

Abstract
The β-thalassemias are inherited anemias that are caused by the absent or insufficient production of the ß chain of hemoglobin. We report 6–8 year follow-up of four adult patients with transfusion-dependent β-thalassemia who were infused with autologous CD34+ cells transduced with the TNS9.3.55 lentiviral globin vector after reduced-intensity conditioning (RIC) in a phase I clinical trial (NCT01639690). Patients were monitored for insertional mutagenesis and the generation of a replication-competent lentivirus (RCL) (safety and tolerability of the infusion product following RIC; primary endpoint) and engraftment of the genetically modified autologous CD34+ cells, the expression of the transduced β-globin gene, and the post-transplant transfusions requirements (efficacy, secondary endpoint). No unexpected safety issues occurred during conditioning and cell product infusion. Hematopoietic gene marking was very stable but low, reducing transfusion requirements in two patients albeit not achieving transfusion independence. Our findings suggest that non-myeloablative conditioning can achieve durable stem cell engraftment but underscore a minimum CD34+ cell transduction requirement for effective therapy. Moderate clonal expansions were associated with integrations near cancer-related genes, suggestive of non-erythroid activity of globin vectors in stem/progenitor cells. These correlative findings highlight the necessity to cautiously monitor patients harboring globin vectors.
Introduction
The β-thalassemias are hereditary anemias caused by over 400 mutations that affect the β-globin gene (HBB), resulting in absent or insufficient production of the β chain of hemoglobin (Hb)1–3(https://www.ithanet.eu/db/ithagenes). The treatment of the severe form of the disease, known as transfusion-dependent β-thalassemia (TDT), requires lifelong transfusions to supply Hb-replete red blood cells (RBC), which the thalassemic bone marrow is unable to produce4,5. This treatment is life saving but fraught with medical complications6–8. The only curative therapy is the transplantation of allogeneic hematopoietic stem cells (HSCs) harboring functional globin genes, but this option is not available to the vast majority of thalassemic patients, for whom a suitably matched related donor cannot be found9. While bone marrow transplantation from a matched related donor carries a low risk of morbidity and mortality10,11, few patients opt for alternative transplant modalities owing to the potentially serious risks associated with matched-unrelated or mismatched transplants12. These medical risks, together with their socio-economic cost13, warrant the pursuit of alternative curative therapies9. Gene therapy strategies in autologous cells offer the prospect of a safe transplant without–the allo-immune complications of graft rejection or graft-versus-host disease14–17.
The goal of globin gene transfer is to enable the patients’ own HSCs to generate RBCs that contain enough Hb to obviate transfusion therapy18,19. We previously demonstrated that globin gene therapy is curative in two mouse models of β-thalassemia intermedia and Cooley’s anemia, using a lentiviral vector encoding the human β-globin gene and selected regulatory elements that enabled therapeutic globin expression20–23. This vector, termed TNS9, corrected the anemia in β-thalassemic mice and produced an average increase of 4–6 g/dL Hb per vector copy in steady-state peripheral blood. These findings were confirmed and extended by others in various models of thalassemia and sickle cell disease (SCD) using variant lentiviral vectors also harboring the HS2, HS3 and HS4 elements of the β-globin locus control region (LCR) to regulate the vector-encoded β-, γ- or mutated β-globin gene (reviewed in 18,19,24,25). To enable the clinical translation of globin gene therapy, we conducted a CD34+ cell mobilization study in five adult patients with TDT, which showed safe and efficient G-CSF-induced hematopoietic stem/progenitor cell (HSPC) mobilization and globin vector transduction efficiency achieving a mean vector copy number (VCN) of 0.2–0.6 in bulk CD34 cells26.
Based on these encouraging results and following a public review by the NIH’s Recombinant DNA Advisory Committee (RAC), (https://osp.od.nih.gov//wp-content/uploads/RAC_Minutes_Jun_2007.pdf), we proceeded to a phase I clinical trial to evaluate the safety and level of globin gene transfer following reduced intensity conditioning (RIC) in adults with TDT. RIC was chosen because (1) this approach had been used previously and successfully in the context of gene therapy and could be sufficient for the engraftment of gene transduced cells and (2) it was a safer first approach. With a median follow-up of 90 months, we report here on the four patients treated on this protocol.
Results
Patient characteristics.
Four patients with TDT were infused with TNS9.3.55-transduced autologous CD34+ cells between November 2012 and May 2015 (Table 1). Patients were enrolled from the Cervello Hospital in Palermo (N=3) and Microcimetie Hospital in Cagliari (N=1). All underwent successful HSPC mobilization, cytoreduction and cell infusion at MSKCC. One patient who had undergone successful mobilization and transduction eventually opted not to proceed to cytoreduction and cell infusion. The four infused patients included 2 females and 2 males, age 18–39 years at the time of infusion. One patient had a β0/β0 genotype and three patients a β0/β+ genotype, two of which bore the severe β+ IVSI-110 mutation. All four patients were on RBC transfusion programs, receiving transfusion volumes of 177–271 ml/kg/year prior to enrollment onto this study (Table 1). One patient was splenectomized at age 9. All patients received chelation with Deferasirox (N=1), Desferrioxamine and Deferiprone (N=1), Deferiprone (N=1) or Desferrioxamine (N=1). All patients had increased liver iron concentrations at enrollment ranging from 11.7–15.8 mg/g dry weight. The median follow-up for these 4 patients is 90 months (range 72–96).
Table 1 |.
Patient Characteristics
| Patient ID | Patient 1 | Patient 2 | Patient 3 | Patient 4 |
|---|---|---|---|---|
| Age at cell infusion (years) | 23 | 18 | 39 | 18 |
| Sex | F | F | M | M |
| Genotype | β0/β+ | β0/β+ | β0/β0 | β0/β+ |
| Mutation | cod39/IVSI-110 | cod39/IVSI-6 | cod39/cod39 | cod39/IVSI-110 |
| Transfusion requirement (ml/kg/year) | 203 | 214 | 177 | 271 |
| Chelation | DFX | DFO + DFP | DFP | DFO |
| Serum ferritin (ng/ml) | 1217 | 2477 | 838 | 1739 |
| Cardiac T2* by MRI (ms) | 41 | 44 | 32 | 35 |
| LIC (mg Fe per g dw) | 14.8 | 15.6 | 11.7 | 15.8 |
| Liver fibrosis | minimal | no | no | no |
| Splenectomy | no | yes | no | no |
| HCV | negative | negative | serology pos, RNA negative | negative |
F, female; M, male; Transfusion requirements prior to infusion expressed as volume in ml per body weight (in kg) per year, mean annual volume over the last 5 years prior to gene therapy; DFX: deferasirox, DFO: desferrioxamine, DFP: deferiprone; ms, milliseconds; LIC, liver iron concentration in mg of Fe (iron) per g dw (dry weight), estimated by T2* liver MRI; Liver fibrosis evaluation: liver biopsie in patient 1 and via fibroscan in patient 2–4.
CD34+ cell collection and transduction.
A median of 16.0×106 CD34+ cells/kg (range 10.3–30.7×106 CD34+ cells/kg) was collected via apheresis after G-CSF mobilization (Table 2), which was well tolerated (Supplementary Table 1). A back up of 2×106 CD34+ cells/kg was cryopreserved for each patient. The mean vector copy number (VCN) in the drug products (DP) ranged from 0.09–0.15 (median 0.15). Patients received a median total TNS9.3.55-transduced dose of 9.0×106 CD34+ cells/kg (range 7.08–11.71×106 CD34+ cells/kg). Cell infusions were administered in two successive aliquots27.
Table 2 |.
Collection, Transduction and Dosage of TNS9.3.55-transduced CD34+ Cells
| Patient ID | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Median |
|---|---|---|---|---|---|
| Number of mobilization cycles | 1 | 1 | 1 | 2 | |
| Number of CD34+ cells collected (×106/kg) | 30.7 | 10.25 | 13.6 | 7.2 11.2 |
16.0 |
| DP VCN (Average VCN in all CFCs) | 0.15 | 0.09 | 0.15 | 0.17 0.12 |
0.15 |
| VCN from liquid EC | 0.37 | 0.20 | 0.28 | 0.39 0.22 |
0.28 |
| CD34+ cell infusion dose (×106/kg) | 11.71 | 7.08 | 10.60 | 3.87 3.50 |
9.0 |
| %CD34+ EOP cells | 99.9 | 98.3 | 97.3 | 99.4 94.3 |
97.8 |
VCN, vector copy number/diploid genome; EC, erythroid culture; CFC, colony-forming cells; EOP, end of production.
Conditioning, engraftment and toxicities.
The cumulative busulfan exposure (39.8 to 59.7 mg*l/h) was in the non-myeloablative range28 for all four patients (Supplementary Table 2). Neutropenia (absolute neutrophil count (ANC) <500/μL) occurred on day 11 (median; range day 10–13) and persisted for 8 days (median; range 5–11 days). Neutrophil engraftment occurred on day 19 (median; range day 16–24). Significant thrombocytopenia (platelet count <20,000/μL) with transfusion dependence occurred in 3 patients on day 11 (median; range 9–12) and lasted for 6 days (median; range 5–14 days) with platelet engraftment on day 19.5 (median; range day 14–25) after transplant (Supplementary Table 2). Patient 2 developed severe metrorrhagia and received platelet transfusions to maintain a platelet count >50,000/μL. The minimal platelet count observed in this patient was 43,000/μL. She received the last platelet transfusion on day 18 (Extended Data Fig. 1).
Non-hematologic busulfan toxicity was limited to grades 0–3 (Supplementary Table 3). Febrile neutropenia and metrorrhagia (grade 1–2) occurred in 3 and 2 patients, respectively. Importantly, no veno-occlusive disease or grade 4–5 serious adverse events (SAEs). were observed. At the time of neutrophil engraftment (day 15), Patient 1 developed symptoms consistent with engraftment syndrome and received methylprednisolone intravenously for 3 consecutive days. G-CSF was administered to Patient 1 and Patient 2 during nadir period. Patients were discharged to the outpatient clinic on days 20–25 (median 22) and returned home to Italy on day 30. All patients had their medical follow-up, and care including transfusions at their home thalassemia center, with intermittent follow-up at MSK for specific exams such as bone marrow aspiration.
Gene marking and multi-lineage engraftment.
The VCN per diploid genome was measured by quantitative PCR in bone marrow and peripheral blood, including sorting of lymphoid (CD3, CD19) and myeloid (CD14, CD15) at multiple time points (Fig. 1). Twelve months after infusion, the median VCN in whole bone marrow (wBM) was 0.04 copies (range 0.02–0.15) and 0.03 (range 0.01–0.08) in peripheral blood mononuclear cells (PBMCs). At last follow-up, the median VCN in PBMCs was 0.03 (range 0.01–0.11). The engraftment patterns were overall similar in the three younger adults and noticeably diminished in Patient 3. The highest VCNs were observed in one of the youngest and only splenectomised patient (0.11 94 months post infusion).
