5, and hERG representative members of the calcium, sodium, and potassium channel families, but hCav1.2 and hNav1.5 are the most commonly profiled ion channels after hERG when assessing cardiac risk. In particular, hCav1.2, hNav1.5, and hERG are the minimum set of ion channels required for reliable prediction of the drug‐included ventricular arrhythmia known as Torsades de Pointes. [61] Fixed concentration (10 μM) data indicate that the most potent compounds from series 2–7 are selective for CatSper over hCav1.2, however only compound 7 a is selective for CatSper over all three of these off‐target ion channels implicated in cardiotoxicity.
Table 9.
Ion channel profiling of most potent hits and reference antagonists.
|
Compound |
Inhibition at 10 μM [%] |
||||
|---|---|---|---|---|---|
|
CatSper[a] |
hSlo3[b] |
hCav1.2[c] |
hNav1.5[d] |
hERG[e] |
|
|
Dofetilide |
NA |
NA |
NA |
NA |
IC50= 16 nM[f] |
|
Nifedipine |
NA |
NA |
IC50= 0.13 nM |
NA |
NA |
|
Verapamil |
NA |
NA |
NA |
IC50= 10 μM |
NA |
|
1 a |
NA |
−2 |
4 |
68 |
92 |
|
2 a |
73 |
28 |
39 |
77 |
100 |
|
3 a |
96 |
42 |
10 |
94 |
100 |
|
4 a |
83 |
75 |
65 |
95 |
85 |
|
5 f |
59 |
17 |
14 |
66 |
99 |
|
6 b |
100 |
34 |
27 |
62 |
97 |
|
7 a |
93 |
62 |
−9 |
9.9 |
58[f] |
NA=not applicable; [a] CatSper data from Figure 7, n=3 independent experiments; [b] HEK293 cell line stably expressed with hSlo3 and LRRC52, assay performed by manual patch‐clamp, n=5 cells; [c] hCAV1.2 tested in stably expressed cell line using a FLIPR assay protocol, n=2 cells; [d] CHO cell line stably expressing hNAV1.5, assay performed by manual patch‐clamp, n=2 cells; [e] HEK293 cell line stably expressed with hERG gene, assay performed by manual patch‐clamp, n=2 cells; [f] n=2 independent experiments.
Conclusion
In this study, a high‐throughput screen was performed using an innovative calcium influx assay in human sperm. From the 72,000 compounds screened, seven hit scaffolds were reconfirmed and validated using endogenous CatSper openers progesterone and PGE1. The SAR of these scaffolds was explored using commercially available analogs. For several hit series (2, 3 and 6), narrow, shallow SAR patterns persisted and only limited increases in potency were observed, while many analogs were inactive. In other hit series (1, 4, 5, 7), SAR patterns emerged that provide opportunities for future chemical biology and drug development efforts.
Promising compounds from each hit series were validated for their effects on CatSper by human sperm electrophysiological and motility assays. Little or no cytotoxicity in both human sperm and somatic cells was observed. These assays confirmed the CatSper‐specific action of these scaffolds, which likely block the CatSper channel directly, rather than via an indirect effect on ABHD2. We also showed that except for 1 a, all tested compounds inhibited hSLO3 currents and that compounds 4 a and 7 a were the most potent inhibitors reducing hSLO3 current to 25 % and 38 % of control, respectively. In addition, we tested the seven hits for inhibition of ion channels hCav1.2, hNav1.5, and hERG and found that compound 7 a is the most selective hit compound with little inhibition of hCav1.2, hNav1.5 and moderate inhibition of hERG at 10 μM concentration. Compound 7 a is therefore an excellent candidate for further study because of its selectivity for both CatSper and Slo3 that could provide a dual inhibitor series with high potential for sperm motility inhibition.
Currently, the field relies predominantly on two ion channel blockers, mibefradil and NNC 55–0396, for pharmacological blockade of CatSper. These structurally closely related compounds likely have substantial off‐target activity based on reported reduction in progressive motility and prominent cytotoxicity in human sperm as well as IMR‐90 fibroblasts. This work substantially increases the chemical diversity of CatSper blockers, which will aid the investigation of this complex ion channel that is crucial for reproduction. These compounds represent promising starting points for chemical probes to understand the biology of reproduction and the development of male contraceptive agents.
Experimental Section
General chemical information
The identity and purity of all repurchased hit compounds and analogs was confirmed by 1H NMR and UPLC/MS. All compounds were >95 % pure except for compound 6 b, which had a purity of 92 %.
