

Abstract
Proteostasis is essential for cellular survival and particularly important for highly specialised post‐mitotic cells such as neurons. Transient reduction in protein synthesis by protein kinase R‐like endoplasmic reticulum (ER) kinase (PERK)‐mediated phosphorylation of eukaryotic translation initiation factor 2α (p‐eIF2α) is a major proteostatic survival response during ER stress. Paradoxically, neurons are remarkably tolerant to PERK dysfunction, which suggests the existence of cell type‐specific mechanisms that secure proteostatic stress resilience. Here, we demonstrate that PERK‐deficient neurons, unlike other cell types, fully retain the capacity to control translation during ER stress. We observe rescaling of the ATF4 response, while the reduction in protein synthesis is fully retained. We identify two molecular pathways that jointly drive translational control in PERK‐deficient neurons. Haem‐regulated inhibitor (HRI) mediates p‐eIF2α and the ATF4 response and is complemented by the tRNA cleaving RNase angiogenin (ANG) to reduce protein synthesis. Overall, our study elucidates an intricate back‐up mechanism to ascertain translational control during ER stress in neurons that provides a mechanistic explanation for the thus far unresolved observation of neuronal resilience to proteostatic stress.
Keywords: ANG, HRI, neuron‐specific, PERK, translational control
Subject Categories: Neuroscience, Translation & Protein Quality
A back‐up translational control mechanism provides neurons with tolerance towards PERK dysfunction.
Introduction
Protein homeostasis or proteostasis is essential for cell function and viability and is continuously challenged by internal and environmental stressors. Central in the resilience to proteostatic stress is reduction in global protein synthesis via the integrated stress response (ISR; Pakos‐Zebrucka et al, 2016). The ISR is regulated by phosphorylation of eukaryotic initiation factor 2α (eIF2α), which is mediated by eIF2α kinases (EIF2AK1–4) that are triggered by different stressors (Donnelly et al, 2013). EIF2AK3—also called protein kinase R‐like endoplasmic reticulum kinase (PERK)—is activated by endoplasmic reticulum (ER) stress as part of the unfolded protein response (UPR; Harding et al, 1999). Phosphorylated eIF2α (p‐eIF2α) inhibits eukaryotic initiation factor 2B (eIF2B), resulting in reduced protein synthesis, paradoxically accompanied by increased translation of specific mRNAs, including activating transcription factor 4 (ATF4; Harding et al, 2000; Scheuner et al, 2001; Harding et al, 2003). Translational control via the ISR is essential for cell viability (Han et al, 2013), yet deficiency of the EIF2AKs differentially affects different cell types and tissues (Yang et al, 1995; Han et al, 2001; Harding et al, 2001; Zhang et al, 2002). In particular, the apparent resilience of neurons to PERK deficiency is surprising in view of the importance of proper ISR control for the function of the highly specialised post‐mitotic neurons (Kapur et al, 2017).
Global deletion of PERK expression results in severe defects in multiple tissues in mice in vivo, in particular the pancreas, resulting in diabetes and perinatal lethality (Harding et al, 2001). Mice treated with a PERK inhibitor also show pancreatic deficiency (Moreno et al, 2013). In agreement with this, Wolcott–Rallison syndrome—a human disease in which PERK function is disrupted—is primarily characterised clinically by juvenile diabetes, combined with renal, hepatic and skeletal problems (Delépine et al, 2000). In contrast, neuron‐specific deletion of PERK leads to overall healthy mice with no morphological or functional defects in the brain (Trinh et al, 2012). Also in patients with Wolcott–Rallison syndrome, cognitive symptoms are only observed in a minority of cases and are considered to be a secondary effect of diabetes (Julier & Nicolino, 2010). Together, this indicates that neurons tolerate PERK dysfunction better than other cell types, which suggests the existence of cell type‐specific mechanisms that secure proteostatic stress resilience. To date, neuronal PERK deficiency has only been studied in the heterogeneous context of the brain, in a mixture of PERK‐deficient neurons, wild‐type neurons and other brain cells, predominantly astrocytes. This makes it impossible to distinguish the cell autonomous neuronal response to proteostatic stress. Hence, the molecular pathways that convey neuron‐specific proteostatic stress resilience are unknown.
Here, we employed PERK‐deficient neuron and astrocyte monocultures to investigate the mechanisms underlying neuron‐specific ER stress resilience in the absence of PERK. We confirm that PERK deficiency incapacitates ER stress‐induced translational control in astrocytes, as previously reported for other cell types. Surprisingly, we demonstrate that PERK‐deficient neurons retain the capacity to reduce global protein synthesis during ER stress. Selective ATF4 upregulation and the ATF4 transcriptional response are decreased and therefore uncoupled from the preserved reduction in protein synthesis. Importantly, we identified that two pathways, one mediated by haem‐regulated inhibitor (HRI)/eIF2α and another by the RNase angiogenin (ANG), jointly convey this neuron‐specific shift to secure translational control during ER stress.
Results
ER stress‐induced reduction in protein synthesis is preserved in PERK‐deficient neurons
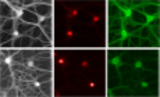
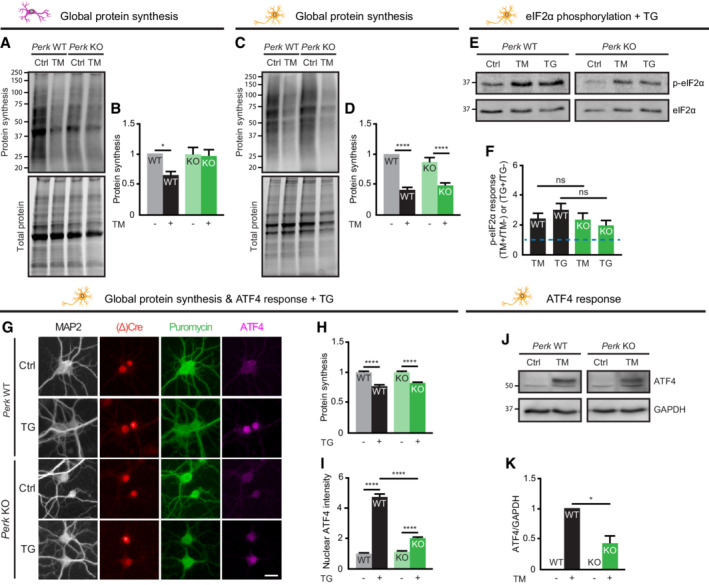
To study cell type‐specific PERK signalling in the major cell types of the brain, conditional PERK‐deficient (knockout; KO) neurons and astrocytes were cultured from Perk loxP/loxP mice. Perk WT and Perk KO cells were generated by in vitro transduction with lentiviruses expressing inactive ΔCre‐ or active Cre‐recombinase, respectively. The presence of an EGFP or mCherry tag allowed identification of transduced cells (Fig 1A; Appendix Fig S1A and B). This transduction protocol resulted in virtually complete loss of PERK protein in Perk KO cells (Fig 1B). To investigate the PERK‐mediated ISR in neurons and astrocytes, the phosphorylation of eIF2α (p‐eIF2α) and the downstream responses (Fig 1C) were assessed during ER stress induced by 20–24 h of treatment with tunicamycin (TM). All ISR interventions and experimental readouts used in this study are summarised in Fig EV1.
Figure 1. ER stress‐induced reduction in protein synthesis is preserved in PERK‐deficient neurons.
-
ASchematic overview of timeline and experimental set‐up.
-
BWB showing depletion of PERK in Perk KO astrocytes and neurons.
-
CSchematic representation of the core events of the PERK signalling pathway.
-
D–IeIF2α phosphorylation in WT and Perk KO astrocytes (N = 3) and neurons (N = 6) ± 20–24 h of TM‐induced ER stress. Representative WB (D, G), quantification of absolute phosphorylated eIF2α level (p‐eIF2α/eIF2α) (E, H) and the ER stress‐induced p‐eIf2α response (F, I).
-
J–OProtein synthesis in WT and Perk KO astrocytes (N = 5) and neurons (N = 5) ± 20–24 h of TM‐induced ER stress. Representative images obtained by high‐content microscopy showing puromycinilated proteins detected by immunofluorescence (J, M), quantification of puromycin intensity as a measure for de novo protein synthesis (K, N) and the ER stress‐induced protein synthesis response (L, O). Astrocytic soma (GFAP, white), dendrites (MAP2, white), nuclei ((Δ)Cre, red) and de novo‐synthesised proteins (puromycin, green). Scale bar: 25 μm.
Data information: (E, H, K, N) Data are normalised to untreated WT. (F, I, L, O) Data are related to untreated cells of the same genotype. The baseline level (without ER stress) is depicted by a dashed line. Data are presented as mean ± SEM. N: Biological replicate. Relevant P‐values are indicated: **P < 0.01, ***P < 0.001, ****P < 0.0001 and ns: not significant. Statistical analysis: two‐way ANOVA with Tukey's post‐hoc test (E, H, K, N); Student's t‐test (F, I); and nested t‐test (L, O). See also Fig EV3 and Appendix Fig S1.
Source data are available online for this figure.
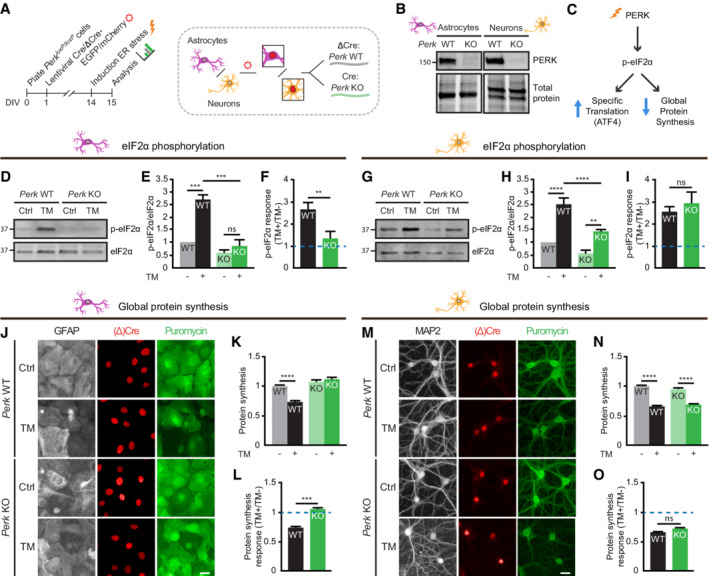
Figure EV1. ISR interventions and experimental readouts used in this study.
Schematic overview of the interventions inhibiting (red) and activating (green) the ISR and experimental readouts (blue text) used in this study. WB: Western blot; IF: Immunofluorescence.
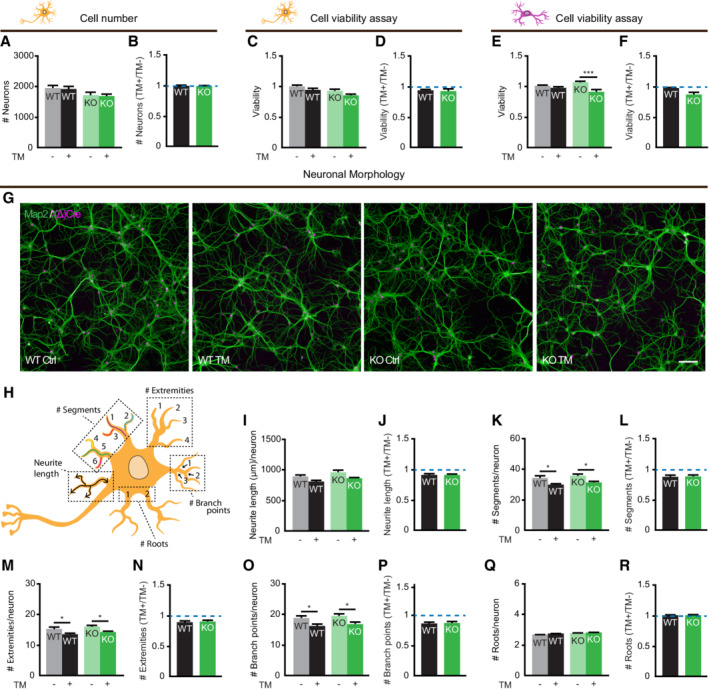
To validate that neurons in vitro are tolerant to PERK dysfunction, as was previously shown in vivo, the effect of ER stress on the cell number and metabolic activity of WT and Perk KO neurons was determined (Fig EV2A–D). These data show that ER stress did not affect the viability of WT nor Perk KO neurons. In contrast, under the same conditions the metabolic activity of Perk KO astrocytes but not WT astrocytes decreased upon ER stress, showing that PERK deficiency reduces the tolerance of astrocytes to ER stress (Fig EV2E). No major difference is detected between the viability of WT and Perk KO astrocytes upon ER stress (Fig EV2F), allowing analysis of astrocytes as non‐neuronal brain cell type in this study. To assess more subtle effects on neuronal degeneration, neuronal morphology was quantitatively determined by high‐content microscopy and automated analysis (Rosato et al, 2019; Fig EV2G–R). ER stress did have a small effect on the morphology of neurons, but no differences between Perk KO and WT neurons were observed. Together, these data indicate that Perk KO neurons retain resilience to ER stress in vitro.
Figure EV2. Neurons in vitro are tolerant to PERK dysfunction.
-
A–FViability of WT and Perk KO neurons (A‐D) and astrocytes (E‐F) ± 20–24 h of TM‐induced ER stress. (A) Absolute cell number and (B) the change in cell number in response to ER stress (N = 7). (C, E) Absolute metabolic activity and (D, F) the change in metabolic activity in response to ER stress, determined by the CellTiter‐Glo Luminescent Cell Viability Assay (Promega) (E‐D, N = 6; E‐F, N = 3). Baseline level (without ER stress) is depicted by dashed line.
-
G–RNeuronal morphology of WT and Perk KO neurons ±20–24 h of TM‐induced ER stress (N = 7). (G) Representative images obtained by high‐content microscopy. Dendrites (MAP2, green), nuclei ((Δ)Cre, magenta). Scale bar: 100 μm. (H) Schematic representation of morphological measures. Absolute morphological measures ((I) neurite length, (K) # segments, (M) # extremities, (O) # branch points and (Q) # roots) and the change in neuronal morphology in response to ER stress (J, L, N, P, R). Baseline level (without ER stress) is depicted by dashed line.
Data information: Data are presented as mean ± SEM. N: Biological replicate. Relevant P‐values are indicated: *P < 0.05 and ***P < 0.001. Statistical analysis: Two‐way ANOVA with Tukey's post‐hoc test (A, C, E, I, K, M, O, Q); nested t‐test (B, D, F, J, L, N, P, R).
Source data are available online for this figure.
In order to study PERK signalling, p‐eIF2α was analysed by Western blot (WB) first. This experiment showed that ER stress significantly increased p‐eIF2α in WT astrocytes (Fig 1D–F) and neurons (Fig 1G–I). Deletion of Perk abolished ER stress‐induced eIF2α phosphorylation in astrocytes (Fig 1D–F). Unexpectedly, ER stress increased eIF2α phosphorylation to the same extent in Perk KO and WT neurons (Fig 1G–I). Because basal p‐eIF2α levels were reduced, the absolute p‐eIF2α levels were 1.8‐fold lower in Perk KO neurons (Fig 1H). This demonstrates that the ER stress‐induced phosphorylation of eIF2α is retained yet rescaled in PERK‐deficient neurons but not astrocytes.
To study the ER stress‐induced responses downstream of p‐eIF2α, de novo protein synthesis rates were assessed first. We employed SUnSET (Schmidt et al, 2009) combined with high‐content microscopy and automated analysis of puromycin immunofluorescence to detect nascent peptides formed during puromycin pulse labelling. This analysis showed that ER stress reduced global protein synthesis by 30% in WT astrocytes (Fig 1J–L) and 35% in WT neurons (Fig 1M–O). Deletion of Perk abolished the ER stress‐induced protein synthesis block in astrocytes, as expected (Fig 1J–L). However, in Perk KO neurons protein synthesis was reduced to the same extent as in WT neurons upon ER stress (Fig 1M–O). Basal protein synthesis rates were not altered (Fig 1N). Bulk analysis of puromycinylated proteins by WB confirmed that the ER stress‐induced reduction in global protein synthesis was not affected in Perk KO neurons (Fig EV3A–D). In addition, similar results were obtained for p‐eIF2α and de novo protein synthesis when thapsigargin (TG), a mechanistically different ER stress inducer, was used (Fig EV3E–H). These results demonstrate that the reduction in protein synthesis observed in Perk KO neurons is not due to subcellular relocalisation of translation and not a non‐specific effect of TM. These data therefore indicate that neurons have a specific adaptation to maintain ER stress‐induced protein synthesis reduction in the absence of PERK.
Figure EV3. ER stress‐induced reduction in protein synthesis is preserved in PERK‐deficient neurons—Related to Figs 1 and 2 .