Fig. 1 |. Multilineage engraftment of transduced cells in peripheral blood and bone marrow.
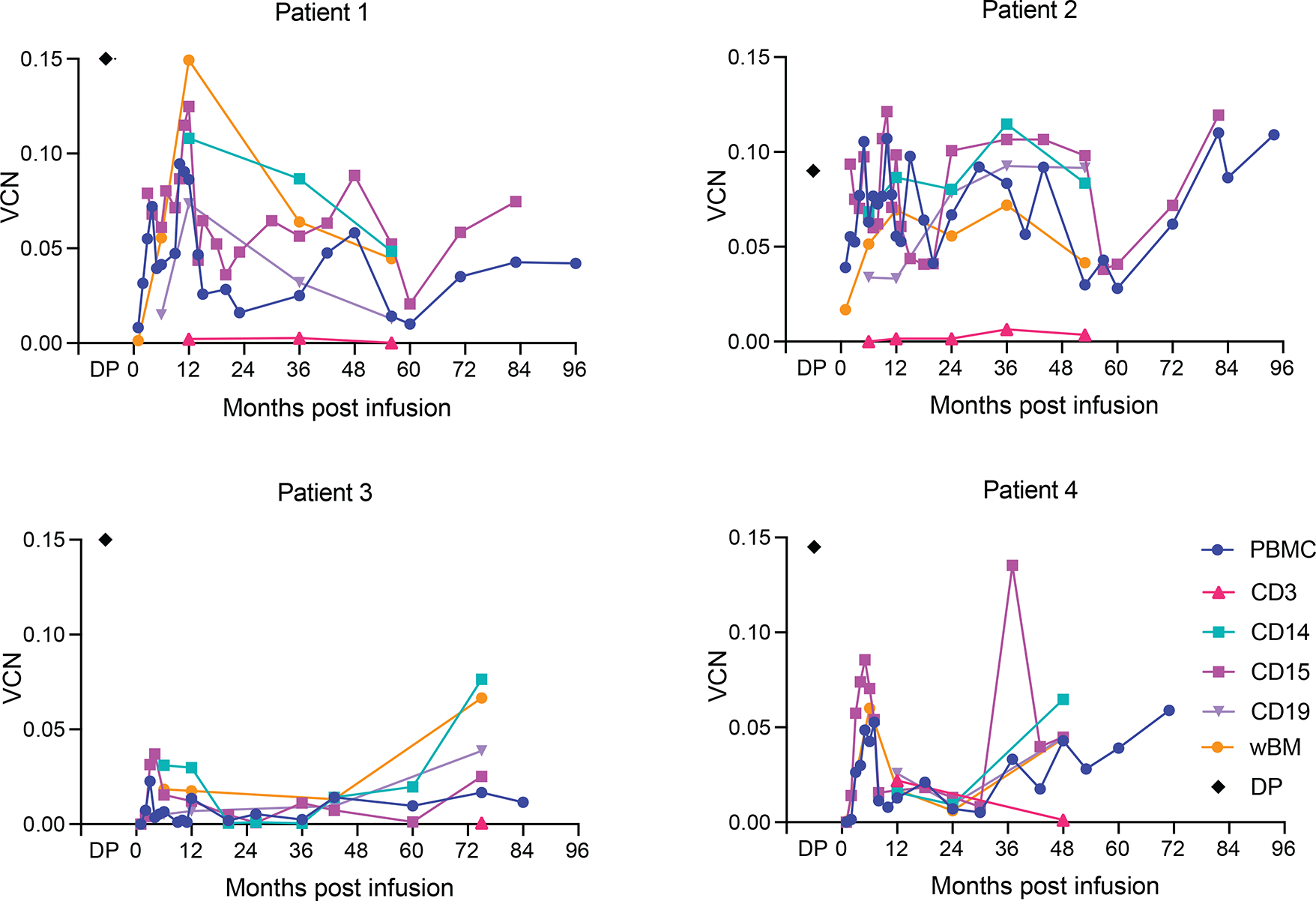
Vector copy number (VCN) per diploid genome in different lymphoid (CD3, CD19) and myeloid (CD14, CD15) lineages sorted from peripheral blood mononuclear cells (PBMC) of patients and whole bone marrow (wBM). Drug product (DP) VCNs were evaluated on infused cells.
Replication competent lentivirus (RCL) assays were performed pre-infusion, followed by testing of all patients at 3, 6 and 12 months post infusion therafter. No RCL was detected at any time point. Based on those results, and according to U.S. Food and Drug Administration (FDA) guidelines, patients no longer need RCL testing and can be followed by a yearly review of medical history for up to 15 years post infusion (https://www.fda.gov/media/113790/download).
Transfusion requirements and transgene expression.
All patients required continued transfusions post-transplant. Transfusion target levels were temporarily brought down post-transplant, then re-adjusted to pre-transplant levels. Based on the latter, transfusion requirements were durably decreased as compared to baseline by 35–57% in Patients 2 and 4. Patient 2 was an 18-year-old female with a β0/β+ genotype. She received 7.08 × 106 transduced CD34+ cells/kg, the lowest number of transduced cells in the cohort. Her cell infusion product had a DP VCN of 0.09 The VCN in PBMCs at last follow-up was 0.11. The mean transfusion requirement dropped from a pre-infusion requirement of 214 ml/kg/Year (range: 198–225 ml/kg/Year) to 101 ml/kg/Year 8 years post-infusion (mean of 93 ml/kg/Year), representing an overall reduction of 57% in transfusion volume (Fig. 2). Patient 4 was also an 18 year-old male with a β0/β+ genotype. He received two cell products for a total count of 7.37 × 106 CD34+ cells/kg (3.87 × 106 CD34+ cells/kg with a DP VCN of 0.17 and 3.5 × 106 CD34+ cells/kg with a DP VCN of 0.12). The VCN in PBMCs at his last follow-up was 0.06. The mean RBC transfusion requirement decreased from 271 ml/kg/Year (range 265–280 ml/Kg/Year) to 183 ml/kg/Year 5 years post infusion with a minimum of 145 ml/Kg/Year (mean 174 ml/kg/Year), accounting for an overall volume reduction of 35% (Fig. 2).
Fig. 2 |. Transfusion requirements and VCN in peripheral blood mononuclear cells.
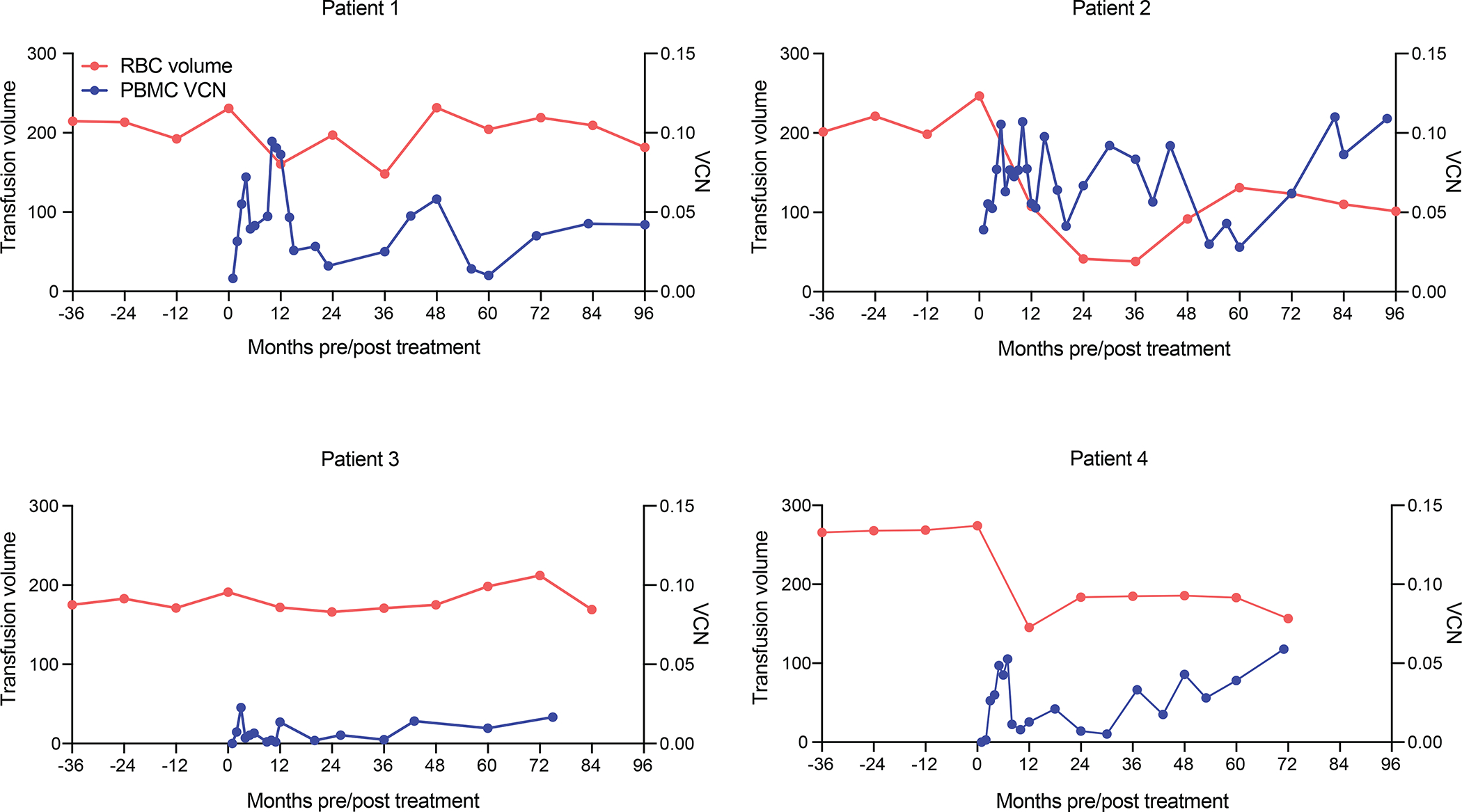
Annual blood transfusion volume (ml/kg/year) and VCN (copy/diploid genome) in peripheral blood mononuclear cells (PBMC) pre- and post-infusion with TNS9.3.55 transduced CD34+ HSPCs.