3‐Amino‐5‐(azepan‐1‐yl)‐6‐chloro‐N‐(diaminomethylene)pyrazine‐2‐carboxamide (1 a): 1H NMR (400 MHz, DMSO‐d6 ) δ 8.05 (br s, 2H), 6.70 (br s, 2H), 4.04 (s, 2H), 3.71 (t, J=6.0 Hz, 4H), 1.77 (p, J=5.4 Hz, 4H), 1.52 (p, J=2.7 Hz, 4H). 96 % purity determined by HPLC. ESI/MS calcd for C12H18ClN7O [M+H]+ 312.13, found 312.13.
4‐(4‐Chlorophenyl)‐1‐(1H‐indol‐3‐ylmethyl)‐4‐piperidinol (2 a): 1H NMR (400 MHz, MeOD) δ 7.68 (d, J=7.9 Hz, 1H), 7.48 (d, J=9.0 Hz, 2H), 7.38 (d, J=8.0 Hz, 1H), 7.32 (d, J=8.0 Hz, 2H), 7.28 (s, 1H), 7.12 (td, J=7.0 Hz, 1H), 7.06 (td, J=7.0 Hz, 1H), 3.85 (s, 2H), 2.92 (ddd, J=11 Hz, 2H), 2.67 (ddd, 2H), 2.11 (ddd, J=13, 13, 4 Hz, 2H), 1.73 (ddd, 2H). 98 % purity determined by HPLC. ESI/MS calcd for C20H21ClN2O [M+H]+ 341.13, found 341.16.
(2,6,8‐Trimethyl‐N‐(2‐phenylethyl)‐4‐quinolinamine (3 a): 1H NMR (400 MHz, CDCl3) δ 7.38–7.32 (m, 2H), 7.29–7.26 (m, 4H), 7.15 (s, 1H), 6.37 (s, 1H), 4.81 (br s, 1H), 3.56 (td, J=7.1, 5.4 Hz, 2H), 3.05 (t, J=7.0 Hz, 2H), 2.72 (s, 3H), 2.62 (s, 3H), 2.42 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 157.5, 149.0, 145. 8, 138.8, 136.6, 132.8, 131.4, 128.8, 126.7, 117.1, 116.0, 99.2, 44.3, 35.1, 26.1, 21.7, 18.5. 99 % purity determined by HPLC. ESI/MS calcd for C20H22N2 [M+H]+ 291.18, found 291.26.
N ,4‐Bis(3‐(trifluoromethyl)phenyl)piperazine‐1‐carboxamide (4 a): 1H NMR (400 MHz, DMSO‐d6 ) 1H NMR (400 MHz, DMSO) δ 8.97 (s, 1H), 7.95 (s, 1H), 7.77 (d, J=8.0 Hz, 1H), 7.48 (t, J=7.8 Hz, 1H), 7.45 (t, J=8.0 Hz, 1H), 7.28 (d, J=8.0 Hz, 2H), 7.24 (s, 1H), 7.11 (d, J=7.6 Hz, 1H), 3.64 (t, J=5.2 Hz, 4H), 3.30 (t, J=5.0, 4H). 97 % purity determined by HPLC. ESI/MS calcd for C19H17F6N3O [M+H]+ 418.13, found 418.17.
1‐(3‐Bromophenyl)‐2‐(2‐imino‐3‐(2‐(piperidin‐1‐yl)ethyl)‐2,3‐dihydro‐1H‐benzo[d]imidazol‐1‐yl)ethan‐1‐ol (HCl salt form) (5 f): 1H NMR (400 MHz, DMSO‐d6 ) δ 10.87 (br s, 1H), 9.18 (s, 2H), 7.88‐7.79 (m, 2H), 7.70 (d, J=7.2 Hz, 1H), 7.62 (d, J=7.7 Hz, 1H), 7.54 (d, J=8.0 Hz, 1H), 7.41–7.35 (m, 3H), 5.94 (d, J=4.3 Hz, 1H), 5.06–4.94 (m, 1H), 4.76 (t, J=7.8 Hz, 2H), 4.32 (d, J=6.5 Hz, 2H), 3.57 (d, J=12.0 Hz, 2H), 3.45–3.39 (m, 2H), 3.10–2.97 (m, 2H), 1.92–1.69 (br m, 5H), 1.51–1.34 (br m, 1H). 99 % purity determined by HPLC. ESI/MS calcd for C22H27BrN4O [M+H]+ 443.14, found 443.16.