-
A–DProtein synthesis in WT and Perk KO astrocytes (A, B, N = 4) and neurons (C, D, N = 7) ± 20–24 h of TM‐induced ER stress. Representative WB (A, C) and quantification (B, D) of puromycinilated proteins as a measure for de novo protein synthesis. Data are normalised to untreated WT.
-
E–IeIf2α phosphorylation (N = 4), protein synthesis (N = 3) and ATF4 expression (N = 3) in WT and Perk KO astrocytes and neurons ±20–24 h of TG‐induced ER stress. Representative WB (E) and quantification (F) of the ER stress‐induced p‐eIf2α response, relative to untreated cells of the same genotype. Baseline level (without ER stress) is depicted by dashed line. (E) All lanes are from the same gel/blot. (G) Representative images obtained by high‐content microscopy showing puromycinilated proteins and ATF4 immunofluorescence. Dendrites (MAP2, white), nuclei ((Δ)Cre, red), de novo‐synthesised proteins (puromycin, green) and ATF4 (magenta). Scale bar: 25 μm. Quantification of (H) puromycin intensity as a measure for de novo protein synthesis and (I) absolute nuclear ATF4 intensity. Data are normalised to untreated WT.
-
J, KRepresentative WB (J) and quantification (K) of ATF4 expression in WT and Perk KO neurons ±20–24 h of TM‐induced ER stress (N = 4). ATF4 level is corrected for GAPDH level. Data are normalised to TM‐treated WT. (J) All lanes are from the same gel/blot.
Data information: Data are presented as mean ± SEM. N: Biological replicate. Relevant P‐values are indicated: *P < 0.05, ****P < 0.0001 and ns: not significant Statistical analysis: two‐way ANOVA with Tukey's post‐hoc test (B D, F, H, I); one‐sample t‐test with a hypothetical value of 1 (K).
Source data are available online for this figure.
ER stress‐induced protein synthesis reduction and the ATF4 response are uncoupled in PERK‐deficient neurons
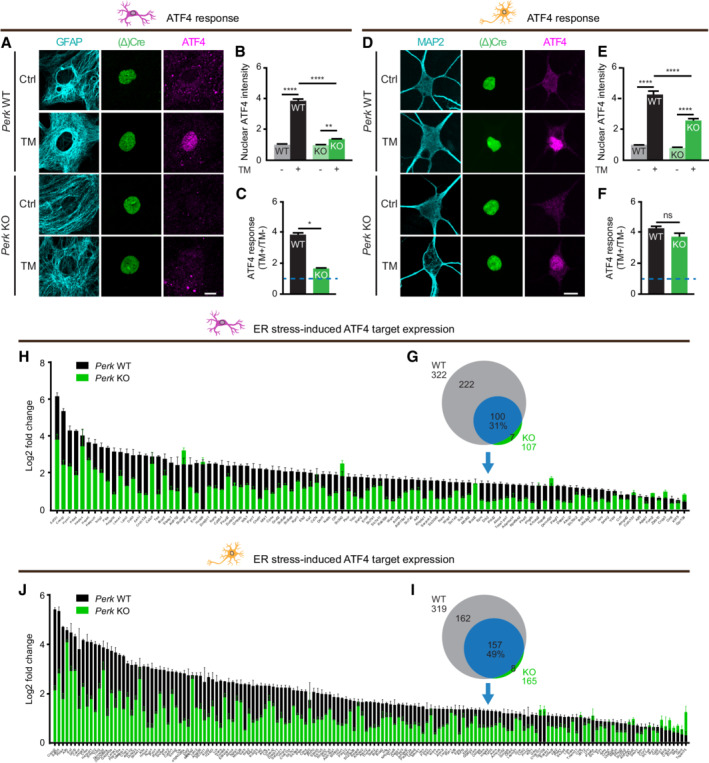
To examine ISR‐induced selective mRNA translation (Fig 1C), the level of the transcription factor ATF4 in the nucleus was assessed by immunofluorescence and confocal imaging. As expected, the ER stress‐induced increase in nuclear ATF4 level was significantly reduced in Perk KO astrocytes (Fig 2A–C). In contrast, in Perk KO neurons, the ER stress‐induced upregulation of ATF4 was preserved (Fig 2D–F). Due to a lower basal level, the absolute ATF4 level during ER stress was 1.5‐fold lower in Perk KO than in WT neurons (Fig 2E). Similar results were obtained with TG‐induced ER stress (Fig EV3G and I). Furthermore, bulk analysis by WB confirmed the rescaled ER stress‐induced regulation of ATF4 in Perk KO neurons (Fig EV3J and K). These results demonstrate that the observed increase in nuclear ATF4 in Perk KO is not a non‐specific effect of TM and is not due to subcellular relocalisation. Hence, the ER stress‐induced ATF4 response is preserved albeit rescaled in PERK‐deficient neurons.
Figure 2. ER stress‐induced ATF4 response is rescaled in PERK‐deficient neurons.
-
A–FATF4 expression in WT and Perk KO astrocytes (N = 3) and neurons (N = 9) ± 20–24 h of TM‐induced ER stress. (A, D) Representative confocal images of ATF4 immunofluorescence. Astrocytic soma (GFAP, cyan), dendrites (MAP2, cyan), nuclei ((Δ)Cre, green) and ATF4 (magenta). Scale bar: 10 μm. (B, E) Quantification of absolute nuclear ATF4 intensity. Data are normalised to untreated WT. (C, F) Quantification of the ER stress‐induced ATF4 response. Data are related to untreated cells of the same genotype. The baseline level (without ER stress) is depicted by a dashed line.
-
G–J(G, I) Venn diagram showing the number of significantly upregulated ATF4 target genes in WT (grey and blue) and Perk KO (green and blue) astrocytes (G, N = 3) and neurons (I, N = 3) upon 20–24 h of TM‐induced ER stress determined by mRNA‐seq. Percentage: fraction of upregulated ATF4 target genes in WT cells that are also upregulated in Perk KO cells. (H, N = 3; J, N = 3) Log2 fold change of ATF4 target genes that are significantly upregulated in both WT and Perk KO cells (blue in Venn diagram).
Data information: Data are presented as mean ± SEM. N: Biological replicate. Relevant P‐values are indicated: *P < 0.05, **P < 0.01, ****P < 0.0001 and ns: not significant. Statistical analysis: two‐way ANOVA with Tukey's post‐hoc test (B, E); and nested t‐test (C, F). See also Fig EV3 and Appendix Fig S2.
Source data are available online for this figure.
To examine the effect of lower ATF4 levels on transcription, we employed mRNA sequencing (mRNA‐seq). This experiment showed that in WT and Perk KO astrocytes, the expression of 3,808 and 997 mRNA transcripts was significantly upregulated by ER stress, respectively. In WT and Perk KO neurons, the expression of 3,897 and 2092 mRNA transcripts was significantly upregulated by ER stress, respectively (Appendix Fig S2). Analysis of the transcriptional targets of ATF4 (Han et al, 2013) showed that only 31% (100/322) of the ER stress‐induced ATF4 target genes identified in WT astrocytes were also significantly upregulated in Perk KO astrocytes (Fig 2G). In addition, the increase in expression of these ATF4 target genes was reduced in Perk KO astrocytes (Fig 2H). In Perk KO neurons, expression of 49% (157/319) of the ATF4 target genes that were upregulated by ER stress in WT neurons was significantly increased (Fig 2I). Like in astrocytes, the ER stress‐induced increase in ATF4 target gene expression was reduced in Perk KO neurons (Fig 2J). These data therefore demonstrate that the reduced ATF4 levels in Perk KO neurons are not sufficient to induce the full transcriptional response. This indicates that the control of ATF4 translation is uncoupled from the control of global protein synthesis during ER stress, because the latter is preserved in PERK‐deficient neurons.
Preservation of ER stress‐induced translational control is a conserved neuron‐specific feature
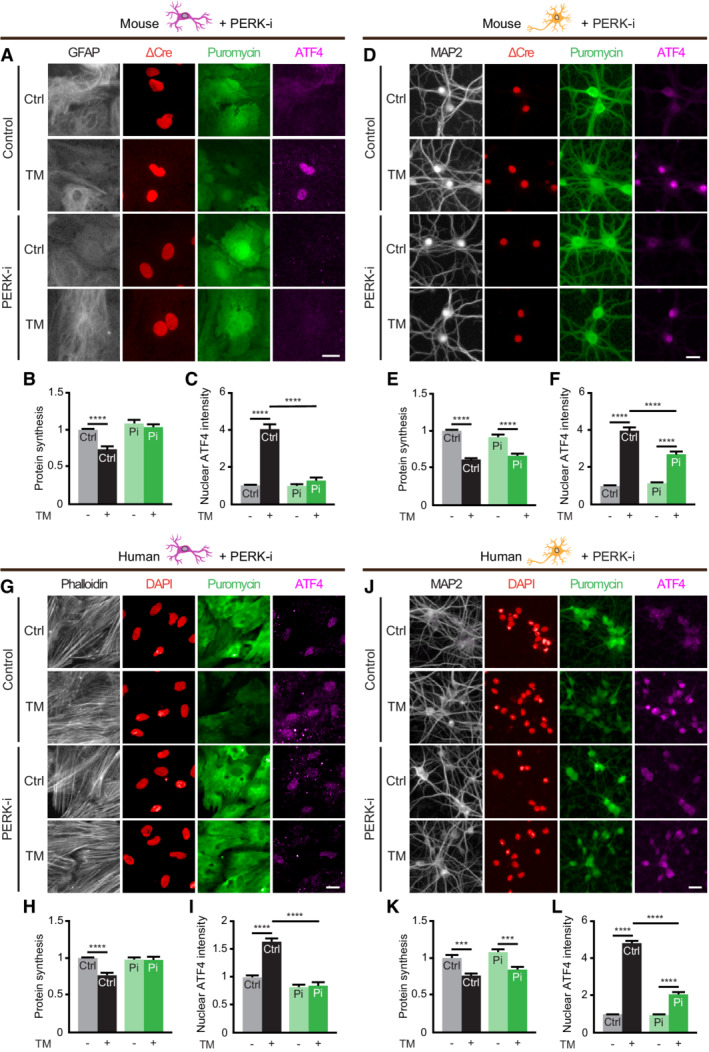
To test whether the compensatory mechanism of Perk KO neurons to retain translational control during ER stress results from adaptation to long‐term PERK deficiency, ER stress was induced in the presence of the pharmacological PERK inhibitor GSK2606414 (PERK‐i). We established a protocol for high‐content microscopy and automated analysis to quantify both puromycinylated proteins and ATF4 levels in the same experiment. PERK‐i treatment abolished both the ER stress‐induced reduction in protein synthesis and ATF4 upregulation in WT astrocytes (Fig 3A–C). In contrast, PERK‐i‐treated WT neurons show preserved ER stress‐induced protein synthesis reduction and decreased induction of p‐eIF2α and ATF4 (Figs 3D–F and EV4A and B). PERK‐i treatment did not affect the ER stress‐induced reduction in protein synthesis or ATF4 upregulation in Perk KO astrocytes and neurons (Fig EV4C–H), excluding that the preservation of ER stress‐induced translational control of Perk KO neurons is caused by low level residual PERK. Collectively, these data show that neurons adapt similar to acute PERK impairment and chronic Perk inactivation.
Figure 3. Preservation of ER stress‐induced translational control is a conserved neuron‐specific feature.
-
A–LProtein synthesis and ATF4 expression in mouse (A‐F) and human (G‐L) astrocytes (A‐C and G‐I) and neurons (D‐F and J‐L) ± 20–24 h of TM‐induced ER stress ± PERK‐i (Pi). (A, D, G, J) Representative images obtained by high‐content microscopy. Astrocytic soma (GFAP/phalloidin, white), dendrites (MAP2, white), nuclei (ΔCre, red), de novo‐synthesised proteins (puromycin, green) and ATF4 (magenta). Scale bar: 25 μm. (B, N = 5; E, N = 5; H, N = 3; K, N = 3) Quantification of puromycin intensity as a measure for de novo protein synthesis. (C, N = 5; F, N = 7; I, N = 4; L, N = 3) Quantification of absolute nuclear ATF4 intensity.
Data information: Data are normalised to untreated WT and presented as mean ± SEM. N: Biological replicate. Relevant P‐values are indicated: ***P < 0.001 and ****P < 0.0001. Statistical analysis: Two‐way ANOVA with Tukey's post‐hoc test. See also Fig EV4.
Source data are available online for this figure.
Figure EV4. Preservation of ER stress‐induced translational control in Perk KO neurons is not caused by low level residual PERK—Related to Fig 3 .
-
A, BeIf2α phosphorylation in WT and Perk KO astrocytes and neurons ±20–24 h of TM‐induced ER stress in the presence or absence of PERK‐i (N = 3). Representative WB (A) and quantification (B) of phosphorylated eIF2α level (p‐eIF2α/eIF2α). Data are normalised to untreated WT. (A) All lanes are from the same gel/blot.
-
C–HProtein synthesis and ATF4 expression in Perk KO astrocytes and neurons ±20–24 h of TM‐induced ER stress in the presence of PERK‐i. (C, F) Representative images obtained by high‐content microscopy. Astrocytic soma (GFAP, white), dendrites (MAP2, white), nuclei (Cre, red), de novo‐synthesised proteins (puromycin, green) and ATF4 (magenta). Scale bar: 25 μm. (D, N = 4; G, N = 4) Quantification of puromycin intensity as a measure for de novo protein synthesis. (E, N = 5; H, N = 4) Quantification of absolute nuclear ATF4 intensity. Data are normalised to untreated Perk KO cells. See for data WT cells Fig 3A and D (representative images are of the same experiment).
Data information: Data are presented as mean ± SEM. N: Biological replicate. Relevant P‐values are indicated: *P < 0.05, ****P < 0.0001 and ns: not significant. Statistical analysis: two‐way ANOVA with Tukey's post‐hoc test.
Source data are available online for this figure.
To determine whether neuron‐specific adaptation to PERK impairment is unique for mouse, neurons derived from human induced pluripotent stem cells (hiPSCs) and human primary astrocytes were investigated. In human astrocytes, PERK‐i treatment abolished both the ER stress‐induced protein synthesis and ATF4 response (Fig 3G–I). In contrast, in PERK‐i‐treated hiPSC‐derived neurons the ER stress‐induced protein synthesis reduction was unaffected and the ATF4 response was reduced (Fig 3J–L). These observations indicate that preservation of the capacity to reduce protein synthesis and uncoupling from the ATF4 response during ER stress are conserved properties of PERK‐deficient neurons.
HRI mediates part of the ER stress‐induced translational control in PERK‐deficient neurons
In order to investigate whether the UPR is involved in the compensatory mechanism of PERK‐deficient neurons during ER stress, the activating transcription factor 6 (ATF6) and Inositol‐requiring enzyme 1 (IRE1) pathways of the UPR were examined by qPCR analysis. Upregulation of Ddit3 (CHOP) and Eif4ebp1 mRNA—transcriptional targets of the PERK pathway via ATF4—was reduced in TM‐treated Perk KO (Appendix Fig S3A and B). Interestingly, also the upregulation of Hspa5 (BiP/GRP78) and splicing of Xbp1, markers for the ATF6 and IRE1 pathway of the UPR, respectively, were reduced in Perk KO neurons (Appendix Fig S3C and D). In addition, treatment with the small‐molecule IRE1 inhibitor 4μ8C (IRE1‐i; van Ziel et al, 2019) had no effect on ER stress‐induced protein synthesis repression and ATF4 upregulation in Perk WT or KO neurons (Appendix Fig S3E–H). These results illustrate the crosstalk between PERK and the other UPR arms, but make it unlikely that the UPR is involved in the adaptation of the PERK pathway.
Next, data obtained by the mRNA‐seq analysis were used to identify an alternative stress‐regulated mechanism. Gene Ontology (GO) analysis showed enrichment of differentially expressed genes (DEGs) in several cellular responses to oxidative stress in TM‐treated Perk KO compared with WT neurons (Fig 4A). Importantly, none of these stress responses were identified when comparing TM‐treated WT and Perk KO astrocytes (Fig 4B). Therefore, we assessed whether ER stress induced the accumulation of reactive oxygen species (ROS) in PERK‐deficient neurons, using CellROX Green reagent that results in a fluorescent nuclear signal in the presence of ROS (Fig 4C), confirmed by treatment with the ROS inducer menadione (Men; Appendix Fig S4A–D). CellROX Green labelling showed that ER stress resulted in increased ROS levels in Perk KO, but not WT neurons (Fig. 4D and 4E), nor WT or Perk KO astrocytes (Fig 4F and G). Similar results were obtained in PERK‐i‐treated neurons and astrocytes (Appendix Fig S4E and F). Immunofluorescence showed no nuclear translocation of nuclear factor erythroid 2‐related factor 2 (NRF2) in WT and PERK KO neurons upon ER stress (Appendix Fig S4G and H). These data indicate that independent of NRF2 regulation, PERK‐deficient neurons accumulate ROS during ER stress.
Figure 4. HRI mediates part of the ER stress‐induced translational control in PERK‐deficient neurons.
