

Abstract
Hypertension is an important risk factor in the pathogenesis of diastolic dysfunction. Growing evidence indicates that glucose metabolism plays an essential role in diastolic dysfunction. TP53-induced glycolysis and apoptosis regulator (TIGAR) has been shown to regulate glucose metabolism and heart failure (HF). In the present study, we investigated the role of TIGAR in diastolic function and cardiac fibrosis during pressure overload (PO)-induced HF. WT mice subjected to transverse aortic constriction (TAC), a commonly used method to induce diastolic dysfunction, exhibited diastolic dysfunction as evidenced by increased E/A ratio and E/E’ ratio when compared to its sham controls. This was accompanied by increased cardiac interstitial fibrosis. In contrast, knockout of TIGAR attenuated PO-induced diastolic dysfunction and interstitial fibrosis. Mechanistically, the levels of glucose transporter Glut-1, and Glut-4, and key glycolytic enzyme phosphofructokinase 1 (PFK-1) were significantly elevated in TIGAR KO subjected to TAC as compared to that of WT mice. Knockout of TIGAR significantly increased fructose 2,6-bisphosphate levels and phosphofructokinase activity in mouse hearts. In addition, PO resulted in a significant increase in perivascular fibrosis and endothelial activation in the WT mice, but not in the TIGAR KO mice. Our present study suggests a necessary role of TIGAR-mediated glucose metabolism in PO-induced cardiac fibrosis and diastolic dysfunction.
Keywords: TIGAR, glucose metabolism, diastolic function, cardiac fibrosis
1. Introduction
Heart failure (HF) is one of the leading causes of hospitalization, morbidity, mortality, and health care burden in the United States (Benjamin et al., 2019). More than half of the HF patients are diagnosed with HF with preserved ejection fraction (HFpEF) (Dhingra et al., 2014; Upadhya, Taffet, Cheng, & Kitzman, 2015). Diastolic dysfunction is one of the major characteristics of HFpEF and patients with reduced EF. Hypertension, diabetes mellitus, obesity, chronic renal failure, left ventricular (LV) hypertrophy, and aging are the major risk factors for the development of HF and diastolic dysfunction (Brouwers et al., 2013; Lam, Donal, Kraigher-Krainer, & Vasan, 2011; Mandinov, Eberli, Seiler, & Hess, 2000; Mohammed et al., 2015; Redfield et al., 2003; Tanno, Kuno, Horio, & Miura, 2012). However, our understanding of its pathophysiological and molecular mechanism is incomplete.
Recent studies demonstrate that impaired glucose metabolism, especially aberrant glycolysis plays s critical role in the development of diastolic dysfunction (Fillmore et al., 2018; Stahrenberg et al., 2010). Our previous studies showed that specific knockout of Sirtuin 3 (SIRT3) in endothelial cells (ECs) reduced EC glycolysis and angiogenesis, and thus led to diastolic dysfunction (He et al., 2017). Fructose 2,6-bisphosphate (F2,6-BP) is a potent allosteric activator of phosphofructokinase 1 (PFK-1), whose level is finely regulated by TP53-induced glycolysis and apoptosis regulator (TIGAR) and 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase isoform 3 (PFKFB3) (De Bock et al., 2013). TIGAR reduces the level of F2,6-BP and thus decreases PFK-1 activity and glycolysis (Bensaad et al., 2006; Green & Chipuk, 2006). Okawa and colleagues recently demonstrated that the ablation of TIGAR attenuated pressure overload (PO)-induced systolic dysfunction and HF-associated damage by preserving glucose oxidation and glycolysis in cardiomyocytes (Okawa et al., 2019). However, the role of TIGAR in diastolic function during PO-induced HF has not been studied.
By using the TIGAR knockout (TIGAR KO) mice, we aimed to further explore the potential roles of TIGAR in regulating glucose metabolism, diastolic function, and cardiac remodeling in the PO-induced HF models. Our data revealed that the wild type (WT) mice subjected to PO exhibited severe diastolic function as evidenced by elevated E/e’ and E/A ratios, together with a significant increase in cardiac fibrosis. Mechanistically, knockout of TIGAR enhanced cardiac glucose metabolism by upregulating the levels of F2,6-BP and PFK-1 activity, together with increased expression of glucose transporters. Our present data suggest a fundamental role of TIGAR in glycolytic metabolism in PO-induced diastolic dysfunction.
2. Materials and Methods
2.1. Mice
Male C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and were used as wild type (WT) controls. Male TIGAR deficient (TIGAR KO) mice (Tang et al., 2018) on the C57BL/6 background were kindly gifted by Dr. Jeffrey Pessin at the Albert Einstein College of Medicine and maintained in the Laboratory Animal Facilities at the University of Mississippi Medical Center (UMMC). All animals were fed laboratory standard chow and water and housed in individually ventilated cages. All protocols were approved by the Institutional Animal Care and Use Committee at UMMC (Protocol ID: 1564) and were in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Pub. No. 85-23, Revised 1996).
2.2. Transverse Aortic Constriction Procedure
4-5 months old WT and TIGAR KO mice were subjected to transverse aortic constriction (TAC) for 8 weeks to induce PO-induced heart failure as previously described with minor modification (Tavakoli, Nemska, Jamshidi, Gassmann, & Frossard, 2017; Zeng, He, & Chen, 2020). Briefly, the mice were anesthetized with a single intraperitoneal injection of ketamine (50 mg/kg) and xylazine (10 mg/kg) in a supine position on a heating pad. Partial thoracotomy to the second rib was performed and a small piece of 6-0 silk suture was placed between the innominate and the left common carotid arteries. A small L-shaped piece of a blunt 27-gauge needle was placed parallel to the transverse aorta. The suture was tied snugly and quickly, and the needle was removed to yield a constriction of 0.4 mm in diameter. The thorax was then closed using 6-0 silk suture and mice recovered in a warming chamber until they were fully awake. The sham procedure is identical, except without the ligation of the aorta. Mice were inspected daily, and postoperative analgesia is administrated by intraperitoneal injection of 1 ml/kg body weight Carprofen (5 mg/ml) every 24 h over 3 days. Echocardiogram was conducted on mice at 8 weeks post-surgery. The animals were then euthanized, and the tissues were harvested for further experiments.
2.3. Echocardiography
Transthoracic echocardiograms were performed on mice using a Vevo 3100 Preclinical Imaging Platform equipped with an MX400 transducer (FUJIFILM Visual Sonics Inc., Canada). The mice were anesthetized by inhalation of 1–1.5% isoflurane mixed with 100% medical oxygen, and the heart rate was maintained between 450 and 500 beats per minute. Transmitral inflow pulsed-wave (PW) Doppler and Tissue Doppler (TD) imaging were used to measure diastolic function. From an apical 4-chamber view, the peak velocity of early (E) and late (A) filling of mitral inflow, isovolumic relaxation time (IVRT), isovolumic contraction time (IVCT), and aortic ejection time (AET) were assessed. The myocardial performance index (MPI) was calculated using the following formula: MPI = (IVRT + IVCT)/AET. In addition, TD images were obtained from the mitral annulus to measure tissue motion velocity in early and late diastole (e’ and a’, respectively) and to calculate E/e’ ratio (Gao, Ho, Vatner, & Vatner, 2011; He et al., 2017; He, Zeng, Roman, & Chen, 2018; Zeng et al., 2020).