2‐(Azepan‐4‐yl)‐4‐(4‐chlorobenzyl)phthalazin‐1(2H)‐one (6 b): 1H NMR (400 MHz, CDCl3) δ 8.45 (d, J=5.9 Hz, 1H), 7.73–7.67 (m, 3H), 7.29–7.27 (d, J=8.4 Hz, 2H), 7.20 (d, J=8.4 Hz, 2H), 5.36 (p, J=7.9 Hz, 1H), 4.27 (s, 2H), 3.47–3.38 (m, 1H), 3.34–3.23 (m, 1H), 3.20–3.09 (m, 2H), 2.48–2.24 (m, 2H), 2.23–2.11 (m, 2H), 2.10–1.81 (m, 3H). 92 % purity determined by HPLC. ESI/MS calcd for C21H22ClN3O [M+H]+ 368.15, found 368.19.
N ‐(2‐(Diethylamino)ethyl)‐1‐(3‐(m–tolyl)‐[1,2,4]triazolo[4,3‐b]pyridazin‐6‐yl)piperidine‐4‐carboxamide (7 a): 1H NMR (400 MHz, acetone‐d6 ) δ 8.40 (s, 1H), 8.36 (d, J=7.8 Hz, 1H), 8.00 (d, J=10.1 Hz, 1H), 7.45 (t, J=7.7 Hz, 1H), 7.38 (d, J=10.2 Hz, 1H), 7.33 (d, J=7.7 Hz, 1H), 6.96 (br s, 1H), 4.36 (d, J=13.4 Hz, 2H), 3.28–3.15 (m, 4H), 2.62–2.48 (m, 7H), 2.46 (s, 3H), 1.99–1.80 (m, 4H), 0.99 (t, J=7.1 Hz, 6H). 13C NMR (101 MHz, MeOD) δ 177.4, 156.9, 148.7, 144.7, 139.6, 131.9, 129.7, 129.1, 127.5, 125.7, 124.9, 116.8, 52.6, 48.2, 47.0, 44.1, 37.7, 29.1, 21.6, 11.5. 98 % purity determined by LC. ESI/MS calcd for C24H33N7O [M+H]+ 436.27, found 436.25.
Isolation of human sperm
Semen samples for the human sperm studies were obtained from volunteers with informed consent. The protocol was approved by the Institutional Review Board of the University of Minnesota: IRB protocol #1102 M96152. Sperm were isolated by centrifugation for FLIPR and cytotoxicity assays or “swim‐up” for CASA and electrophysiology. Sperm samples were subjected to basic semen analysis to establish sperm motility and cell density using a hemocytometer. Centrifugation. Freshly collected semen from male donors was incubated at 37 °C with gentle shaking for up to 60 min to allow complete liquefaction. The sample was washed twice with human tubal fluid (HTF) buffer, followed by centrifugation (800×g, 25 °C, 10 min) and the resulting sperm pellet was re‐suspended in Low/Low (low pH/low K+) buffer (typically 10 mL per plate assayed). Low/Low buffer contained (in mM): 101 NaCl, 4.69 KCl, 0.198 MgSO4 ⋅ 7 H2O, 0.36 KH2PO4, 24.99 NaHCO3, 0.32 sodium pyruvate, 2.78 glucose, 94.08 sodium lactate, 0.2 CaCl2 ⋅ 2 H2O. “Swim‐up”. Human semen was allowed to liquefy at 37 °C for at least 40 min. Concomitantly, conical tubes containing 5 mL of HAMs−F10 (Millipore Sigma, N2147, sodium bicarbonate free) were warmed to 37 °C at a 45° angle. After liquefaction, 1 mL of sample was layered beneath the buffer in each tube and the tubes were incubated at 37 °C under 5 % CO2 for 1 h. The top 2 mL of buffer from each tube was then removed and combined. Sperm prepared using the swim‐up method were used further only if motility was confirmed. For both methods, if a cell density of≥10x106 cell/mL was not achieved, the sample was centrifuged at 400×g for 7 min, and the pellets were resuspended in the appropriate buffer volume to achieve≥10×106 cells/mL.