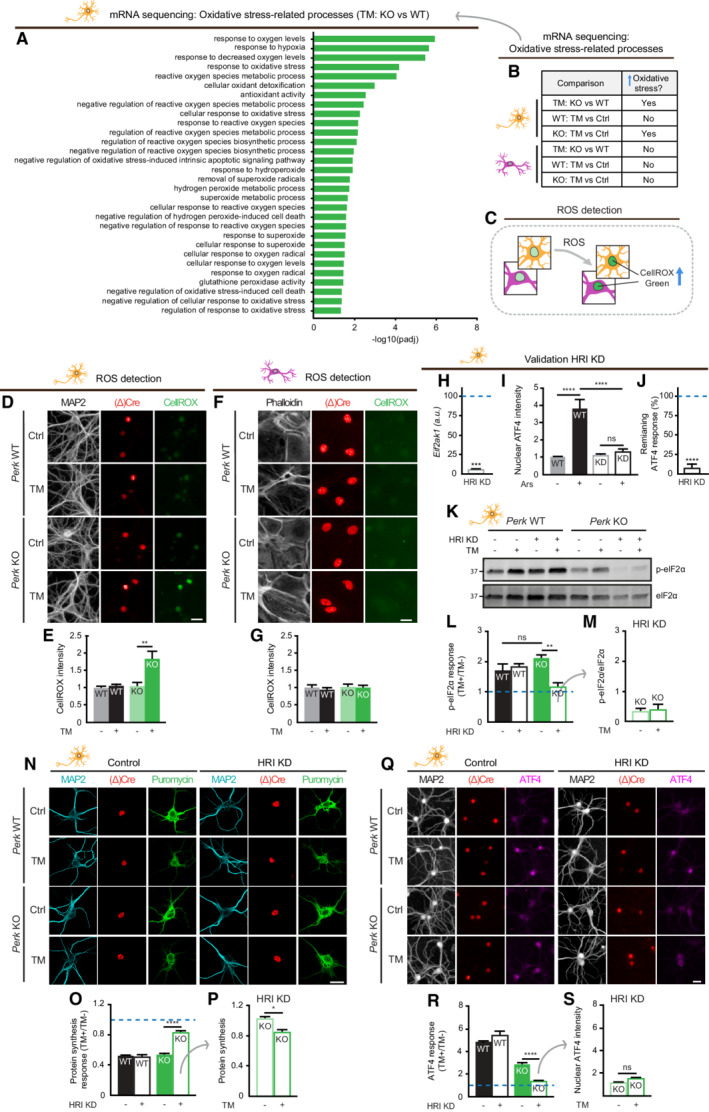
-
AOxidative stress‐related processes identified by GO analysis comparing TM‐treated Perk KO and WT neurons (N = 3).
-
BOxidative stress‐related processes are only enriched in TM‐treated Perk KO neurons. Source data ((adjusted) P‐values, gene IDs and gene names) are available online.
-
CSchematic representation of the method to quantify ROS accumulation by nuclear CellROX green intensity.
-
D–GROS accumulation in WT and Perk KO neurons (N = 3) and astrocytes (N = 3) ± 20–24 h of TM‐induced ER stress. (D, F) Representative images obtained by high‐content microscopy. Dendrites (MAP2, white), astrocytic soma (phalloidin, white), nuclei ((Δ)Cre, red) and ROS (CellROX green, green). Scale bar: 25 μm. (E, G) Quantification of CellROX green intensity.
-
HmRNA expression of Eif2ak1 (HRI) in HRI KD neurons determined by RT–qPCR, normalised to WT transduced with shRNA control (represented by a dashed line) (N = 3).
-
IQuantification of an increase in nuclear ATF4 intensity in WT neurons ± HRI KD ± 20 h of sodium arsenite (Ars) treatment (N = 3). Representative images are shown in Appendix Fig S5.
-
JThe remaining ATF4 response (%) upon HRI KD, related to ATF4 response of WT transduced with shRNA control (100%, represented by a dashed line) (N = 3). Calculated from data in (I): .
-
K–MeIF2α phosphorylation in HRI KD WT and Perk KO neurons (N = 3) ± 20–24 h of TM‐induced ER stress. Representative WB (K), quantification of the ER stress‐induced p‐eIf2α response (L) and absolute phosphorylated eIF2α level (p‐eIF2α/eIF2α) (M).
-
N–SProtein synthesis (N = 3) and ATF4 expression (N = 4) in HRI KD WT and Perk KO neurons ±20–24 h of TM‐induced ER stress. Representative images obtained by (N) confocal and (Q) high‐content microscopy. Dendrites (MAP2, white/cyan), nuclei ((Δ)Cre, green/red), de novo‐synthesised proteins (puromycin, green), ATF4 (magenta). Scale bar: 25 μm. Quantification of the ER stress‐induced protein synthesis response (O) and puromycin intensity as a measure for de novo protein synthesis (P). Quantification of the ER stress‐induced ATF4 response (R) and absolute nuclear ATF4 intensity (S).
Data information: (E, G, M, P, S) Data are normalised to untreated WT. (L, O, R) Data are related to untreated cells of the same genotype and normalised to WT without HRI KD. Baseline level (without ER stress) is depicted by a dashed line. Data are presented as mean ± SEM. N: Biological replicate. Relevant P‐values are indicated: *P < 0.05, **P < 0.01, ****P < 0.0001 and ns: not significant. Statistical analysis: Two‐way ANOVA with Tukey's post‐hoc test (E, G, I, L, O, R); one‐sample t‐test with a hypothetical value of 100 (H,J); Student's t‐test (M); and nested t‐test (P, S). See also Fig EV5 and Appendix Figs S2 and S4.
Source data are available online for this figure.
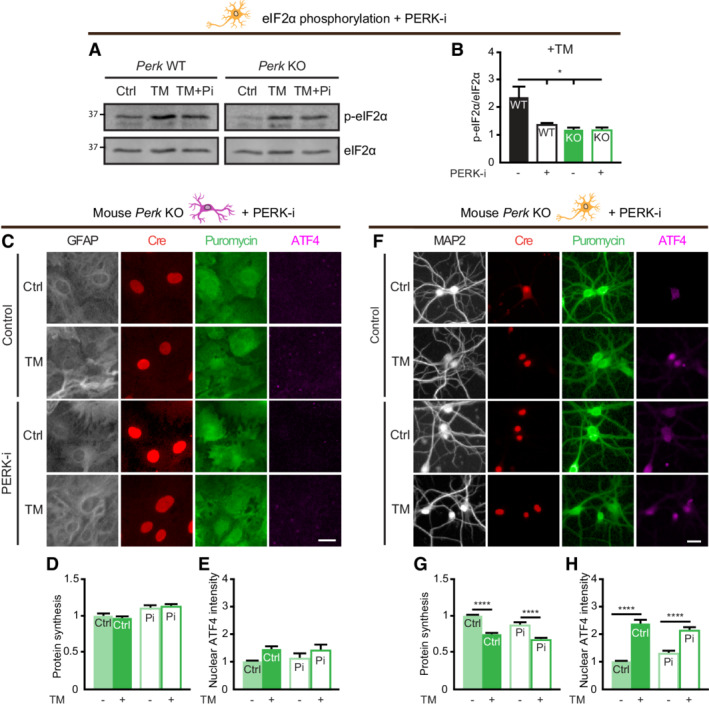
Since ROS trigger the ISR via Eif2ak1/HRI activation, the involvement of HRI in the adaptation of PERK‐deficient neurons was studied using lentiviral shRNA‐mediated targeting. This approach abrogated mRNA expression (Fig 4H) and the function of HRI, demonstrated by the abolishment of sodium arsenite (Ars)‐induced ATF4 upregulation (Fig 4I; Appendix Fig S5; 92% reduction Fig 4J). Interestingly, WB analysis demonstrated that knockdown (KD) of HRI abrogated the ER stress‐induced eIF2α phosphorylation in Perk KO, but not WT neurons (Fig 4K–M). Confocal analysis showed that HRI KD inhibited the reduction in protein synthesis during ER stress in Perk KO, but not WT neurons (Fig 4N–P). In addition, HRI KD abolished the ER stress‐induced increase in ATF4 in Perk KO, but not WT neurons (Fig 4Q–S). In accordance, treatment with the pharmacological HRI inhibitor hemin (HRI‐i) reduced the ER stress‐induced upregulation of ATF4 in Perk KO neurons and not in WT neurons (Fig EV5A–C). Interestingly, although HRI KD abolished the ER stress‐induced eIF2α phosphorylation and upregulation of ATF4 in Perk KO neurons, it did not completely abrogate the reduction in protein synthesis (Fig 4M and S vs. P). Partial KD of the remaining two eIF2α kinases, Eif2ak2/ protein kinase R (PKR) and Eif2ak4/ general control nonderepressible 2 (GCN2; Fig EV5D–I), did not differentially affect ER stress‐induced ATF4 upregulation in Perk KO neurons (Fig EV5J–L). GCN2 slightly reduced the ER stress‐induced ATF4 upregulation, but in both WT and Perk KO neurons (Fig EV5L). Functional KD was confirmed by treatment with PKR activator BEPP and the GCN2 activator Halofuginone (Halo), demonstrating that ATF4 upregulation was reduced by 63% in PKR KD neurons and abolished in the GCN2 KD neurons, respectively (Fig EV5E and F, and H, and I). Therefore, it is unlikely that these eIF2α kinases contribute to the compensatory mechanism of PERK‐deficient neurons during ER stress. WB analysis showed that neither PERK deletion nor ER stress affected the HRI protein levels (Appendix Fig S6A and B), suggesting that the compensatory effect of HRI in PERK‐deficient neurons is mediated by changes in the activity of HRI rather than the levels. Together, these data demonstrate that the rescaled p‐eIF2α and ATF4 responses of PERK‐deficient neurons are fully mediated by HRI and the global protein synthesis response partially.
Figure EV5. HRI mediates part of the ER stress‐induced translational control in PERK‐deficient neurons—Related to Fig 4 .
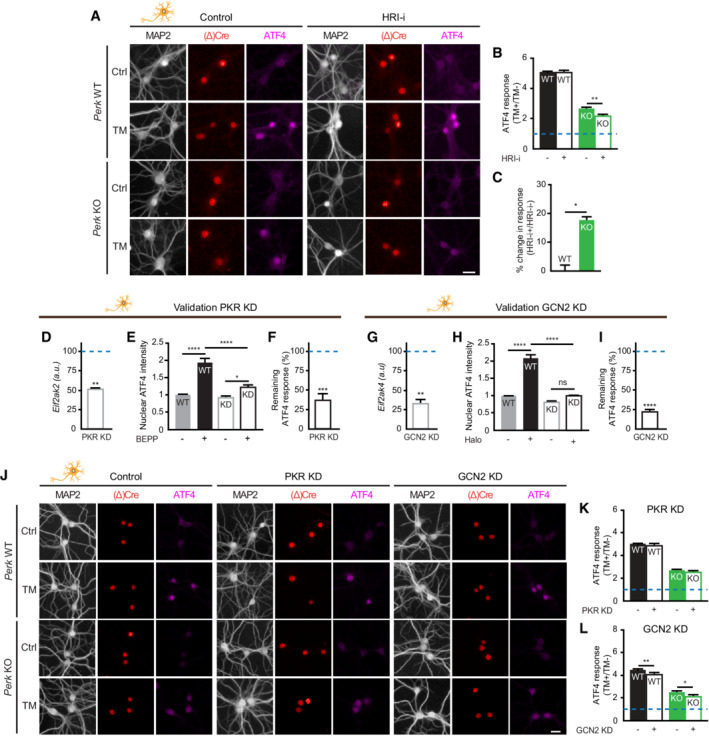
-
A–CATF4 expression in WT and Perk KO neurons ±20–24 h of TM‐induced ER stress in the absence or presence of HRI‐I (N = 3). (A) Representative images obtained by high‐content microscopy. Dendrites (MAP2, white/cyan), nuclei ((Δ)Cre, green/red), ATF4 (magenta). Scale bar: 25 μm. (B) Quantification of the ER stress‐induced ATF4 response. Data are normalised to WT without HRI‐i. Baseline level (without ER stress) is depicted by dashed line. (C) Quantification showing the percentage change in the ATF4 response induced by HRI‐i. Calculated from data in (B): .
-
D–IValidation of PKR and GCN2 KD. (D, G) mRNA expression of (D) Eif2ak2 (PKR) in PKR KD neurons and (G) Eif2ak4 (GCN2) in GCN2 KD neurons, determined by RT–qPCR (N = 3). Data are normalised to WT transduced with a scrambled construct (represented by a dashed line). (E, H) Quantification of the increase in nuclear ATF4 intensity in WT neurons ± PKR KD ± 6 h of BEPP treatment (N = 3) or in WT neurons ± PKR KD ± 6 h of Halofuginone (Halo) treatment (N = 4). (F, N = 3; I, N = 4) The remaining ATF4 response (%) upon PKR KD and GCN2 KD, respectively. Related to ATF4 response of WT transduced with shRNA control (100%, represented by a dashed line). (F) Calculated from data in (E): . (I) Calculated from data in (H): .
-
J–LEffects of PKR and GCN2 KD in WT and Perk KO neurons ±20–24 h of TM‐induced ER stress. (J) Representative images of ATF4 immunofluorescence obtained by high‐content microscopy and quantification of the ER stress‐induced ATF4 response in PKR KD (K) and GCN2 KD (L) neurons (N = 5). Dendrites (MAP2, white/cyan), nuclei ((Δ)Cre, green/red) and ATF4 (magenta). Scale bar: 25 μm. Data are related to untreated (scrambled) cells of the same genotype. Baseline level (without ER stress) is depicted by a dashed line.
Data information: Data are presented as mean ± SEM. N: Biological replicate. Relevant P‐values are indicated: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 and ns: not significant. Statistical analysis: Two‐way ANOVA with Tukey's post‐hoc test (B, E, H, K, L); nested t‐test (C); and one‐sample t‐test with a hypothetical value of 100 (D, F, G, I).
Source data are available online for this figure.
To investigate the dependence of the adapted ISR on p‐eIF2α, overexpression of eIF2αWT or a dominant negative phospho‐deficient mutant (eIF2αS51A) was employed. This experiment showed that eIF2αS51A inhibited the ER stress‐induced reduction in protein synthesis and ATF4 increase in both WT and Perk KO neurons (Fig 5A–F). This finding indicated that both ER stress‐induced global and selective translational regulation in PERK‐deficient neurons are at least partially p‐eIF2α‐dependent.
Figure 5. ER stress‐induced ATF4 response in PERK‐deficient neurons is fully p‐eIF2α‐dependent.
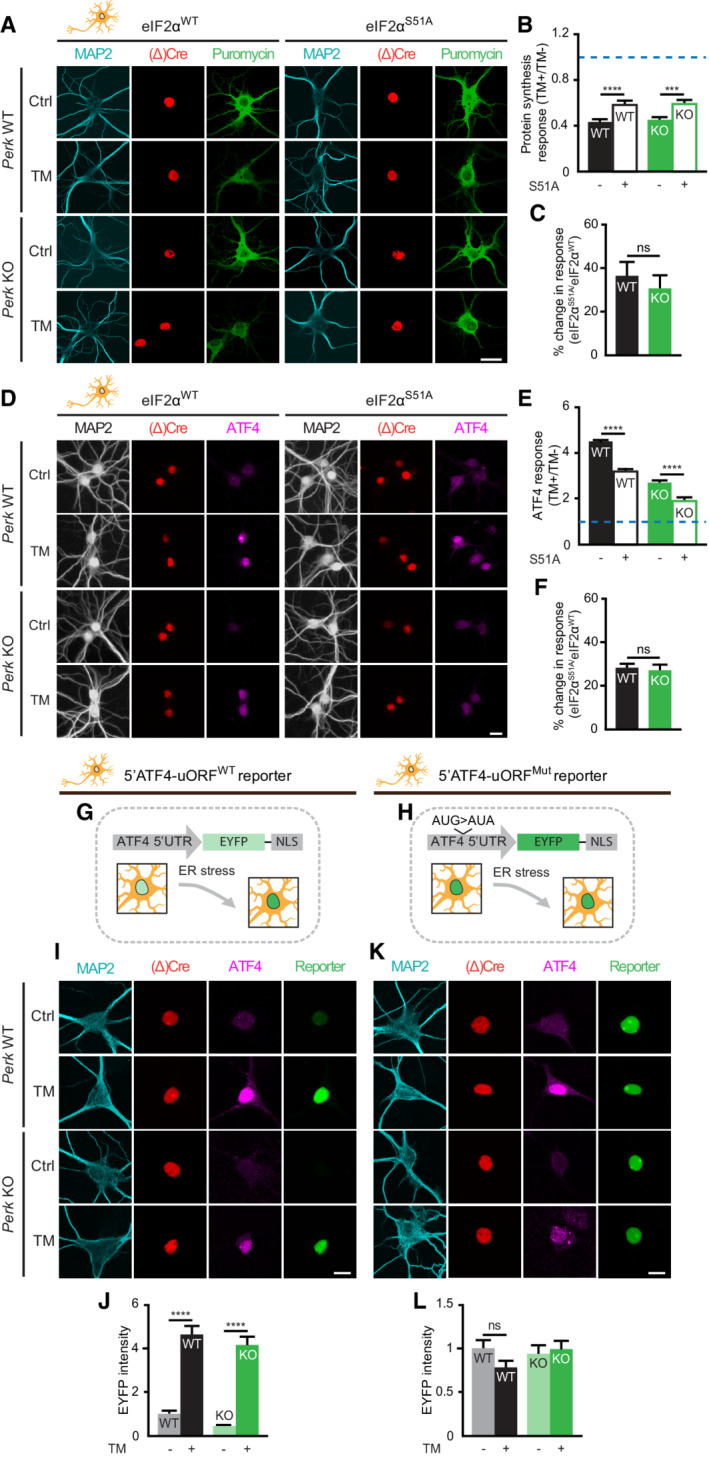
-
A–FProtein synthesis (A‐C, N = 3) and ATF4 expression (D‐F, N = 5) in WT and Perk KO neurons overexpressing eIF2αWT or phospho‐deficient eIF2αS51A ± 20–24 h of TM‐induced ER stress. Representative images obtained by (A) confocal and (D) high‐content microscopy. Dendrites (MAP2, white/cyan), nuclei ((Δ)Cre, red), de novo‐synthesised proteins (puromycin, green) and ATF4 (magenta). Scale bar: 25 μm. Quantification of the ER stress‐induced protein synthesis (B) and ATF4 response (E). Data are normalised to WT eIF2αWT. Quantification showing the percentage change in the ER stress‐induced protein synthesis (C) and ATF4 (F) response induced by eIF2αS51A compared with eIF2αWT. (C) Calculated from data in (B): . (F) Calculated from data in (E): .
