

Abstract
Despite the long-standing observation of vast neuronal loss in Alzheimer’s disease (AD) our understanding of how and when neurons are eliminated is incomplete. While previous investigation has focused on apoptosis, several novel forms of cell death (i.e. necroptosis, parthanatos, ferroptosis, cuproptosis) have emerged that require further investigation. This review aims to collect evidence for different modes of neuronal cell death in AD and to also discuss how these different forms of cell death may impact the neuroinflammatory environment that prevails in the AD brain. Improved understanding of how neurons die may help to delineate disease pathogenesis, provide insights toward treatment, and aid in the development of improved animal models of AD.
Keywords: Alzheimer’s disease, cell death, neuroinflammation, apoptosis, necrosis, necroptosis, pyroptosis, ferroptosis, parthanatos, microglia, phagocytosis, amyloid beta, neurodegenerative disease, multiple sclerosis, neuroimmunology, disease-associated microglia
Introduction: The Shrinking Brain
Alzheimer’s disease (AD) is the most common form of dementia affecting 1 in 9 individuals over the age of 65 in the United States [1]. It is characterized by extracellular deposition of amyloid-β (Aβ) plaques and intraneuronal, tau-containing neurofibrillary tangles (NFT), both of which are thought to provoke neuroinflammation, neuronal dysfunction, and progressive neuronal loss. Despite the significant healthcare, economic, and social burden that AD imposes, there remains no effective treatment to slow disease progression.
Pathological examination of patients with AD suggests accelerated loss of brain mass and neuronal loss even in mild AD [2]. Despite this longstanding observation, the specific forms of cell death that contribute to neurodegeneration in AD are not fully defined. Answering the question of how and when neurons die is significant as it will help to uncover therapeutic targets to counter neurodegeneration. Several reasons likely underlie the lack of knowledge in this area. For one, neuronal death in AD patients and in models of disease is a gradual process; thus, histopathological assessment can only provide a snapshot of unfolding cell death. Additionally, knowledge of how cells die, and how to recognize different modes of cell death, is rapidly evolving as evidenced by the recent identification of multiple novel cell death pathways including necroptosis [3], ferroptosis [4], parthanatos [5], and cuproptosis [6]. Whether these recently identified forms of cell death contribute to AD is an active area of research. The goals of this review are to describe, in brief, the involvement of known forms of neuronal death, especially non-apoptotic forms, in AD and to further discuss their contributions to neuroinflammation.
How Alzheimer’s Disease Kills Neurons
Apoptosis
Apoptosis was the first form of regulated cell death to be described [7] and is the mode of cell death that has been most extensively explored in AD. Neuronal apoptosis in AD has been extensively studied and reviewed elsewhere [8–10] and, therefore, is not the focus in this review. Notably, however, given how potent Aβ has been reported to be in driving neuronal apoptosis in cell culture studies [11–15] and following intracerebral injections [16], it is somewhat surprising that more widespread neuronal apoptosis is not observed in post-mortem tissue samples of AD patients [17]. This may be due to increased resistance to apoptosis by mature neurons. Neurons are post-mitotic and, except for a few special circumstances, are not able to be replaced once lost. Homeostatic, mature neurons are largely resistant to apoptosis due to upregulation of anti-apoptotic Bcl-2 family proteins and downregulation of both the pro-apoptotic protein Bax and several pro-apoptotic BH3-only proteins [18]. Given the resistance of neurons to apoptotic cell death, consummate apoptosis may be a relatively infrequent event but, nonetheless, may contribute to neuronal loss in AD over time. Alternatively, several other forms of cell death may account for the widespread neuronal loss in AD.
Necrosis and Necroptosis
Necrosis is a form of cell death that results from overwhelming external injury (e.g. heat, pathogens, ischemia, etc.). It is characterized by organelle swelling (oncosis), rupture of the plasma membrane, and the release of pro-inflammatory, intracellular contents. This description of a passive necrosis can be contrasted with a regulated form of necrotic cell death known as necroptosis. Necroptosis is carried out by the effector proteins RIPK1 and RIPK3, and the executioner protein MLKL, which once activated oligomerizes and forms MLKL pores in the plasma membrane leading to cell lysis (Figure 1) [19]. In the absence of Caspase-8 activity, which directs cells towards apoptotic cell death, necroptosis can be activated by death receptors (TNFR, Fas, TRAIL-R) and pattern recognition receptors (PRRs) (e.g. TLR3 and TLR4), among other triggers [20].
Figure 1: Modes of programmed cell death that contribute to neurodegenerative disease.