As we could not distinguish the vector-encoded and endogenous β chains, and because of continued transfusions, we turned to hematopoietic colony analysis to assess vector function. Hematopoietic colonies were established 1 year after cell infusion from bone marrow harvests. The paucity of hematopoietic colonies harboring a single vector copy and the limited material retrieved from individual colonies (needed for both VCN and HPLC assays) made these studies technically challenging and exceedingly labor-intensive, only allowing a limited study in one subject (Patient 2). In a small analysis, the β/α expression ratio determined by HPLC increased from a mean of 0.11 to 0.39 in BFU-Es harboring a single copy of the integrated vector (Extended Data Fig. 2), corresponding to a β chain output of 55% (0.28/0.50) relative to a normal endogenous allele, in line with our earlier assessments26. We further attempted to evidence correction of erythropoiesis in bone marrow samples from Patient 2. The pathologic accumulation of polychromatophylic erythroblasts and marked deficit of orthochromatic erythroblasts and reticulocytes relative to normal bone marrow29 improved at month 24, contemporaneous with a reduction in transfusion requirements (Extended Data Fig. 3). In all four patients, we found that the VCN was slightly higher in Glycophorin+ bone marrow cells compared to the CD45+ fraction (Extended Data Fig. 4).
Analysis of integration site distribution and clonal abundance.
We analyzed integration site distributions and assessed possible insertional mutagenesis associated with outgrowth of gene-modified clones. We focused our analyses on whole blood samples available over 5.75 years or more (up to 7 years for Patient 1). No integration site that mapped uniquely on the genome showed clonal abundance exceeding 20% at any time point (Fig. 3a). A targeted study of integration sites in genomic repeat sequences also did not uncover clones exceeding 20% abundance at any time point. Population structure of gene modified cells was quantified using Gini coefficients, Shannon index, and UC50 (Extended Data Fig. 5 and Supplementary Table 4). Populations of transduced cells were generally highly diverse.
Fig. 3 |. Long-term clonal abundance and succession in all patients.
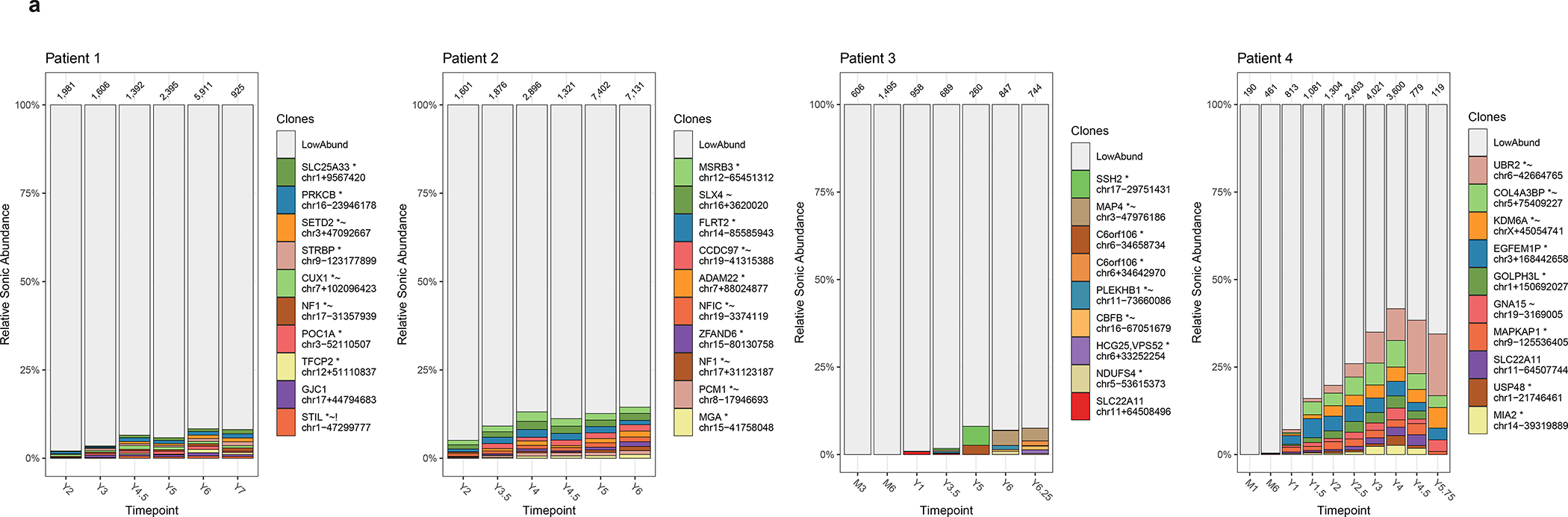
a, Longitudinal relative abundance of the 10 most abundant cell clones in whole blood as marked by lentiviral integration sites. The different colors (horizontal bars) indicate the top 10 most abundant cell clones while the remaining sites are binned as low abundance (LowAbund; grey). The x-axis indicates time points after infusion and the y-axis is scaled by proportion of the total cells sampled. The total number of genomic fragments used to identify integration sites are listed atop of each plot.
A key to the sites, named for the nearest gene, is shown on the right side of the graphs. The nearest genes possess additional annotations: *site is within a transcription unit; ~ site is within 50kb of human cancer-related genes.
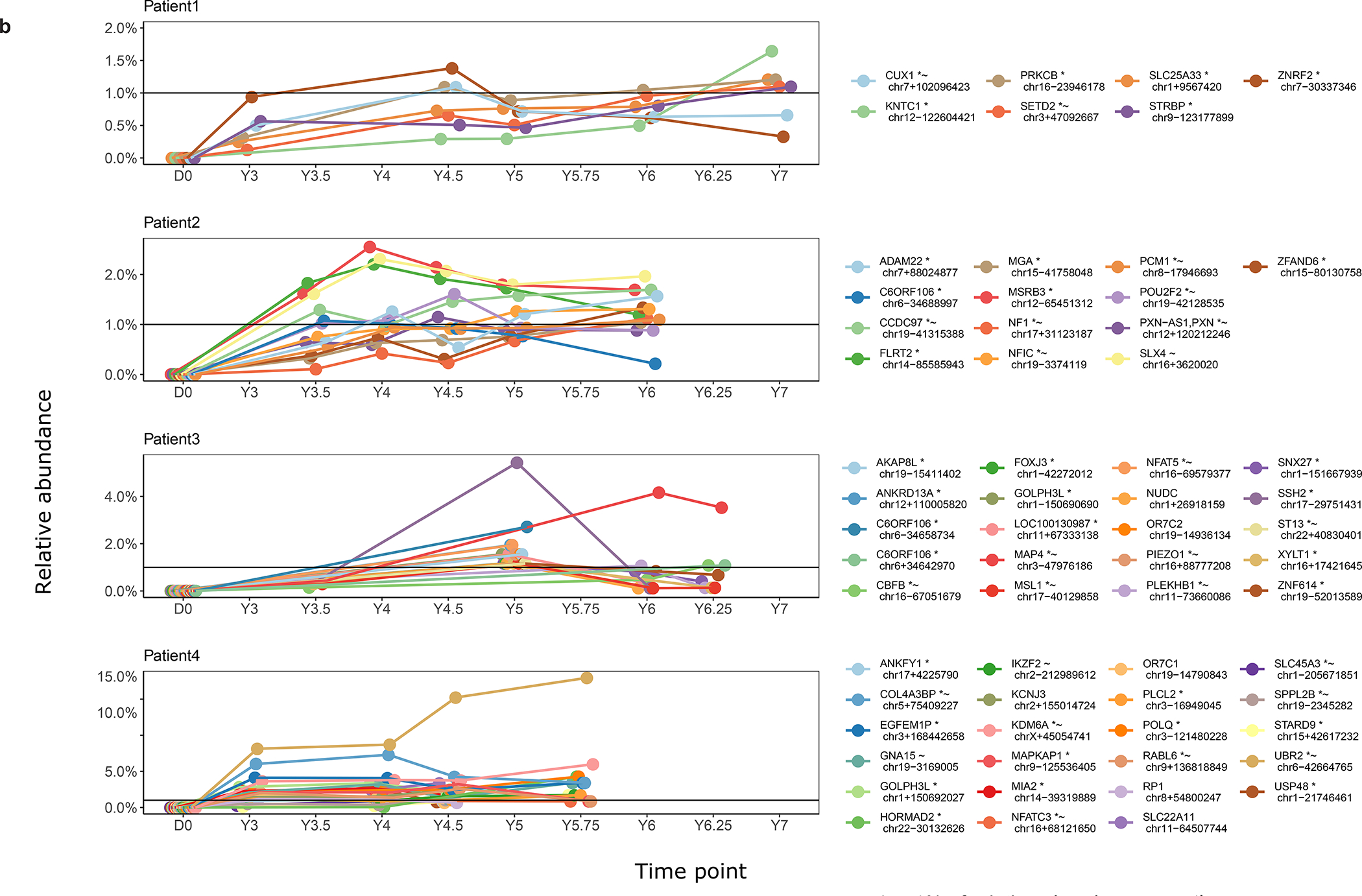
b, Cell clones surpassing 1% of relative abundance over time. Longitudinal representation of relative abundance of cell clones in whole blood as marked by lentiviral integration sites.
Some clones were expanded in relative abundance (Fig. 3b), raising the question of whether insertional mutagenesis might have contributed. We thus compared integration site distributions in the initial transduction product to distributions at late time points (~6 years) after transduction, taking account of clonal abundance. The frequency of integration sites were elevated near cancer-associated genes (Supplementary report, Supplementary Table 5), regardless of which of several lists of cancer-associated genes were used. For patient 4, the most expanded clone (17% of total at the last time point), harbored an integration site near the cancer-associated gene UBR230. More broadly, genes associated with extracellular exosome, protein serine/threonine kinase activity, endosome and signal transduction were modestly enriched in samples from patients ~6 years after transduction (Supplementary Table 6; all p values significant only before correction for multiple comparisons).
We also assessed clustering of vector integration sites as a potential indication of insertional mutagenesis. Clusters selectively present in post-infusion samples potentially mark locations where insertional mutagenesis promoted cell growth and survival. Conversely, clusters present in pre-infusion samples may mark genes that favor growth in culture but that are less important for growth after transplantation. We identified clusters of integration sites using scan statistics as described in31, comparing pooled pre-transplant samples to pooled ~year 6 samples (Supplementary Table 7). One notable cluster was seen in a 13 kilobase interval on Chromosome 17 (Extended Data Fig. 6) encoding the first intron of the STAT3 gene (6 integration sites detected at year 6, zero detected pre-cell infusion, 4 of which were in the same transcriptional orientation as STAT3). HIV integration in the first intron of STAT3 in the same transcriptional orientation was previously reported to be associated with clonal proliferation in T cells32 and a B cell lymphoma in an HIV+ subject 33.
Further clusters of integration sites were seen in additional genes of potential interest, including ASH1L, encoding a histone lysine methyltransferase important in growth control in multiple cell types including the hematopoetic cell lineage, and DNMT1, encoding a DNA methyl transferase important in gene regulation. Thus these data, together with the observed expansion of the UBR2-associated clone (Fig. 3b), suggest potential examples of insertional mutagenesis influencing subsequent cell growth34, though all to date are clinically benign.