2.4. Histology and Immunofluorescence
Left ventricles were fixed with 10% neutral buffered formalin, processed, embedded in paraffin, and sectioned at 5-μm thickness. Picrosirius red staining was used to evaluate the degree of cardiac interstitial and perivascular fibrosis. Microscopic photos of 5-10 fields were randomly selected and taken by using the Nikon Eclipse 80i microscope for each mouse. The fibrotic fraction was calculated as the ratio of Picrosirius red-stained area to total myocardial area. Fibrosis around the coronary arteries was semi-automatically quantified and expressed as perivascular fibrosis index using the following formula: perivascular fibrosis index = perivascular area (μm2)/vessel wall area (μm2). Vascular remodeling index was calculated as the square root of ratio of vessel wall area to the lumen area.
Cryostat sections also were stained with either Alexa Fluor™ 488 conjugated Isolectin B4 (1:50; IB4, Invitrogen, OR), Alexa Fluor™ 488 conjugated wheat germ agglutinin (5μg/mL, WGA, Invitrogen, OR), or anti-VCAM-1 (1:400, #39036, Cell Signaling, CO) primary antibody, followed by incubation with anti-rabbit Cy™3 conjugated secondary antibody (Jackson ImmunoResearch, PA). 5 to 10 fields per section per mouse were taken by using the Nikon Eclipse 80i microscope, coupled with an X-Cite® 120 Fluorescence Illumination system (Nikon Instruments, NY). The cross-sectional area of cardiomyocyte and VCAM-1 positive area were measured by ImageJ software.
2.5. Immunoblot Analysis
Protein extractions from heart ventricular samples were prepared in lysis buffer with protease/phosphatase inhibitor cocktail (A32961, Thermo Fisher Scientific, NY). Lysates were separated by SDS-PAGE under reducing conditions, transferred to a PVDF membrane, and analyzed by immunoblotting. The PVDF membranes were probed with primary antibodies PFK-1 (sc-377346, Santa Cruz, TX), Glut-1 (NB110-39113, Novus Biologicals, CO), Glut-4 (#2213, Cell Signaling, MA), VCAM-1 (#39036, Cell Signaling, MA), GAPDH (#2118, Cell Signaling, MA), or β-tubulin (#86298, Cell Signaling, MA). The membranes were then washed and incubated with an anti-rabbit (31460) or anti-mouse (31430) secondary antibody conjugated with horseradish peroxidase (1:10000, Thermo Fisher Scientific, NY). Densitometries were analyzed in the Image Lab software 6.0 (Bio-Rad, CA).
2.6. Tissue F2,6-BP Assay
Tissue F2,6-BP concentration was determined as previously described (Van Schaftingen, Lederer, Bartrons, & Hers, 1982). Briefly, samples of tissues were weighed and homogenized in NaOH (0.05 M). The resulting mixture was heated for 20 min at 80°C. After cooling, the samples were neutralized with 1M acetic acid in the presence of 20 mM Hepes, and then centrifuged. Samples were incubated at 37 °C for 5 min in the following assay mixture: 50 mM Tris, 5 mM Mg2+, 1 mM fructose-6-phosphate (Sigma #F3627), 0.15 mM NADH (Sigma #N4505), excessive PPi-dependent PFK-1 (enriched from potato tubers), 0.2U/mL aldolase (Sigma #A2714), 8U/mL triosephosphate isomerase (Sigma #T2507) and 1U/mL glycerol-3-phosphate dehydrogenase (Sigma #10127752001). After the 5 min pre-incubation time, 0.5 mM pyrophosphate was added to start the reaction, and the rate of change in OD340 nm every 30 seconds was followed for 5 min in a Bio-Rad xMark microplate spectrophotometer (Bio-Rad). Data are expressed as the fold change compared to the WT controls.
2.7. Phosphofructokinase Activity Assay
Tissue PFK-1 activity was determined as previously described (Deng et al., 2008). Briefly, samples of tissues were weighted and homogenized in lysis buffer followed by sonication and centrifugation. The reaction was performed using 4 μg of total protein in a 96-well plate containing 80 μL of the reaction buffer (50 mM Tris–HCl, pH 7.5, 5 mM MgCl2, 5mM ATP (Sigma #A6419), 0.2 mM NADH, 100 mM KCl, 5 mM Na2HPO4, 0.1 mM AMP (Sigma #A2252), 5 mM fructose-6-phosphate, 5U/mL triosephosphate isomerase, 1U/mL aldolase, and 10U/mL glycerol-3-phosphate dehydrogenase. Absorbance at 340 nm was read at 37°C every 30 seconds for a period of 30 min in a Bio-Rad xMark microplate spectrophotometer. Data are expressed as the change in absorbance at 340 nm/min/mg of protein.
2.8. Statistical Analysis
Data are presented as mean ± SEM. The assumptions of normality in both comparison groups were determined by normality and long-normality test. Statistical significance was determined by using two-way ANOVA followed by Tukey’s post-hoc test for multiple comparisons in GraphPad Prism 8 software (San Diego, CA). P<0.05 was considered statistically significant.
3. Results
3.1. Knockout of TIGAR attenuates pressure overload induced fibrosis and hypertrophy
Cardiac fibrosis is the key factor that contributes to the development of diastolic dysfunction (Katholi & Couri, 2011; Mohammed et al., 2015; Ogata et al., 2004; Reed et al., 2011). We then examined the cardiac fibrosis by Picrosirius red staining. As shown in Figure 1A and 1B, eight weeks after subjected to TAC, WT mice exhibited significantly greater interstitial fibrosis when compared to that of the WT sham mice, whereas ablation of TIGAR attenuated PO-induced interstitial fibrosis. The whole heart sections showed that the WT mice exhibited remarkable cardiac hypertrophy after TAC, but not the TIGAR KO mice (Figure 1A). Immunofluorescent staining showed that WT mice exhibited a significantly greater number of TGF-β1-positive cells in the myocardium, but it was not seen in the TIGAR KO mice (Figure 1C). In addition, wheat germ agglutinin (WGA) staining revealed a significant increase in the cross-sectional area of cardiomyocytes after TAC in the WT mice than that of the TIGAR KO mice (Figure 1D).
Figure 1.