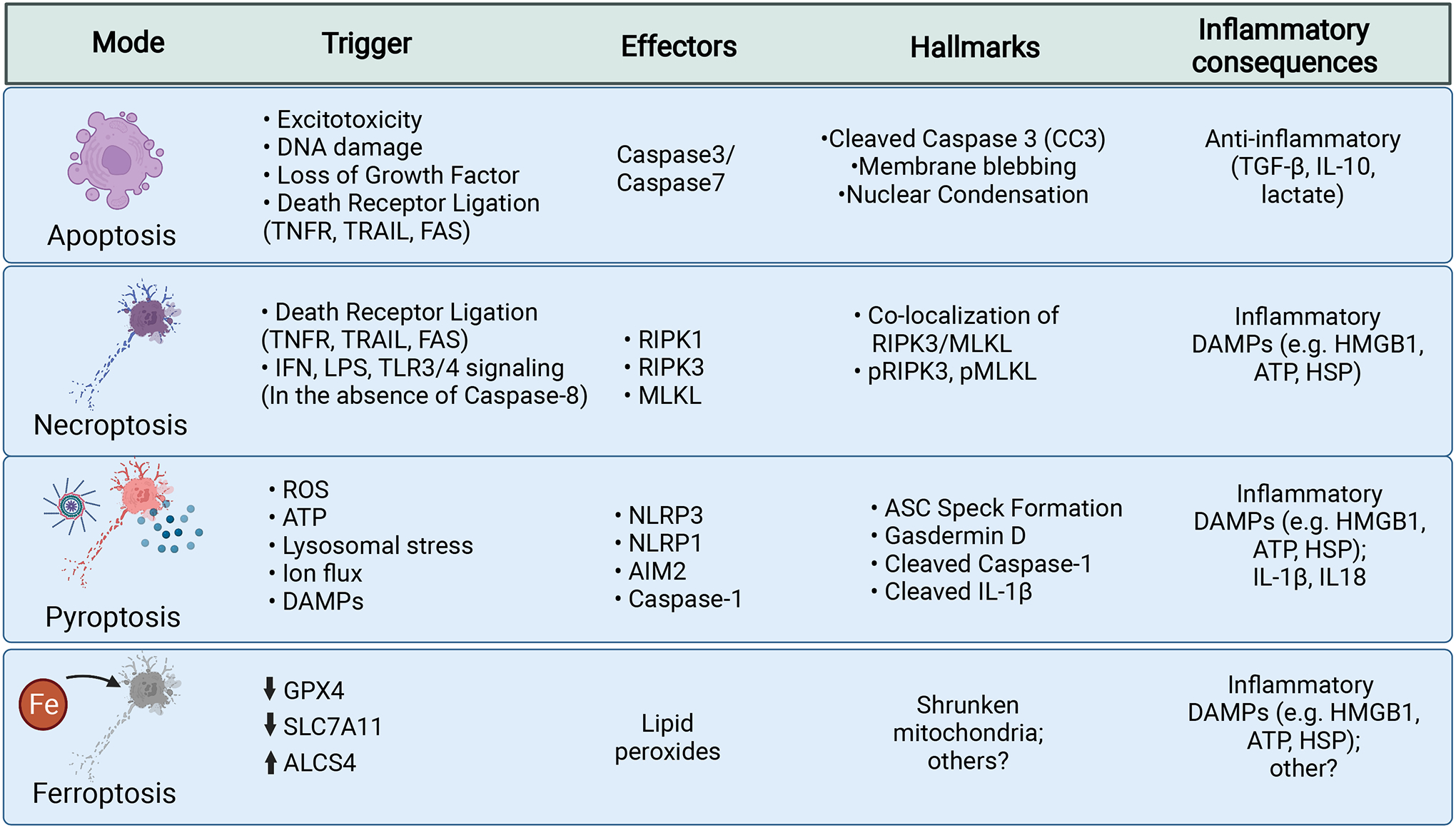
Cell death mechanisms are listed here including triggers for cell death, the molecular effectors, hallmarks of cell death, and the associated inflammatory consequences.
Apoptosis is a silent, or non-inflammatory, form of cell death that leads to cellular clearance without release of intracellular components. It is triggered by diverse stimuli and is enacted through two broad pathways: intrinsic and extrinsic apoptosis. These pathways converge on cleavage of Caspase-3. Cleaved Caspase-3 activates proteins to manifest the hallmarks of nuclear condensation (pyknosis), DNA fragmentation (karyorrhexis), cytoplasmic condensation, and membrane blebbing. Apoptosis leads to the release of TGF-β, IL-10, and lactate, which maintain homeostasis and provide anti-inflammatory signals.
Necroptosis can be initiated by several signals in the absence of Caspase-8. Canonically, necroptosis takes place downstream of death receptors (TNFR, TRAIL, FAS). These receptors recruit RIPK1, which is autophosphorylated and goes on to phosphorylate and activate RIPK3, which then acts to phosphorylate MLKL. pMLKL is then able to oligomerize into a pore like structure at the plasma membrane leading to loss of cell polarity and eventual lysis. Necroptosis is an inflammatory form of cell death that is associated with the release of intracellular contents that can act as DAMPs.