Discussion
We report here on the safety and efficacy of globin gene therapy for β-thalassemia using the TNS9.3.55 lentiviral vector, with a median follow-up of 90 months. Autologous CD34+ cell products were administered after RIC in four adult patients with severe β-thalassemia genotypes. No SAEs or unexpected safety issues related to the cell product or the conditioning were observed (Supplementary Table 3). Late effects included gonadal failure. Cumulative Busulfan AUC exposures ranged from 39.8 to 59.7 mg*l/h, within the non-myeloablative range28. All other recent and ongoing globin gene therapy trials in patients with TDT rely on myeloablative conditioning, using either busulfan or treosulfan-tiotepa35–37.
RIC aims to minimize transplant-related morbidity and mortality related to severe short-term toxicities, such as prolonged cytopenia, mucositis and, in case of busulfan conditioning, VOD. It is well established that the risk of VOD decreases when lowering busulfan administration38–40. None of our 4 patients developed VOD, in contrast to 7 cases reported from globin gene therapy trials relying on myeloablative busulfan conditioning (Supplementary Table 8). Sub-myeloablative conditioning also shortened hospitalization and time to platelet engraftment (Supplementary Table 8), which was similar to that recorded in the GLOBE trial (14–25 and 10–24 days, respectively). The time to platelet engraftment is particularly important in thalassemia patients who are often refractory to platelet transfusion and at risk for severe bleeding while in aplasia41. RIC is especially appealing in patients with underlying health conditions such as patients with hemoglobinopathies. RIC has been shown to result in fewer median hospital days and a lower overall cost compared to myeloablative conditioning42,43.
Alkylating agents also expose to the long-term risk of secondary malignancies. While few secondary malignancies have been reported in thalassemia patients who underwent allogeneic HSCT following myeloablative conditioning44, recent reports of two participants who developed MDS/AML after lentiviral vector-mediated globin gene therapy for SCD (NCT02140554) raised concern regarding the risk of myeloid neoplasms in patients with SCD45. Several potential causes, such as insertional mutagenesis45, alkylating agents46 or a generally increased risk for myeloid neoplasms in SCD due to the constant hematopoietic hyperplasia46–49 have been proposed to increase the chance of somatic mutations and result in transformation of myeloid precursors.
Long-term engraftment of up to 8 years follow-up was remarkably stable in all patients, albeit with a moderate discrepancy between the DP VCN and the VCN in the engrafted cells (Fig. 1). Gene marking stabilizing at a lower level than the VCN measured in the cell infusion product is common in gene therapy trials. An ongoing Phase I/II study in SCD recently demonstrated in three patients that RIC allows for successful engraftment of HSCPs transduced with a lentiviral vector encoding a modified γ-globin gene50. Interestingly, a third patient who was given better transduced CD34+ HSPCs showed 2–4X higher gene marking of PBMCs after 6 months, supporting our interpretation that it is the transduction efficiency rather than the conditioning intensity that precluded achieving transfusion independence in our cohort.
Therapeutic efficacy of globin gene therapy for β-thalassemia aims to achieve durable transfusion independence, which we did not reach in these 4 patients. The cell product size (median: 9.0 × 106 CD34+ cells/kg, Table 2) and rapid engraftment after conditioning (Extended Data Fig. 1) argue against cell dosing being limiting. The vector function was robust, based on extensive preclinical evaluation20–22,26 and limited measurements of vector performance in BFU-E and bone marrow erythropoiesis (Extended Data Figs. 2, 3b and 4). These findings argue, as discussed below, that rate limiting transduction was therefore the most likely explanation for not achieving transfusion independence in this cohort.
Three other phase I/II clinical trials investigating the globin lentiviral vectors BB305 and GLOBE have recently reported on clinical outcomes and provided VCN measurements. Both vectors are variants of TNS9.3.55, with GLOBE omitting the HS4 LCR element. In HGB-204/205, 15 of the 22 patients infused with BB305-transduced autologous CD34+ cells became transfusion independent35. In the GLOBE trial, 4 of the 6 pediatric patients became transfusion independent, but none of the three adult patients51,52. The patients in whom transfusion therapy was suspended all received a cell product with a VCN >0.6 in the HGB trials and >0.7 in the GLOBE study. We did not achieve the same level of transduction, owing to impurities in the vector lot that precluded increasing the multiplicity of infection (our vector stock, produced earlier in time, was not chromatography purified), only achieving a median DP VCN of 0.15.
Based on VCN measurements in bone marrow and PBMC, the lowest gene marking associated with transfusion independence was 0.8 in the GLOBE study52 and 0.3 in the BB305 trials35. In our patients, the highest in vivo VCN reached in PBMCs was 0.11. In the two subjects that sustained a VCN >0.05 for extended periods, transfusion requirements were diminished but not abrogated.
The minimum VCN requirement however depends on the severity of the β chain deficit, determined by the patient’s genotype. In the HGB-204/−205 studies, 12 of the 13 patients with a non-β0/β0 genotype stopped transfusions after treatment, while only 3 of the 9 patients with a severe genotype (β0/β0 or IVSI-110/ IVSI-110) discontinued transfusions. Three out of the four patients in our trial had a severe genotype: one patient with β0/β0 mutations and two patients with a β0/β+ genotype of equivalent severity (IVSI-110). Patient 2 had a milder β0/β+ genotype (IVSI-6) and showed the best response. The combined data from the HGB, GLOBE and MSK studies suggest that a minimum PBMC VCN of at least 0.3 for vectors encoding HS2-HS3-HS4 LCR elements is required in patients with milder TDT to achieve transfusion independence and that a VCN of 0.05–0.3 will only result in a reduction of transfusion needs in subjects with severe genotypes. The GLOBE study suggests that a VCN >0.8 may be necessary for HS2-HS3 vectors.
Additional factors other than VCN, β chain output per VC and genotype likely determine response to therapy. In the GLOBE trial, the only patients who achieved transfusion independence were the pediatric patients. Of three subjects who received a cell product with a VCN of 0.7, only the pediatric patient but not the two adults achieved transfusion independence. In our trial, Patient 2 who had the best response, was one of the youngest patients (age 18 years). In addition to the patient’s age, splenectomy may be another contributing factor. The patient with the best clinical response in our trial was the one who was previously splenectomised. Nearly half the patients in the HGB-204/−205 studies were also splenectomised. It is conceivable that diminished splenic entrapment may result in improved engraftment, which may also be enhanced by intra-osseous cell infusion52.
The regular monitoring of vector integration sites in peripheral blood samples showed a polyclonal profile without extensive (>20%) clonal dominance. However, several clones accounting for >1% of circulating genetically modified cells were detected in all patients (Fig. 3b).
Integration site mapping revealed significant skewing towards cancer-related genes, as has been previously observed in persisting clones of either hematopoietic or T cell origin34. The most prominent clone, accounting for up to 17.5% of all genetically marked cells in patient 4 after 5.75 years, harbored a vector integration in proximity to UBR2, which encodes an E3 ubiquitin ligase implicated—in hyperproliferation, chromosome instability, and hypersensitivity to DNA damage-inducing reagents and found to be overexpressed in multiple cancers30. Clustering of integration sites was observed in the first intron of STAT3, a location previously implicated in insertional mutagenesis and clonal expansion in studies of HIV32,33. Integrations reported in 6 subjects from the HGB and GLOBE trials showed a lower frequency of integrations near cancer-related genes, but those were reported at earlier time points (1.5–2 years post infusion) relatives to ours after 4–7 years (Supplementary Table 9).
A case of insertional mutagenesis53 has been previously documented in a thalassemia patient treated with the HPV569 vector, a precursor to BB30554. In this patient, an expanded clone harboring the globin vector integrated in the HMGA2 locus showed a a 10,000-fold increase in expression of HMGA2 transcripts in erythroblasts, resulting from a combination of deregulated post-transcriptional regulation and increased transcription53. HMGA2 over-expression was later shown to expand mouse lineage negative bone marrow HSCs as well as human umbilical cord stem and progenitor cells in mice transplanted with these constructs55–57. Some LCR elements, in particular HS1 and HS2, are active in hematopoietic progenitor cells58,59, which provides a potential mechanism for the trans-activation of cancer-related genes. Further studies in murine hematopoietic chimeras will be useful to assess the non-erythroid transcriptional activity of regulatory LCR elements contained in globin vectors While our associations with clonal expansion are correlative, UBR2 and STAT3, like HMGA2, may thus have been activated by the non-erythroid transcriptional activity of globin vector elements, accounting for their favored capture in our integration site analysis.
In summary, our studies establish that a DP VCN of 0.15 is not sufficient to achieve transfusion independence in TDT. Other studies have shown that a DP VCN of at least 0.3 is needed in patients with mild genotypes. Our study, which is the only one to not have relied on myeloablative conditioning to maximize CD34+ cell engraftment, further suggests that reduced-intensity conditioning is sufficient to achieve stable, long-term HSC engraftment in TDT patients, and could be potentially curative provided that higher VCNs are achieved. This approach should be prospectively evaluated. Finally, our analyses of integration site distributions emphasize the importance and urgency of carefully monitoring clonal expansions in patients with TDT or SCD who have been treated with lentiviral globin vectors.
Methods
Study design and oversight.
This is an open-label phase I clinical trial (NCT01639690) for the treatment of TDT with autologous CD34+ hematopoietic stem and progenitor cells (HSPCs) transduced with TNS9.3.55, a lentiviral vector encoding the normal human β-globin gene. This protocol was reviewed and allowed by the NIH RAC in 2007 and approved by MSK’s Institutional Review Board (IRB) in 2010, and by the Food and Drug Administration (FDA) in 2012. Patients were enrolled from 19 September 2012 (Patient 1) to 29 January 2014 (Patient 4). The clinical trial was initiated in 2012. Study enrollment, mobilization, collection and transduction of peripheral blood HSPCs, patient and conditioning, and transplantation were performed at MSKCC. Patients are followed at their center of origin at the Cervello Hospital in Palermo and Microcimetie Pediatric Hospital in Cagliari, Italy.
The primary endpoints of this phase I trial are the safety and tolerability of (1) the infused autologous CD34+ hematopoietic cells transduced with lentiviral vector TNS9.3.55, as defined by the insertional oncogenesis and the generation of a replication-competent lentivirus (RCL), and of (2) the low dose non-myeloablative conditioning regimen.
Secondary endpoints included the level of engraftment of the genetically modified autologous CD34+ cells, the expression of the transduced ß-globin gene, and the post-transplant transfusions requirements. Clinical information and in vivo biological material from all patients treated in this trial were collected and analyzed during the years 2012–2021 in adherence to informed consent. Primary and secondary endpoints are further detailed in Supplementary Table 10.
Patients.