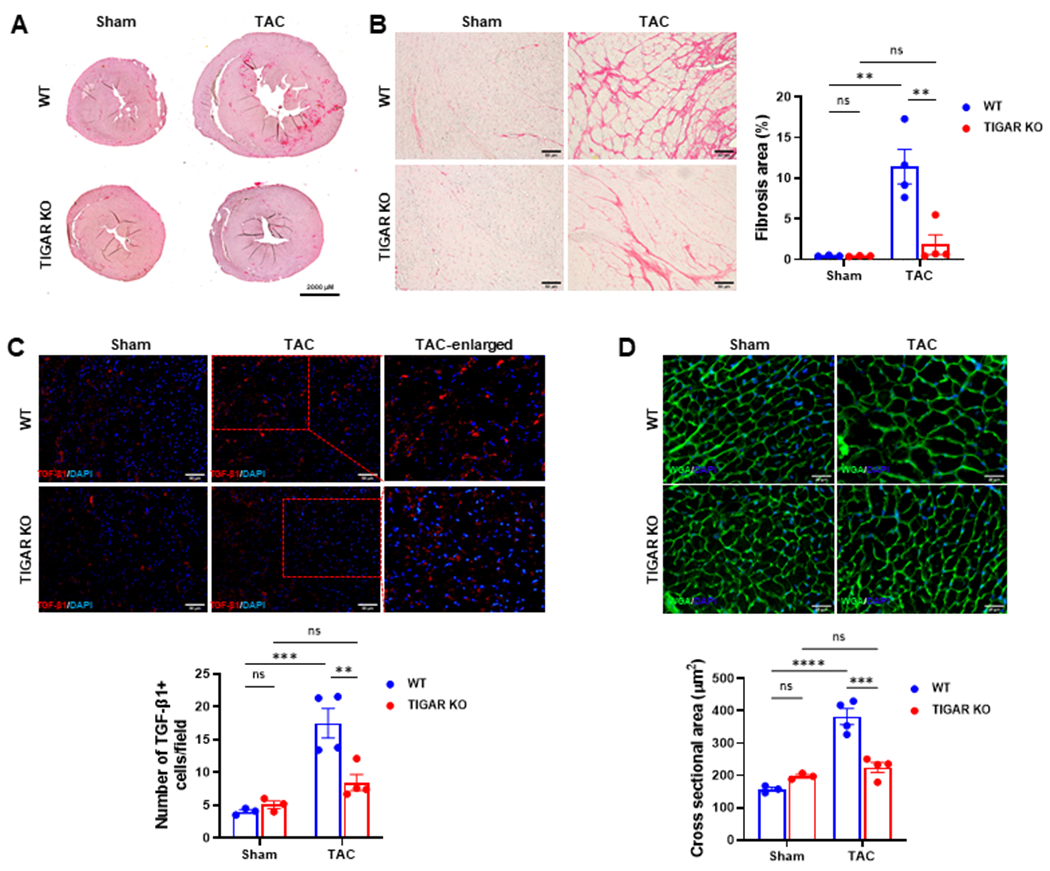
Knockout of TIGAR attenuates pressure overload induced fibrosis. A, The representative images of Picrosirius red-stained whole heart section. Bar=2000 μM. B, The representative images of Picrosirius red-stained paraffin-embedded heart sections and quantification of the percentage of interstitial fibrosis area in the indicated groups (n=3–4). Bar=50 μm. C, The representative images of TGF-β1-stained frozen heart sections and quantification of the number of TGF-β1-positive cells (red)/field in the indicated groups (n=3–4). 5–15 randomly selected fields per LV sec-tion of each mouse was measured. Bar=50 μm. D, The representative images of wheat germ agglutinin–stained frozen heart sections in the WT and TIGAR KO mice. Cardiomyocyte hypertrophy was assessed by measuring cross-sectional areas in the indicated groups. Bar=25 μm. n=3–4. **p<0.01, ***p<0.001, ****p<0.0001.
3.2. Knockout of TIGAR attenuates pressure overload induced diastolic dysfunction
TAC induced PO in mice is a commonly used model to study cardiac remodeling and dysfunction, including diastolic dysfunction. We first investigated the effect of TIGAR knockout on the diastolic function during PO-induced heart failure. Echocardiographic measurements are presented in Table 1. TAC resulted in a significant increase in E in the WT mice and thus led to a dramatic increase in E/A ratio (Figure 2A and 2B, greater than 2, 2.11±0.09 versus 1.37±0.08 in the sham control). In addition, e’ was significantly decreased in the WT mice subjected to TAC, which resulted in an increase in E/e’ ratio (Figure 2A–C), suggesting an elevation of left atrial pressure and the development of diastolic dysfunction. In contrast, knockout of TIGAR diminished PO-induced elevation of E/A and E/e’ ratios and attenuated diastolic dysfunction (Figure 2). In addition, echocardiographic measurements showed that TAC caused a significant decrease in LV ejection fraction (EF) (27.22 ± 3.80% of WT mice vs 60.62.9 ± 2.00% in sham mice). Whereas, knockout of TIGAR significantly improved LV function after TAC, as indicated by significantly less reduction of LV EF and fractional shortening (Table 1). Overall, there was no significant difference in survival between the WT and TIGAR KO mice after TAC (Figure 2D).
Table 1.
Echocardiographic measurements.
| Parameters | WT | TIGAR KO | ||
|---|---|---|---|---|
|
| ||||
| Sham (n=6) | TAC (n=8) | Sham (n=6) | TAC (n=8) | |
| HR (bpm) | 459±4 | 464±7 | 471±3 | 459±6 |
| EF (%) | 60.62±2.00 | 27.22±3.80* | 60.41±1.64 | 46.02±1.82†# |
| FS (%) | 32.14±1.43 | 12.90±1.93* | 31.89±1.11 | 22.74±1.10†# |
| LV mass (mg) | 111.20±7.80 | 237.30±15.16* | 104.4±3.78 | 124.4±6.42† |
| E (cm/s) | 80.49±2.85 | 105.68±1.33* | 77.16±1.21 | 81.58±3.15† |
| A (cm/s) | 60.44±5.35 | 51.98±1.61 | 61.88±2.88 | 61.16±2.50 |
| e’ (cm/s) | 2.51±0.12 | 1.68±0.16* | 2.57±0.08 | 2.15±0.04† |
| a’ (cm/s) | 2.43±0.16 | 1.35±0.16* | 2.48±0.18 | 1.96±0.09† |
| IVRT (ms) | 15.11±0.55 | 15.87±1.53 | 13.73±0.25 | 15.86±0.28# |
| IVCT (ms) | 8.65±0.50 | 10.04±0.47 | 9.13±0.52 | 10.22±0.37 |
| AET (ms) | 50.16±1.67 | 58.17±2.34* | 48.79±1.25 | 58.25±1.50# |
| MPI | 0.47±0.01 | 0.45±0.03 | 0.47±0.02 | 0.45±0.02 |
Data are shown as mean ± SEM. EF, ejection fraction; FS, fractional shortening; IVRT, isovolumic relaxation time; IVCT, isovolumic contraction time; ET, aortic ejection time; MPI, myocardial performance index.
p<0.05 vs. WT sham;
p<0.05 vs. TIGAR KO sham;
p<0.05 vs. WT+TAC.
Figure 2.
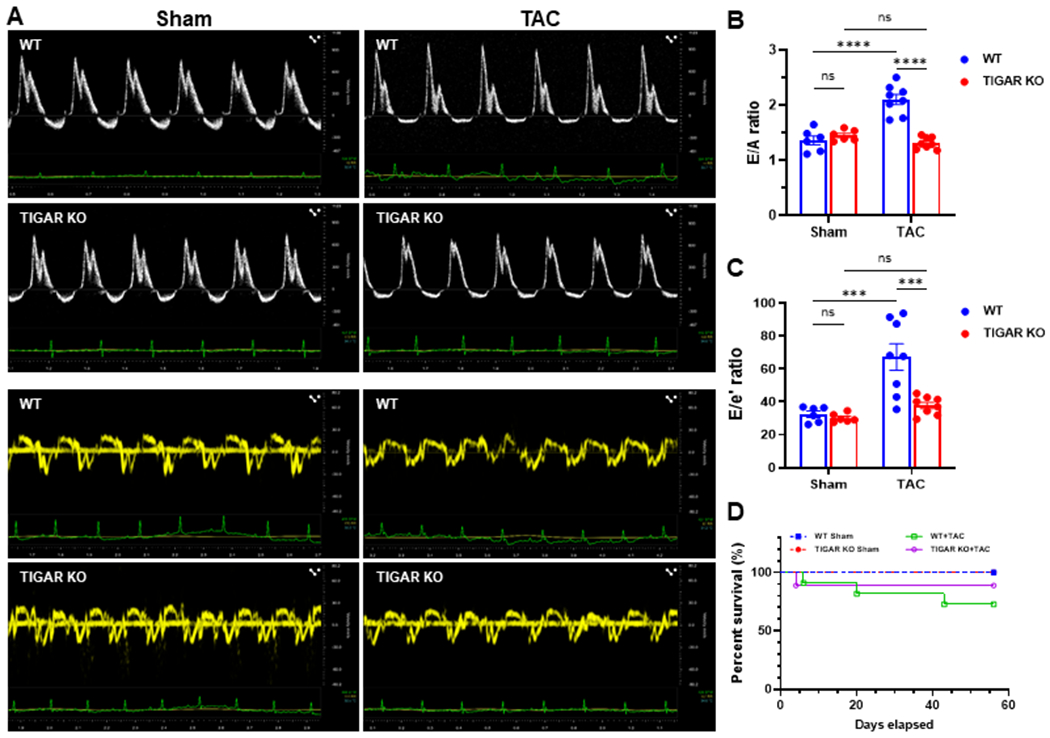
Knockout of TIGAR preserves diastolic function. A, The representative pulsed-wave Doppler and tissue Doppler images from an apical 4-chamber view of WT and TIGAR KO mice subjected to either sham or TAC procedure for eight weeks. B, The ratio of the peak velocity of early (E) to late (A) filling of mitral inflow (E/A) in the indicated groups (n=6–8). C, The ratio of E to the tissue motion velocity in early diastole (e’) was calculated in the indicated groups (n=6–8). D, Kaplan-Meier survival curve. There was no significant difference in survival between the WT and TIGAR KO mice after TAC. ***p<0.001, ****p<0.0001.