Pyroptosis similarly results in cell lysis via membrane pore formation. It is initiated by the integration of two signals. The first primes the inflammasome by producing inactive substrates. The second signal is generated by intracellular pattern recognition receptors (e.g. NLRP3, NLRP1, AIM2) that are activated by diverse triggers and go on to form inflammasome complexes. Activation of these sensors leads to recruitment of the adaptor ASC and the activation of Caspase-1 activation. Active Caspase-1 cleaves substrates including pro-IL-1β, pro-1L-18, and Gasdermin D to their active forms. Active Gasdermin D oligomerizes and forms pores in the plasma membrane, leading to release of cytokines and intracellular components, loss of membrane integrity, and eventual cell lysis. Pyroptosis is an inflammatory cell death that is associated with the release of proinflammatory cytokines and intracellular DAMPs.
Ferroptosis results from the build-up of toxic lipid hydroperoxides that form through lipids undergoing Fenton chemistry reactions with iron and interactions with other free-radicals, and also by the activity of lipoxygenase enzymes. Hydroperoxides lead to the generation of additional free-radical species that can damage lipids, proteins, and nucleic acids; although the ultimate executioner of cell death in unknown. GPX4 provides cellular defense against ferroptosis by neutralizing lipid hydroperoxides into lipid alcohols. Reductions in GPX4 or SLC7A11 activity, which provides reduced glutathione necessary for GPX4 activity, sensitizes cells to ferroptosis. Increases in the specific lipid substrates susceptible to peroxidation by enzymes such as ALCS4 can also sensitize cells. Ferroptosis is likely associated with release of intracellular contents that act as DAMPs. While, there are currently no widely used markers of ferroptosis, it has been noted that mitochondria in cells undergoing ferroptosis exhibit a fragmented/shrunken appearance, which can be evaluated through electron microscopy.
Early investigations using electron microscopy provided evidence that primary cortical neurons underwent necrotic, and not apoptotic, death when exposed to Aβ peptides. However, given what we know today it should be mentioned that this early study would not have been able to differentiate between various forms of lytic cell death [21]. Intriguingly, Tanaka and colleagues showed more recent evidence that neuronal necrosis plays a role in dementia and that it peaks at the stage of mild cognitive impairment (MCI) before the manifestation of clinical AD. They found elevated levels of high mobility group box-1 (HMGB1), a damage-associated molecular pattern (DAMP) released from necrotic cells, in the cerebrospinal fluid of MCI patients. This group also reported that YES-associated protein (YAP) was sequestered in intra-neuronal Aβ aggregates in MCI and AD tissue samples, and they hypothesize that loss of YAP activity leads to Hippo-dependent necrosis. This hypothesis was further supported by the observation that overexpression of YAP in 5xFAD mice rescued cognitive impairments and endoplasmic reticulum ballooning in neurons [22].
Multiple lines of evidence also support a role for necroptosis in AD. Yang and colleagues demonstrated reduced cell death in Aβ oligomer-treated immortalized hippocampal (HT22) cells upon treatment with necrostatin-1 (Nec-1), a pharmacological inhibitor of RIPK1, and subsequent necroptosis. Notably, they also showed a reduction in cortical plaque numbers in the APP/PS1 model of AD after i.v. administration of Nec-1 [23]. Studies from AD patients similarly point toward a role for necroptosis. For instance, Caccamo and colleagues observed increased RIPK1 and MLKL protein in post-mortem tissue of AD patients. They also showed increased colocalization of RIPK3 and MLKL, as evidence of necrosome formation, and additionally, necrosome formation was inversely correlated with brain weight and mini-mental status examination (MMSE) scores [24]. Subsequent studies have demonstrated similar phenotypes. For instance, increased expression of the active, phosphorylated forms of RIPK3 and MLKL were found in hippocampal tissue of AD patients, and pRIPK3 and pMLKL-positive neuronal expression was inversely associated with the density of total neurons in the hippocampus [25]. Furthermore, necrosome components have also been shown to localize to neurons exhibiting granulovacuolar degeneration (GVD) and shown to be associated with NFT pathology in hippocampal tissue from AD patients [26]. These studies point towards a prominent role for necroptosis in AD and suggest that necroptosis inhibition may offer a strategy to limit AD pathogenesis.
Pyroptosis
Pyroptosis is an inflammatory form of cell death that is triggered by the assembly of multi-protein complexes known as inflammasomes (Figure 1). Inflammasomes activate Caspase-1 and allow for secretion of pro-inflammatory IL-1 family cytokines. They additionally activate the protein Gasdermin-D, which oligomerizes at the plasma membrane leading to pore formation, the release of cellular contents, and eventual cell lysis [27]. This potent, inflammatory cell death can alert the immune system to infection and damage and plays a protective role in the defense against specific pathogens [28]. In the brain, microglia express inflammasome components to the greatest extent, but expression in neurons has also been reported [29] during development [30], in infection [31], in brain hemorrhage [32], and in the context of neurodegeneration (e.g. Huntington’s disease [33]).