-
G–LExpression of p‐eIF2α sensitive (5′ATF4‐uORFWT) and p‐eIf2α insensitive (5′ATF4‐uORFMut) EYFP translational reporters in WT and Perk KO neurons ±20–24 h of TM‐induced ER stress (N = 3). (G, H) Schematic depiction of the reporters. (I, K) Representative confocal images. Dendrites (MAP2, cyan), nuclei ((Δ)Cre, red), endogenous ATF4 (magenta) and ATF4 reporter (green). Scale bar: 10 μm. (J, L) Quantification of EYFP intensity, normalised to untreated WT.
Data information: Data are presented as mean ± SEM. N: Biological replicate. Relevant P‐values are indicated: ***P < 0.001, ****P < 0.0001 and ns: not significant. Statistical analysis: two‐way ANOVA with Tukey's post‐hoc test (B, E, J, L) and nested t‐test (C, F).
Source data are available online for this figure.
Overexpression of WT or mutant eIF2α was toxic in neurons; therefore, full inhibition of the ISR was not achieved using this approach (Fig 5C and F). To address the p‐eIF2α dependence of ATF4 translation more directly, an ATF4 translation reporter was used in which the ATF4 5’ UTR drives expression of EYFP fused to a nuclear localisation signal (5′ATF4‐uORFWT; Fig 5G; van Ziel et al, 2020). Comparison with a uORF mutant 5′UTR reporter that is insensitive to p‐eIF2α (5′ATF4‐uORFMut; Fig 5H; Lu et al, 2004) allows assessment whether the residual ER stress‐induced ATF4 increase is caused by p‐eIF2α‐dependent translation. It is important to note that this experimental set‐up uses the accumulation of EYFP, which has a much higher t1/2 (approximately 15 h; Danhier et al, 2015) than ATF4 (<60 min; Ameri et al, 2004; Frank et al, 2010); therefore, the data are not a reflection of ATF4 accumulation. As expected, 5′ATF4‐uORFWT showed enhanced nuclear EYFP signal in response to ER stress (like endogenous ATF4 expression) in both WT and Perk KO neurons (Fig 5I and J). In contrast, 5′ATF4‐uORFMut was not responsive to ER stress in WT nor Perk KO neurons (Fig 5K and L). Therefore, the residual ER stress‐induced ATF4 response in PERK‐deficient neurons is fully p‐eIF2α‐dependent.
ER stress‐induced protein reduction in PERK‐deficient neurons is partially eIF2‐independent
To further investigate the involvement of p‐eIF2α in the reduction of protein synthesis, the small‐molecule ISR inhibitor (ISRIB) was used (Sidrauski et al, 2013; Zyryanova et al, 2021). ISRIB interferes with the ISR downstream of p‐eIF2α by preventing its inhibitory effect on eIF2B. Interestingly, whereas ISRIB inhibited the ER stress‐induced translational repression in WT neurons, it did not in Perk KO neurons (Fig 6A–C). In contrast, treatment with ISRIB reduced the ER stress‐induced upregulation of ATF4 to the same extent in WT and Perk KO neurons (Fig 6D–F), in line with the full HRI/p‐eIF2α dependence of the ATF4 response. To further investigate whether a change at the level of eIF2B underlies part of the adapted ISR in PERK‐deficient neurons, we cultured neurons and astrocytes derived from a mouse model representative for leucodystrophy vanishing white matter (VWM). These cells are homozygous for the Arg191His mutation in the ε subunit of the eIF2B complex (2b5 ho ; Dooves et al, 2016). Cells heterozygous for the mutation (2b5 he ) served as control (Dooves et al, 2016). Evaluation of de novo protein synthesis and nuclear ATF4 levels showed no difference between the ER stress‐induced ISR of 2b5 he and 2b5 ho astrocytes (Fig 6G–J; Wisse et al, 2018) and neurons (Fig 6K–N). Inhibition of PERK activity by treatment with PERK‐i inhibited ER stress‐induced protein synthesis reduction and ATF4 upregulation in both 2b5 he and 2b5 ho astrocytes (Fig 6G–J), albeit slightly less efficient in the latter. In contrast, ER stress‐induced reduction in protein synthesis was unaffected by PERK‐i treatment in 2b5 he and 2b5 ho neurons (Fig 6K and L) and the ATF4 response reduced (Fig 6M and N). These data showed that the adaptation that preserves ER stress‐mediated protein synthesis reduction in PERK‐deficient neurons was not sensitive to ISRIB or eIF2B mutations and therefore independent of eIF2B. Together, the data indicate that in addition to an HRI/eIF2α‐dependent part, an eIF2‐independent mechanism is part of the neuron‐specific translational adaptation to PERK deficiency.
Figure 6. ER stress‐induced reduction in protein synthesis in PERK‐deficient neurons is partially eIF2‐independent.
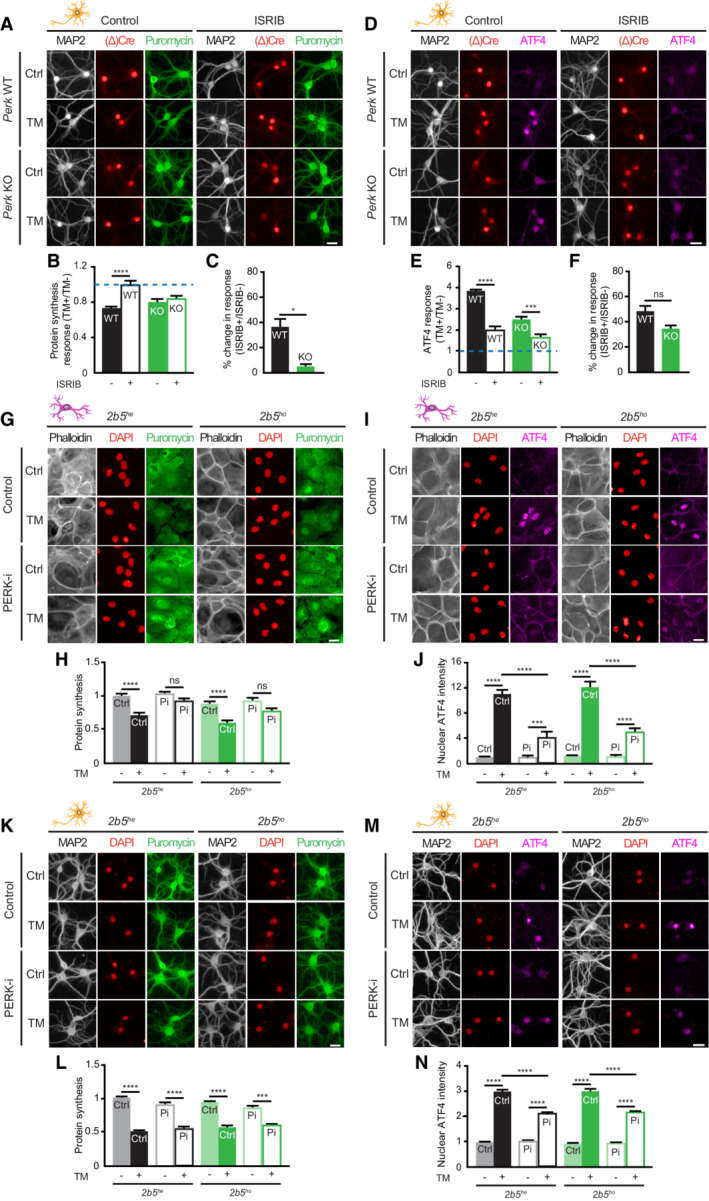
-
A–FProtein synthesis and ATF4 expression in ISRIB‐treated WT and Perk KO neurons ±20–24 h of TM‐induced ER stress (N = 4). (A, D) Representative images obtained by high‐content microscopy. Dendrites (MAP2, white), nuclei ((Δ)Cre, red), de novo‐synthesised proteins (puromycin, green) and ATF4 (magenta). Scale bar: 25 μm. Quantification of the ER stress‐induced protein synthesis (B) and ATF4 (E) response. Data are normalised to WT without ISRIB. Baseline level (without ER stress) is depicted by dashed line. Quantification showing the percentage change in the ER stress‐induced protein synthesis (C) and ATF4 (F) response induced by ISRIB. (C) Calculated from data in (B): . (F) Calculated from data in (E): .
-
G–NProtein synthesis and ATF4 expression in 2b5 he and 2b5 ho astrocytes and neurons ±20–24 h of TM‐induced ER stress in the presence or absence of PERK‐i (Pi) (N = 3). (G, I, K, M) Representative images obtained by high‐content microscopy. Astrocytic soma (phalloidin, white), dendrites (MAP2, white), nuclei (DAPI, red), de novo‐synthesised proteins (puromycin, green) and ATF4 (magenta). Scale bar: 25 μm. (H, L) Quantification of puromycin intensity as a measure for de novo protein synthesis and (J, N) absolute nuclear ATF4 intensity. Data are normalised to untreated WT.
Data information: Data are presented as mean ± SEM. N: Biological replicate. Relevant P‐values are indicated: *P < 0.05, ***P < 0.001, ****P < 0.0001 and ns: not significant. Statistical analysis: Two‐way ANOVA with Tukey's post‐hoc test (B, E); nested t‐test (C, F); three‐way ANOVA with Tukey's post‐hoc test (H, J, L, N).
Source data are available online for this figure.
PERK‐deficient neurons activate angiogenin‐mediated protein synthesis reduction
Next, the involvement of two major eIF2‐independent stress‐regulated translational pathways was studied: mTOR signalling (reviewed in Ben‐Sahra & Manning, 2017) and stress granule formation (reviewed in Riggs et al, 2020). Activation of mTOR complex 1 (mTORC1) is accompanied by phosphorylation of mTOR kinase at serine 2448 and regulates protein synthesis in an eIF2‐independent manner by phosphorylation of its downstream targets S6 kinase (S6K) and eIF4E‐binding protein (4E‐BP). WB analysis was employed to determine the levels of p‐mTOR, p‐S6K and p‐4E‐BP1 (Appendix Fig S7A–E). The levels of 4E‐BP1 and p‐4E‐BP1 were reduced in Perk KO neurons, since 4E‐BP1 is a target of the ATF4 transcriptional response that was reduced in Perk KO (Appendix Figs S3B and S6C). However, ER stress‐induced alterations in the phosphorylation of mTOR signalling factors were not observed in Perk KO neurons (Appendix Fig S7A–E). Moreover, treatment with Torin1—a potent selective ATP‐competitive inhibitor of mTOR (Appendix Fig S7F)—did not affect ER stress‐induced protein synthesis reduction in WT or Perk KO neurons (Appendix Fig S7G and H). Next, the formation of stress granules was studied by quantifying the clustering of the immunofluorescence signal of the stress granule marker Rab‐GTPase‐activating protein SH3‐domain‐binding protein (G3BP). Stress granules were observed in very few neurons upon 1 h TG treatment, as previously shown (Wiersma et al, 2019; Appendix Fig S6I and J). However, TM‐induced ER stress for 6 or 24 h did not induce the formation of stress granules in WT or Perk KO cells (Appendix Fig S7I and K). Therefore, mTOR signalling and stress granule assembly are not involved in the translational adaptation of PERK‐deficient neurons.
In addition to the well‐established activation of the ISR by ROS via HRI, oxidative stress was demonstrated to activate an additional translational response that is mediated via transfer RNA (tRNA) cleavage by the endoribonuclease ANG (Thompson et al, 2008; Yamasaki et al, 2009). Analysis of the oxidative stress‐related DEGs (Fig 4A) in TM‐treated Perk KO neurons showed that Ang is one of the most highly regulated genes (Fig 7A). Expression of Ang mRNA was significantly increased upon ER stress in Perk KO neurons but not astrocytes (Fig 7B and C). WB analysis confirmed that the ANG protein levels were increased in Perk KO but not WT neurons during ER stress (Fig 7D and E). It was previously shown that ANG‐mediated tRNA cleavage is facilitated by oxidative stress‐induced tRNA unfolding (Mishima et al, 2014). Unfolding of tRNAs is accompanied by exposure of the common 1‐methyladenosine (m1A) methylation, which can be determined by m1A immunodetection in situ (Itoh et al, 1988; Mishima et al, 2014). Immunofluorescence and high‐content microscopy showed that the m1A intensity significantly decreased upon ER stress in WT, but not Perk KO neurons (Fig 7F and G). These data indicate that in PERK‐deficient neurons during ER stress, ANG levels are increased and tRNAs are in a more unfolded state.
Figure 7. PERK‐deficient neurons activate angiogenin‐mediated protein synthesis reduction.
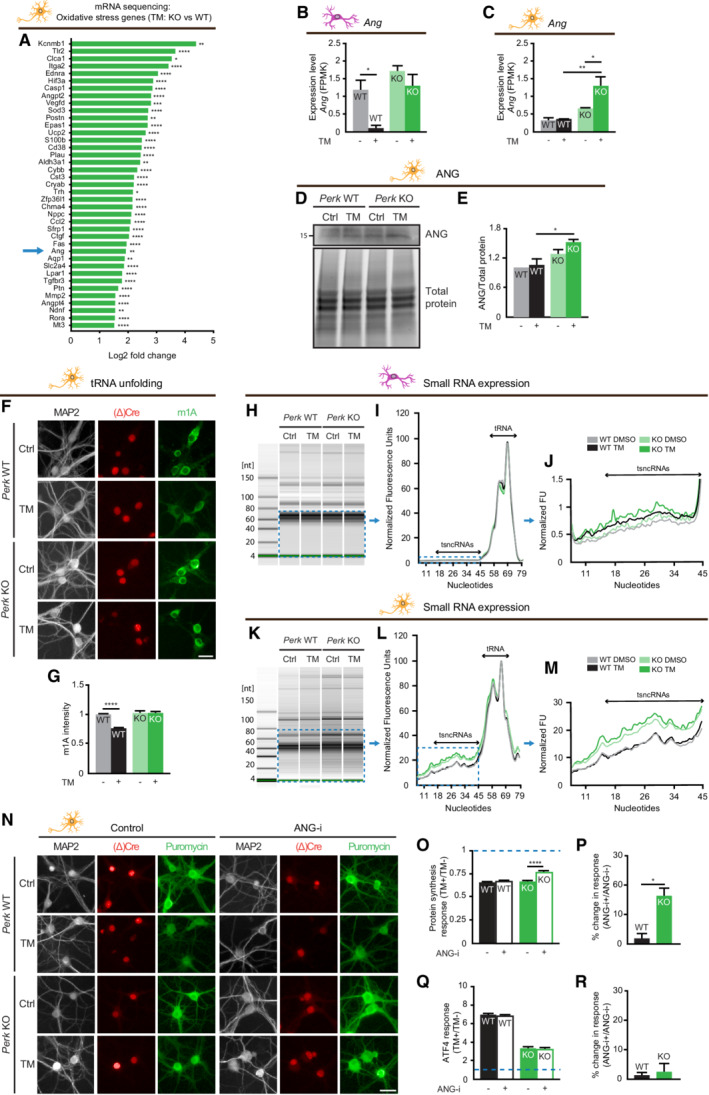
- A
-
B, CAng mRNA level in WT and Perk KO neurons and astrocytes ±20–24 h of TM‐induced ER stress, determined by mRNA‐seq (N = 3). FPMK: Fragments per kilobase of transcript per Million.