Patients were screened for the inclusion criteria as specified on the IRB approved protocol and eligible patients signed a consent form. All volunteers were evaluated with a medical history, physical examination. Baseline laboratory tests, including complete blood count and differential, reticulocyte count, iron, total iron-binding capacity (TIBC), transferrin saturation, ferritin, coagulation profile, blood chemistry studies (electrolytes and renal function tests), ABO blood typing, and testing for infectious disease markers were performed. Female subjects of childbearing age underwent a serum pregnancy test. Adverse events during and after therapy were assessed according to the National Institutes of Health Common Terminology Criteria for Adverse Events Version 4.0.
Patients eligible on this protocol were 18 years or older, of any gender or ethnic background, with a diagnosis of β-thalassemia major of any genotype, with a transfusion requirement of > 100 ml/kg/year for at least 2 years. Patients with HLA matched siblings were excluded. Patient’s eligibility included adequate organ function, with a Karnofsky performance score of > 70%. The inclusion and exclusion criteria can be found at ClincalTrials.gov. Informed consent was obtained for all patients. While the Recombinant DNA Advisory Committee supported enrollment of patients age 15 and above the FDA requested initial enrollment of adult patients only (https://osp.od.nih.gov//wp-content/uploads/RAC_Minutes_Jun_2007.pdf).
Trial procedures.
HSPC collection was performed via apheresis after G-CSF mobilization, as previously described 26 with a goal of a minimum of 8 × 106 CD34+ cells/kg. CD34+ HSPCs were selected using the CliniMACS Plus system 60, pre-simulated and transduced with cGMP grade TNS9.3.55 vector stocks at the MSKCC Cell Therapy and Cell Engineering Facility. After busulfan conditioning, patients were infused with two graft aliquots, the first aliquot on day 0 and the second on day 2. The cell infusion product consisted in autologous TNS9.3.55 transduced CD34+cells. A non-selected back-up cell dose of a minimum of 2 × 106 CD34+ cells/kg was cryopreserved and stored for rescue in case of graft failure. All treatment-related adverse events occurred as expected in an autologous HSCT setting and were managed according to institutional standards. Additional details regarding the trial procedures are listed at ClinicalTrial.gov (NCT01639690).
Mobilization and apheresis.
The entire procedure of mobilization and apheresis was performed in the outpatient clinic. Patients received G-CSF at a dose of 10 μg/kg once daily subcutaneously for six days. Patients underwent specific monitoring of side effects and adverse events and complete blood counts on days 1, 3, 5 and 6 of mobilization. Apheresis was performed via two peripheral veins when available (n=4) or via a central venous catheter when intravenous access was insufficient to allow for the insertion of large bore intravenous access catheter (n=1). Each patient underwent leukapheresis with a collection goal of 8 × 106 CD34+ cells/kg. Leukaphereses were performed on the mornings of the 5th and 6th day of G-CSF treatment on a continuous flow cell separator according to institutional standards utilizing acid citrate dextrose for anti-coagulation and calcium prophylaxis to prevent citrate toxicity. The volume of blood processed per leukapheresis session was approximately 3 times the total blood volume as tolerated by the patient. CD34+ cells were positively selected using a CliniMacs Plus system as per Standard Operating Procedures in the Cell Therapy Laboratory at MSKCC. The final number of collected CD34+ HSPC per patient was measured by cell counting and fluorescence-activated cell sorting (FACS) analysis.
Busulfan Regimen.
Patients received pharmacokinetic-adjusted, single-agent intravenous busulfan conditioning. Patients 1, 2 and 3 received intravenous busulfan at a planned non-myeloablative dosing of 2 mg/kg/dose Q 12 hours × 4 doses (planned total dose of 8 mg/kg over two days), with a target first dose area-under-the curve (AUC) of 2,800 μM*min. Patient 4 received 0.8 mg/kg/dose Q 6 hours × 14 doses (planned total dose of 11.2mg/kg over 4 days), with a target first dose AUC of 1,100 μM*min. Busulfan dosing was adjusted in Patient 3 and 4 according to the first-dose pharmacokinetic analysis to achieve the targeted AUC. The cumulative busulfan exposure (mg*l/h) was calculated by the sum of the daily extrapolated AUC measurements and indicated that all four patients had received non-myeloablative conditioning.
Engraftment criteria.
Neutrophil engraftment was defined as absolute neutrophil count (ANC) exceeding 500/μL for 3 consecutive days. The first of these 3 consecutive days was considered the day of engraftment. Platelet engraftment was defined as the first of three consecutive days with a platelet count exceeding 20,000/μL without platelet transfusion support in the past 7 consecutive days.
Post-transplant transfusions requirements.
Pre-transplant, all patients were transfused according to the standard clinical practice for thalassemia at their Thalassemia Center taking into consideration individual clinical conditions. The target Hb transfusion levels were 9 for patients 1, 2, and 4, based on their institutional standards, and were 10 for patient 3, also according to their standards. The transfusions history pre- and post-infusion documented each pre-transfusion Hb, individual transfusions, milliliters of packed red blood cells infused and actual patients body weight. The transfusion volume per year was recorded as ml blood/kg/year and the mean daily decrease in Hb (%) was calculated. Patients were screened for deletional and non-deletional hereditary persistence of fetal hemoglobin (HPFH). As characteristically seen during bone marrow regeneration after hematopoietic stem cell transplantation61, a moderate, transient increase in HbF was observed in all patients in the first 3–14 months post infusion (see Supplementary Table 11). Of note, the results of the Hb transfusion reduction described in this manuscript are based on the last follow-up and HbF at pre-infusion levels for all patients.
Post-transplant, the target Hb transfusion level was temporarily decreased in all patients as illustrated in Supplementary Table 12, but eventually changed back to the pre-transplant Hb transfusion levels. The results of the Hb transfusion reduction in the manuscript were based on the Hb levels equivalent to the pre-transplant ones.
Globin lentiviral vector.
The TNS9.3.55 lentiviral vector encodes the wild-type β-globin chain under the erythroid-specific transcriptional control of the human β-globin promoter and 3 segments of the locus control region (LCR) known as HS2, HS3 and HS420. Clinical grade TNS9.3.55 vector stock was produced under current good manufacturing practice (cGMP) conditions at the Center for Biomedicine and Genetics (CBG, Duarte, CA). TNS9.3.55 vector stocks were titrated on a quality-controlled cell bank of HeLa cells at various dilutions (in the range of 1/5 to 1/12500) of the vector. Titers were calculated using consecutive dilutions that yield linear data. The HeLa titer of the cGMP vector lot was 3.5 × 108 transduction units (TU) /ml.
Transduction and vector copy number (VCN) quantification.
CD34+ HPCs were cultured for 18–24hrs in serum-free X-VIVO 10 supplemented with human stem cell factor, Flt-3 ligand, thrombopoietin and interleukin-3. Fractions were subsequently cultured for 14–16 days in liquid erythroid cultures (EC) or hematopoietic colony assays for VCN quantification by Q-PCR using the Applied Biosystems 7500 real-time PCR system.
RCL analysis.
RCL assays were performed at the Indiana University Vector Production Facility IUVPF according to U.S. Food and Drug Administration (FDA) guidelines by PCR as previously described 62.
CD34+ HSPC transduction.
Selected CD34+ HSPCs were released to MSKCC Cell Therapy and Cell Engineering Facility where they were either pre-simulated immediately or after overnight storage at 4°C for 18–24hrs in serum-free X-VIVO 10 supplemented with 100 ng/ml human stem cell factor (SCF), human Flt-3 ligand (Flt-3L), human thrombopoietin (TPO) and 20 ng/ml human IL-3 (IL-3). Prior to prestimulation, a fraction of the cells was also tested for colony formation potential in methylcellulose colony formation unit (CFU) assays. CD34+ HSPCs underwent two rounds of transduction 18–24 hrs apart using GMP TNS9.3.55 vector stocks. Cells were subsequently cultured in serum-free X-VIVO 10 with the same cytokines. On day 0 and at the end of the cultures CD34+ HSPCs were inoculated into methylcellulose for colony formation. Transduced CD34+ HSPCs were cryopreserved on day 2 or day 3. Aliquot were subsequently differentiated for 14–16 days to the erythroid lineage or plated for colony differentiation before extracting genomic DNA for VCN measurement.
Erythroid differentiation and colony forming unit assay.
CD34+ cell were seeded at 0.5 – 1 × 106/mL in a 6 well plate containing Alpha MEM (Sigma), FBS (Stem Cell Technologies), Glutamine (Gibco), BSA (Sigma), human erythropoietin (Amgen), β-mercaptoethanol (Gibco), dexamethasone (American Regent Laboratories), holo-transferrin (American Regent Laboratories) and human recombinant SCF (Sigma). Cells were counted and fed from day 4 to 10 with the same EC differentiation medium. This differentiation process was used for in vitro modeling of erythropoiesis and VCN determination. For CD34+ cells pre- and post-transduction, CD34+ cells were plated at 500 cells per plate in MethoCultH4435 Enriched (StemCell Technology). BFU-E, CFU-M, CFU-G and CFU-GM were distinguished and counted after 14 – 16 days as per as per manufacturers’ directions.
Vector copy number quantification.
Genomic DNA (gDNA) was extracted using the Gentra Puregene kit (QIAGEN, Valencia, CA) as per manufacturers’ directions. 100 ng gDNA was used for real time PCR reaction. For gDNA extraction from CFCs, isolated colonies were aspirated with a pipette tip under the microscope, washed in PBS and suspended into 25 ul of proteinase K (Roche) containing lysis buffer (14mM Tris HCl, pH=8.3; 2.5 mM MgCl2; 105 mM KCl; 0.3 mg/mL Gelatin; 0.45% Tween-20; 0.45% NP40; and 0.2 mg/mL proteinase K) in 96-well plates. Plates were incubated at 55°C for 60 min, 95°C for 15min to end the reaction and kept at 4°C until use within 48 hrs. Similarly to Charrier et al. 63, we found that vector frequency is underestimated in CFUs when compared to liquid EC cultures from which gDNA is extracted with Puregene kit. DP VCN in the cell infusion product was assessed in all CFCs. Quantification of the TNS9.3.55 lentiviral vector copy number was performed using the Applied Biosystems 7500 real-time PCR system. Average TNS9.3.55 vector copy number per cell was calculated by normalizing to the endogenous ALB gene. Analysis was performed using the 7500 System SDS software v1.4.0 (Applied Biosystems).
Bone marrow studies.
The bone marrow studies, agreed to by participating subjects on a voluntary basis after month 12, ceased after month 60. We are grateful to the patients for their benevolent marrow collections and understand their cessation after 5 years on the study (except patient 3 who returned once to New York City for a bone marrow aspiration at month 75).
HPLC analysis.