High‐throughput calcium influx assay
Sperm isolated by centrifugation were resuspended in Low/Low buffer containing 10 μM Fluo‐4‐AM and, when present, 1 mM probenecid to prevent dye efflux for a dye‐loading period of 30 min in the dark at RT. Following a final wash, the sample was resuspended in Low/Low buffer and 10 μL of dye‐loaded sperm (2–5×106 cells/mL) was dispensed using a MultiDrop Combi (Thermo Scientific) into black, clear‐bottom 384‐well assay plates (Corning 3683). A separate 384‐well compound microplate contained test compound at 2.5x final concentration in Low/Low buffer prepared using a Labcyte Echo® 550 acoustic dispenser. A separate 384‐well buffer microplate was also prepared containing buffers to either induce opening of the CatSper channel (activation buffer) or not (control buffer), corresponding to high and low controls, respectively. Following transfer of the plates to a FLIPR Tetra (Molecular Devices), the assay plate fluorescence was monitored at 470–495 nm (excitation) and 515–575 nm (emission) at 2.5 s intervals. Baseline measurements were taken for 0.5 min, followed by addition of 10 μL test compound from the compound plate using the FLIPR 384‐pipette head. After a 2 min compound binding period (phase 1), 5 μL from the buffer plate was transferred to the sperm assay plate and fluorescence monitored for an additional 2–4 min (phase 2).
For primary HTS data analysis, the resulting RFU values were loaded into ActivityBase (IDBS v.8.0) and % inhibition values for compounds at 10 μM (n=1) were calculated at 240 s post activation relative to the high and low controls. Compounds producing a % inhibition between 60 and 110 % were identified as hits using SARview (IDBS, v.7.3). IC50 values for hits cherry‐picked from the HTS library DMSO stocks were determined from 8‐point, 5‐fold serial dilutions of compound covering a range of 62.5 μM to 0.8 nM in duplicate. Percent inhibition was calculated with traces zeroed at the compound addition step. FLIPR traces for compounds showing well‐defined sigmoidal log concentration‐response curves with IC50 values of <55 μM were examined to eliminate those showing substantial increases or decreases in RFU during the first phase of the trace following compound addition, e. g., due to autofluorescence molecules or fluorescence quenchers, respectively. In parallel, an assessment of these compounds based on properties such as undesirable chemical functionalities, chemical tractability, hydrophobicity, and synthetic feasibility was performed.
Progesterone‐induced calcium uptake assay and competition assays
The progesterone activation assay was identical to the HTS assay, except that progesterone (3 μM) in Low/Low buffer was added to the assay plate instead of activation buffer. Repurchased compounds were tested in 8‐point, half‐log serial dilutions in duplicate covering a range of 32 nM to 100 μM. For the progesterone competition studies, progesterone dose response experiments (9.5 to 30 μM) were performed in the presence of four concentrations of test compound. Calcium‐dependent fluorescence was monitored in the FLIPR as described for the HTS assay, except that phases 1 and 2 were both 5 min.
Electrophysiology
Electrophysiology experiments using human sperm were performed as described previously. [62] Briefly, a gigaohm seal between the patch pipette and human spermatozoon was formed at the cytoplasmic droplet using a flame polished pipette with intrinsic resistances of 12 to 18 MΩ. Seals were formed in high saline (HS) solution containing (in mM): 130 NaCl, 5 KCl, 1 MgSO4, 2 CaCl2, 5 glucose, 1 sodium pyruvate, 10 lactic acid, and 20 HEPES, pH 7.4 adjusted with NaOH. The osmolarity of the solution was confirmed to be between 317 to 320 mOsm/liter. Transition to the whole‐cell mode was performed by applying 1 ms, 450 mV pulses. Access resistance was between 30 to 90 MΩ. ICatSper was elicited by a voltage ramp of −80 mV to +80 mV from a holding potential of 0 mV. Cells were stimulated every 3 s. Data were sampled at 5 kHz and filtered at 1 kHz. HS solution was used to record baseline current. Pipettes recording monovalent CatSper currents were filled with CsDVF medium containing (in mM): 130 Cs‐methanesulfonate, 70 HEPES, 3 EGTA, 2 EDTA, 0.5 TrisHCl, pH 7.4 adjusted with CsOH. Bath divalent free (DVF) solution for recording of monovalent CatSper currents contained (in mM): 140 Cs‐methanesulfonate, 40 HEPES, 1 EDTA, pH 7.4 adjusted with CsOH. Upon break‐in to whole cell mode, bath solution was changed to CsDVF to measure the control ICatSper current, followed by a switch to CsDVF containing indicated compound concentration for ∼8–10 s to measure the inhibited ICatSper current. Osmolarities of the bath and pipette solutions were approximately 321 mOsm/liter and 335 mOsm/liter, respectively. All experiments were performed at RT.