-
D, ERepresentative WB and quantification of ANG protein level (N = 3).
-
F, GtRNA unfolding in WT and Perk KO neurons ±20–24 h of TM‐induced ER stress (N = 4). (F) Representative images obtained by high‐content microscopy of 1‐methyladenosine (m1A) immunofluorescence to determine tRNA unfolding. Dendrites (MAP2, white), nuclei ((Δ)Cre, red), unfolded tRNA (m1A, green). Scale bar: 25 μm. (G) Quantification of m1A intensity, normalised to untreated WT.
-
H–MAbundance of small RNAs in WT and Perk KO astrocytes (N = 3) and neurons (N = 6) ± 20–24 h of TM‐induced ER stress, determined by high‐resolution automated electrophoresis. (H, K) Representative gel image. Electropherogram (I, L) and zoom (J, M) showing quantified fluorescence units (FU) of small RNAs, size ranges of tRNAs and tsncRNAs are indicated. Shown is the mean of the biological replicates.
-
N–RProtein synthesis and ATF4 expression in WT and Perk KO neurons ±20–24 h of TM‐induced ER stress in the presence or absence of ANG‐I (N = 3). (N) Representative images obtained by high‐content microscopy. Dendrites (MAP2, white), nuclei ((Δ)Cre, red) and de novo synthesis of proteins (puromycin, green). Scale bar: 25 μm. Quantification of the ER stress‐induced protein synthesis (O) and ATF4 response (Q). Data are normalised to WT without ANG‐i. Baseline level (without ER stress) is depicted by dashed line. Quantification showing the percentage change in the ER stress‐induced protein synthesis (P) and ATF4 response (R) induced by ANG‐i. (P) Calculated from data in (O): . (R) Calculated from data in (Q): .
Data information: Data are presented as mean ± SEM. N: Biological replicate. Relevant P‐values are indicated: *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. Statistical analysis: two‐way ANOVA with Tukey's post‐hoc test (B, C, E, G, O, Q); nested t‐test (P, R).
Source data are available online for this figure.
To investigate whether the generation of tRNA fragments was induced by ER stress in Perk KO neurons, high‐resolution automated electrophoresis suitable for the detection of the size range of tRNA‐derived small non‐coding RNA (tsncRNAs; 16–48 nucleotides) was used. In astrocytes, there was no difference in the levels of small RNAs in this size range between WT and Perk KO astrocytes. In addition, these were not affected by ER stress treatment irrespective of the genotype (Fig 7H–J). In contrast, small RNAs were more abundant in Perk KO than in WT neurons. Moreover, ER stress further increased the abundance of small RNAs in Perk KO neurons but not WT neurons (Fig 7K–M). This indicates that ER stress increases the level of small RNAs in Perk KO neurons, but not astrocytes.
In order to directly study whether ANG is involved in the ER stress‐induced reduction in protein synthesis in Perk KO neurons, ANG activity was targeted by the small‐molecule inhibitor NSC‐65828 (ANG‐i; Kao et al, 2002). ANG inhibition reduced the ER stress‐induced translational repression in Perk KO, but not in WT neurons (Fig 7N–P). In addition, ER stress‐induced ATF4 upregulation in WT neurons was not affected by treatment with ANG‐i in WT nor Perk KO neurons (Fig 7Q and R), in agreement with the uncoupling of global protein synthesis from the ATF4 response. These data show that ANG activity mediates an eIF2α‐independent mechanism that complements the HRI/eIF2α pathway to preserve the ER stress‐induced protein synthesis block in PERK‐deficient neurons.
Discussion
Here, we show that PERK‐deficient neurons shift cellular signalling to retain the capacity to control translation during ER stress, which explains the neuronal tolerance to PERK deficiency (Fig EV2; Trinh et al, 2012; Stone et al, 2019). In contrast, PERK deficiency incapacitates ER stress‐induced translational control in astrocytes, as previously shown in a variety of other cell types (Harding et al, 2000; Jiang & Wek, 2005; Whitney et al, 2009; Liu et al, 2010; Saito et al, 2011; Teske et al, 2011; Gupta et al, 2012; Tsukumo et al, 2014, Hou et al, 2015, Guthrie et al, 2016; Shimizu et al, 2020). Quantification of metabolic activity by bulk analysis showed that PERK‐deficient astrocytes are less tolerant to ER stress than WT astrocytes (Fig EV2E). It is important to note that, unlike post‐mitotic neurons, astrocytes retain the capacity to divide in culture, which could lead to an underestimation of the effect of PERK deficiency on the survival of astrocytes during ER stress.
We show that eIF2α phosphorylation and the ATF4 response are rescaled in PERK‐deficient neurons: the ER stress‐induced increase is the same as in WT neurons, but the absolute levels are lower. In contrast, the control of global protein synthesis is fully retained, indicating that ER stress‐induced protein synthesis reduction is uncoupled from the ATF4 response. ER stress‐induced translational control in PERK‐deficient neurons is partially driven by HRI/eIF2α following the canonical ISR pathway. In addition, protein synthesis reduction, but not the ATF4 response, is complemented by eIF2‐independent translational control by ANG RNase. Together, the HRI/eIF2α and ANG RNase pathways explain the full preservation of the protein synthesis response in PERK‐deficient neurons.
The neuron‐specific signalling shift we report here is induced upon both genetic inactivation of PERK expression and acute pharmacological inhibition of PERK function, indicating that this shift is not a developmental or long‐term adaptation to chronic PERK deficiency, but a cell type‐specific response to compensate for impairments in a key proteostatic pathway. Moreover, human iPSC‐derived neurons display the same response to PERK inhibition as mouse neurons, indicating that it is a conserved principle. Neurons are long‐lived post‐mitotic cells that continue to develop extensive specialised properties during their lifetimes, giving individual cells unique properties that cannot readily be replaced. Reduction in protein synthesis during ER stress is essential for cell viability (Harding et al, 2000; Han et al, 2013). In addition, ISR regulation has been implicated in synaptic plasticity (reviewed in Costa‐Mattioli et al, 2009; Trinh & Klann, 2013). Loss of specialised neurons or unique properties by impaired ISR regulation would therefore come at a high cost and warrant the existence of a back‐up programme specifically in neurons.
Inactivation of PERK was previously reported to decrease the basal p‐eIF2α levels in the brain (Ma et al, 2013). This is consistent with the rescaled eIF2α phosphorylation and ATF4 response we observed in PERK‐deficient neurons. Inactivation of ATF4 in adult mice is not lethal, indicating that reduced ATF4 levels are relatively well tolerated (Tanaka et al, 1998; Masuoka & Townes, 2002). In contrast, overexpression of ATF4 or its target CHOP results in cell death (Marciniak et al, 2004; Han et al, 2013). The preservation of protein synthesis reduction and not the ATF4 response in PERK‐deficient neurons is therefore in line with the essential role of stress‐induced protein synthesis reduction for cell survival (Han et al, 2013). It is important to note that our cell culture system is not well suited for large‐scale direct biochemical measures of selective translation. We used nuclear accumulation of ATF4 as proxy and did not study other potentially important targets of selective ISR translation. Our study provides the basis for dedicated studies to profile cell type‐specific selective translation in the brain to better address the role of specific mRNAs and proteins.
RNA‐seq analysis identified that ER stress induced cell type‐specific regulation of processes related to oxidative stress in PERK‐deficient neurons. Indeed, ER stress leads to the accumulation of ROS in PERK‐deficient neurons but not astrocytes. This may be the result of increased generation of ROS during ER stress specifically in neurons. PERK is directly connected to the major ROS‐generating processes in the cell; to oxidative protein folding in the ER via its role in translational regulation (Tu & Weissman, 2004; Sevier & Kaiser, 2008); and to mitochondrial oxidative phosphorylation (Lebeau et al, 2018; Balsa et al, 2019). Hence, ER stress may affect ROS generation differently in PERK‐deficient cells. However, it is currently unknown whether and how these processes are regulated in a cell type‐specific manner. Interestingly, PERK is actively contributing to mitochondria ER contact sites, a function that is independent of its kinase activity (Verfaillie et al, 2012). This presents an ISR‐independent effect of PERK deficiency that may be relevant to study in neurons.
Alternatively, the ROS accumulation in PERK‐deficient neurons may be caused by a decreased antioxidant response. PERK activates antioxidant responses via ATF4 (Lewerenz & Maher, 2009) and NRF2 (reviewed in Tonelli et al, 2018). However, because the ATF4 response is reduced in both PERK‐deficient neurons and astrocytes this cannot directly explain the ROS accumulation specifically in PERK‐deficient neurons. In addition, in line with the epigenetic inactivation of neuronal NRF2 (Bell et al, 2015), our data do not show evidence for NRF2 activation in neurons during ER stress. This indicates that loss of ATF4‐ or NRF2‐mediated antioxidant regulation does not directly underlie selective ROS accumulation in PERK‐deficient neurons. It is important to note that astrocytes are more resistant to oxidative stress than neurons (Lassmann & van Horssen, 2016; Chen et al, 2020) and may therefore have an overall higher capacity to buffer ROS, irrespective of PERK. This may partly explain the differential ROS accumulation in PERK‐deficient neurons. Interestingly, we identified that the neuron‐specific translational control shift in PERK‐deficient neurons is mediated by two ROS‐activated pathways: HRI (Lu et al, 2001; McEwen et al, 2005; Abdel‐Nour et al, 2019; Alvarez‐Castelao et al, 2020; Guo et al, 2020) and ANG (Mishima et al, 2014; Lai et al, 2016; Mesitov et al, 2017; Chen et al, 2018; Elkordy et al, 2018).
HRI is fully responsible for the rescaled ATF4 response via the canonical eIF2α‐dependent pathway. However, the protein synthesis response is only partially mediated by HRI. eIF2αS51A inhibits the reduction in protein synthesis, confirming that it is partially eIF2α‐dependent (Fig 5A–C). Interestingly, ISRIB treatment did not affect the protein synthesis reduction in Perk KO neurons (Fig 6A–C), which is unexpected in view of its partial eIF2α dependence. This cannot be explained by a different sensitivity of PERK‐deficient neurons for the compound, as the ATF4 response is completely inhibited by ISRIB (Fig 6D–F). Global protein synthesis was reported to respond less strongly to ISRIB than to the ATF4 response (Sidrauski et al, 2013; Halliday et al, 2015). Therefore, it is likely that the ineffectiveness of ISRIB to inhibit the HRI/eIF2α‐mediated component of the protein synthesis response is due to detection limitations. In any case, our data demonstrate that the mechanism underlying the uncoupling of the protein synthesis reduction from the ATF4 response is not affected by ISRIB or the Arg191His mutation in the ε subunit of eIF2B and therefore eIF2‐independent.
The oxidative stress‐responsive protein ANG was identified as a crucial factor that complements HRI to fully maintain translational control upon ER stress. ANG is an endoribonuclease that cleaves tRNAs in the anticodon loop, thereby generating tRNA fragments induced by stress (tiRNAs; Yamasaki et al, 2009). We show that small RNAs in the size range of tiRNAs are increased by ER stress in PERK‐deficient neurons. The limited knowledge about specific tRNA cleavage products in general and in neurons in particular makes targeted analysis currently not feasible. Moreover, because only a very minor fraction of the tRNA pool is cleaved by ANG (Yamasaki et al, 2009) and required for downstream effects (Ivanov et al, 2011), the RNA yield within the culture protocols used in this study is insufficient for profiling to identify specific tiRNAs. ANG has been shown to reduce protein synthesis via 3 different mechanisms. tiRNAs from G‐quadruplex structures that bind and inhibit eIF4G (Ivanov et al, 2011; Lyons et al, 2017, 2020). In addition, ANG can remove the 3′CCA from tRNAs, which is essential for tRNA aminoacylation (Czech et al, 2013). Finally, tiRNAs may induce stress granules (Ivanov et al, 2011), but these are not required for tiRNA‐mediated translational repression (Lyons et al, 2016). Our data do not show evidence for stress granule formation in PERK‐deficient neurons, suggesting that a stress granule‐independent mechanism of ANG‐mediated protein synthesis reduction is involved.
Oxidative stress loosens the tertiary structure of tRNAs, thereby facilitating cleavage by ANG (Mishima et al, 2014). Our data show that ER stress reduces m1A intensity in WT neurons in situ, indicating reduced exposure of the m1A tRNA modification. This structural change is likely related to the different use and recycling of tRNAs during reduced protein synthesis (Yang et al, 2006; Voorhees & Ramakrishnan, 2013; Zhou et al, 2020). Importantly, this stress‐induced decrease in m1A exposure is not observed in PERK‐deficient neurons, suggesting that the tRNA structure is more unfolded, which is in line with the observed presence of ROS. Therefore, in PERK‐deficient neurons, ER stress enhances both ANG expression, the susceptibility of tRNAs to cleavage and the formation of small RNA fragments, supporting activation of ANG‐mediated translational control. Indeed, inhibition of ANG activity decreased the ER stress‐induced protein synthesis response in PERK‐deficient, but not WT neurons (Fig 7N–P), confirming the direct involvement of ANG in the cellular signalling shift.
Aberrant activation of PERK has been implicated in neurodegenerative diseases (reviewed in Scheper & Hoozemans, 2015). The resulting chronic ISR activation was proposed to result in a collapse of neuronal protein synthesis, which in turn contributes to neurodegeneration (Halliday et al, 2017). Preclinical studies show impressive amelioration of neurodegenerative phenotypes in mouse models upon genetic or pharmacological PERK inhibition (Moreno et al, 2012, 2013; Radford et al, 2015; Mercado et al, 2018). However, our results demonstrate that inhibition of PERK does not affect the neuronal capacity to reduce protein synthesis, indicating that the therapeutic effect of PERK inhibition is not mediated via the rescue of neuronal protein synthesis. It is possible that the positive effects on neurodegeneration are mediated by lowering the stress‐induced ATF4 levels in PERK‐deficient neurons, which we demonstrate in this study. Alternatively, a cell non‐autonomous effect, for example via cells that are fully susceptible to PERK inhibition such as astrocytes, may play a role. A recent study reported that targeting p‐eIF2α specifically in astrocytes was sufficient to ameliorate neurodegeneration in a prion disease model (Smith et al, 2020), which would be supportive of the latter mechanism. Targeting the ISR downstream of p‐eIF2α also showed protective effects on neurodegenerative phenotypes (e.g. Das et al, 2015; Halliday et al, 2015). Given the neuron‐specific shift in cellular signalling upon PERK impairment we report here, it is not unlikely that neurons are also resilient to other ISR interventions. Interestingly, neurons expressing a VWM‐associated eIF2B mutation also retained the capacity to shift cellular signalling upon PERK impairment (Fig 6K–N). Future studies on targeting of the ISR for neurological disease should take cell type‐specific responses to the interventions into account.
Interestingly, activity‐reducing ANG mutations have been identified in amyotrophic lateral sclerosis (ALS), Parkinson's disease (PD) and Alzheimer's disease (AD; reviewed in Prehn & Jirström, 2020). Supplementation of ANG activity ameliorates neurodegenerative phenotypes in preclinical ALS and PD models (Kieran et al, 2008; Steidinger et al, 2013; Crivello et al, 2018). Together with our results that indicate the involvement of ANG in the neuron‐specific translational control during proteostatic stress, this indicates that ANG has an important role in neuronal proteostasis.
In conclusion, our data demonstrate the remarkable resilience of neurons to counter proteostatic challenges. We identified HRI and ANG as novel crucial factors to preserve stress‐regulated translational control in neurons. These results are relevant for a better understanding of the pathomechanism that connects proteostatic stress to neurodegeneration.
Materials and Methods
Animals
Perk loxP/loxP mice purchased from the Jackson Laboratory (Stock No: 023066) and Eif2b5 ho /Eif2b5 he mice (previously described in Dooves et al (2016)) were used for timed mating. Animals were housed and bred according to institutional and Dutch governmental guidelines, and experiments were approved by the ethical committee of the Vrije Universiteit/Amsterdam UMC (license number: DEC‐FGA 11–03 and AVD112002017824).