Erythroid colonies were generated and collected as described above. Half the material was processed for genomic DNA extraction and the other half washed twice with PBS, and lysed in water by 3 rapid freeze-thaw cycles, followed by a centrifugation at 16,000 g. Supernatants were recovered and stored in liquid nitrogen before HPLC analysis. HPLC analysis was performed as previously described 64.
Bone marrow flow cytometry.
Bone marrow erythroblasts from Patient 2 were analyzed at three consecutive timepoints for surface expression of glycophorin A (GPA), α4 integrin and band 3 as previously described29.
The following flurophore-conjugated antibodies were used: anti-human APC-α4 integrin (clone MZ18–24A9, Miltenyi Biotec, #130–124-008, 1:100), anti-human PE-glycophorin A (GPA or CD235a; clone JC159, Dako Cytomation, #R7078, 1:100). Mouse monoclonal antibodies against the extracellular regions of human band 3 were generated by the New York Blood Center (1:100).
Data was collected using the FACSDiva software version 7.0 (BD) on a FACSCanto™ flow cytometer and analyzed using the FlowJo software version 7.6.5.
Integration site analysis.
Integration site analysis was carried out using ligation-mediated PCR essentially as described65,66. DNA sample were fragmented by sonication, then DNA adaptors ligated to the free DNA ends. Two rounds of PCR were then carried out, amplifying from the adaptor to the LTR of the lentiviral vector. All samples were worked up in quadruplicate to suppress the effects of PCR jackpotting. Sequences were aligned to the hg38 draft human genome assembly. Clonal abundance was quantified using the SonicAbundance method67; in this method, the points of linker ligation associated with each unique integration site are quantified and used as an abundance estimate—this is more accurate than counting sequence reads, where abundance is subject to distortion during PCR steps. For the analysis of association with cancer associate genes, all cells sampled were used for the statistical comparison (sonic abundance), and not the counts of unique sites, to better reflect the clonal structure. Sequence samples studied and their sonic abundance, number of unique sites, and population metrics are in the Supplementary Report on the analysis of the frequency of integration near cancer related genes (database fisher report). All sequences acquired in this study are available under the NCBI SRA accession id: PRJNA70203.
Statistical analysis.
The reported means, medians and ranges are provided for descriptive purpose. The individual transfusion requirement (in ml of blood per kg bodyweight per year) were calculated before (mean volume per year over 5 years before treatment and after). The transfusion requirement follow up was updated as of 12/31/2020. Statistics of integration site analysis are outlined in Results.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. All requests for raw and analyzed data and materials are promptly reviewed by the corresponding author to verify if the request is subject to any intellectual property or confidentiality obligations. Patient-related data not included in the paper were generated as part of clinical trials and may be subject to patient confidentiality. Any data and materials that can be shared will be released via a material transfer agreement.
The following databases were used for For oncogenes definitions: the “Bushman lab oncogenes database” (http://www.bushmanlab.org/links/genelists, v5 June 2021) and four levels of The Cancer Genome Atlas (TCGA) version of the OncoVar database (https://oncovar.org, v1.2 August 2020).
Sequence data were deposited in the NCBI’s Sequence Read Archive (SRA BioProject PRJNA705203). For integration site analysis relative to Gene ontology the “GO.db” Bioconductor annotation data package v3.13.0 was used.
Code Availability Statement:
Source code for manuscript analysis has been deposited in an archived format in the Zenodo coade base (DOI: 10.5281/zenodo.4569099).
Extended Data
Extended Data Fig. 1 |. Cell count recovery after conditioning and infusion of TNS9.3.55 transduced CD34+ HSPCs.
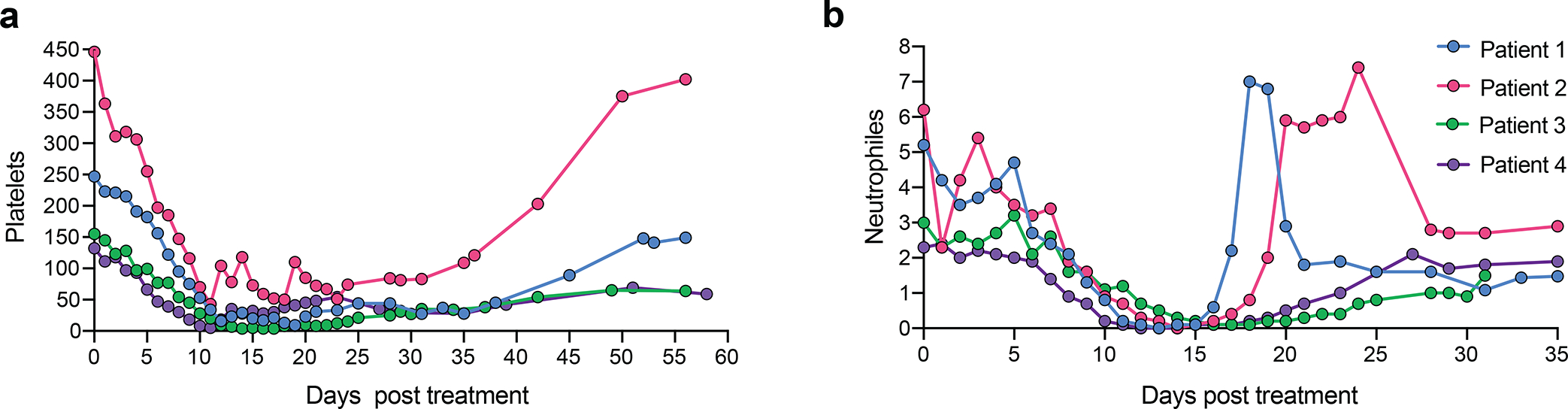
a, Platelet count (×109/l) after treatment. b, Absolute neutrophil count (×109/l) after treatment. Granulocyte colony stimulating factor (G-CSF) was administered for Patient 1 (day 13–16) and Patient 2 (day 16–18) during aplasia. Patient 1 received methylprednisolone intravenously for 3 consecutive days (day 16–18) for treating the engraftment syndrome.
Extended Data Fig. 2 |. β-globin expression in Patient 2 at 12 months post treatment.
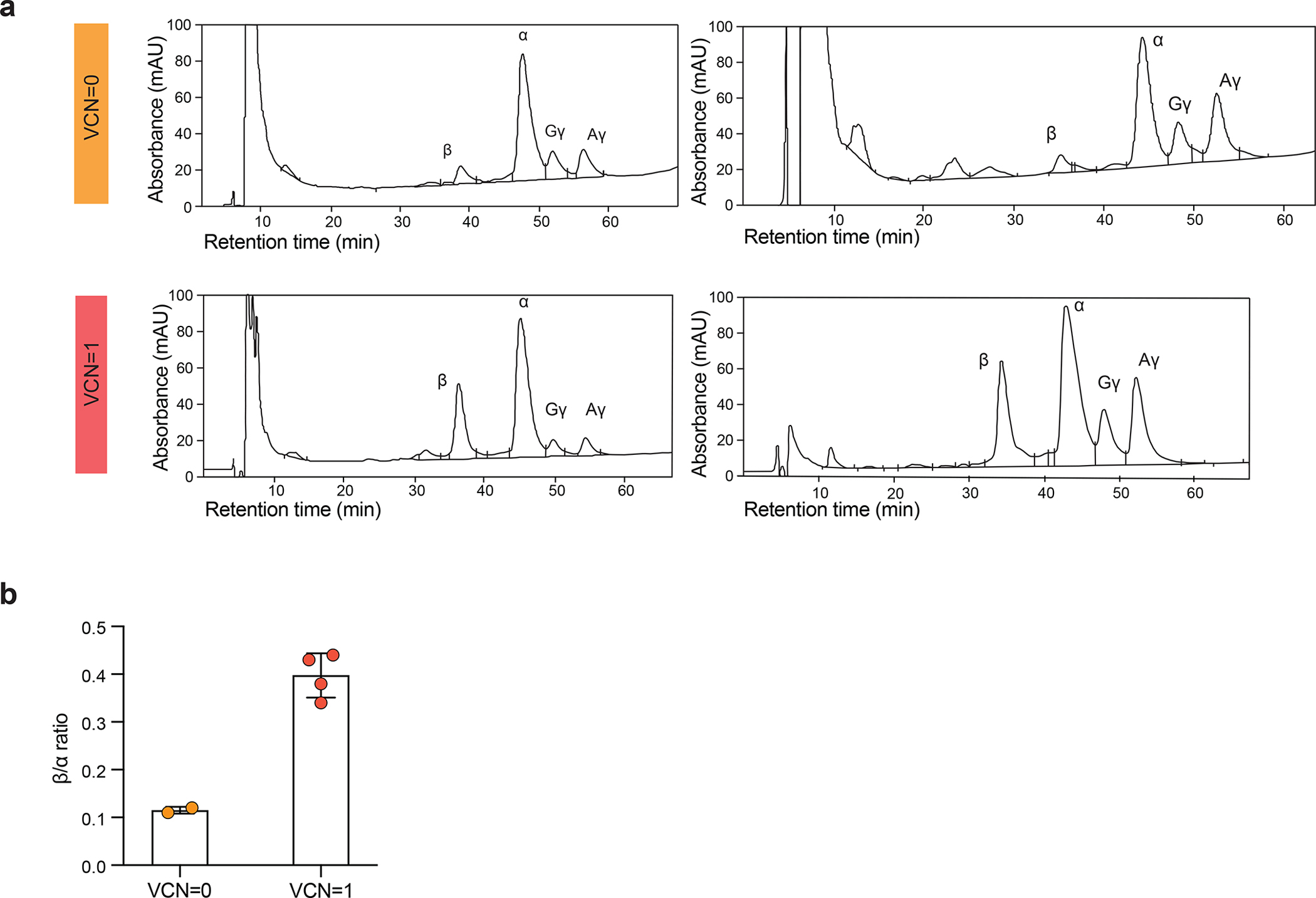
a, HPLC chromatograms illustrating globin production of eythroid cells from four individual BFU-Es derived from bone marrow obtained from Patient 2 at 12 months post infusion.
Top chromatograms: two representative examples from two individual, non-transduced BFU-E from Patient 2; lower chromatograms: two representative examples from two individual, transduced BFU-E from Patient 2. b, β-globin to α-globin ration in erythroids derived from untransduced and transduced HSCs obtained from Patient 2 at 12 months post infusion.
The β/α expression ratio determined by HPLC in single BFU-Es increased from a mean of 0.11 to 0.38 in BFU-Es harboring a single copy of the integrated vector, representing a gain of 0.27.
Extended Data Fig. 3 |. Erythropoietic maturation in bone marrow (Patient 2).

