

Abstract
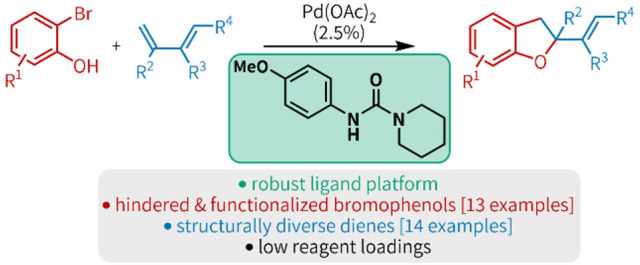
We disclose a palladium and urea ligand-mediated heteroannulation of 2-bromophenols and 1,3-dienes. This method addresses synthetic challenges present in the palladium-catalyzed heteroannulation of bifunctional reagents and olefins by engaging a diverse scope of coupling partners under a unified set of reaction conditions. Our recently developed urea ligand platform outperforms phosphine ligands to generate the dihydrobenzofuran motif in a convergent manner.
Dihydrobenzofuran (DHB) and other furan derivatives are prevalent core scaffolds in natural products1−3 and as therapeutics,4,5 organic materials,6,7 and agrochemicals.8 There are a number of modern approaches for the preparation of DHBs9 (Figure 1a), including phenol alkylation,10 intramolecular carbene insertion,11 benzofuran reduction,10 and ring contraction.12 Although these methods are well established, most are intramolecular and require reagents that are prepared with significant synthetic overhead, factors that make it arduous to build representative small molecule DHB libraries.
Figure 1.

(A) Traditional approaches to access DHB cores. (B) A convergent approach to DHBs: Pd-catalyzed heteroannulation reactions of olefins and bifunctional reagents.
An attractive alternative approach to the synthesis of DHBs is through a multicomponent annulation where both C–C and C–O bonds are formed in one step (Figure 1b). This convergent strategy allows for greater variation in reagents, making it more amenable to the preparation of diverse libraries of DHB-containing compounds. One method demonstrating these qualities is palladium-mediated coupling of functionalized phenols with olefins.13,14 Pioneering studies have made important advances to establish the viability of this transformation, but the synthetic utility remains limited. Existing methods typically have a narrow substrate scope with respect to one or both coupling partners–activated alkenes or linear 1,3-dienes are usually required13d−13f – and are intolerant of steric encumbrance in either. Reaction conditions can be substrate dependent, and large excess of the olefin (3–5 equiv.) is common in the absence of a directing group or tethering of the nucleophile to the olefin.13d,13g,13h Herein, we report a urea ligand-enabled heteroannulation reaction under palladium catalysis that provides access to structurally and functionally diverse DHB products.
Very recently, our group has advanced urea-derived ligands as an alternative ligand platform for palladium catalysis.15 We found that urea ligands, which are small and kinetically labile, are effective for the palladium-catalyzed heteroannulation of N-tosyl-bromoanilines and 1,3-dienes. In addition to the potential to uncover complementary reactivity and selectivity to traditional ligands for palladium, these ligands possess practical features that make them attractive: they are readily prepared from widely available and inexpensive amine precursors, and are bench stable and robust to a variety of conditions. Given these features and our success in heteroannulation reactions forming indolines, we sought to extend our urea-enabled methodology to the use of bromophenols as bifunctional reagents. By using trisubstituted urea ligands, we can now access the analogous DHB core. Our urea-enabled method engages a diverse scope of bromophenols and dienes under a unified set of reaction conditions. This method is a convergent approach for generating a representative library of functionalized DHBs.