Primary mouse astrocyte and neuron cultures
Neuron and astrocyte cultures were prepared from newborn (P1) mice. Cerebral cortices were dissected and disposed of meninges in Hanks' Balanced Salt Solution (Sigma‐Aldrich) supplemented with 10 mM HEPES (Gibco) (Hanks–HEPES). For astrocyte cultures, cortices were digested with 3% papain (Worthington Biochemical Corporation) in DMEM containing 4.5 g/l glucose and UltraGlutamine I (Lonza) supplemented with 0.2 mg/ml L‐cysteine, 1 mM CaCl2 and 0.5 mM EDTA (all Sigma‐Aldrich) for 45 min at 37°C. After this, cortices were washed with DMEM supplemented with 10% heat‐inactivated fetal bovine serum (HI‐FBS; Gibco), 2.5 mg/ml bovine serum albumin (BSA; Acros Organics) and 2.5 mg/ml trypsin inhibitor (Sigma‐Aldrich) for 15 min at 37°C, and triturated with fire‐polished glass pipettes in DMEM (Lonza) supplemented with 10% HI‐FBS, 1% penicillin/streptomycin (pen‐strep; both Gibco) and 1% non‐essential amino acid solution (NEAA; Sigma‐Aldrich) (DMEM+). Dissociated cells were cultured in DMEM+ in T75 flasks for 6–8 days until reaching a confluency of 80%. Next, the flask was washed with Dulbecco's phosphate‐buffered saline (DPBS; Gibco) and astrocytes were dissociated with 0.05% Trypsin–EDTA (Gibco) for 1–3 min at 37°C. Astrocytes were resuspended, counted and plated in DMEM+ at a density of 50.000 and 1.000–3.000 astrocytes/well in 12‐ and 96‐well plates, respectively. The next day, DMEM+ was changed to Neurobasal medium (NB) supplemented with 2% B‐27, 1.8% HEPES, 0.25% GlutaMAX and 0.1% pen‐strep (all Gibco) (NB+), and at days in vitro (DIV) 6 or 7, 50% of the medium was refreshed.
For neuronal cultures, cortices were digested with 0.25% trypsin (Gibco) in Hanks–HEPES for 20 min at 37°C. After this, cortices were washed three times with Hanks–HEPES and triturated with fire‐polished glass pipettes in DMEM+. Dissociated cells were centrifuged for 5 min at 800 revolutions per minute (rpm) and resuspended, counted and plated in NB+. Neurons were plated at a density of 300.000, 25.000 and 15.000 neurons/well in 6‐, 12‐ and 96‐well plates, respectively. At DIV 3, 2 μM 1‐β‐D‐arabinofuranosylcytosine (AraC) or 1 μM palbociclib (both from Sigma‐Aldrich) was added to prevent the growth of glial cells, and at DIV 6 or 7, 50% of the NB+ medium was refreshed. All plates were coated with 5 μg/ml poly‐L‐ornithine and 2.5 mg/ml laminin (both from Sigma‐Aldrich) in DPBS. Cultures were maintained at 37°C/5% CO2, and all experiments were performed at DIV 14–15.
Primary human astrocyte and neuron cultures
Primary human foetal astrocytes (ScienCell #1800) were maintained in 1:100 Geltrex in DMEM/F12 (both from Gibco)‐coated plates in astrocyte medium supplemented with astrocyte growth supplement, 2% HI‐FBS and 1% pen‐strep (all ScienCell). The medium was refreshed twice a week, and cells were passaged once a week by dissociation with Accutase (Sigma‐Aldrich) and used until passage number 7. For experiments, astrocytes were plated at a density of 3,000 cells/well in coated (1:100 Geltrex in DMEM/F12) 96‐well plates in NB medium supplemented with 2% B27 + VitA, 1% GlutaMAX, 1% pen‐strep (all Gibco), 10 ng/ml NT3, 10 ng/ml BDNF (both from Peprotech) (hNB+) and 0.5% HI‐FBS (Gibco). Cultures were maintained at 37°C/5% CO2, and experiments were performed 6–7 days after plating.
Human induced pluripotent stem cells (hiPSCs) stably expressing doxycycline‐inducible rtTA/Ngn2 (previously generated in Frega et al (2017)) were maintained on Vitronectin (Stem Cell Technologies)‐coated plates in TeSR‐E8 medium (Stem Cell Technologies) supplemented with TeSR‐E8 supplement (Stem Cell Technologies), 0.5% pen‐strep (Gibco), 50 μg/ml G418 (Sigma‐Aldrich) and 0.5 μg/ml puromycin (Sigma‐Aldrich). Colonies were fed daily and passaged once a week by dissociation with Gentle Cell Dissociation Reagent (Stem Cell Technologies) and replating in the presence of 10 μM Rho‐associated, coiled‐coil containing protein kinase inhibitor (RI; Selleckchem) for 24 h to promote cell survival, and maintained until passage number 16. For experiments, hiPSCs were plated at a density of 4,000 cells/well in coated (1:100 Geltrex in DMEM/F12) 96‐well plates in TesR‐E8 medium supplemented with 4% TeSR‐E8 supplement (both from Stem Cell Technologies), 0.5% pen‐strep (Gibcol), 10 ng/ml NT3, 10 ng/ml BDNF (both from Peprotech), 10 μM RI and 2 μg/ml doxycycline (Sigma) to induce the Ngn2 transgene. The day after plating (day 2), the medium was changed to DMEM/F12 supplemented with 1% N2 supplement, 1% NEAA, 1% pen‐strep (all from Gibco), 10 ng/ml NT3, 10 ng/ml BDNF (both Peprotech) and 2 μg/ml doxycycline (Sigma‐Aldrich); and at day 3, the medium was changed to hNB+ medium supplemented with 2 μg/ml doxycycline (Sigma‐Aldrich). At day 4, cells were treated with 2 μM AraC (Sigma‐Aldrich) for 24 h by refreshing 50% of the hNB+ medium supplemented with 2 μg/ml doxycycline and 4 μM AraC. At day 5, 50% of the medium was refreshed with hNB+, and at day 10, 17 and 24, 50% of the medium was refreshed with hNB+ supplemented with 0.5% HI‐FBS (Gibco). Cultures were maintained at 37°C/5% CO2, and the experiments were performed 29–30 days after plating.
Lentiviral constructs and transductions
Inactive ΔCre‐ or active Cre‐recombinase, fused to EGFP or mCherry, and a nuclear localisation sequence (NLS) of nucleoplasmin in the N‐terminus were recloned from Kaeser et al (2011). Plasmids for short hairpin RNA (shRNA) constructs of EIF2AK1/2/4 and the scrambled control were obtained from the MISSION shRNA library (Sigma‐Aldrich; TRCN0000028788, TRCN0000027041, TRCN0000028842). The eIF2αWT and eIF2αS51A overexpression constructs were a gift from David Ron (eIF2a 1 and 2, Addgene plasmids #21807 and #21808). The ATF4 translational reporter (5′ATF4‐uORFWT) was previously described in van Ziel et al (2020), and the 5′ATF4‐uORFMut construct was generated by replacing the AUG start codon of the second open reading frame (ORF) to AUA. All generated constructs were subcloned into pLenti vectors under the synapsin or CMV promoter using the Gateway system (Invitrogen), and lentiviral particles were produced as described previously (Naldini et al, 1996). Briefly, pLenti vectors were cotransfected with pMD2.G (Addgene, #12259) lentiviral envelope and pCMV∆R8.2 (Addgene, #12263) lentiviral packaging into HEK293T cells cultured in DMEM+ medium, using polyethylenimine (PEI; Polysciences). The next day, the medium was changed for Opti‐MEM I (Gibco) supplemented with 1% pen‐strep (Gibco). Forty hours after transfection, the conditioned medium was harvested and centrifuged at 1,000 g for 5 min at RT to remove cell debris. The supernatant was transferred to Amicon Ultra‐15 Centrifugal Filter Units (Merck Millipore, 100 kDa cut‐off) and centrifuged at 4,000 g for 30 min at RT. The concentrated virus was diluted in DPBS (Gibco) and filtered through a 0.2‐μm filter (VWR). Lentiviral particles were stored at −80°C until use. Cells were transduced with ΔCre/Cre at DIV 1, shRNA constructs and eIF2αWT/eIF2αS51A at DIV 10 and the ATF4 translational reporters at DIV 3.
Cell treatments
To induce ER stress, cells were treated with 5 μg/ml tunicamycin (TM; Sigma‐Aldrich) or 1 μM thapsigargin (TG; Santa Cruz Biotechnology) for indicated times. HRI activation was triggered by 5 μM sodium arsenite (Sigma‐Aldrich) treatment for 20 h, PKR activation by 20 μM BEPP (Sigma‐Aldrich) treatment for 6 h and GCN2 activation by 10 nM Halofuginone (Cayman Chemical Company) treatment for 20 h, and ROS was generated by 100 μM menadione (Men; Sigma‐Aldrich) treatment for 4 h. Treatment with 0.5 μM PERK inhibitor GSK2606414 (Sigma‐Aldrich), 40 μM IRE1α inhibitor 4μ8C (Tocris), 75 μM angiogenin inhibitor NSC‐65828 [8‐amino‐5‐(4′‐hydroxybiphenyl‐4ylazo)naphthalene‐2‐sulfonate] (National Cancer Institute (NCI)) and 5 μM mTOR inhibitor Torin1 (Tocris) was started 1 h before induction of ER stress. Treatment with 200 nM ISR inhibitor ISRIB (Selleck Chemicals) was started 2 h before fixation, and treatment with 5 μM HRI inhibitor hemin chloride (Santa Cruz Biotechnology) was started 3 h before fixation. Compounds were dissolved in H2O (sodium arsenite), 100 mM NaOH (hemin chloride) or DMSO (rest). For all treatments, equal volumes of solvents were added to the controls. To determine ROS levels, cells were incubated with 5 μM CellROX green reagent (Thermo Fisher) 30 min before fixation.
Antibodies
Primary antibodies used in this study for Western blot (WB) analysis or immunofluorescence (IF) were as follows: PERK (C33E10) (1:1000; Cell Signaling Technology; Cat# 3192, RRID:AB_2095847), pospho‐eIF2α (Ser51) (1:500; Cell Signaling Technology; Cat# 9721, RRID:AB_330951), eIF2α (1: 1000; Abcam; Cat# ab5369, RRID:AB_304838), puromycin (WB: 1:2500, IF: 1:500; Millipore; Cat#MABE343, RRID: AB_2566826), ATF4 (WB: 1:500, IF: 1:250; Cell Signalling Technology; Cat#11815, RRID: AB_2616025), GFAP (1:500; Sigma‐Aldrich; Cat# G3893, RRID:AB_477010), MAP2 (1:500; Abcam; Cat# ab5392, RRID:AB_2138153), ANG I (1:500; Santa Cruz Biotechnology; Cat# sc‐74,528, RRID:AB_2227157), 1‐methyladenosine (m1A) (1:500; MBL International; Cat# D345‐3, RRID:AB_2728758), GAPDH (1:2000; Millipore; Cat# MAB374, RRID:AB_2107445), NRF2 (1:250; Novus; Cat# NBP1‐32822, RRID:AB_10003994), HRI (1:750; Merck Millipore; Cat# 07–728, RRID:AB_441964), phospho‐mTOR (59.Ser 2448) (1:250; Santa Cruz Biotechnology; Cat# sc‐293,133, RRID:AB_2861149), mTOR (1:250; Santa Cruz Biotechnology; Cat# sc‐517,464), phospho‐p70 S6 Kinase (Thr389) (1:1000; Cell Signaling Technology; Cat# 9205, RRID:AB_330944), p70 S6 Kinase (1:1000; Cell Signaling Technology; Cat# 2708, RRID:AB_390722), phospho‐4E‐BP1 (Thr37 / Thr46) (1:1000; Cell Signaling Technology; Cat# 2855, RRID:AB_560835), 4E‐BP1 (53H11) (1:1000; Cell Signaling Technology; Cat# 9644, RRID:AB_2097841) and G3BP (1:100; BD Biosciences; Cat# 611126, RRID:AB_398437). Alexa Fluor 405, 488, 546 or 647 secondary antibodies for IF were from Thermo Fisher Scientific (1:1000; Cat# A‐11001, RRID:AB_2534069; Cat# A‐11003, RRID:AB_2534071; Cat# A‐21235, RRID:AB_2535804; Cat# A‐11008, RRID:AB_143165; Cat# A‐11010, RRID:AB_2534077; Cat# A‐21244, RRID:AB_2535812; Cat# A‐11039, RRID:AB_2534096; Cat# A‐11040, RRID:AB_2534097; Cat# A‐21449, RRID:AB_2535866) or abcam (Cat# ab175675, RRID:AB_2810980). Horseradish peroxidase (HRP)‐conjugated secondary antibodies for WB were from Agilent DAKO (1:2000; Cat# P0448, RRID:AB_2617138; Cat# P0447, RRID:AB_2617137), and IRDye 680RD and 800CW secondary antibodies were from LI‐COR Biosciences (1:5000; Cat# 925–32210, RRID:AB_2687825; Cat# 926–68,073, RRID:AB_10954442).
Viability assay
The CellTiter‐Glo Luminescent Cell Viability Assay (Promega) was used to determine the viability of neurons. Cells were cultured in white 96‐well plates (Greiner 655083), and the assay was performed according to the manufacturer's protocol. Luminescence was recorded using the SpectraMax i3 Multi‐Mode Detection Platform controlled by Softmax Pro 6.4 software (both from Molecular Devices).
Labelling of de novo‐synthesised proteins
To evaluate protein synthesis rates, surface sensing of translation (SUnSET) was performed as described by Schmidt et al (2009). Briefly, cells were incubated with 2 μM puromycin (InvivoGen) 15 min prior to fixation or 30 min before harvesting lysates. Puromycinylated proteins were detected with the anti‐puromycin antibody by WB or IF analysis.
Western blot analysis
Neurons cultured in 6‐well plates and astrocytes cultured in 12‐well plates were washed with ice‐cold PBS, and cell lysates were prepared by incubation and scraping in lysis buffer consisting of 1% Triton X‐100 (Thermo Fisher Scientific) or 1% SDS (VWR) in PBS supplemented with cOmplete protease inhibitor cocktail and PhosSTOP phosphatase inhibitors (both Roche) pooling 6 wells for HRI WB and 2–3 wells per condition for all other WB. Nuclear lysates were passed through a 21G needle. Whole‐cell lysates were cleared by centrifugation for 10 min at 14,000 g at 4°C. The total protein concentration in the supernatant was determined with the Pierce BCA protein assay kit (BCA) protein assay (Thermo Scientific), and equal protein concentrations in loading buffer containing SDS were boiled for 5 min at 98°C. Samples were separated on 10% or 4–15% gradient Mini‐Protean TGX stain‐free precast polyacrylamide gels (Bio‐Rad), and the total amount of loaded protein on the gel was visualised using the Gel Doc EZ System (Bio‐Rad) and analysed with Image Lab 6.0 software (Bio‐Rad). Next, semi‐dry electroblotting was used to transfer the samples onto 0.2 μM nitrocellulose or PVDF (only for ANG) membranes using the Trans‐Blot Turbo transfer system and kit (all from Bio‐Rad). Membranes were blocked in blocking buffer, consisting of 5% BSA (Acros Organics) or 5% milk (Sigma‐Aldrich) in TBS containing 0.05% Tween‐20 (Sigma‐Aldrich) (TBS‐T) for 1 h at room temperature (RT). Subsequently, primary and HRP‐conjugated or IRDye secondary antibody incubations were performed overnight at 4°C and for 2 h at RT, respectively. All antibodies were diluted in blocking buffer. Between and after the antibody incubations, cells were washed three times for 10 min with TBS‐T. Membranes that were incubated with HRP‐conjugated secondary antibodies (Agilent DAKO) were developed with SuperSignal West Femto substrate (Thermo Scientific) or Lumi‐Light Western blotting substrate (Roche) for 5 min. Chemiluminescence and immunofluorescence of labelled proteins were visualised with the Odyssey Imaging System (LI‐COR) for appropriate times and analysed with Image Studio 5.2 software (LI‐COR).
Immunofluorescence
Cells cultured in 96‐well plates or 12‐well plates containing 18‐mm glass coverslips were fixed at RT in two steps of 15–20 min with 1.85% (added to culture medium) and 3.7% formaldehyde (Electron Microscopy Sciences) in PBS. After washing with PBS, mouse cells were permeabilised with 0.5% Triton X‐100 (Thermo Fisher Scientific) in PBS for 5 min and blocked with blocking solution consisting of 2% normal goat serum (NGS) and 0.1% Triton X‐100 (both from Thermo Fisher Scientific) in PBS for 30 min at RT. Next, incubations with appropriate primary and secondary antibodies were performed overnight at 4°C and for 2 h at RT, respectively. Between and after the antibody incubations, cells were washed three times for 5 min in PBS. If applicable, cells were incubated with 5 μg/ml DAPI (Carl Roth) for 10 min after the secondary antibody incubations. Human cells were blocked and permeabilised in blocking solution consisting of 5% NGS, 0.3% Triton X‐100 (both from Thermo Fisher Scientific) and 0.1% BSA (Acros Organics) in PBS for 3 h at RT. Primary antibody incubations were performed for 1 h at RT and overnight at 4°C, and secondary antibody incubations were performed for 3 h at RT. Between and after the antibody incubations, cells were washed three times for 15 min in PBS. If applicable, cells were incubated with 2.5 μg/ml DAPI (Carl Roth) for 15 min during the second wash after the secondary antibody incubations. Phalloidin stainings (Thermo Fisher Scientific/abcam) were performed during the secondary antibody incubations. Coverslips were subsequently mounted on microscope slides with Mowiol 4–88 (Sigma‐Aldrich). The plates and microscope slides were stored at 4°C until further analysis. All antibodies were diluted in blocking solution.