Computer‐aided sperm analysis
Viable human spermatozoa were selected by swim‐up. Samples were discarded if there was a loss of motility following the final centrifugation step determined by computer‐assisted sperm analysis (CASA, HTM‐IVOS sperm analysis system, version 12.3, Hamilton Thorne Biosciences, Beverly, MA). CASA analysis including determination of average path velocity (VAP, μm/s), straight‐line velocity (VSL, μm/s,) and curvilinear velocity (VCL, μm/s). From these measurements, linearity of progression [LIN=(VSL/VCL) ×100] and straightness [STR=(VSL/VAP) ×100] were determined and used to calculate total and progressive motility and to determine the population of cells displaying hyperactivated motility (%HAM). For capacitation experiments, cells were suspended in HAM's‐F10 containing 5 % (w/v) BSA, 15 mM NaHCO3, and 100 nM progesterone or PGE1 and incubated for 3.5 h at 37 °C in 5 % CO2 either in the presence of compound or DMSO vehicle. Data is given as the respective percent of the whole population and reflects the average of≥3 independent experiments±SEM. All experiments were performed at 37 °C and a minimum of 10 fields of view were analyzed containing at least 200 cells total for each condition.
Cytotoxicity assays
Cell viability was measured in both human sperm and IMR‐90 fibroblasts using AlamarBlue®. Human sperm prepared using the centrifugation method were brought up in Low/Low buffer. IMR‐90 lung fibroblast cells purchased from ATCC were cultured for 2 weeks in EMEM supplemented with 10 % FBS and split 2–4 days prior to the experiment. IMR‐90 cells were detached using trypsin‐EDTA (0.25 %/0.53 mM) and re‐suspended in culture medium. For both cell types, 20 μL of cell suspension was dispensed into black, clear‐bottom 384‐well assay plates (Corning 3683) containing 20 μL of test compounds at 2.2x in Low/Low buffer. Compounds were tested at 8 concentrations using half‐log dilutions in duplicate, with 100 μM to 32 nM final compound concentrations. AlamarBlue reagent (4 μL) was added and the assay plate was incubated at 37 °C. Fluorescence readings were taken at 15 min intervals over 3 h for sperm or 30 min intervals over 7 h for IMR‐90 cells using a SpectraMax® M2e plate reader (Molecular Devices) at 560/590 nm (ex/em).
Data analysis
IC50 values for calcium influx and cytotoxicity studies were determined using Prism 7.0.5 (GraphPad, San Diego, CA, USA). Statistical analysis of the CASA and electrophysiology data was conducted by one‐way ANOVA followed by Dunnett's multiple comparisons test to identify statistically significant differences using Prism.
Methods for SLO3 inhibition
Cell culture: HEK‐293 cells stably expressing SLO3 and LRRC52 were provided by Jerod Denton (Vanderbilt University) and maintained in DMEM complete, composed of Dulbecco's Modified Eagle Medium supplemented with 700 μg/mL Zeocine, 10 % FBS, and 1 % Penicillin‐Streptomycin (Thermo Fisher Scientific). Cells were grown on plastic Petri dishes and flasks at 37 °C and 5 % CO2 in a Galaxy 170 S incubator (Eppendorf). Prior to patching, flasks at ∼80 % confluence were rinsed with Hank's balanced salt solution followed by disassociation with trypsin‐EDTA (Thermo Fisher Scientific). Suspended cells were diluted in DMEM complete and distributed to 9.2 cm2 Petri dishes coated with 1 μL 2 % Matrigel® Basement Membrane Matrix (Corning) in Minimum Essential Medium (Thermo Fisher Scientific).