In our initial studies, we examined the effect of various ligands on the desired heteroannulation of 2-bromophenol 1a and diene 2a (Figure 2a). Without any ligand, the reaction afforded the desired product 3aa in 49% yield; reactivity in the absence of exogenous ligand was poorer for more challenging branched dienes.16 Although we did not observe inhibition by phosphines,17 there was generally no discernible ligand effect across a range of phosphines. The lack of any effect is unexpected–we confirmed via 31P NMR that strong phosphine binding occurs under conditions relevant to catalysis–and elucidating the nature of this phenomenon will be the subject of future study.16,18 Product yield noticeably improved when urea was used; with 4a as ligand, 3aa was isolated in 68% yield. With this result in hand, we explored the effect that substituting the urea has on reactivity. Although we had previously found monosubstituted ureas to be optimal, in this case trisubstituted urea 4d performed comparably to 4b (57% vs 54%); disubstituted urea 4c showed no ligand effect, and tetrasubstituted urea 4g inhibited the reaction. Introducing a para-OMe group to the N-aryl substituent, as in 4e, further improved product yield (65%); electron-withdrawing groups did not affect reactivity (4f) and no clear electronic trend was observed. Further investigation of substituent effects revealed that piperidine is the most effective group; other cyclic and acyclic amines perform worse (4h–k). Ultimately, 4e maintained good reactivity across a wider range of substrates than any other phosphine or urea ligand investigated, including 4a (see Figure 4 [3aj]).16 A 2:1 ligand/palladium ratio was optimal; product yield dropped significantly with 1:1 4e/Pd(OAc)2.16
Figure 2.

(A) Ligand structure–reactivity relationships in Pd-catalyzed heteroannulation of 1a and diene 2a. (B) Steric profile of trisubstituted urea 4d (Vbur = buried volume).
Figure 4.

Diene scope. Yields and product ratios correspond to isolated products and are an average of three runs. Conditions: 1a (0.5 mmol), 2 (0.75 mmol), Pd(OAc)2 (2.5 mol %), 4e (5 mol %), NaOtBu (0.55 mmol), 90:10 PhMe/anisole (0.25 M), 110 °C, 24 h. Legend: (a) 2.0 equiv. of diene used. (b) Reaction ran for 48 h. (c) Inseparable mixture of diastereomers.
To better understand the binding properties of trisubstituted ureas, we conducted DFT studies on model complexes. To discern the preferred urea binding mode, we modeled a PdCl2 (ureate) complex using urea 4d.(16,19) As with monosubstituted ureas, coordination through N is significantly favored over O-coordination (−11.4 kcal/mol).15,19e We then compared the buried volume (%Vbur) of trisubstituted ureas with monosubstituted ureas. These Vbur calculations were performed on a post-migratory insertion Pd complex using bromophenol, isoprene, and one bound urea ligand (Figure 2b). Although 4d (Figure 2b) is considerably larger than 4b in any conformation (%Vbur = 33 vs 17),15,17 it is concentrated in one region of the complex rather than equally distributed as seen in phosphine ligand complexes.16,19d This steric profile leaves much of the active site open and thus minimizes repulsive interactions between the ligand and other groups on the metal. These calculated properties are consistent with our previous findings.15
We next explored the bromophenol scope (Figure 3). Under our optimized conditions, diene 2b and bromophenol 1a coupled to afford 3ab in 74% yield (0.5 mmol) and 64% yield at gram scale. Substrates bearing alkyl substitution were effective regardless of the substituent position, even when adjacent to either the bromide or phenol (3bb, 3eb). Substitution adjacent to the oxygen ring is common in DHB-based natural bioactive molecules,1,2 and previous methods have not tolerated these substitution patterns.13d,13e No clear electronic trend was observed for substitution para to the bromide (3fb–gb). When para to the phenol, electron-donating (3hb) and weakly withdrawing substituents (3jb) were compatible, but no product was observed with substrates bearing strongly withdrawing substituents such as CF3 (3ib). Bromophenols bearing halogen substituents such as fluorine afforded product in good yields (3kb); likewise, esters are well tolerated (3mb, 65%). Nitrogen functionality is attractive but is a challenging substrate in this type of reaction because of the propensity for nitrogen to competitively coordinate palladium. We observed modest reactivity with substrates containing tertiary amines (3lb, 34%), and good reactivity with a pyridinol-based substrate (3nb, 65%). Ketones, aldehydes, amides, nitrile, and nitro groups are not well tolerated; this is due to electron-withdrawing effects in some cases and poor solubility in others.16
Figure 3.

Bromophenol scope. Yields and product ratios correspond to isolated products, and are an average of three runs at 0.5 mmol scale and two runs at gram scale. Conditions: 1 (0.5 mmol), 2b (0.75 mmol), Pd(OAc)2 (2.5 mol %), 4e (5 mol %), NaOtBu (0.55 mmol), 90:10 PhMe/anisole (0.25M), 110 °C, 24 h.