Inflammasome signaling and pyroptosis have been strongly implicated in AD using animal models. In early studies utilizing primary cell culture, Halle and colleagues showed that the NLRP3 inflammasome was activated in microglia as a result of Aβ-induced lysosomal stress [34]. Further investigations showed that whole-body knock-out of constituent inflammasome proteins significantly ameliorated pathology in the APP/PS1 model of AD [35]. More recently, it was demonstrated that tau-hyperphosphorylation and aggregation are dependent on the NLRP3 inflammasome in mice treated with intracerebral Aβ [36]. While mounting evidence indicates pivotal roles for microglial NLRP3 inflammasome activation and pyroptosis in AD, much less is currently known about the involvement of neuron-specific pyroptosis in neurodegenerative disease pathogenesis. However, a study by Tan and colleagues has shed some light on this by providing evidence that the NLRP1 inflammasome drives neuronal pyroptosis in the APP/PS1 mouse model of AD. They showed colocalization of NLRP1 components with neurons as well as a reduction in neuronal loss and improved cognition after administration of NLRP1 siRNA in the cortex and hippocampus. Additionally, they reported that in vitro treatment of neurons with Aβ oligomers incites NLRP1-mediated pyroptosis [37]. A potential role for the NLRP1 inflammasome is also supported by evidence of NLRP1 genetic variants that are associated with AD [38]. Despite these advancements, further investigation is still required to fully characterize the role of neuron-specific pyroptosis. Current evidence supports several mechanisms by which AD pathology might drive activation of inflammasomes in neurons including lysosomal stress, ion flux, cell-death related DAMPs, and reactive oxygen species [39]. Another potential trigger of neuronal inflammasome activation and pyroptosis could be the hallmark of aggregated, hyperphosphorylated tau within neurons, as Tau has also been shown to activate the NLRP3 inflammasome in microglia [40].
Other forms of cell death (Ferroptosis, Parthanatos, Cuproptosis)
Other recently described forms of cell death may also contribute to neuronal loss in AD. Ferroptosis is an iron-dependent form of cell death that is driven by accumulation of reactive lipid peroxides (Figure 1) [41]. Intriguingly, the central regulator of ferroptosis, glutathione peroxidase-4 (GPX4), which converts toxic lipid peroxides into non-toxic lipid alcohols [42–44], was recently highlighted in an unbiased CRISPR screen to identify proteins necessary for neuronal survival under conditions of oxidative stress, which is common in many neurodegenerative diseases [45]. Supporting its role in neuronal viability, inducible neuron-specific deletion of GPX4 under the control of the Thy1 promoter in mice was shown to provoke rapid degeneration of motor neurons, paralysis, and death [46]. Likewise, it was also found that targeted deletion of GPX4 in Camk2α-expressing neurons leads to hippocampal neurodegeneration and cognitive deficits [47]. Moreover, ferroptosis has been implicated in other models of neurodegenerative disease including those used to study multiple sclerosis [48], stroke [49], amyotrophic lateral sclerosis [50,51], and Parkinson’s disease [52,53]. In further support of a role for ferroptosis in AD, it has also recently been shown that global over-expression of GPX4 in the 5xFAD mouse model limits both cognitive decline and the loss of cortical layer 5 neurons [54]. While increasing evidence suggests a potential role for ferroptosis in AD, additional studies are needed to further define specific markers of ferroptosis that can be deployed to measure its activation in AD patient samples [55].
Parthanatos is a form of cell death driven by the overactivation of poly (ADP-ribose)-polymerase-1 (PARP-1). More specifically, hyperactive PARP-1 leads to the synthesis of PAR polymers which can cause mitochondrial membrane depolarization and the release of apoptosis inducing factor (AIF). AIF can then translocate to the nucleus where it plays a role in initiating DNA fragmentation and cell death [56,57]. PARP activity is stimulated by DNA damage and plays physiological roles in DNA damage repair [58]. Parthanatos may contribute to AD pathogenesis as increased DNA damage [59,60] and diminished DNA repair activity [61,62] have been observed in tissue from AD patients. Moreover, increased activity of PARP-1 has also been observed in the brains of AD patients [63] and AD mouse models [64]. Despite these connections that point towards a potential role for parthanatos in AD, this has not been studied in great detail to date and, thus, requires further investigation.
Cuproptosis has recently been proposed as a novel mode of cell death by which aggregation of lipoylated proteins and proteotoxic stress resulting from excessive copper accumulation leads to cellular demise [6]. While reports measuring copper in the AD brain are somewhat contradictory, there is evidence that increased labile copper may be associated with oxidative tissue damage in the brains of AD patients [65]; therefore, future studies exploring the role of cuproptosis in neurons and AD are warranted.
Neuronal cell death as a driver of CNS inflammation
In the relationship between neurons and inflammation in AD, it is convention to conceptualize inflammation as an actor upon neurons. Despite this convention, however, it is also the case that neurons can play an active role in spurring inflammation. This is the case with inflammatory forms of neuronal death that signal existential threats to neurons and glia (Figure 2). This bi-directional understanding of neuroinflammation in AD situates neurons as recipients and instigators of inflammation and may provide a fuller picture of the inflammatory landscape present in CNS disease. Moreover, characterizing the specific modes of cell death might aid in the understanding of how neurons may generate inflammation.
Figure 2: Neuronal cell death as a driver of neuroinflammation in AD.