3.3. Knockout of TIGAR elevates glycolysis related enzyme and glucose transporters
To explorer the molecular events by which TIGAR regulates cardiac remodeling, we examined the glycolysis-related proteins. The expression of PFK-1 was significantly reduced in the WT mice subjected to TAC, but it was upregulated in the TIGAR KO mice when compared to the WT mice +TAC (Figure 3A & 3B). In addition, the expression of Glut-1 was decreased in both WT and TIGAR KO mice after TAC, but the level of Glut-1 was significantly higher in the TIGAR KO TAC mice than that in the WT TAC mice (Figure 3A & 3C). The expression of Glut-4 was also decreased in the WT mice subjected to TAC, whereas it was unchanged in the TIGAR KO mice and its level was significantly higher than that of the WT mice (Figure 3A & 3D). These data suggest an enhanced glycolysis and glucose uptake in the hearts of TIGAR KO mice during PO-induced HF.
Figure 3.
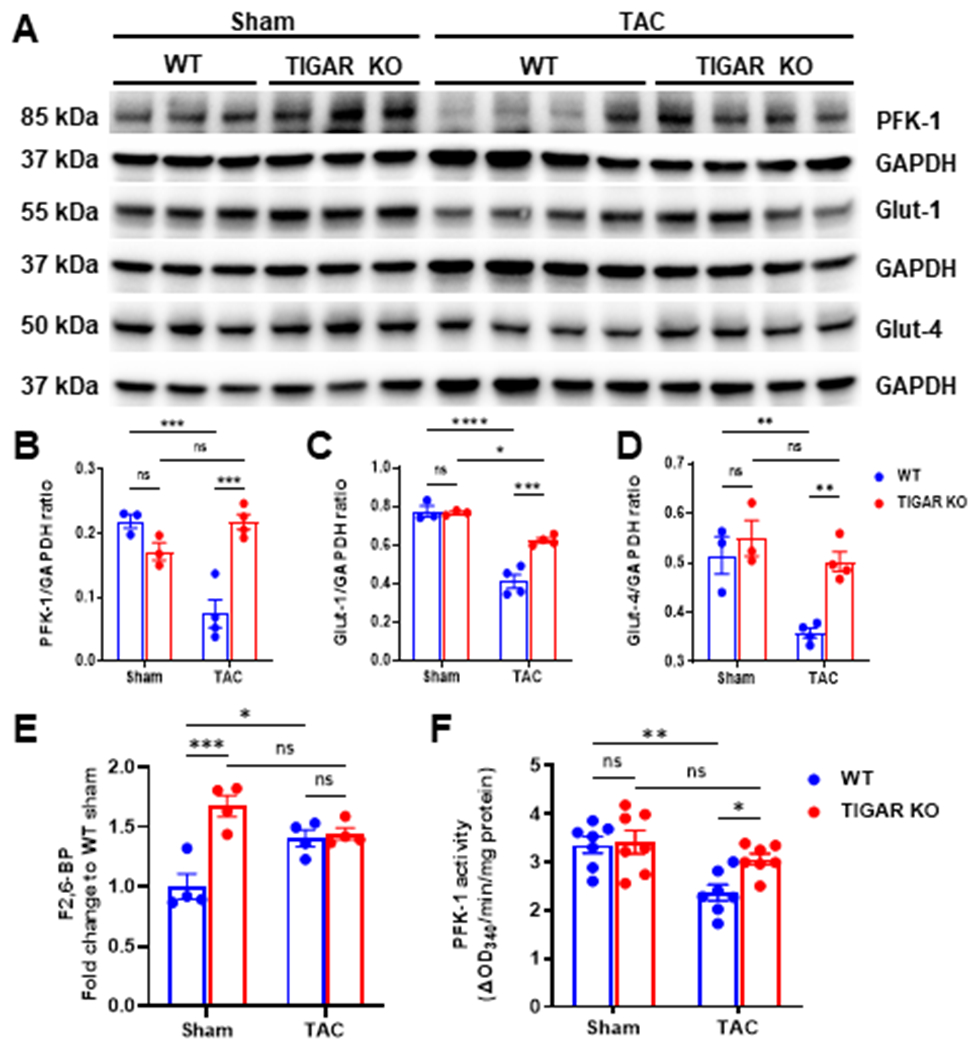
The ablation of TIGAR increases glucose metabolism-related proteins, the level of F2,6-BP, and PFK-1 activity. A-D, Representative immunoblots and quantitative analysis of PFK-1 (key glycolytic enzyme), Glut-1, Glut-4 (glucose transporters), and corresponding GAPDH in the indicated groups. E, Cardiac F2,6-BP level was determined by the coupled-enzymatic assay and expressed as the fold change to the WT sham group. n=4. F, Cardiac PFK-1 activity was determined by the coupled-enzymatic assay and expressed as the OD340nm/min/mg protein. n=7. *p<0.05, **p<0.01, ***p<0.001. ****p<0.0001.
3.4. Knockout of TIGAR increases the level of F2,6-BP and PFK-1 activity
Given that the expression of glycolysis related proteins (PFK-1 and glucose transporters) was changed, we further measured the level of glycolytic intermediate and PFK-1 activity. By using a coupled-enzymes method, we found that knockout of TIGAR significantly increased in the level of F2,6-BP (~1.7-fold) in mouse hearts when compared to that of the WT sham mice (Figure 3E). WT mice subjected to TAC showed increased levels of F2,6-BP (~1.4-fold) in the hearts. Due to the higher basal level of F2,6-BP, TIGAR KO mice subjected to TAC did not further increase F2,6-BP as compared to the TIGAR KO sham control (Figure 3E). Moreover, PFK-1 activity in the WT mice + TAC was significantly reduced when compared to the sham controls, but it was significantly increased in TIGAR KO mice + TAC (Figure 3F). These data suggest that knockout of TIGAR enhanced glycolytic function by increasing the level of F2,6-BP and PFK-1 activity.