Terminal erythroid differentiation begins with proerythroblasts differentiating into basophilic, then polychromatic, then orthochromatic erythroblasts that enucleate to become reticulocytes. Each distinct stage of terminal human erythroid differentiation can be distinguished using a combination of cell surface markers for glycophorine A (GPA), band 3 and α4 integrin. a, Representative flow cytometry plot of band 3 vs α4-integrin of GPA+ cells in normal erythopoesis: proerythrobalsts (I), early basophilic (II), late basophilic (III), polychromatic (IV), and orthochromatic erythroblasts (V) and reticulocytes (VI). The box plot represents the quantitation of the proportion of cells at each distinct stage of maturation after normalization based on total nucleated erythroid cells (I-V) as 100% as described in ref29. The left panel is adapted from ref29.
b, Bone marrow erythroblasts from Patient 2 were analyzed by flow cytometry at 6, 12 and 24 months post infusion stained with GPA, α4-integrin, and band 3. Plot of band 3 vs α4-integrin of GPA+ cells represents the quantitation of distinct stages of maturation of erythroblasts as described in a. c, Quantitation of the proportion of cells at each distinct stage of maturation after normalization to total nucleated erytroid cells (I-V).
Extended Data Fig. 4 |. Engraftment of transduced cells in bone marrow.
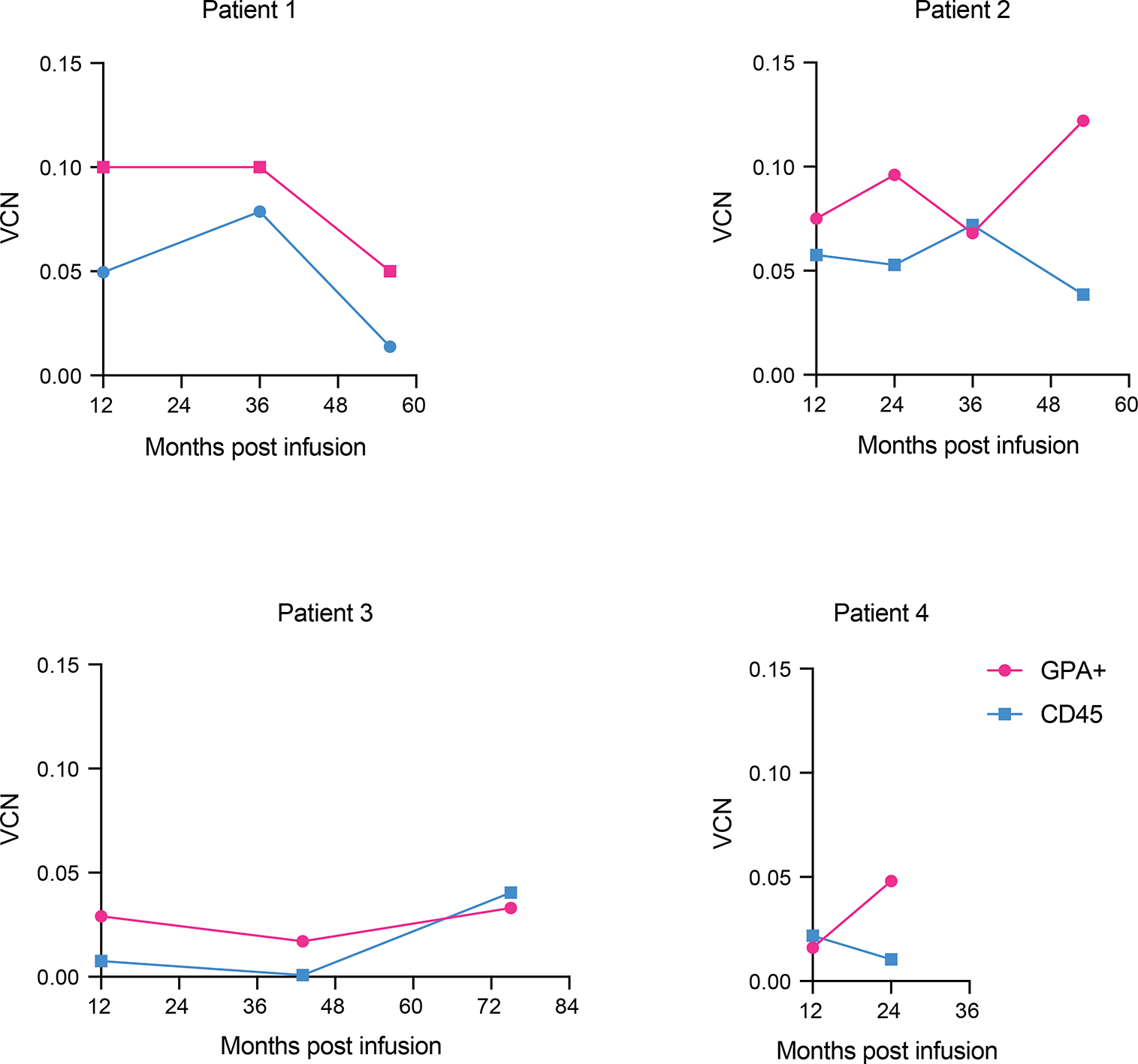
Vector copy number (VCN) in erythroid glycophorine A+ (GPA+) cells and CD45+ cells sorted from bone marrow of patients.
Extended Data Fig. 5 |. Gini and Shannon Index values.

Timepoints in months.
Extended Data Fig. 6 |. Annotation of the genes STAT3 and STAT5A on chromosome 17.
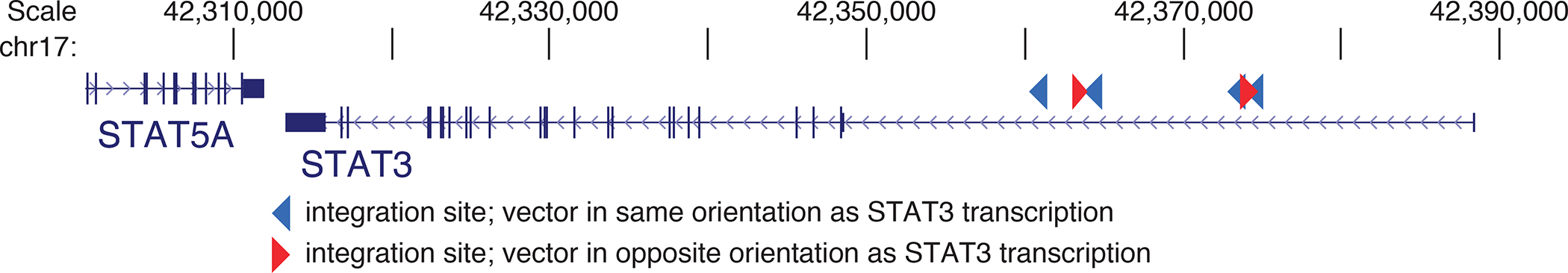
Illustration of a cluster of transgene integrations in the first intron of STAT3. Six integration sites were detected at year six, zero were detected pre-transplant. Four out of six integration sites detected were in the same transcriptional orientation as STAT3.
Supplementary Material
Acknowledgements
The authors would first like to thank the patients and families of those included in the trial and further acknowledge the expert care provided to patients by staff of the Department of Pediatrics at Memorial Sloan Kettering Cancer Center. We thank Dr Rosaria Ctristantielli, Ms Gertrude Gunset and Ms Eden Bechard for their further assistance in making our patients’ journeys and their follow-up pleasant and efficient. This clinical trial was supported by the Stavros Niarchos Foundation (MS), the Memorial Hospital Research fund (FB), the Leonardo Giambrone Foundation (MS) and the Cooley’s Anemia Foundation (MS) for transduction, biosafety and clinical costs, and Errant Gene Therapy (MS) for TNS9.3.55 vector lot, produced at the Center for Biomedicine and Genetics, Duarte, CA. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. We also thank Scott Avecilla, Qing He, Clare Taylor, Mitsu Fink, Teresa Wasielewska, Shirley Bartido, Yongzeng Wang and the members of the Cell Therapy and Cell Engineering Facility who, for seven years, have assisted in the manufacturing and monitoring of the CD34+ cell infusion products for our patients.
Footnotes
Competing Interest Statement
The authors declare no competing financial interest. The TNS9.3.55 vector technology has been granted to Errant Gene Therapy without financial compensation.