High‐content microscopy and automated analysis
Single focal plane images from cells cultured in black 96‐well culture plates (Greiner 655,090) were acquired using a CellInsight CX7 High‐Content Screening (HCS) Platform (Thermo Fisher Scientific) controlled by HCS Studio Cell Analysis software (Thermo Fisher Scientific). Images were acquired with a 10x or 20x objective in widefield mode. Per well, the maximum of 25 fields (10x objective) or 64 fields (20x objective) were acquired to cover the majority of the well. The nuclear fluorescence of the ΔCre/Cre constructs or DAPI was used for automatic focusing. Images were analysed with Columbus 2.5 software (PerkinElmer) and in‐house‐developed scripts. Nuclear regions of interest (ROI) were based on the intensity of the ΔCre/Cre construct or DAPI, dendrites were selected using a MAP2‐based ROI detected by the CSIRO neurite detection method, astrocytic soma were selected using a GFAP‐ or phalloidin‐based ROI, and neuronal soma were selected using a MAP2‐based ROI (excluding dendrites). Transduction efficiency of Cre‐recombinase was based on the nuclear EGFP or mCherry intensity in a DAPI‐based ROI. Cells were considered transduced when the nuclear intensity was higher than the mean + 2x the standard deviation (SD) of the background intensity. Cell number was determined by quantification of the nuclei. Morphological measures of dendrites (neurite length, no. of segments, no. of extremities, no. of segments and no. of branch points) were obtained by the CSIRO neurite analysis module (Rosato et al, 2019). ATF4 intensity was quantified by subtracting the mean fluorescence intensity in a 3‐pixel ring region around the nucleus from the mean intensity in the nucleus. Nuclei touching the border of the image field were excluded for analysis. Mean puromycin intensity was quantified in dendrites of neurons or in the soma of astrocytes, mean m1A intensity in the soma (including nucleus) and mean NRF2 intensity in both the soma and nucleus. Mean CellROX intensity was quantified in nuclei, and a background correction was performed by subtracting the mean nuclear intensity of equally treated cells that were not loaded with CellROX. All technical replicates (different wells) were used for statistical analysis. Zooms of the obtained images are shown. Scale bars are shown in the figures. It is important to note that neuronal nuclei are smaller than astrocytic nuclei in vitro and that the morphology of human cells differs from mouse cells.
Confocal microscopy and image analysis
Microscope slides were imaged using a Nikon Eclipse Ti confocal microscope equipped with a 60x oil immersion objective (NA = 1.4), controlled by NIS‐Elements 4.30 software (Nikon). Z‐stacks of 5 images with a step size of 1 μm were acquired. Acquisition settings were kept equal for all images within each experiment. Maximum intensity projections are shown and were analysed. Mean intensity of endogenous ATF4 immunofluorescence and ATF4 translational EYFP reporters was quantified in a ΔCre/Cre construct‐based nuclear ROI, and mean puromycin intensity was quantified in a MAP2‐based dendritic ROI using ImageJ software (National Institutes of Health). Stress granule formation in the soma and dendrites—using a MAP2‐based ROI—was quantified by analysing the standard deviation (SD) of the intensity of G3BP immunofluorescence signal using the CellProfiler 3.1.9 software (Broad Institute).
RNA isolation
Neurons cultured in 6‐well plates and astrocytes cultured in 12‐well plates were washed with ice‐cold PBS, and cell lysates were prepared by incubation and scraping in TRI Reagent solution (Invitrogen), pooling 2 wells per condition. Cell lysates were loaded on Phase Lock Gel‐Heavy tubes (Quantabio) and mixed 1:5 with chloroform (VWR Chemicals). The organic and aqueous phases were separated by centrifugation at 12,000 g for 15 min at 4°C. The aqueous phase was mixed 1:1 with Isopropanol (VWR Chemicals) and 150 mg/ml glycoblue (Invitrogen) and centrifuged at 12,000 g for 10 min at 4°C to precipitate RNA. Precipitated RNA was washed with 75% ethanol (Sigma‐Aldrich) and centrifuged at 7,500 g for 5 min. After removal of ethanol, the pellet was air‐dried and dissolved in RNase‐free H2O.
RNA spectrophotometry and electrophoresis
RNA concentration and purity were determined using the NanoDrop 2000 spectrophotometer (Thermo Scientific) or the TapeStation 4200 system with RNA ScreenTape, ladder and sample buffer (all Agilent) according to the manufacturer's protocol. To analyse small RNA, the Bioanalyzer 2100 system with the small RNA kit (both from Agilent) was used according to the manufacturer's protocol. Electropherograms were analysed with 2100 Expert Software (Agilent). Data were normalised to the tRNA peak. Per condition, 6 (neurons) or 3 (astrocytes) independent samples were analysed.
cDNA synthesis and Real‐Time quantitative PCR (RT–qPCR)
Synthesis of cDNA was performed using the SensiFAST cDNA Synthesis Kit (Bioline) according to the manufacturer's protocol. The RNA input (0.5–1 μg) was equal for each sample within an experiment; Eef1a1 qPCR was used to correct for mRNA input. RT–qPCR was performed using the LightCycler 480 system (Roche). Per sample, 1 μl cDNA was loaded in triplicate into a 384‐well plate and qPCR mixtures were prepared using the SensiFAST Probe No‐ROX Kit (Bioline) and Universal ProbeLibrary probes (Roche) or the SensiFAST SYBR No‐ROX Kit (Bioline), according to the manufacturer's protocols. Used primers and Universal probes are provided in Table 1. For the SYBR Green reactions, a melting curve was obtained afterwards by continuous acquisition and a temperature increase to 95°C. Results were analysed using the LightCycler 480 software (version 1.5.0.39).
Table 1.
Primers and probes used for qPCR.
| Target gene | Primer sequence 5′‐3′ | Probe/ SYBR Green |
|---|---|---|
| Ddit3 | fw: CCACCACACCTGAAAGCAG | #33 |
| rev: TCCTCATACCAGGCTTCCA | ||
| Eef1a1 | fw: ACACGTAGATTCCGGCAAGT | #31 |
| rev: AGGAGCCCTTTCCCATCTC | ||
| Eif2ak1 | fw: ATCCCAGTACGATGCCAAGT | SYBR Green |
| rev: TCCCGAATGGCTGAAAGA | ||
| Eif2ak2 | fw: CAGAGCCTGCACCTGAAGTA | #15 |
| rev: GCCATTTTTCCTCCCAGTG | ||
| Eif2ak4 | fw: CTACCCAAGGAAACAATCTTACA | SYBR Green |
| rev: CACAGTTGTGTTAAATGCCTGTT | ||
| Eif4ebp1 | fw: GATGAGCCTCCCATGCAA | #21 |
| rev: CCATCTCAAATTGTGACTCTTCA | ||
| Hspa5 | fw: GCCAACTGTAACAATCAAGGTCT | #15 |
| rev: TGACTTCAATCTGGGGAACTC | ||
| Xbp1s | fw: TCCGCAGCAGGTGCAG | SYBR Green |
| rev: CCAACTTGTCCAGAATGCCC | ||
| Xbp1u | fw: GCAGCACTCAGACTATGTG | SYBR Green |
| rev: CCAACTTGTCCAGAATGCCC |
mRNA sequencing
Transcriptome analysis was performed by Novogene (UK) Company Limited, Cambridge, United Kingdom. Quantity and quality of isolated RNA were assessed by agarose gel electrophoresis, spectrophotometry using the NanoPhotometer (IMPLEN) and the RNA Nano 6000 assay kit of the Bioanalyzer 2100 system (Agilent). Only qualified samples were used for mRNA sequencing. First, sequencing libraries were generated, using 1 μg RNA per sample and the NEBNext Ultra TM RNA Library Prep Kit for Illumina (New England BioLabs), according to the manufacturer's protocol, and index codes were added to attribute sequences. PCR products were purified using the AMPure XP system, and library quality was assessed using the Bioanalyzer 2100 system (Agilent). Clustering of the index‐coded samples was performed using a cBot Cluster Generation System and PE Cluster Kit cBot‐HS (Illumina), according to the manufacturer's protocol. Finally, the library preparations were sequenced on a NovaSeq 6000 platform (Illumina) and paired‐end reads were generated. Raw data were processed through fastp, and clean data were mapped to the reference mouse genome (ensembl_mus_musculus_grcm38_p6_gca_000001635_8) using HISAT2 software. FeatureCounts software was used to count the read numbers mapped of each gene (readcounts), and fragments per kilobase of transcript sequence per million base pairs sequenced (FPKM) of each gene were calculated. Differential expression analysis between two conditions (three biological replicates per condition) was performed using the obtained readcounts and DESeq2 R package, calculating the log2FoldChange, P‐values and adjusted P‐values (Padj) using Benjamini and Hochberg's approach for controlling the false discovery rate. Gene Ontology (GO) enrichment analysis of differentially expressed genes (DEGs) was performed using the ClusterProfiler R package.
Statistics
Unless Unless stated otherwise in the figure or figure legends, the data in this study were visualised in one of the following ways: (1) normalised raw data—These graphs show the data for all conditions used in the experiment (Y‐axis: p‐eIF2α/eIF2α, protein synthesis or nuclear ATF4 intensity); (2) the response to ER stress—In these graphs, the effect of ER stress on p‐eIF2α, protein synthesis or ATF4 upregulation is quantified by dividing the raw data of the condition + ER stress by the condition – ER stress of the same genotype/treatment (Y‐axis: p‐eIF2α/eIF2α response, protein synthesis response or nuclear ATF4 intensity response (TM+/TM−)); (3) change in the response to ER stress by an intervention—In these graphs, the % change in the ISR response was calculated to more precisely determine the effect of interventions in WT versus PERK‐deficient cells (Y‐axis: % change in response (intervention+/intervention−)). The used formulas are as follows:
The specific formulas for different experiments are defined in the figure legends. Per experiment (N, biological replicate), the data were normalised to untreated WT cells or cells of the same genotype/treatment (defined in figure legends). All technical replicates (n, different wells or cells, defined in Source data) were used for statistical analysis.
GraphPad Prism 8.0.1 software was used to perform statistical analysis. Shapiro–Wilk test was used to assess distribution normality. Nested t‐test (for experiments with technical replicates) and two‐tailed Student's t‐test (for experiments without technical replicates) were used for the statistical analyses of two groups (unpaired). Two‐way and three‐way ANOVAs followed by Tukey's multiple comparison post‐hoc test were used for analysis of more than two groups. For graphs with only one condition, the one‐sample t‐test was used with a hypothetical value of 100 (qPCR) or 1 (ATF4 WB). Source data (individual datapoints, mean, SEM, SD, N and n, P‐values, statistical tests and used methods) are available. Sample sizes were based on pilot experiments. No blinding was done. Experiments in which the positive or negative control failed were excluded. Relevant P‐values are also indicated in the figures: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 and ns: not significant. P < 0.05 was considered statistically significant.
Data availability
mRNA sequencing data are available in Gene Expression Omnibus (GEO) GSE200742 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE200742).
Author contributions
Kimberly Wolzak: Conceptualization; data curation; formal analysis; validation; investigation; visualization; methodology; writing—original draft; writing—review and editing. Anna Nölle: Data curation; formal analysis; validation; investigation; visualization; methodology; writing—review and editing. Margherita Farina: Data curation; validation; investigation; methodology; writing—review and editing. Truus EM Abbink: Resources; writing—review and editing. Marjo S van der Knaap: Resources; writing—review and editing. Matthijs Verhage: Conceptualization; supervision; writing—review and editing. Wiep Scheper: Conceptualization; formal analysis; supervision; funding acquisition; writing—original draft; project administration; writing—review and editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
KW and WS conceptualized the study, wrote the manuscript, and analysed and interpreted the data. KW performed the experiments. AN and MF conceptualised the study, and performed and analysed pilot experiments. AN contributed to (p‐)eIF2α and ATF4 Western blots. TA and MvdK contributed the 2b5 mice. MV provided extensive feedback during the project and writing of the manuscript. All authors have approved the final version.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix S1
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
The authors thank Robbert Zalm for cloning and producing viral particles; Kevin Batenburg and Nicole Breeuwsma for primary human astrocyte and neuron cultures; Fabian Bangel and Joost Hoetjes for isolating RNA, qPCR experiments and overall technical support; and Joke Wortel for animal breeding. The authors also thank Chantal Scheepbrouwer (Amsterdam UMC, Department of Neurosurgery) and Susan Kenter (Amsterdam UMC, Department of Human Genetics) for help with analysis of small RNAs. For discussions and critical reading of the manuscript, the authors thank the Molecular Neurodegeneration Group (VU University Amsterdam, Department of Functional Genomics). This study was supported by: Alzheimer Nederland (grant WE.03‐2017‐10), Janssen Pharmaceutica (Stellar Neurodegeneration Project grant) and European Commission (Joint Programming Initiative Neurodegenerative Diseases/JPco‐fuND, ZonMW #733051062), all to WS.