3.5. Knockout of TIGAR attenuates perivascular fibrosis and endothelial activation
Our previous study demonstrated that ablation of TIGAR preserved coronary microvascular density and flow reserve after TAC (He, Zeng, Cantrell, & Chen, 2021). The development of endothelial dysfunction and perivascular fibrosis were associated with the progression of diastolic dysfunction (Ma, Zhao, Gao, Zhou, & Fan, 2000; Reed et al., 2011). To investigate the potential mechanisms of diastolic dysfunction, we further examined the vascular remodeling and endothelial activation after PO. As shown in Figure 4A, WT mice subjected to TAC exhibited dramatic perivascular fibrosis when compared to its sham control and the TIGAR KO mice + TAC. Intriguingly, WT mice subjected to TAC showed a small decrease in the arteriole wall thickness, whereas TIGAR KO mice exhibited a significant increase in the vessel wall thickness (Figure 4A). In addition, the levels of VCAM-1 in the capillaries were upregulated in the WT mice + TAC, but not in the TIGAR KO mice + TAC (Figure 4B & 4C), suggesting an inflammatory activation of the endothelium in the WT mice whereas ablation of TIGAR attenuates the endothelial activation.
Figure 4.
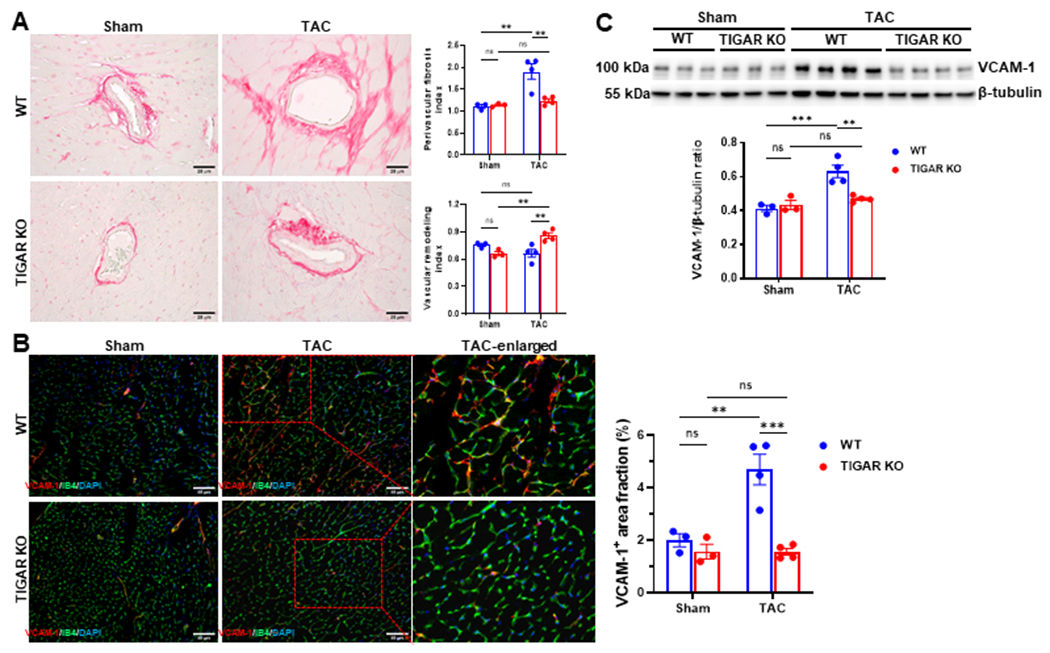
The ablation of TIGAR attenuates perivascular fibrosis and vascular activation. A, The representative images of Picrosirius red-stained paraffin-embedded heart sections showing coronary arteries and perivascular fibrosis in the indicated groups. Bar=25 μm. Right panel: Quantification of perivascular fibrosis index and vascular remodeling in the indicated groups (n=3–4). B, The representative images of VCAM-1 and IB4 double-stained frozen heart sections and quantification of the area fraction of VCAM-1-positive cells (red)/field in the indicated groups (n=3–4). A minimum of 10 randomly selected fields per LV section of each mouse was measured. Bar=50 μm. C, Representative immunoblot and quantitative analysis of VCAM-1 in the indicated groups. n=3–4. *p<0.05, **p<0.01, ***p<0.001.
4. Discussion
Accumulating investigations indicate that diastolic dysfunction is closely associated with metabolic remodeling, histological derangements, and coronary vascular dysfunction that led to a gradual decline in energy metabolism, blood flow, and supply of nutrients to maintain cardiac function. In the present study, for the first time, we demonstrate that knockout of TIGAR blunts PO-induced diastolic dysfunction via increasing the level of F2,6-BP, PFK activity, and glucose transporters in mouse hearts. This was associated with decreased interstitial and perivascular fibrosis. Knockout of TIGAR also attenuated PO-induced vascular remodeling and endothelial activation.
Hypertension or aortic valvar stenosis related PO is the most common cause of HF (Okawa et al., 2019), but the underlying mechanisms remain poorly understood. Compiling evidence demonstrated that the degree of diastolic dysfunction was correlated with the severity of myocardial fibrosis in both animal and clinical studies (Burlew & Weber, 2002; Muller-Brunotte et al., 2007; Reed et al., 2011). Fibrosis as an endpoint of many pathological processes may alter the myocardial relaxation properties. Consistent with the previous study (Okawa et al., 2019), we found that PO induced severe interstitial fibrosis and hypertrophy in the WT mice, but not in the TIGAR KO mice. However, they did not investigate the effect of TIGAR on diastolic function. Interestingly, WT mice subjected to TAC exhibited an E/A ratio greater than 2 (versus ~1.37 in the sham control), which in clinical settings suggests a restrictive filling pattern and increased left atrial pressure. Along with the dramatic increased E/e’ ratio, these results indicate the development of diastolic dysfunction in the WT mice after TAC. Our study further supports the notion that cardiac fibrosis may be a cause of diastolic dysfunction and indicates a potential role of TIGAR in cardiac remodeling and diastolic function.
Glucose metabolism was significantly associated with the prevalence of diastolic dysfunction (Stahrenberg et al., 2010). The failing heart is more dependent on glycolysis for generating ATP (McGarrah, Crown, Zhang, Shah, & Newgard, 2018; Sankaralingam & Lopaschuk, 2015; Wende, Brahma, McGinnis, & Young, 2017), due to decreases in fatty acids and glucose oxidation (Doenst, Nguyen, & Abel, 2013; Taegtmeyer, 2004). With further progression of HF, inadequate compensation by glycolysis leads to energy deficits, and increasing glycolysis can ultimately lead to substrate depletion (Nascimben et al., 2004; Neubauer, 2007). Accumulating evidence has demonstrated that HF is mainly due to profound metabolic remodeling (Ashrafian, Frenneaux, & Opie, 2007; Doenst et al., 2013; Gibb & Hill, 2018; Kundu et al., 2015; Wende, Brahma, et al., 2017). Studies have shown that glycolysis plays an essential role in maintaining normal heart function (McGarrah et al., 2018; Sankaralingam & Lopaschuk, 2015; Wende, Brahma, et al., 2017). During PO-induced cardiac remodeling and diastolic dysfunction, cardiomyocytes were subjected to a significant metabolic shift from fatty acid oxidation (FAO) to glycolysis (Doenst et al., 2013; Taegtmeyer, 2004). If the reduced FAO was not sufficiently compensated by enough increase in glucose oxidation, it will lead to cardiac hypertrophy and fibrosis during PO because glucose was preferentially utilized for biomass synthesis via pentose phosphate pathway (PPP) rather than ATP production (Umbarawan et al., 2018). Inhibition of glycolysis has been shown to cause greater impairment of diastolic function (Kagaya et al., 1995; Kusuoka & Marban, 1994), whereas increased glycolytic substrate had a protective effect against diastolic dysfunction (Eberli, Weinberg, Grice, Horowitz, & Apstein, 1991). Therefore, knockout of TIGAR attenuated the PO-induced diastolic dysfunction may partially be explained by enhancing cardiac glucose utilization and reducing the PPP flux. Previous studies also showed that reduction of F2,6-BP level and PFK-1 activity led to more profound cardiac hypertrophy, fibrosis, and dysfunction in response to PO (Donthi et al., 2004; Wang et al., 2013). Elevation of glycolysis through activation of F2,6-BP and PFK1 is considered as an adaptive response to PO-induced HF (Tran & Wang, 2019; Wang et al., 2013). Our data show that the level of F2,6-BP and PFK-1 were significantly reduced in the WT mice subjected to PO, but not in the TIGAR KO mice. Thus, knockout of TIGAR and improving glycolysis may provide a protective effect against HF. Studies reveal that the increase in glycolysis at the onset of HF is associated with an adaptive mechanism to protect the heart from PO (Donthi et al., 2004; Wang et al., 2013). In contrast, chronic activation of glycolysis may lead to decompensation and progression of HF (Tran & Wang, 2019). Our data showed that the basal level of F2,6-BP was significantly increased in the hearts of TIGAR KO mice, which may provide early protection during PO. Although the level of F2,6-BP was also elevated in WT mice after 8 weeks of TAC, this increase may be due to decompensation and progression of HF in response to PO. Taken together, these data suggest that the heart is more dependent on the glycolytic function during PO-induced diastolic dysfunction.