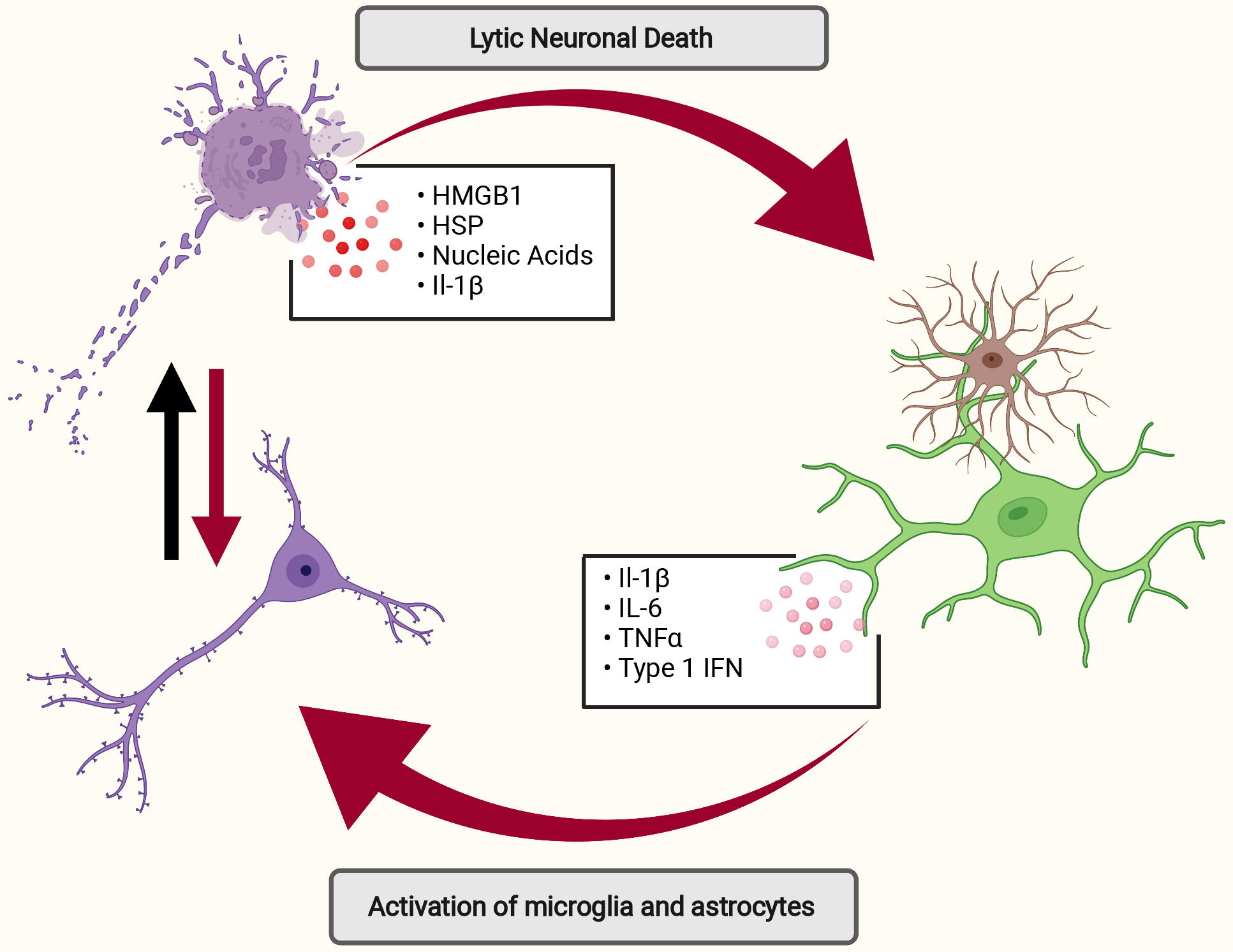
Lytic neuronal cell death is associated with the release of pro-inflammatory DAMPs (e.g. HMGB1, HSP, and nucleic acids) which can incite and perpetuate inflammatory responses in glial cells or directly contribute to neuronal dysfunction (red arrows-path of inflammation). Conversely, inflammation may sensitize neurons to cell death and increase neuronal death (black arrow). Specific forms of neuronal death may be more inflammatory (i.e. pyroptosis requires activation of inflammasome complexes that cleave and activate pro-inflammatory IL-1 family cytokine) or less inflammatory through neuronal apoptosis, which is associated with the release of anti-inflammatory factors (e.g. TGF-β, IL-10, and lactate). Aβ, amyloid beta; AD, Alzheimer’s disease; DAMPs, damage-associated molecular patterns; HMGB1, High Mobility Group; HSP, heat shock protein.