Our heteroannulation also engages structurally and functionally diverse dienes (Figure 4), with only a slight excess of diene required (1.5 equiv. vs 3–5 equiv.).13d,13f Linear conjugated dienes bearing both electron-rich and electron-poor aryl substitution give products 3ac–d in 72% and 70% yield, respectively. Unprotected and benzoate-protected primary alcohols were compatible with this methodology (3ae–af). Dienes with sensitive functionality such as a furan ring reacted smoothly, affording product in 64% yield (3ah). Various heterocycles including thiophene and phthalimide are also effective (3ag, 3ai). Additionally, branched dienes (2j–l), including those with sensitive functional groups (2k, 2l) were good coupling partners in our methodology. Although a single product was observed with linear dienes, branched dienes gave a mixture of regioisomers with good selectivity (3/3′ ∼ 85:15). In contrast to linear dienes, a singular phosphine ligand enhanced reactivity with myrcene (3aj), but it provided no advantage over urea 4e. Sterically encumbered dienes effectively engaged in the reaction, affording 3am and 3an in good yield. The inclusion of the 1,2-disubstituted diene in 3am allows for generation of a fully substituted carbon at the 2-position of the dihydrofuran ring.
We have shown that urea-enabled, palladium-catalyzed heteroannulation can be applied to the synthesis of functionalized DHBs. In contrast to existing methods, our method engages structurally diverse dienes under a unified set of reaction conditions, with broad functional group tolerance. Moreover, this chemistry can be performed with low reagent loadings and is robust to ambient conditions, making this an attractive approach for the synthesis of these core structures. Current efforts in our lab are focused on better understanding the impact that nucleophile identity has on ligand requirements for these reactions, as well as continuing to expand this methodology for the preparation of diverse heterocyclic scaffolds.
Acknowledgments
The authors thank the University of Rochester for financial support. Analytical data were obtained from the CENTC Elemental Analysis Facility at the University of Rochester (NSF CHE-0650456) and the University of Rochester Medical Center Mass Spectrometry Resource Laboratory. We thank Caitlyn P. McNichol (University of Rochester) for checking the experimental procedure for preparation of compound 3db in Figure 3, and Jakub Vaith (University of Rochester) for his assistance with the DFT and %Vbur calculations.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.2c02301.
Additional experimental details, experimental procedures, computational studies, compound characterization, NMR spectra for all new compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Chaturvedula V. S. P.; Schilling J. K.; Kingston D. G. I. New Cytotoxic Coumarins and Prenylated Benzophenone Derivatives from the Bark of Ochrocarpos punctatus from the Madagascar Rainforest. J. Nat. Prod. 2002, 65, 965–972. 10.1021/np020030a. [DOI] [PubMed] [Google Scholar]
- Nguyen M. T. T.; Nguyen N. T.; Nguyen K. D. H.; Dau H. T. T.; Nguyen H. X.; Dang P. H.; Le T. M.; Nguyen Phan T. H.; Tran A. H.; Nguyen B. D.; Ueda J.-y.; Awale S. Geranyl Dihydrochalcones from Artocarpus altilis and Their Antiausteric Activity. Planta Med. 2014, 80, 193–200. 10.1055/s-0033-1360181. [DOI] [PubMed] [Google Scholar]
- Harinantenaina L.; Brodie P. J.; Slebodnick C.; Callmander M. W.; Rakotobe E.; Randrianasolo S.; Randrianaivo R.; Rasamison V. E.; TenDyke K.; Shen Y.; Suh E. M.; Kingston D. G. I. Antiproliferative Compounds from Pongamiopsis pervilleana from the Madagascar Dry Forest. J. Nat. Prod. 2010, 73, 1559–1562. 10.1021/np100430r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanam H.; Shamsuzzaman Bioactive Benzofuran derivatives: A review. Eur. J. Med. Chem. 2015, 97, 483–504. 10.1016/j.ejmech.2014.11.039. [DOI] [PubMed] [Google Scholar]