Electrophysiology: Voltage‐clamp recordings were performed using an Axopatch 200B amplifier (Molecular Devices). Recordings were filtered at 2 kHz with the amplifier internal filter and digitized at 10 kHz using a Digidata 1550B digitizer (Molecular Devices). Recording pipettes were pulled from borosilicate glass with tip resistances of 2–3 MΩ after filling with pipette solution containing (in mM): 140 KOH, 10 MES, 1 EGTA, 10 HEPES, pH 7.4 with KOH. Bath solutions contained (in mM): 135 NaOH, 5 KOH, 1 MgCl2, 10 MES, 10 HEPES, 5 TEA, pH 7.4 with NaOH. In order to exclude endogenous potassium channels, 5 mM TEA was added to perfusion. Test compounds were prepared daily from a 10 mM stock solution (in DMSO). Data were analyzed with pClamp 10.6 (Molecular Devices). Statistical analysis was performed with OriginPro 7.5 (OriginLab Corporation). Mean values are presented as mean± SD in text and mean± SEM in figures. Paired Student's t tests were used to compare whole‐cell currents of cells before and after treatment with test compounds. P<0.05 was considered statistically significant.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We gratefully acknowledge the support from NIH/NICHD grants U01HD076542 and HHSN275201300017 C (to G.I.G) and R33HD099742 (to C.M.S) and R01HD069631 (to C.M.S). We thank Dr. Stanley Thayer of the University of Minnesota for the use of electrophysiology equipment and Dr. Polina Lishko of UC Berkeley for sperm electrophysiology training. We thank Dr. Henry Wong for coordinating sperm donations.
E. J. Carlson, R. Francis, Y. Liu, P. Li, M. Lyon, C. M. Santi, D. J. Hook, J. E. Hawkinson, G. I. Georg, ChemMedChem 2022, 17, e202000499.
Contributor Information
Prof. Jon E. Hawkinson, Email: hawkinso@umn.edu.
Prof. Gunda I. Georg, Email: georg@umn.edu.
References
- 1. Lancet 1990, 336, 955–959.1977002 [Google Scholar]
- 2. de la Torre B., Noren S., Hedman M., Diczfalusy E., Contraception 1979, 20, 377–396. [DOI] [PubMed] [Google Scholar]
- 3. Long J. E., Lee M. S., Blithe D. L., Clin. Chem. 2019, 65, 153–160. [DOI] [PubMed] [Google Scholar]
- 4. Roth M. Y., Amory J. K., Semin. Reprod. Med. 2016, 34, 183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Long J. E., Lee M. S., Blithe D. L., J. Clin. Endocrinol. Metab. 2021, 106, e2381-e2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Quill T. A., Ren D., Clapham D. E., Garbers D. L., Proc. Natl. Acad. Sci. USA 2001, 98, 12527–12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ren D., Navarro B., Perez G., Jackson A. C., Hsu S., Shi Q., Tilly J. L., Clapham D. E., Nature 2001, 413, 603–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shu F., Zhou X., Li F., Lu D., Lei B., Li Q., Yang Y., Yang X., Shi R., Mao X., J. Assist. Reprod. Genet. 2015, 32, 1643–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Williams H. L., Mansell S., Alasmari W., Brown S. G., Wilson S. M., Sutton K. A., Miller M. R., Lishko P. V., Barratt C. L., Publicover S. J., Martins da Silva S., Hum. Reprod. 2015, 30, 2737–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smith J. F., Syritsyna O., Fellous M., Serres C., Mannowetz N., Kirichok Y., Lishko P. V., Proc. Natl. Acad. Sci. USA 2013, 110, 6823–6828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Avenarius M. R., Hildebrand M. S., Zhang Y., Meyer N. C., Smith L. L., Kahrizi K., Najmabadi H., Smith R. J., Am. J. Hum. Genet. 2009, 84, 505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xia J., Reigada D., Mitchell C. H., Ren D., Biol. Reprod. 2007, 77, 551–559. [DOI] [PubMed] [Google Scholar]
- 13. Suarez S. S., Hum. Reprod. Update 2008, 14, 647–657. [DOI] [PubMed] [Google Scholar]
- 14. Navarro B., Kirichok Y., Clapham D. E., Proc. Natl. Acad. Sci. USA 2007, 104, 7688–7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ho H. C., Suarez S. S., Reproduction 2001, 122, 519–526. [DOI] [PubMed] [Google Scholar]