Apoptosis is typically an anti-inflammatory form of cell death. The apoptotic program leads to the release of anti-inflammatory factors such as lactate, IL-10, and TGF- β [66]. The result of apoptosis, however, can vary depending on the tissue and disease context. For instance, there is evidence that neuronal apoptosis may in fact disrupt the homeostatic microglial phenotype [67]. Other forms of cell death have more direct consequences for neuroinflammation. Lytic cell death as seen in necrosis, necroptosis, pyroptosis, and ferroptosis leads to the release of intracellular components that act as DAMPs to incite further inflammation. These include HMGB1, heat shock proteins, and nucleic acids which signal through PRRs (e.g. TLR4, AIM2) [68] to perpetuate neuritic damage and trigger aberrant microglial activation [69]. Neurons that undergo pyroptotic cell death also lead to the release of potent inflammatory cytokines such as IL-1β [70]. IL-1β plays diverse roles in the brain with effects that include the activation of microglia and the loss of neuronal synapses [71]. Additionally, activation of necroptosis has also been associated with activation of the NLRP3 inflammasome and release of IL-1β [72]. Moreover, lack of Caspase-8 activity downstream of death receptor engagement, as is found in necroptotic cell death, has also been shown to disinhibit inflammatory pathways [19]. The unique consequences of cell death and mechanism of the execution in ferroptosis have yet to be unraveled, although given its lytic nature it is likely that ferroptosis may lead to the release of intracellular contents and DAMPs that can initiate inflammation [73]. Finally, parthanatos can lead to increased inflammation as a result of unchecked PARP-1 activation. However, while evidence exists to suggest that PARP-1 activation leads to pro-inflammatory responses in microglia [74], the consequences of PARP-1 stimulation on inflammatory cytokine production by neurons are less well defined and this remains an important area of future investigation.
Non-apoptotic cell death is associated with loss of homeostasis and often contributes to local inflammation through the release of DAMPs. Given the strong evidence for several forms of non-apoptotic neuronal cell death in AD, it is likely that neurons play integral roles in both the initiation and propagation of inflammatory responses during AD progression. Therefore, intervening in neuronal cell death may quell the neuroinflammatory environment that contributes to damage in AD.
Conclusion
How neurons are lost in AD is not well defined. It is apparent that several different forms of cell death may contribute to neuronal loss either independently or simultaneously. The study of neuronal cell death is only one thread in a much larger conversation surrounding the pathogenesis of AD. Recent research in the field has focused on the role that the brain resident macrophage, microglia, play in AD progression. This microglial focus largely stems from recent studies in AD patients indicating that many of the genetic risk factors for late-onset AD are strongly or, in some cases, exclusively expressed in microglia [75]. This has greatly improved our understanding of the contributions of immune signaling in disease. Concurrently, however, our understanding of how cells die and the overlapping pathways of cell death that can contribute to dysfunction, has evolved. Renewed interest and understanding of how neurons are dying in AD, and other neurodegenerative diseases, will help to provide new insights into neurodegenerative disease pathoetiology and may lead to novel therapeutic interventions. Improved understanding of how glia impact pathways leading to neuronal death and how glia survive in AD will also be an important area of future investigation.
Table 1:
Evidence for different mechanisms of neuronal death in AD
| Cell Death | Authors | Model System | Finding |
|---|---|---|---|
| Apoptosis | [13] Loo et al. 1993 | Primary mouse neuron | In vitro exposure of hippocampal neurons to Aβ peptides led to ultrastructural features of apoptosis in electron microscopy imaging studies. |
| [17] Stadelmann et al. 1998 | Human tissue | Tissue samples from patients with AD and ponstosubicular neuron necrosis (PSNN), included as a positive control for neuronal apoptosis, were compared to evaluate if neurons in the AD brain exhibit microstructural features of apoptosis. Many of the neurons in the AD samples showed DNA fragmentation but very few showed associated hallmarks of apoptosis. | |
| [14] Ivins, et al. 1999 | Primary neurons | Caspase-8 inhibitor prevented Aβ-induced cell death in neurons; induction of dominant negative FADD also protected neurons. This indicates a possible role for extrinsic apoptosis in AD. | |
| [12] Yao, Nguyen, and Pike 2005 | Primary rat neurons | Aβ significantly reduced the expression of the antiapoptotic molecules Bcl-w and Bcl-xL in primary rat neurons. Overexpression of Bcl-w rescued neuronal cell death upon exposure to Aβ. | |
| [76] Rohn et al. 2008 | 3xTG | Overexpression of Bcl-2 decreased caspase-9 cleavage, reduced NFT and plaque formation, and improved place recognition memory in 3xTG mice. | |