- Naik R.; Harmalkar D. S.; Xu X.; Jang K.; Lee K. Bioactive benzofuran derivatives: Moracins A–Z in medicinal chemistry. Eur. J. Med. Chem. 2015, 90, 379–393. 10.1016/j.ejmech.2014.11.047. [DOI] [PubMed] [Google Scholar]
- Belmonte-Vázquez J. L.; Avellanal-Zaballa E.; Enríquez-Palacios E.; Cerdán L.; Esnal I.; Bañuelos J.; Villegas-Gómez C.; López Arbeloa I.; Peña-Cabrera E. Synthetic Approach to Readily Accessible Benzofuran-Fused Borondipyrromethenes as Red-Emitting Laser Dyes. J. Org. Chem. 2019, 84, 2523–2541. 10.1021/acs.joc.8b02933. [DOI] [PubMed] [Google Scholar]
- Makino K.; I T.; Kubo Y. A benzofuran[b]-fused BODIPY as an efficient sensitizer for photocatalytic hydrogen production. Sustain. Energy Fuels 2021, 5, 3676–3686. 10.1039/D1SE00387A. [DOI] [Google Scholar]
- Drijfhout F. P.; Morgan E. D. Terrestrial Natural Products as Antifeedants. Comp. Nat. Prod. II: Chem. Biol. 2010, 4, 457–501. [Google Scholar]
- For a comprehensive review on methods to synthesize dihydrobenzofurans, see:; Sheppard T. D. Strategies for the Synthesis of 2,3-Dihydrobenzofurans. J. Chem. Res. 2011, 35, 377–385. 10.3184/174751911X13096980701749. [DOI] [Google Scholar]
- a For examples of direct phenol alkylation, see:Dinda S. K.; Das S. K.; Panda G. Application of Phenolate Ion Mediated Intramolecular Epoxide Ring Opening in the Enantioselective Synthesis of Functionalized 2,3-Dihydrobenzofurans and 1-Benzopyrans. Synthesis 2009, 11, 1886–1896. [Google Scholar]; b Jiang H.; Sugiyama T.; Hamajima A.; Hamada Y. Asymmetric Synthesis of 2-Substituted Dihydrobenzofurans and 3-Hydroxydihydrobenzopyrans through the Enantioselective Epoxidation of O-Silyl-Protected ortho-Allylphenols. Adv. Synth. Catal. 2011, 353, 155–162. 10.1002/adsc.201000505. [DOI] [Google Scholar]
- Davies H. M. L.; Grazini M. V. A.; Aouad E. Asymmetric Intramolecular C–H Insertions of Aryldiazoacetates. Org. Lett. 2001, 3, 1475–1477. 10.1021/ol0157858. [DOI] [PubMed] [Google Scholar]
- For examples of rearragements leading to the formation of dihydrobenzofurans, see:; a Hashmi A. S. K.; Rudolph M.; Bats J. W.; Frey W.; Rominger F.; Oeser T. Gold-Catalyzed Synthesis of Chroman, Dihydrobenzofuran, Dihydroindole, and Tetrahydroquinoline Derivatives. Chem.—Eur. J. 2008, 14, 6672–6678. 10.1002/chem.200800210. [DOI] [PubMed] [Google Scholar]; b Khanna M. S.; Singh O. V.; Garg C. P.; Kapoor R. P. Regioselective Synthesis of Methyl 2,3-Dihydro-2-aryl Benzofuran-3-Carboxylates Using Thallium(III) Nitrate. Synth. Commun. 1993, 23, 585–590. 10.1080/00397919308009816. [DOI] [Google Scholar]
- For palladium-mediated heteroannulations of phenols and olefins, see:; a Ni H.-Q.; Cooper P.; Engle K. M. Recent advances in palladium-catalyzed (hetero)annulation of C-C bonds with ambiphilic organo(pseudo)halides. Chem. Commun. 2021, 57, 7610–7624. 10.1039/D1CC02836G. [DOI] [PMC free article] [PubMed] [Google Scholar]; b McDonald R. I.; Liu G.; Stahl S. S. Palladium(II)-Catalyzed Alkene Functionalization via Nucleopalladation: Stereochemical Pathways and Enantioselective Catalytic Applications. Chem. Rev. 2011, 111, 2981–3019. 10.1021/cr100371y. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Yang J.; Mo H.; Jin X.; Cao D.; Wu H.; Chen D.; Wang Z. Vinylogous Elimination/Heck Coupling/Allylation Domino Reactions: Access to 2-Substituted 2,3-Dihydrobenzofurans and Indolines. J. Org. Chem. 2018, 83, 2592–2600. 