- 16. Gwathmey T., Blackmore P. F., Mahony M. C., J. Androl. 2000, 21, 534–540. [PubMed] [Google Scholar]
- 17. Burkman L. J., Arch. Androl. 1984, 13, 153–165. [DOI] [PubMed] [Google Scholar]
- 18. Pilikian S., Adeleine P., Czyba J. C., Ecochard R., Guerin J. F., Mimouni P., Int. J. Androl. 1991, 14, 167–173. [DOI] [PubMed] [Google Scholar]
- 19. Hillensjo T., Sjogren A., Strander B., Andino N., Acta Endocrinol. 1985, 108, 407–413. [DOI] [PubMed] [Google Scholar]
- 20. Lishko P. V., Botchkina I. L., Kirichok Y., Nature 2011, 471, 387–391. [DOI] [PubMed] [Google Scholar]
- 21. Strunker T., Goodwin N., Brenker C., Kashikar N. D., Weyand I., Seifert R., Kaupp U. B., Nature 2011, 471, 382–386. [DOI] [PubMed] [Google Scholar]
- 22. Brenker C., Goodwin N., Weyand I., Kashikar N. D., Naruse M., Krahling M., Muller A., Kaupp U. B., Strunker T., EMBO J. 2012, 31, 1654–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brenker C., Zhou Y., Muller A., Echeverry F. A., Trotschel C., Poetsch A., Xia X. M., Bonigk W., Lingle C. J., Kaupp U. B., Strunker T., eLife 2014, 3, e01438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Su L., Cheng C. Y., Mruk D. D., Int. J. Biochem. Cell Biol. 2010, 42, 1864–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tash J. S., Attardi B., Hild S. A., Chakrasali R., Jakkaraj S. R., Georg G. I., Biol. Reprod. 2008, 78, 1127–1138. [DOI] [PubMed] [Google Scholar]
- 26. Lue Y., Sinha Hikim A. P., Wang C., Leung A., Baravarian S., Reutrakul V., Sangsawan R., Chaichana S., Swerdloff R. S., J. Androl. 1998, 19, 479–486. [PubMed] [Google Scholar]
- 27. Chang Z., Qin W., Zheng H., Schegg K., Han L., Liu X., Wang Y., Wang Z., McSwiggin H., Peng H., Yuan S., Wu J., Wang Y., Zhu S., Jiang Y., Nie H., Tang Y., Zhou Y., Hitchcock M. J. M., Tang Y., Yan W., Nat. Commun. 2021, 12, 1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shang E., Nickerson H. D., Wen D., Wang X., Wolgemuth D. J., Development 2007, 134, 3507–3515. [DOI] [PubMed] [Google Scholar]
- 29. Amory J. K., Muller C. H., Shimshoni J. A., Isoherranen N., Paik J., Moreb J. S., D. W. Amory, Sr. , Evanoff R., Goldstein A. S., Griswold M. D., J. Androl. 2011, 32, 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Noman M. A. A., Kyzer J. L., Chung S. S. W., Wolgemuth D. J., Georg G. I., Biol. Reprod. 2020, 103, 390–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Carlson A. E., Burnett L. A., del Camino D., Quill T. A., Hille B., Chong J. A., Moran M. M., Babcock D. F., PLoS One 2009, 4, e6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang L., Keyser B. M., Tagmose T. M., Hansen J. B., Taylor J. T., Zhuang H., Zhang M., Ragsdale D. S., Li M., J. Pharmacol. Exp. Ther. 2004, 309, 193–199. [DOI] [PubMed] [Google Scholar]
- 33. Tamburrino L., Marchiani S., Minetti F., Forti G., Muratori M., Baldi E., Hum. Reprod. Update 2014, 29, 418–428. [DOI] [PubMed] [Google Scholar]
- 34. Rennhack A., Schiffer C., Brenker C., Fridman D., Nitao E. T., Cheng Y. M., Tamburrino L., Balbach M., Stolting G., Berger T. K., Kierzek M., Alvarez L., Wachten D., Zeng X. H., Baldi E., Publicover S. J., Kaupp U. B., Strunker T., Br. J. Pharmacol. 2018, 175, 3144–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Carlson E. J., Georg G. I., Hawkinson J. E., Mol. Pharmacol. 2022, 101, 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mannowetz N., Miller M. R., Lishko P. V., Proc. Natl. Acad. Sci. USA 2017, 114, 5743–5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miller M. R., Kenny S. J., Mannowetz N., Mansell S. A., Wojcik M., Mendoza S., Zucker R. S., Xu K., Lishko P. V., Cell Rep. 2018, 24, 2606–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brenker C., Rehfeld A., Schiffer C., Kierzek M., Kaupp U. B., Skakkebæk N. E., Strünker T., Hum. Reprod. Update 2018, 33, 1915–1923. [DOI] [PubMed] [Google Scholar]