| [16] Kudo et al. 2012 | C57B6; hippocampal slice culture | Aβ increased Bim but decreased Bcl-2 levels and led to the activation of Bax and neuronal cell death in both hippocampal slice culture and in vivo. Inhibition of Bax through Bax-inhibiting peptide or Bax gene knockout prevented Aβ-induced neuronal cell death. | |
| [15] Robbins et al. 2018 | hiPSC neuron culture | Aβ peptide induced Caspase-3 cleavage in hiPSC neurons. Clusterin KO prevented reductions in neurite length upon Aβ exposure. | |
| [77] Zhang et al. 2021 | 5xFAD | Increased BAD protein levels in AD tissue samples and in 5xFAD mice. Disruption of Bad alleles rescued spatial and leaning deficits in 5xFAD. | |
| Pyroptosis | [34] Halle et al. 2008 | Primary mouse neuron | Showed that primary microglia activate the NLRP3 inflammasome in response to fibrillar Aβ. This process is dependent on microglial phagocytosis, lysosomal stress, and cathepsin B release, which lead to subsequent NLRP3 activation. |
| [35] Heneka et al. 2013 | APP/PS1 | Nlrp3−/−APP/PS1 and Casp1−/−APP/PS1 mice were both protected from loss of spatial memory and other sequelae associated with AD. These mice demonstrated reduced brain Caspase-1 and interleukin-1β activation as well as enhanced amyloid-β clearance. Increased cleaved Caspase-1 was found in AD/MCI patient tissue. | |
| [37] Tan et al. 2014 | APP/PS1 / primary culture | Aβ1–42 increased NLRP1 expression in primary cortical neurons, and siRNA-mediated knockdown of Nrlp1 increased neuronal viability after treatment with Aβ. siRNA targeted ablation of Nlrp1 in APP/PS1 mice increased neuronal survival in the hippocampus and cortex. | |
| [36] Ising et al. 2019 | APP/PS1 | Loss of NLRP3 inflammasome function reduced tau hyperphosphorylation and aggregation. Tau activated the NLRP3 inflammasome and intracerebral injection of fibrillar amyloid-beta-containing brain homogenates induced tau pathology in an NLRP3-dependent manner. | |
| Necrosis | [21] Behl et al. 1994 | Rat primary cortical neurons, PC12 cells | Early investigations using electron microscopy provided evidence that primary cortical neurons undergo necrosis when exposed to Aβ peptides; no apoptotic bodies or nuclear fragmentation was seen, while organellar damage and vacuolization was apparent. |
| [22] Tanaka et al. 2020 | Human tissue/hiPSC | The necrosis-associated protein HMGB1 was increased in the CSF of MCI, but not AD patients. Post-mortem tissue demonstrated increased staining of the necrosis marker pSer46-MARCKS in MCI but not AD cases compared to healthy controls. In vivo imaging revealed instability of endoplasmic reticulum (ER). Genome-edited human AD iPS cell-derived neurons exhibited decreased nuclear Yes-associated protein (YAP) due to the sequestration into cytoplasmic Aβ-aggregates, supporting the feature of YAP-dependent necrosis. | |
| Necroptosis | [24] Caccamo et al. 2017 | Human tissue | Elevated RIPK1 and MLKL protein expression, as well as increased colocalization of RIPK3 and MLKL were found in post-mortem tissue of AD patients indicating increased necroptosis. Necrosome formation was shown to be inversely correlated with brain weight and cognitive scores. Genes regulated by RIPK1 overlapped significantly with multiple independent AD transcriptomic signature. Treatment of 5xFAD mice with the RIPK1 inhibitor Nec-1 reduced attenuated neuronal degeneration. |
| [23] Yang et al. 2019 | HT22 cells/ APP/PS1 | Nec-1 was shown to directly disaggregate Aβ fibrils and oligomers, suggesting a cell-extrinsic function of the small-molecule. Nec-1 also prevented Aβ oligomer-induced cell death in both BV2 and HT22 cells. Finally, i.v. administration of Nec-1 (bi-weekly), for 4 weeks in 8 month old APP/PS1 mice reduced cortical plaque number. | |
| [26] Koper et al. 2020 | Human tissue | Activated necrosome components (RIPK1, RIPK3, MLKL) were detected in neurons proximal to GVD lesions and necrosome markers colocalized with GVD biomarkers (pTDP-43 and CK1δ). GVD neurons with activated necroptosis components inversely correlated with neuronal density in the early affected CA1 region of the hippocampus and in the late affected frontal cortex layer III. | |
| [25] Jayaraman et al. 2021 | Human tissue | CA1 pyramidal neurons from AD patients displayed increased levels of activated necroptotic proteins (e.g. pRIPK3 and pMLKL). The density of pRIPK3+ and pMLKL+ neurons also correlated inversely with total neuron density and showed significant sexual dimorphism within the AD cohort. Exposure of human iPSC-derived glutamatergic neurons to TNF increased necroptotic cell death when apoptosis was inhibited. | |
| Ferroptosis | [51] Chen et al. 2022 | 5xFAD | 5xFAD exhibited elevated levels of the lipid peroxidation end-product 4-HNE. Global overexpression of GPX4 rescued spatial learning deficits and prevented the loss of neurons in layer 5 of the cortex. |
Acronym Key: Aβ, amyloid beta; AD, Alzheimer’s disease; ER, endoplasmic reticulum; FADD, Fas-Associated Death Domain; GPX4, glutathione peroxidase-4; HMGB1, High Mobility Group Box-1; MCI, mild cognitive impairment; MLKL, Mixed lineage kinase domain-like protein; MMSE, mini-mental status examination; Nec-1, necrostatin-1; PSNN, ponstosubicular neuron necrosis; RIPK1/3, Receptor-interacting serine/threonine-protein kinase 1/3; YAP, Yes-associated protein.