References
- Abdel‐Nour M, Carneiro LAM, Downey J, Tsalikis J, Outlioua A, Prescott D, Da Costa LS, Hovingh ES, Farahvash A, Gaudet RG et al (2019) The heme‐regulated inhibitor is a cytosolic sensor of protein misfolding that controls innate immune signaling. Science 364: eaaw4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez‐Castelao B, Dieck ST, Fusco CM, Donlin‐Asp PG, Perez JD, Schuman EM (2020) The switch‐like expression of heme‐regulated kinase 1 mediates neuronal proteostasis following proteasome inhibition. Elife 9: e52714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameri K, Lewis CE, Raida M, Sowter H, Hai T, Harris AL (2004) Anoxic induction of ATF‐4 through HIF‐1‐independent pathways of protein stabilization in human cancer cells. Blood 103: 1876–1882 [DOI] [PubMed] [Google Scholar]
- Balsa E, Soustek MS, Thomas A, Cogliati S, García‐Poyatos C, Martín‐García E, Jedrychowski M, Gygi SP, Enriquez JAPP (2019) ER and Nutrient Stress Promote Assembly of Respiratory Chain Supercomplexes Through PERK/eIF2α Axis. Mol Cell 74: 877–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell KFS, Al‐Mubarak B, Martel MA, McKay S, Wheelan N, Hasel P, Márkus NM, Baxter P, Deighton RF, Serio A et al (2015) Neuronal development is promoted by weakened intrinsic antioxidant defences due to epigenetic repression of Nrf2. Nat Commun 6: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐Sahra I, Manning BD (2017) mTORC1 signaling and the metabolic control of cell growth. Curr Opin Cell Biol 45: 72–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Lai UH, Zhu L, Singh A, Ahmed M, Forsyth NR (2018) Reactive oxygen species formation in the brain at different oxygen levels: The role of hypoxia inducible factors. Front Cell Dev Biol 6: 132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Qin C, Huang J, Tang X, Liu C, Huang K, Xu J, Guo G, Tong A, Zhou L (2020) The role of astrocytes in oxidative stress of central nervous system: A mixed blessing. Cell Prolif 53: e12781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa‐Mattioli M, Sossin WS, Klann E, Sonenberg N (2009) Translational Control of Long‐Lasting Synaptic Plasticity and Memory. Neuron 61: 10–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crivello M, O'Riordan SL, Woods I, Cannon S, Halang L, Coughlan KS, Hogg MC, Lewandowski SA, Prehn JHM (2018) Pleiotropic activity of systemically delivered angiogenin in the SOD1G93A mouse model. Neuropharmacology 133: 503–511 [DOI] [PubMed] [Google Scholar]
- Czech A, Wende S, Mörl M, Pan T, Ignatova Z (2013) Reversible and Rapid Transfer‐RNA Deactivation as a Mechanism of Translational Repression in Stress. PLoS Genet 9: e1003767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danhier P, Krishnamachary B, Bharti S, Kakkad S, Mironchik Y, Bhujwalla ZM (2015) Combining Optical Reporter Proteins with Different Half‐lives to Detect Temporal Evolution of Hypoxia and Reoxygenation in Tumors. Neoplasia 17: 871–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das I, Krzyzosiak A, Schneider K, Wrabetz L, D'Antonio M, Barry N, Sigurdardottir A, Bertolotti A (2015) Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 348: 239–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delépine M, Nicolino M, Barrett T, Golamaully M, Mark Lathrop G, Julier C (2000) EIF2AK3, encoding translation initiation factor 2‐α kinase 3, is mutated in patients with Wolcott‐Rallison syndrome. Nat Genet 25: 406–409 [DOI] [PubMed] [Google Scholar]
- Donnelly N, Gorman AM, Gupta S, Samali A (2013) The eIF2α kinases: their structures and functions. Cell Mol Life Sci 70: 3493–3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooves S, Bugiani M, Postma NL, Polder E, Land N, Horan ST, Van Deijk ALF, Van De Kreeke A, Jacobs G, Vuong C et al (2016) Astrocytes are central in the pathomechanisms of vanishing white matter. J Clin Invest 126: 1512–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkordy A, Mishima E, Niizuma K, Akiyama Y, Fujimura M, Tominaga T, Abe T (2018) Stress‐induced tRNA cleavage and tiRNA generation in rat neuronal PC12 cells. J Neurochem 146: 560–569 [DOI] [PubMed] [Google Scholar]
- Frank CL, Ge X, Xie Z, Zhou Y, Tsai LH (2010) Control of activating transcription factor 4 (ATF4) persistence by multisite phosphorylation impacts cell cycle progression and neurogenesis. J Biol Chem 285: 33324–33337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frega M, Van Gestel SHC, Linda K, Van Der Raadt J, Keller J, Van Rhijn JR, Schubert D, Albers CA, Kasri NN (2017) Rapid neuronal differentiation of induced pluripotent stem cells for measuring network activity on micro‐electrode arrays. J Vis Exp 2017: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Aviles G, Liu Y, Tian R, Unger BA, Lin YHT, Wiita AP, Xu K, Correia MA, Kampmann M (2020) Mitochondrial stress is relayed to the cytosol by an OMA1–DELE1–HRI pathway. Nature 579: 427–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Giricz Z, Natoni A, Donnelly N, Deegan S, Szegezdi E, Samali A (2012) NOXA contributes to the sensitivity of PERK‐deficient cells to ER stress. FEBS Lett 586: 4023–4030 [DOI] [PubMed] [Google Scholar]
- Guthrie LN, Abiraman K, Plyler ES, Sprenkle NT, Gibson SA, McFarland BC, Rajbhandari R, Rowse AL, Benveniste EN, Meares GP (2016) Attenuation of PKR‐like ER Kinase (PERK) signaling selectively controls endoplasmic reticulum stress‐induced inflammation without compromising immunological responses. J Biol Chem 291: 15830–15840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday M, Hughes D, Mallucci GR (2017) Fine‐tuning PERK signaling for neuroprotection. J Neurochem 142: 812–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday M, Radford H, Sekine Y, Moreno J, Verity N, Le Quesne J, Ortori CA, Barrett DA, Fromont C, Fischer PM et al (2015) Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis 6: e1672‐9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M et al (2013) ER‐stress‐induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15: 481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han AP, Yu C, Lu L, Fujiwara Y, Browne C, Chin G, Fleming M, Leboulch P, Orkin SH, Chen JJ (2001) Heme‐regulated eIF2α kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J 20: 6909–6918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D (2000) Regulated Translation Initiation Controls Stress‐Induced Gene Expression in Mammalian Cells. Mol Cell 6: 1099–1108 [DOI] [PubMed] [Google Scholar]
- Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD, Ron D (2001) Diabetes mellitus and exocrine pancreatic dysfunction in Perk‐/‐ mice reveals a role for translational control in secretory cell survival. Mol Cell 7: 1153–1163 [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D (2000) Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell 5: 897–904 [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic‐ reticulum‐resident kinase. Nature 397: 271–274 [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R et al (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11: 619–633 [DOI] [PubMed] [Google Scholar]
- Hou X, Liu Y, Liu H, Chen X, Liu M, Che H, Guo F, Wang C, Zhang D, Wu J et al (2015) PERK silence inhibits glioma cell growth under low glucose stress by blockage of p‐AKT and subsequent HK2's mitochondria translocation. Sci Rep 5: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Mizugaki M, Ishida N (1988) Preparation of a monoclonal antibody specific for 1‐methyladenosine and its application for the detection of elevated levels of 1‐Methyladenosine in urines from cancer patients. Jpn J Cancer Res 79: 1130–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov P, Emara MM, Villen J, Gygi SP, Anderson P (2011) Angiogenin‐induced tRNA fragments inhibit translation initiation. Mol Cell 43: 613–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang HY, Wek RC (2005) Phosphorylation of the α‐subunit of the eukaryotic initiation factor‐2 (eIF2α) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J Biol Chem 280: 14189–14202 [DOI] [PubMed] [Google Scholar]
- Julier C, Nicolino M (2010) Wolcott‐Rallison syndrome. Orphanet J Rare Dis 5: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser PS, Deng L, Wang Y, Dulubova I, Liu X, Rizo J, Südhof TC (2011) RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ‐domain interaction. Cell 144: 282–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao RYT, Jenkins JL, Olson KA, Key ME, Fett JW, Shapiro R (2002) A small‐molecule inhibitor of the ribonucleolytic activity of human angiogenin that possesses antitumor activity. Proc Natl Acad Sci U S A 99: 10066–10071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur M, Monaghan CE, Ackerman SL (2017) Regulation of mRNA translation in neurons—a matter of life and death. Neuron 96: 616–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieran D, Sebastia J, Greenway MJ, King MA, Connaughton D, Concannon CG, Fenner B, Hardiman O, Prehn JHM (2008) Control of motoneuron survival by angiogenin. J Neurosci 28: 14056–14061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai K, Luo C, Zhang X, Ye P, Zhang Y, He J, Yao K (2016) Regulation of angiogenin expression and epithelial‐mesenchymal transition by HIF‐1α signaling in hypoxic retinal pigment epithelial cells. Biochim Biophys Acta 1862: 1594–1607 [DOI] [PubMed] [Google Scholar]
- Lassmann H, van Horssen J (2016) Oxidative stress and its impact on neurons and glia in multiple sclerosis lesions. Biochim Biophys Acta 1862: 506–510 [DOI] [PubMed] [Google Scholar]
- Lebeau J, Saunders JM, Moraes VWR, Madhavan A, Madrazo N, Anthony MCWR (2018) The PERK Arm of the Unfolded Protein Response Regulates Mitochondrial Morphology during Acute Endoplasmic Reticulum Stress. Cell Rep 22: 2827–2836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewerenz J, Maher P (2009) Basal levels of eIF2α phosphorylation determine cellular antioxidant status by regulating ATF4 and xCT expression. J Biol Chem 284: 1106–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Huangfu W, Kumar KGS, Qian J, James P, Hamanaka RB, Grigoriadou C, Aldabe R, Alan J, Fuchs SY (2010) Virus‐induced unfolded protein response attenuates anti‐viral defenses via phosphorylation‐dependent degradation of the Type I interferon receptor. Cell Host Microbe 5: 72–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Han A‐P, Chen J‐J (2001) Translation initiation control by heme‐regulated eukaryotic initiation factor 2α kinase in erythroid cells under cytoplasmic stresses. Mol Cell Biol 21: 7971–7980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu PD, Harding HP, Ron D (2004) Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol 167: 27–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons SM, Achorn C, Kedersha NL, Anderson PJ, Ivanov P (2016) YB‐1 regulates tiRNA‐induced Stress Granule formation but not translational repression. Nucleic Acids Res 44: 6949–6960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons SM, Gudanis D, Coyne SM, Gdaniec Z, Ivanov P (2017) Identification of functional tetramolecular RNA G‐quadruplexes derived from transfer RNAs. Nat Commun 8: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons SM, Kharel P, Akiyama Y, Ojha S, Dave D, Tsvetkov V, Merrick W, Ivanov P, Anderson P (2020) eIF4G has intrinsic G‐quadruplex binding activity that is required for tiRNA function. Nucleic Acids Res 48: 6223–6233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Trinh MA, Wexler AJ, Bourbon C, Gatti E, Pierre P, Cavener DR, Klann E (2013) Suppression of eIF2α kinases alleviates Alzheimer's disease‐related plasticity and memory deficits. Nat Neurosci 16: 1299–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D (2004) CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 18: 3066–3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuoka HC, Townes TM (2002) Targeted disruption of the activating transcription factor 4 gene results in severe fetal anemia in mice. Blood 99: 736–745 [DOI] [PubMed] [Google Scholar]
- McEwen E, Kedersha N, Song B, Scheuner D, Gilks N, Han A, Chen JJ, Anderson P, Kaufman RJ (2005) Heme‐regulated inhibitor kinase‐mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J Biol Chem 280: 16925–16933 [DOI] [PubMed] [Google Scholar]
- Mercado G, Castillo V, Soto P, López N, Axten JM, Sardi SP, Hoozemans JJM, Hetz C (2018) Targeting PERK signaling with the small molecule GSK2606414 prevents neurodegeneration in a model of Parkinson's disease. Neurobiol Dis 112: 136–148 [DOI] [PubMed] [Google Scholar]
- Mesitov MV, Soldatov RA, Zaichenko DM, Malakho SG, Klementyeva TS, Sokolovskaya AA, Kubatiev AA, Mironov AA, Moskovtsev AA (2017) Differential processing of small RNAs during endoplasmic reticulum stress. Sci Rep 7: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishima E, Inoue C, Saigusa D, Inoue R, Ito K, Suzuki Y, Jinno D, Tsukui Y, Akamatsu Y, Araki M et al (2014) Conformational change in transfer RNA is an early indicator of acute cellular damage. J Am Soc Nephrol 25: 2316–2326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA et al (2013) Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion‐infected mice. Sci Transl Med 5: 206ra138 [DOI] [PubMed] [Google Scholar]
- Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J, Dinsdale D, Ortori CA et al (2012) Sustained translational repression by eIF2α‐P mediates prion neurodegeneration. Nature 485: 507–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D (1996) In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272: 263–267 [DOI] [PubMed] [Google Scholar]
- Pakos‐Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM (2016) The integrated stress response. EMBO Rep 17: 1374–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prehn JHM, Jirström E (2020) Angiogenin and tRNA fragments in Parkinson's disease and neurodegeneration. Acta Pharmacol Sin 41: 442–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radford H, Moreno JA, Verity N, Halliday M, Mallucci GR (2015) PERK inhibition prevents tau‐mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol 130: 633–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs CL, Kedersha N, Ivanov P, Anderson P (2020) Mammalian stress granules and P bodies at a glance. J Cell Sci 133: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosato M, Stringer S, Gebuis T, Paliukhovich I, Li KW, Posthuma D, Sullivan PF, Smit AB, van Kesteren RE (2019) Combined cellomics and proteomics analysis reveals shared neuronal morphology and molecular pathway phenotypes for multiple schizophrenia risk genes. Mol Psychiatry 26: 784–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito A, Ochiai K, Kondo S, Tsumagari K, Murakami T, Cavener DR, Imaizumi K (2011) Endoplasmic reticulum stress response mediated by the PERK‐eIF2α‐ATF4 pathway is involved in osteoblast differentiation induced by BMP2. J Biol Chem 286: 4809–4818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheper W, Hoozemans JJM (2015) The unfolded protein response in neurodegenerative diseases: a neuropathological perspective. Acta Neuropathol 130: 315–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner‐Weir S, Kaufman RJ (2001) Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 7: 1165–1176 [DOI] [PubMed] [Google Scholar]
- Schmidt EK, Clavarino G, Ceppi M, Pierre P (2009) SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods 6: 275–277 [DOI] [PubMed] [Google Scholar]
- Sevier CS, Kaiser CA (2008) Ero1 and redox homeostasis in the endoplasmic reticulum. Biochim Biophys Acta 1783: 549–556 [DOI] [PubMed] [Google Scholar]
- Shimizu T, Taguchi A, Higashijima Y, Takubo N, Kanki Y, Urade Y, Wada Y (2020) PERK‐mediated suppression of microRNAs by sildenafil improves mitochondrial dysfunction in heart failure. iScience 23: 101410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C, Acosta‐Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, Gamache K, Gallagher CM, Ang KKH, Wilson C et al (2013) Pharmacological brake‐release of mRNA translation enhances cognitive memory. Elife 2013: 1–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HL, Freeman OJ, Butcher AJ, Holmqvist S, Humoud I, Schätzl T, Hughes DT, Verity NC, Swinden DP, Hayes J et al (2020) Astrocyte unfolded protein response induces a specific reactivity state that causes non‐cell‐autonomous neuronal degeneration. Neuron 105: 855–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steidinger TU, Slone SR, Ding H, Standaert DG, Yacoubian TA (2013) Angiogenin in Parkinson disease models: role of Akt phosphorylation and evaluation of AAV‐mediated angiogenin expression in MPTP treated mice. PLoS One 8: 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone S, Yue Y, Stanojlovic M, Wu S, Karsenty G, Lin W (2019) Neuron‐specific PERK inactivation exacerbates neurodegeneration during experimental autoimmune encephalomyelitis. JCI Insight 4: e124232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Tsujimura T, Takeda K, Sugihara A, Maekawa A, Terada N, Yoshida N, Akira S (1998) Targeted disruption of ATF4 discloses its essential role in the formation of eye lens fibres. Genes Cells 3: 801–810 [DOI] [PubMed] [Google Scholar]
- Teske BF, Wek SA, Bunpo P, Cundiff JK, McClintick JN, Anthony TG, Wek RC (2011) The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol Biol Cell 22: 4390–4405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson DM, Lu C, Green PJ, Parker R (2008) tRNA cleavage is a conserved response to oxidative stress in eukaryotes. RNA 14: 2095–2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonelli C, Chio IIC, Tuveson DA (2018) Transcriptional regulation by Nrf2. Antioxid Redox Signal 29: 1727–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh MA, Kaphzan H, Wek RCC, Pierre P, Cavener DRR, Klann E (2012) Brain‐specific disruption of the eIF2α kinase PERK decreases ATF4 expression and impairs behavioral flexibility. Cell Rep 1: 676–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh MA, Klann E (2013) Translational control by eIF2α kinases in long‐lasting synaptic plasticity and long‐term memory. Neurobiol Learn Mem 105: 93–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukumo Y, Tsukahara S, Furuno A, Iemura SI, Natsume T, Tomida A (2014) TBL2 is a novel PERK‐binding protein that modulates Stress‐Signaling and cell survival during endoplasmic reticulum stress. PLoS One 9: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu BP, Weissman JS (2004) Oxidative protein folding in eukaryotes: mechanisms and consequences. J Cell Biol 164: 341–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A et al (2012) PERK is required at the ER‐mitochondrial contact sites to convey apoptosis after ROS‐based ER stress. Cell Death Differ 19: 1880–1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorhees RM, Ramakrishnan V (2013) Structural basis of the translational elongation cycle. Annu Rev Biochem 82: 203–236 [DOI] [PubMed] [Google Scholar]
- Whitney M, Jefferson L, Kimball S (2009) ATF4 is necessary and sufficient for ER stress‐induced upregulation of REDD1 expression. Biochem Biophys Res Commun 379: 451–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiersma VI, van Ziel AM, Vazquez‐Sanchez S, Nölle A, Berenjeno‐Correa E, Bonaterra‐Pastra A, Clavaguera F, Tolnay M, Musters RJP, van Weering JRT et al (2019) Granulovacuolar degeneration bodies are neuron‐selective lysosomal structures induced by intracellular tau pathology. Acta Neuropathol 138: 943–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisse LE, Ter Braak TJ, Van De Beek MC, Van Berkel CGM, Wortel J, Heine VM, Proud CG, Van Der Knaap MS, Abbink TEM (2018) Adult mouse eIF2Bɛ Arg191His astrocytes display a normal integrated stress response in vitro . Sci Rep 8: 2–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki S, Ivanov P, Hu GF, Anderson P (2009) Angiogenin cleaves tRNA and promotes stress‐induced translational repression. J Cell Biol 185: 35–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XL, Otero FJ, Ewalt KL, Liu J, Swairjo MA, Köhrer C, Rajbhandary UL, Skene RJ, McRee DE, Schimmel P (2006) Two conformations of a crystalline human tRNA synthetase‐tRNA complex: implications for protein synthesis. EMBO J 25: 2919–2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YL, Reis LFL, Paylovic J, Aguzzi S, Schäfer R, Kumar A, Williams BRG, Aguet M, Weissmann C (1995) Deficient signaling in mice devoid of double‐stranded RNA‐dependent protein kinase. EMBO J 14: 6095–6106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, McGrath BC, Reinert J, Olsen DS, Lei L, Gill S, Wek SA, Vattem KM, Wek RC, Kimball SR et al (2002) The GCN2 eIF2α kinase is required for adaptation to amino acid deprivation in mice. Mol Cell Biol 22: 6681–6688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Tanzawa T, Lin J, Gagnon MG (2020) Structural basis for ribosome recycling by RRF and tRNA. Nat Struct Mol Biol 27: 25–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ziel AM, Largo‐Barrientos P, Wolzak K, Verhage M, Scheper W (2019) Unconventional secretion factor GRASP55 is increased by pharmacological unfolded protein response inducers in neurons. Sci Rep 9: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ziel AM, Wolzak K, Nölle A, Hoetjes PJ, Berenjeno‐Correa E, van Anken E, Struys EA, Scheper W (2020) No evidence for cell‐to‐cell transmission of the unfolded protein response in cell culture. J Neurochem 152: 208–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zyryanova AF, Kashiwagi K, Rato C, Harding HP, Crespillo‐Casado A, Perera LA, Sakamoto A, Nishimoto M, Yonemochi M, Shirouzu M et al (2021) ISRIB blunts the integrated stress response by allosterically antagonising the inhibitory effect of phosphorylated eIF2 on eIF2B. Mol Cell 81: 88–103.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]