- 39. Brenker C., Schiffer C., Wagner I. V., Tüttelmann F., Röpke A., Rennhack A., Kaupp U. B., Strünker T., Proc. Natl. Acad. Sci. USA 2018, 115, E344–E346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chung J. J., Navarro B., Krapivinsky G., Krapivinsky L., Clapham D. E., Nat. Commun. 2011, 2, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Moffat J. G., Vincent F., Lee J. A., Eder J., Prunotto M., Nat. Rev. Drug Discovery 2017, 16, 531–543. [DOI] [PubMed] [Google Scholar]
- 42. Mehlin C., Crittenden C., Andreyka J., BioTechniques 2003, 34, 164–166. [DOI] [PubMed] [Google Scholar]
- 43. Quesney L. F., Andermann F., Lal S., Prelevic S., Neurology 1980, 30, 1169–1174. [DOI] [PubMed] [Google Scholar]
- 44. Millan M. J., Dekeyne A., Rivet J. M., Dubuffet T., Lavielle G., Brocco M., J. Pharmacol. Exp. Ther. 2000, 1063–1073. [PubMed] [Google Scholar]
- 45. Sian J., Youdim M., Riederer P., Gerlach M., in Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed. (Eds.: Siegel G., Agranoff B., Albers R., Fisher A., Uhler M.), Lippincott-Raven, Philadelphia, 1999. [Google Scholar]
- 46. Topliss J. G., J. Med. Chem. 1972, 15, 1006–1011. [DOI] [PubMed] [Google Scholar]
- 47. Swanson D. M., Dubin A. E., Shah C., Nasser N., Chang L., Dax S. L., Jetter M., Breitenbucher J. G., Liu C., Mazur C., Lord B., Gonzales L., Hoey K., Rizzolio M., Bogenstaetter M., Codd E. E., Lee D. H., Zhang S.-P., Chaplan S. R., Carruthers N. I., J. Med. Chem. 2005, 48, 1857–1872. [DOI] [PubMed] [Google Scholar]
- 48. Bowman J. D., Merino E. F., Brooks C. F., Striepen B., Carlier P. R., Cassera M. B., Antimicrob. Agents Chemother. 2014, 58, 811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shteinikov V. Y., Barygin O. I., Gmiro V. E., Tikhonov D. B., Eur. J. Pharmacol. 2019, 844, 183–194. [DOI] [PubMed] [Google Scholar]
- 50. Smith-Maxwell C., Begenisich T., J. Gen. Physiol. 1987, 90, 361–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Danko M., Smith-Maxwell C., McKinney L., Begenisich T., Biophys. J. 1986, 49, 509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bernstein J. A., Curr. Med. Res. Opin. 2007, 10, 2441–2452. [DOI] [PubMed] [Google Scholar]
- 53. Park M.-H., Lee S. H., Chu D. H., Won K. H., Choi B. H., Choe H., Jo S.-H., J. Pharmacol. Sci. 2013, 123, 67–77. [DOI] [PubMed] [Google Scholar]
- 54. Miller M. R., Mannowetz N., Iavarone A. T., Safavi R., Gracheva E. O., Smith J. F., Hill R. Z., Bautista D. M., Kirichok Y., Lishko P. V., Science 2016, 352, 555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ghanbari H., Keshtgar S., Kazeroni M., Iran. J. Med. Sci. 2018, 43, 18–25. [PMC free article] [PubMed] [Google Scholar]
- 56. Allouche-Fitoussi D., Bakhshi D., Breitbart H., Mol. Reprod. Dev. 2018, 85, 543–556. [DOI] [PubMed] [Google Scholar]
- 57. Santi C. M., Martínez-López P., de la Vega-Beltrán J. L., Butler A., Alisio A., Darszon A., Salkoff L., FEBS Lett. 2010, 584, 1041–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zeng X. H., Yang C., Kim S. T., Lingle C. J., Xia X. M., Proc. Natl. Acad. Sci. USA 2011, 108, 5879–5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Geng Y., Ferreira J. J., Dzikunu V., Butler A., Lybaert P., Yuan P., Magleby K. L., Salkoff L., Santi C. M., J. Biol. Chem. 2017, 292, 8978–8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kumar S., Ying Y. K., Hong P., Maddaiah V. T., Arch. Androl. 2000, 44, 93–101. [DOI] [PubMed] [Google Scholar]
- 61. Tran P. N., Sheng J., Randolph A. L., Baron C. A., Thiebaud N., Ren M., Wu M., Johannesen L., Volpe D. A., Patel D., Blinova K., Strauss D. G., Wu W. W., PLoS One 2020, 15, e0241362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lishko P., Clapham D. E., Navarro B., Kirichok Y., Methods Enzymol. 2013, 525, 59–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information