10.1021/acs.joc.7b02986. [DOI] [PubMed] [Google Scholar]; d Larock R. C.; Berrios-Pena N.; Narayanan K. Palladium-catalyzed heteroannulation of 1,3-dienes by functionally substituted aryl halides. J. Org. Chem. 1990, 55, 3447–3450. 10.1021/jo00298a011. [DOI] [Google Scholar]; e Chen S.-S.; Meng J.; Li Y.-H.; Han Z.-Y. Palladium-Catalyzed Enantioselective Heteroannulation of 1,3-Dienes by Functionally Substituted Aryl Iodides. J. Org. Chem. 2016, 81, 9402–9408. 10.1021/acs.joc.6b01611. [DOI] [PubMed] [Google Scholar]; f Borrajo-Calleja G. M.; Bizet V.; Mazet C. Palladium-Catalyzed Enantioselective Intermolecular Carboetherification of Dihydrofurans. J. Am. Chem. Soc. 2016, 138, 4014–4017. 10.1021/jacs.6b02158. [DOI] [PubMed] [Google Scholar]; g Ni H.-Q.; Kevlishvili I.; Bedekar P. G.; Barber J. S.; Yang S.; Tran-Dubé M.; Romine A. M.; Lu H.-X.; McAlpine I. J.; Liu P.; Engle K. M. Anti-selective [3 + 2] (Hetero)annulation of non-conjugated alkenes via directed nucleopalladation. Nat. Commun. 2020, 11, 6432. 10.1038/s41467-020-20182-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Nakhla J. S.; Kampf J. W.; Wolfe J. P. Intramolecular Pd-Catalyzed Carboetherification and Carboamination. Influence of Catalyst Structure on Reaction Mechanism and Product Stereochemistry. J. Am. Chem. Soc. 2006, 128, 2893–2901. 10.1021/ja057489m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For an alternative oxidative phenol coupling approach, see:; a Van Dyck S. M. O.; Lemière G. L. F.; Jonckers T. H. M.; Dommisse R. Synthesis of 4-O-Methylcedrusin. Selective Protection of Catechols with Diphenyl Carbonate. Molecules 2000, 5, 153–161. 10.3390/50200153. [DOI] [Google Scholar]; b Sharma U.; Naveen T.; Maji A.; Manna S.; Maiti D. Palladium-Catalyzed Synthesis of Benzofurans and Coumarins from Phenols and Olefins. Angew. Chem., Int. Ed. 2013, 52, 12669–12673. 10.1002/anie.201305326. [DOI] [PubMed] [Google Scholar]; c Blum T. R.; Zhu Y.; Nordeen S. A.; Yoon T. P. Photocatalytic Synthesis of Dihydrobenzofurans by Oxidative [3 + 2] Cycloaddition of Phenols. Angew. Chem., Int. Ed. 2014, 53, 11056–11059. 10.1002/anie.201406393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaith J.; Rodina D.; Spaulding G. C.; Paradine S. M. Pd-Catalyzed Heteroannulation Using N-Arylureas as a Sterically Undemanding Ligand Platform. J. Am. Chem. Soc. 2022, 144, 6667–6673. 10.1021/jacs.2c01019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See the Supporting Information for more details.
- Phosphine ligands are not advantageous over ureas with branched substrates (see the Supporting Information).
- Borrajo-Calleja G. M.; Bizet V.; Besnard C.; Mazet C. Mechanistic Investigation of the Pd-Catalyzed Intermolecular Carboetherification and Carboamination of 2,3-Dihydrofuran: Similarities, Differences, and Evidence for Unusual Reaction Intermediates. Organometallics. 2017, 36, 3553–3563. 10.1021/acs.organomet.7b00483. [DOI] [Google Scholar]
- The SMD(acetone)-MN15-def2TZVPP computational level was used in the Gaussian 16 package, Revision A.03.; a Yu H. S.; He X.; Li S. L.; Truhlar D. G. MN15: A Kohn–Sham global-hybrid exchange–correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions. Chem. Sci. 2016, 7, 5032–5051. 10.1039/C6SC00705H. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]; c Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]; d Falivene L.; Cao Z.; Petta A.; Serra L.; Poater A.; Oliva R.; Scarano V.; Cavallo L. Towards the online computer-aided design of catalytic pockets. Nat. Chem. 2019, 11 (10), 872–879. 10.1038/s41557-019-0319-5. [DOI] [PubMed] [Google Scholar]; e Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16, Revision A.03; Gaussian, Inc.; Wallingford, CT, 2016. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.