References
- 1.Taher AT, Weatherall DJ & Cappellini MD Thalassaemia. Lancet 391, 155–167 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Weatherall DJ, and Clegg JB. The Thalassemia Syndrome, (Blackwell Scientific, Oxford, 1981). [Google Scholar]
- 3.Kountouris P, et al. IthaGenes: an interactive database for haemoglobin variations and epidemiology. PLoS One 9, e103020 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orkin S, and Nathan DG. Hematology of Infancy and Childhood, (W. B. Saunders, Philadelphia, PA, 1998). [Google Scholar]
- 5.Stamatoyannopoulos G The molecular basis of blood diseases, (W.B. Saunders, Philadelphia, 2001). [Google Scholar]
- 6.Borgna-Pignatti C, et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica 89, 1187–1193 (2004). [PubMed] [Google Scholar]
- 7.Ladis V, et al. Survival in a large cohort of Greek patients with transfusion-dependent beta thalassaemia and mortality ratios compared to the general population. Eur J Haematol 86, 332–338 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Mancuso A, Sciarrino E, Renda MC & Maggio A A prospective study of hepatocellular carcinoma incidence in thalassemia. Hemoglobin 30, 119–124 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Sadelain M, et al. Therapeutic options for patients with severe beta-thalassemia: the need for globin gene therapy. Hum Gene Ther 18, 1–9 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Lucarelli G, Isgro A, Sodani P & Gaziev J Hematopoietic stem cell transplantation in thalassemia and sickle cell anemia. Cold Spring Harb Perspect Med 2, a011825 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baronciani D, et al. Hematopoietic Cell Transplantation in Thalassemia and Sickle Cell Disease: Report from the European Society for Blood and Bone Marrow Transplantation Hemoglobinopathy Registry: 2000–2017. Blood 132, 168 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Fitzhugh CD, Abraham A & Hsieh MM Alternative Donor/Unrelated Donor Transplants for the beta-Thalassemia and Sickle Cell Disease. Adv Exp Med Biol 1013, 123–153 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weatherall DJ The challenge of haemoglobinopathies in resource-poor countries. Br J Haematol 154, 736–744 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Mansilla-Soto J, Riviere I, Boulad F & Sadelain M Cell and Gene Therapy for the Beta-Thalassemias: Advances and Prospects. Hum Gene Ther 27, 295–304 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrari G, Thrasher AJ & Aiuti A Gene therapy using haematopoietic stem and progenitor cells. Nat Rev Genet (2020). [DOI] [PubMed] [Google Scholar]
- 16.Magrin E, Miccio A & Cavazzana M Lentiviral and genome-editing strategies for the treatment of beta-hemoglobinopathies. Blood 134, 1203–1213 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Drysdale CM, et al. Hematopoietic-Stem-Cell-Targeted Gene-Addition and Gene-Editing Strategies for beta-hemoglobinopathies. Cell Stem Cell 28, 191–208 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Persons DA & Tisdale JF Gene therapy for the hemoglobin disorders. Semin Hematol 41, 279–286 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Sadelain M Recent advances in globin gene transfer for the treatment of beta-thalassemia and sickle cell anemia. Curr Opin Hematol 13, 142–148 (2006). [DOI] [PubMed] [Google Scholar]
- 20.May C, et al. Therapeutic haemoglobin synthesis in beta-thalassaemic mice expressing lentivirus-encoded human beta-globin. Nature 406, 82–86 (2000). [DOI] [PubMed] [Google Scholar]
- 21.May C, Rivella S, Chadburn A & Sadelain M Successful treatment of murine beta-thalassemia intermedia by transfer of the human beta-globin gene. Blood 99, 1902–1908 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Rivella S, May C, Chadburn A, Riviere I & Sadelain M A novel murine model of Cooley anemia and its rescue by lentiviral-mediated human beta-globin gene transfer. Blood 101, 2932–2939 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Lisowski L & Sadelain M Locus control region elements HS1 and HS4 enhance the therapeutic efficacy of globin gene transfer in beta-thalassemic mice. Blood 110, 4175–4178 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perumbeti A & Malik P Therapy for beta-globinopathies: a brief review and determinants for successful and safe correction. Ann N Y Acad Sci 1202, 36–44 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Quek L & Thein SL Molecular therapies in beta-thalassaemia. Br J Haematol 136, 353–365 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Boulad F, et al. Safe mobilization of CD34+ cells in adults with beta-thalassemia and validation of effective globin gene transfer for clinical investigation. Blood 123, 1483–1486 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Felfly H & Trudel M Successful correction of murine sickle cell disease with reduced stem cell requirements reinforced by fractionated marrow infusions. Br J Haematol 148, 646–658 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Bartelink IH, et al. Association of busulfan exposure with survival and toxicity after haemopoietic cell transplantation in children and young adults: a multicentre, retrospective cohort analysis. Lancet Haematol 3, e526–e536 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu J, et al. Isolation and functional characterization of human erythroblasts at distinct stages: implications for understanding of normal and disordered erythropoiesis in vivo. Blood 121, 3246–3253 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Villa E, et al. The E3 ligase UBR2 regulates cell death under caspase deficiency via Erk/MAPK pathway. Cell Death Dis 11, 1041 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berry CC, Ocwieja KE, Malani N & Bushman FD Comparing DNA integration site clusters with scan statistics. Bioinformatics 30, 1493–1500 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoon JK, et al. HIV proviral DNA integration can drive T cell growth ex vivo. Proc Natl Acad Sci U S A 117, 32880–32882 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Katano H, et al. Integration of HIV-1 caused STAT3-associated B cell lymphoma in an AIDS patient. Microbes Infect 9, 1581–1589 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bushman FD Retroviral Insertional Mutagenesis in Humans: Evidence for Four Genetic Mechanisms Promoting Expansion of Cell Clones. Mol Ther 28, 352–356 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thompson AA, et al. Gene Therapy in Patients with Transfusion-Dependent beta-Thalassemia. N Engl J Med 378, 1479–1493 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Schneiderman J, et al. Interim Results from the Phase 3 Hgb-207 (Northstar-2) and Hgb-212 (Northstar-3) Studies of Betibeglogene Autotemcel Gene Therapy (LentiGlobin) for the Treatment of Transfusion-Dependent β-Thalassemia. Biology of Blood and Marrow Transplantation 26, S87–S88 (2020). [Google Scholar]
- 37.Frangoul H, et al. Safety and Efficacy of CTX001 in Patients with Transfusion-Dependent β-Thalassemia and Sickle Cell Disease: Early Results from the Climb THAL-111 and Climb SCD-121 Studies of Autologous CRISPR-CAS9-Modified CD34+ Hematopoietic Stem and Progenitor Cells. Blood 136, 3–4 (2020).32614960 [Google Scholar]
- 38.Grochow LB, et al. Pharmacokinetics of busulfan: correlation with veno-occlusive disease in patients undergoing bone marrow transplantation. Cancer Chemother Pharmacol 25, 55–61 (1989). [DOI] [PubMed] [Google Scholar]
- 39.Lawson R, Staatz CE, Fraser CJ & Hennig S Review of the Pharmacokinetics and Pharmacodynamics of Intravenous Busulfan in Paediatric Patients. Clin Pharmacokinet 60, 17–51 (2021). [DOI] [PubMed] [Google Scholar]
- 40.Strouse C, et al. Risk Score for the Development of Veno-Occlusive Disease after Allogeneic Hematopoietic Cell Transplant. Biol Blood Marrow Transplant 24, 2072–2080 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marktel S, et al. Platelet transfusion refractoriness in highly immunized beta thalassemia children undergoing stem cell transplantation. Pediatr Transplant 14, 393–401 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Saito AM, et al. Lower costs associated with hematopoietic cell transplantation using reduced intensity vs high-dose regimens for hematological malignancy. Bone Marrow Transplant 40, 209–217 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Svahn BM, Alvin O, Ringden O, Gardulf A & Remberger M Costs of allogeneic hematopoietic stem cell transplantation. Transplantation 82, 147–153 (2006). [DOI] [PubMed] [Google Scholar]
- 44.La Nasa G, et al. Long-term health-related quality of life evaluated more than 20 years after hematopoietic stem cell transplantation for thalassemia. Blood 122, 2262–2270 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Jones RJ & DeBaun MR Leukemia after gene therapy for sickle cell disease: insertional mutagenesis, busulfan, both or neither. Blood (2021). [DOI] [PubMed] [Google Scholar]
- 46.Hsieh MM, et al. Myelodysplastic syndrome unrelated to lentiviral vector in a patient treated with gene therapy for sickle cell disease. Blood Adv 4, 2058–2063 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Y, et al. Myeloid neoplasms in the setting of sickle cell disease: an intrinsic association with the underlying condition rather than a coincidence; report of 4 cases and review of the literature. Mod Pathol 32, 1712–1726 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Seminog OO, Ogunlaja OI, Yeates D & Goldacre MJ Risk of individual malignant neoplasms in patients with sickle cell disease: English national record linkage study. J R Soc Med 109, 303–309 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brunson A, et al. Increased risk of leukemia among sickle cell disease patients in California. Blood 130, 1597–1599 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grimley M, et al. Early Results from a Phase 1/2 Study of Aru-1801 Gene Therapy for Sickle Cell Disease (SCD): Manufacturing Process Enhancements Improve Efficacy of a Modified Gamma Globin Lentivirus Vector and Reduced Intensity Conditioning Transplant. Blood, 20–21 (2020). [Google Scholar]
- 51.Scaramuzza S, et al. Clinical outcomes from a phase I/II gene therapy trial for patients affected by severe transfusion dependent beta-thalassemia: two years follow up. Mol Ther 28, 169 (2020). [Google Scholar]
- 52.Marktel S, et al. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent ss-thalassemia. Nat Med 25, 234–241 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Cavazzana-Calvo M, et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature 467, 318–322 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bank A, Dorazio R & Leboulch P A phase I/II clinical trial of beta-globin gene therapy for beta-thalassemia. Ann N Y Acad Sci 1054, 308–316 (2005). [DOI] [PubMed] [Google Scholar]
- 55.Ikeda K, Mason PJ & Bessler M 3’UTR-truncated Hmga2 cDNA causes MPN-like hematopoiesis by conferring a clonal growth advantage at the level of HSC in mice. Blood 117, 5860–5869 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murakami Y, et al. Deregulated expression of HMGA2 is implicated in clonal expansion of PIGA deficient cells in paroxysmal nocturnal haemoglobinuria. Br J Haematol 156, 383–387 (2012). [DOI] [PubMed] [Google Scholar]
- 57.Yu KR, et al. HMGA2 regulates the in vitro aging and proliferation of human umbilical cord blood-derived stromal cells through the mTOR/p70S6K signaling pathway. Stem Cell Res 10, 156–165 (2013). [DOI] [PubMed] [Google Scholar]
- 58.Bottardi S, Ross J, Pierre-Charles N, Blank V & Milot E Lineage-specific activators affect beta-globin locus chromatin in multipotent hematopoietic progenitors. EMBO J 25, 3586–3595 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jimenez G, Griffiths SD, Ford AM, Greaves MF & Enver T Activation of the beta-globin locus control region precedes commitment to the erythroid lineage. Proc Natl Acad Sci U S A 89, 10618–10622 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schumm M, et al. Isolation of highly purified autologous and allogeneic peripheral CD34+ cells using the CliniMACS device. J Hematother 8, 209–218 (1999). [DOI] [PubMed] [Google Scholar]
- 61.Alter BP, Rappeport JM, Huisman TH, Schroeder WA & Nathan DG Fetal erythropoiesis following bone marrow transplantation. Blood 48, 843–853 (1976). [PubMed] [Google Scholar]
- 62.Yang S, et al. A simple and effective method to generate lentiviral vectors for ex vivo gene delivery to mature human peripheral blood lymphocytes. Hum Gene Ther Methods 23, 73–83 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Charrier S, et al. Quantification of lentiviral vector copy numbers in individual hematopoietic colony-forming cells shows vector dose-dependent effects on the frequency and level of transduction. Gene Ther 18, 479–487 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shear HL, et al. Transgenic mice expressing human fetal globin are protected from malaria by a novel mechanism. Blood 92, 2520–2526 (1998). [PubMed] [Google Scholar]
- 65.Berry CC, et al. INSPIIRED: Quantification and Visualization Tools for Analyzing Integration Site Distributions. Mol Ther Methods Clin Dev 4, 17–26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sherman E, et al. INSPIIRED: A Pipeline for Quantitative Analysis of Sites of New DNA Integration in Cellular Genomes. Mol Ther Methods Clin Dev 4, 39–49 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Berry CC, et al. Estimating abundances of retroviral insertion sites from DNA fragment length data. Bioinformatics 28, 755–762 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. All requests for raw and analyzed data and materials are promptly reviewed by the corresponding author to verify if the request is subject to any intellectual property or confidentiality obligations. Patient-related data not included in the paper were generated as part of clinical trials and may be subject to patient confidentiality. Any data and materials that can be shared will be released via a material transfer agreement.
The following databases were used for For oncogenes definitions: the “Bushman lab oncogenes database” (http://www.bushmanlab.org/links/genelists, v5 June 2021) and four levels of The Cancer Genome Atlas (TCGA) version of the OncoVar database (https://oncovar.org, v1.2 August 2020).
Sequence data were deposited in the NCBI’s Sequence Read Archive (SRA BioProject PRJNA705203). For integration site analysis relative to Gene ontology the “GO.db” Bioconductor annotation data package v3.13.0 was used.