In addition, glucose uptake plays a critical role in regulating glycolysis, cardiac hypertrophy, and diastolic function (Shao & Tian, 2015; Wende, Kim, et al., 2017). Glut-1 is mainly expressed in the cardiac ECs, and is used to transport exogenous glucose across the ECs (Huang et al., 2012), whereas cardiomyocytes take up exogenous glucose primarily via Glut-4 (Huang et al., 2012). Glucose first crosses the EC via Glut-1 and is then transported into cardiomyocytes via Glut-4 (Abel, 2004; Huang et al., 2012; Szablewski, 2017). Endothelial Glut-1 is necessary for the glucose transport from ECs to cardiomyocytes, which govern glucose utilization. We showed that disruption of endothelial Glut-1 and glucose uptake in ECs sensitizes PO-induced heart failure (Zeng et al., 2020). The present study demonstrates that the expression of Glut-1 and Glut-4 following TAC was significantly decreased in the WT mice, implicating that glucose transportation was impaired in the WT mice and may contribute to the impaired glycolytic flux, while the ablation of TIGAR attenuates the decrease in the expression of glucose transporters. Moreover, the levels of VCAM-1 were upregulated in the WT mice after TAC, suggesting that PO caused an increased inflammatory activation in the vascular endothelium. The knockout of TIGAR significantly reduced the level of VCAM-1 under PO. Taken together, TIGAR knockout appears to attenuate diastolic dysfunction, at least in part, by keeping up the substrate supply and reduction of endothelial activation in mice.
In summary, our study demonstrated a necessary role of glycolysis in PO-induced diastolic dysfunction (Figure 5). Mechanistically, knockout of TIGAR attenuated myocardial fibrosis and led to higher levels of PFKFB3 and PFK-1, as well as increased F2,6-BP production and PFK activity. There were associated with increased expression of glucose transporters and reduced endothelial inflammatory activation. Therefore, novel therapeutic targets that regulate myocardial glucose metabolism and improve fibrosis and vascular homeostasis should be beneficial in the prevention and treatment of PO-induced diastolic dysfunction.
Figure 5.
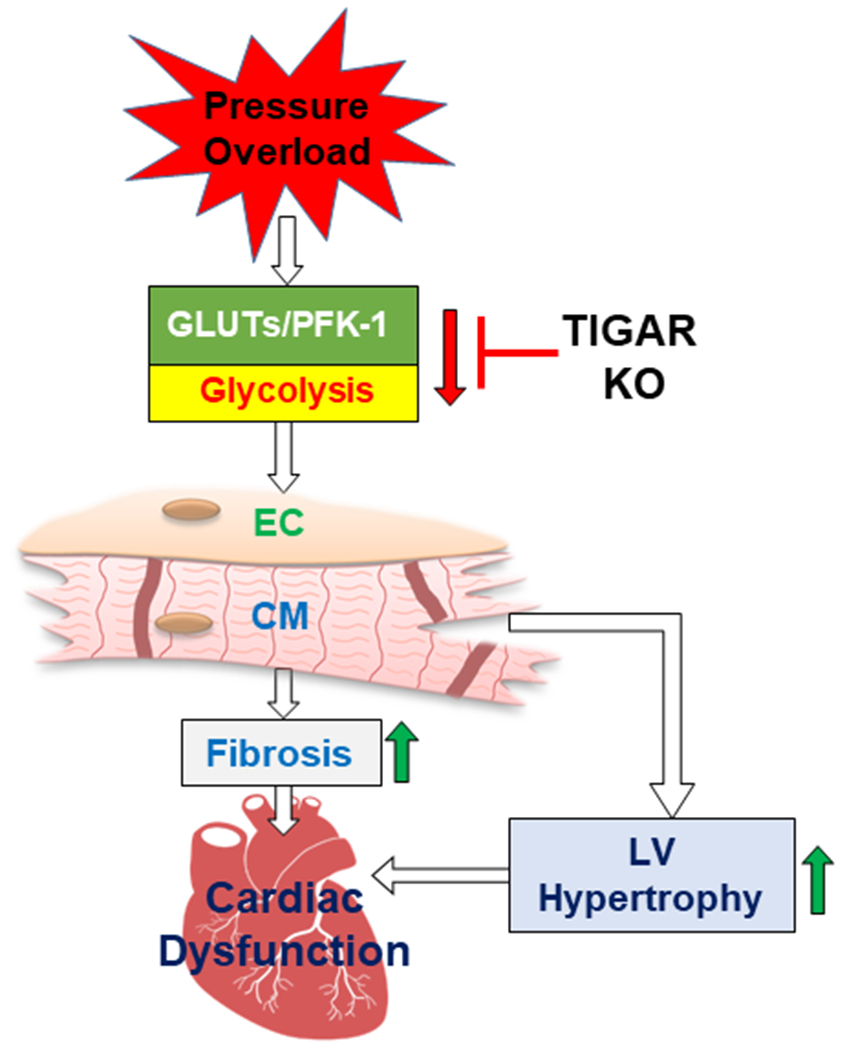
Proposed mechanism of which TIGAR deficiency preserves diastolic function in the development of heart failure. TAC-induced pressure overload impairs myocardial glycolytic function via disrupting glucose transporters and glycolytic enzyme activity, which lead to cardiomyocyte hypertrophy, fibrosis, and eventually diastolic and contractile dysfunction. Knockout of TIGAR improves myocardial glycolytic function, attenuates pathological remodeling, and preserves cardiac function. EC: endothelial cell; CM: cardiomyocyte.
Supplementary Material
Acknowledgments:
We are very grateful to Dr. Jeffrey Pessin at the Albert Einstein College of Medicine for providing the TIGAR KO mice. We also appreciate Mr. Joshua Jefferson for the histological preparation of the tissue samples.
Funding:
This work was supported by the National Heart, Lung, and Blood Institute (R01HL102042, JX Chen) and National Institute of General Medical Sciences and National Heart, Lung, and Blood Institute (R01HL151536, JX Chen), the National Institute of General Medical Sciences of the National Institutes of Health under Award Number P20GM104357 (H.Z).
Footnotes
Conflict of Interests: The authors declare that they have no conflict of interest.
Data Availability Statement:
The data presented in this study are available on request from the corresponding author.
References
- Abel ED (2004). Glucose transport in the heart. Front Biosci, 9, 201–215. doi: 10.2741/1216 [DOI] [PubMed] [Google Scholar]
- Ashrafian H, Frenneaux MP, & Opie LH (2007). Metabolic mechanisms in heart failure. Circulation, 116(4), 434–448. doi: 10.1161/CIRCULATIONAHA.107.702795 [DOI] [PubMed] [Google Scholar]
- Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, . . . Stroke Statistics, S. (2019). Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation, 139(10), e56–e528. doi: 10.1161/CIR.0000000000000659 [DOI] [PubMed] [Google Scholar]
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, . . . Vousden KH. (2006). TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell, 126(1), 107–120. doi: 10.1016/j.cell.2006.05.036 [DOI] [PubMed] [Google Scholar]
- Brouwers FP, de Boer RA, van der Harst P, Voors AA, Gansevoort RT, Bakker SJ, . . . van Gilst WH. (2013). Incidence and epidemiology of new onset heart failure with preserved vs. reduced ejection fraction in a community-based cohort: 11-year follow-up of PREVEND. Eur Heart J, 34(19), 1424–1431. doi: 10.1093/eurheartj/eht066 [DOI] [PubMed] [Google Scholar]
- Burlew BS, & Weber KT (2002). Cardiac fibrosis as a cause of diastolic dysfunction. Herz, 27(2), 92–98. doi: 10.1007/s00059-002-2354-y [DOI] [PubMed] [Google Scholar]
- De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, . . . Carmeliet P. (2013). Role of PFKFB3-driven glycolysis in vessel sprouting. Cell, 154(3), 651–663. doi: 10.1016/j.cell.2013.06.037 [DOI] [PubMed] [Google Scholar]
- Deng H, Yu F, Chen J, Zhao Y, Xiang J, & Lin A (2008). Phosphorylation of Bad at Thr-201 by JNK1 promotes glycolysis through activation of phosphofructokinase-1. J Biol Chem, 283(30), 20754–20760. doi: 10.1074/jbc.M800024200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhingra A, Garg A, Kaur S, Chopra S, Batra JS, Pandey A, . . . Agarwal SK. (2014). Epidemiology of heart failure with preserved ejection fraction. Curr Heart Fail Rep, 11(4), 354–365. doi: 10.1007/s11897-014-0223-7 [DOI] [PubMed] [Google Scholar]
- Doenst T, Nguyen TD, & Abel ED (2013). Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res, 113(6), 709–724. doi: 10.1161/CIRCRESAHA.113.300376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donthi RV, Ye G, Wu C, McClain DA, Lange AJ, & Epstein PN (2004). Cardiac expression of kinase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase inhibits glycolysis, promotes hypertrophy, impairs myocyte function, and reduces insulin sensitivity. J Biol Chem, 279(46), 48085–48090. doi: 10.1074/jbc.M405510200 [DOI] [PubMed] [Google Scholar]
- Eberli FR, Weinberg EO, Grice WN, Horowitz GL, & Apstein CS (1991). Protective effect of increased glycolytic substrate against systolic and diastolic dysfunction and increased coronary resistance from prolonged global underperfusion and reperfusion in isolated rabbit hearts perfused with erythrocyte suspensions. Circ Res, 68(2), 466–481. doi: 10.1161/01.res.68.2.466 [DOI] [PubMed] [Google Scholar]
- Fillmore N, Levasseur JL, Fukushima A, Wagg CS, Wang W, Dyck JRB, & Lopaschuk GD (2018). Uncoupling of glycolysis from glucose oxidation accompanies the development of heart failure with preserved ejection fraction. Mol Med, 24(1), 3. doi: 10.1186/s10020-018-0005-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Ho D, Vatner DE, & Vatner SF (2011). Echocardiography in Mice. Curr Protoc Mouse Biol, 1, 71–83. doi: 10.1002/9780470942390.mo100130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb AA, & Hill BG (2018). Metabolic Coordination of Physiological and Pathological Cardiac Remodeling. Circ Res, 123(1), 107–128. doi: 10.1161/CIRCRESAHA.118.312017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, & Chipuk JE (2006). p53 and metabolism: Inside the TIGAR. Cell, 126(1), 30–32. doi: 10.1016/j.cell.2006.06.032 [DOI] [PubMed] [Google Scholar]
- He X, Zeng H, Cantrell AC, & Chen JX (2021). Regulatory role of TIGAR on endothelial metabolism and angiogenesis. J Cell Physiol, 236(11), 7578–7590. doi: 10.1002/jcp.30401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Zeng H, Chen ST, Roman RJ, Aschner JL, Didion S, & Chen JX (2017). Endothelial specific SIRT3 deletion impairs glycolysis and angiogenesis and causes diastolic dysfunction. J Mol Cell Cardiol, 112, 104–113. doi: 10.1016/j.yjmcc.2017.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Zeng H, Roman RJ, & Chen JX (2018). Inhibition of prolyl hydroxylases alters cell metabolism and reverses pre-existing diastolic dysfunction in mice. Int J Cardiol, 272, 281–287. doi: 10.1016/j.ijcard.2018.08.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Lei L, Liu D, Jovin I, Russell R, Johnson RS, . . . Giordano FJ (2012). Normal glucose uptake in the brain and heart requires an endothelial cell-specific HIF-1alpha-dependent function. Proc Natl Acad Sci U S A, 109(43), 17478–17483. doi: 10.1073/pnas.1209281109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagaya Y, Weinberg EO, Ito N, Mochizuki T, Barry WH, & Lorell BH (1995). Glycolytic inhibition: effects on diastolic relaxation and intracellular calcium handling in hypertrophied rat ventricular myocytes. J Clin Invest, 95(6), 2766–2776. doi: 10.1172/JCI117980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katholi RE, & Couri DM (2011). Left ventricular hypertrophy: major risk factor in patients with hypertension: update and practical clinical applications. Int J Hypertens, 2011, 495349. doi: 10.4061/2011/495349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu BK, Zhong M, Sen S, Davogustto G, Keller SR, & Taegtmeyer H (2015). Remodeling of glucose metabolism precedes pressure overload-induced left ventricular hypertrophy: review of a hypothesis. Cardiology, 130(4), 211–220. doi: 10.1159/000369782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusuoka H, & Marban E (1994). Mechanism of the diastolic dysfunction induced by glycolytic inhibition. Does adenosine triphosphate derived from glycolysis play a favored role in cellular Ca2+ homeostasis in ferret myocardium? J Clin Invest, 93(3), 1216–1223. doi: 10.1172/JCI117075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam CS, Donal E, Kraigher-Krainer E, & Vasan RS (2011). Epidemiology and clinical course of heart failure with preserved ejection fraction. Eur J Heart Fail, 13(1), 18–28. doi: 10.1093/eurjhf/hfq121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma LN, Zhao SP, Gao M, Zhou QC, & Fan P (2000). Endothelial dysfunction associated with left ventricular diastolic dysfunction in patients with coronary heart disease. Int J Cardiol, 72(3), 275–279. doi: 10.1016/s0167-5273(99)00203-x [DOI] [PubMed] [Google Scholar]
- Mandinov L, Eberli FR, Seiler C, & Hess OM (2000). Diastolic heart failure. Cardiovasc Res, 45(4), 813–825. doi: 10.1016/s0008-6363(99)00399-5 [DOI] [PubMed] [Google Scholar]
- McGarrah RW, Crown SB, Zhang GF, Shah SH, & Newgard CB (2018). Cardiovascular Metabolomics. Circ Res, 122(9), 1238–1258. doi: 10.1161/CIRCRESAHA.117.311002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, & Redfield MM (2015). Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation, 131(6), 550–559. doi: 10.1161/CIRCULATIONAHA.114.009625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Brunotte R, Kahan T, Lopez B, Edner M, Gonzalez A, Diez J, & Malmqvist K (2007). Myocardial fibrosis and diastolic dysfunction in patients with hypertension: results from the Swedish Irbesartan Left Ventricular Hypertrophy Investigation versus Atenolol (SILVHIA). J Hypertens, 25(9), 1958–1966. doi: 10.1097/HJH.0b013e3282170ada [DOI] [PubMed] [Google Scholar]
- Nascimben L, Ingwall JS, Lorell BH, Pinz I, Schultz V, Tornheim K, & Tian R (2004). Mechanisms for increased glycolysis in the hypertrophied rat heart. Hypertension, 44(5), 662–667. doi: 10.1161/01.HYP.0000144292.69599.0c [DOI] [PubMed] [Google Scholar]
- Neubauer S (2007). The failing heart--an engine out of fuel. N Engl J Med, 356(11), 1140–1151. doi: 10.1056/NEJMra063052 [DOI] [PubMed] [Google Scholar]
- Ogata T, Miyauchi T, Sakai S, Takanashi M, Irukayama-Tomobe Y, & Yamaguchi I (2004). Myocardial fibrosis and diastolic dysfunction in deoxycorticosterone acetate-salt hypertensive rats is ameliorated by the peroxisome proliferator-activated receptor-alpha activator fenofibrate, partly by suppressing inflammatory responses associated with the nuclear factor-kappa-B pathway. J Am Coll Cardiol, 43(8), 1481–1488. doi: 10.1016/j.jacc.2003.11.043 [DOI] [PubMed] [Google Scholar]
- Okawa Y, Hoshino A, Ariyoshi M, Kaimoto S, Tateishi S, Ono K, . . . Matoba S. (2019). Ablation of cardiac TIGAR preserves myocardial energetics and cardiac function in the pressure overload heart failure model. Am J Physiol Heart Circ Physiol, 316(6), H1366–H1377. doi: 10.1152/ajpheart.00395.2018 [DOI] [PubMed] [Google Scholar]
- Redfield MM, Jacobsen SJ, Burnett JC Jr., Mahoney DW, Bailey KR, & Rodeheffer RJ (2003). Burden of systolic and diastolic ventricular dysfunction in the community: appreciating the scope of the heart failure epidemic. JAMA, 289(2), 194–202. doi: 10.1001/jama.289.2.194 [DOI] [PubMed] [Google Scholar]
- Reed AL, Tanaka A, Sorescu D, Liu H, Jeong EM, Sturdy M, . . . Sutliff RL. (2011). Diastolic dysfunction is associated with cardiac fibrosis in the senescence-accelerated mouse. Am J Physiol Heart Circ Physiol, 301(3), H824–831. doi: 10.1152/ajpheart.00407.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaralingam S, & Lopaschuk GD (2015). Cardiac energy metabolic alterations in pressure overload-induced left and right heart failure (2013 Grover Conference Series). Pulm Circ, 5(1), 15–28. doi: 10.1086/679608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao D, & Tian R (2015). Glucose Transporters in Cardiac Metabolism and Hypertrophy. Compr Physiol, 6(1), 331–351. doi: 10.1002/cphy.c150016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahrenberg R, Edelmann F, Mende M, Kockskamper A, Dungen HD, Scherer M, . . . Wachter R. (2010). Association of glucose metabolism with diastolic function along the diabetic continuum. Diabetologia, 53(7), 1331–1340. doi: 10.1007/s00125-010-1718-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szablewski L (2017). Glucose transporters in healthy heart and in cardiac disease. Int J Cardiol, 230, 70–75. doi: 10.1016/j.ijcard.2016.12.083 [DOI] [PubMed] [Google Scholar]
- Taegtmeyer H (2004). Cardiac metabolism as a target for the treatment of heart failure. Circulation, 110(8), 894–896. doi: 10.1161/01.CIR.0000139340.88769.D5 [DOI] [PubMed] [Google Scholar]
- Tang Y, Kwon H, Neel BA, Kasher-Meron M, Pessin JB, Yamada E, & Pessin JE (2018). The fructose-2,6-bisphosphatase TIGAR suppresses NF-kappaB signaling by directly inhibiting the linear ubiquitin assembly complex LUBAC. J Biol Chem, 293(20), 7578–7591. doi: 10.1074/jbc.RA118.002727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanno M, Kuno A, Horio Y, & Miura T (2012). Emerging beneficial roles of sirtuins in heart failure. Basic Res Cardiol, 107(4), 273. doi: 10.1007/s00395-012-0273-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavakoli R, Nemska S, Jamshidi P, Gassmann M, & Frossard N (2017). Technique of Minimally Invasive Transverse Aortic Constriction in Mice for Induction of Left Ventricular Hypertrophy. J Vis Exp(127), e56231. doi: 10.3791/56231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran DH, & Wang ZV (2019). Glucose Metabolism in Cardiac Hypertrophy and Heart Failure. J Am Heart Assoc, 8(12), e012673. doi: 10.1161/JAHA.119.012673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbarawan Y, Syamsunarno M, Koitabashi N, Yamaguchi A, Hanaoka H, Hishiki T, . . . Iso T. (2018). Glucose is preferentially utilized for biomass synthesis in pressure-overloaded hearts: evidence from fatty acid-binding protein-4 and −5 knockout mice. Cardiovasc Res, 114(8), 1132–1144. doi: 10.1093/cvr/cvy063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhya B, Taffet GE, Cheng CP, & Kitzman DW (2015). Heart failure with preserved ejection fraction in the elderly: scope of the problem. J Mol Cell Cardiol, 83, 73–87. doi: 10.1016/j.yjmcc.2015.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Schaftingen E, Lederer B, Bartrons R, & Hers HG (1982). A kinetic study of pyrophosphate: fructose-6-phosphate phosphotransferase from potato tubers. Application to a microassay of fructose 2,6-bisphosphate. Eur J Biochem, 129(1), 191–195. doi: 10.1111/j.1432-1033.1982.tb07039.x [DOI] [PubMed] [Google Scholar]
- Wang J, Xu J, Wang Q, Brainard RE, Watson LJ, Jones SP, & Epstein PN (2013). Reduced cardiac fructose 2,6 bisphosphate increases hypertrophy and decreases glycolysis following aortic constriction. PLoS One, 8(1), e53951. doi: 10.1371/journal.pone.0053951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wende AR, Brahma MK, McGinnis GR, & Young ME (2017). Metabolic Origins of Heart Failure. JACC Basic Transl Sci, 2(3), 297–310. doi: 10.1016/j.jacbts.2016.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wende AR, Kim J, Holland WL, Wayment BE, O’Neill BT, Tuinei J, . . . Abel ED. (2017). Glucose transporter 4-deficient hearts develop maladaptive hypertrophy in response to physiological or pathological stresses. Am J Physiol Heart Circ Physiol, 313(6), H1098–H1108. doi: 10.1152/ajpheart.00101.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, He X, & Chen JX (2020). Endothelial Sirtuin 3 Dictates Glucose Transport to Cardiomyocyte and Sensitizes Pressure Overload-Induced Heart Failure. J Am Heart Assoc, 9(11), e015895. doi: 10.1161/JAHA.120.015895 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data presented in this study are available on request from the corresponding author.