Papers of Interest.
Chen et al. 2022 – This paper implicates a role for ferroptosis in a mouse model of Aβ amyloidosis. Chen and colleagues show that global overexpression of the ferroptosis regulator GPX4 limits neuronal cell death and rescues some behavioral phenotypes in 5xFAD mice.
Hambright et al. 2017 – This study was the first to explore the role of GPX4 in forebrain neurons. The rapid degeneration of neurons lacking GPX4, driven by the Camk2a promoter, suggests that neurons are particularly susceptible to changes to lipid-redox status. The resulting cognitive deficits found in their model parallel those found in other models of neurodegenerative disease suggesting a possible role for GPX4 driven redox homeostasis and ferroptosis in neurodegeneration.
Tian et al. 2021 – Tian and colleagues performed genome-wide CRISPR interference and CRISPR activation screens in human neurons to evaluate how the selective loss or activation of discrete proteins impacts neuronal survival. Their screen identified GPX4 as a pivotal regulator of neuronal viability under in vitro oxidative stress conditions.
Tsvetkov et al. 2022 – This work identifies cuproptosis as a novel form of programmed cell death. They show that excess copper binds to lipoylated proteins including the subunit of pyruvate dehydrogenase, dihydrolipoamide S-acetyltransferase (DLAT), which may lead to toxic gain of function or toxic protein aggregation. They found that cells with increased mitochondrial respiration were more sensitive to cuproptosis. This paper introduced a novel mechanism of cell death that warrants further investigation in AD and other neurodegenerative diseases.
Caccamo et al. 2017 – This 2017 paper was the first to suggest that necroptosis may play a role in an animal model of AD. Here they found that necroptosis activation levels correlate with disease severity in postmortem AD brains. They also demonstrate that over-expression of constitutively active MLKL in an Aβ-driven mouse model of AD leads to more severe cognitive deficits and neuronal loss.
Tan et al. 2014 – Despite the numerous papers implicating the NLRP3 inflammasome in AD, this paper was the first to directly provide evidence for neuronal cell death by pyroptosis stimulated by the NLRP1 inflammasome. This suggests that inhibition of pyroptosis may be a viable therapy for AD with several potential targets.
ACKNOWLEDGEMENTS
We apologize to authors whose work could not be referenced in this review due to space limitations. We thank members of the Lukens lab and the Center for Brain Immunology and Glia (BIG) for valuable discussions. Graphical illustrations were made using BioRender (https://biorender.com/). This work was supported by The National Institutes of Health/National Institute of Aging (1RF1AG071996-01; awarded to J.R.L.), The National Institutes of Health/National Institute of Neurological Disorders and Stroke (R01NS106383; awarded to J.R.L.), The Alzheimer’s Association (ADSF-21-816651; awarded to J.R.L.), the Cure Alzheimer’s Fund (awarded to J.R.L.), and The Owens Family Foundation (Awarded to J.R.L.). A.M. was supported by a Medical Scientist Training Program Grant (5T32GM007267-38).
Abbreviations:
- Aβ
amyloid beta
- AD
Alzheimer’s disease
- AIF
apoptosis inducing factor
- DAMPs
damage-associated molecular patterns
- ER
endoplasmic reticulum
- FADD
Fas-Associated Death Domain
- GPX4
glutathione peroxidase-4
- HMGB1
High Mobility Group Box-1
- MCI
mild cognitive impairment
- MLKL
Mixed lineage kinase domain-like protein
- MMSE
mini-mental status examination
- Nec-1
necrostatin-1
- NFT
neurofibrillary tangles
- PARP-1
poly (ADP-ribose)-polymerase-1
- PRRs
pattern recognition receptors
- PSNN
ponstosubicular neuron necrosis
- RIPK1/3
Receptor-interacting serine/threonine-protein kinase 1/3
- TNFR
Tumor necrosis factor receptor
- TRAIL
tumor-necrosis factor related apoptosis-inducing ligand
- YAP
Yes-associated protein
Footnotes
COMPETING INTERESTS
The authors declare no competing financial interests.
References
- 1.Alzheimer’s Disease Facts and Figures. Edited by. Alzheimers Dement 2022;18.: Alzheimer’s Association.; 2022. [DOI] [PubMed] [Google Scholar]
- 2.Price JL, Ko AI, Wade MJ, Tsou SK, McKeel DW, Morris JC: Neuron Number in the Entorhinal Cortex and CA1 in Preclinical Alzheimer Disease. Archives of Neurology 2001, 58:1395–1402. [DOI] [PubMed] [Google Scholar]
- 3.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J: Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nature Chemical Biology 2005, 1:112–119. [DOI] [PubMed] [Google Scholar]
- 4.Dixon Scott J, Lemberg Kathryn M, Lamprecht Michael R, Skouta R, Zaitsev Eleina M, Gleason Caroline E, Patel Darpan N, Bauer Andras J, Cantley Alexandra M, Yang Wan S, et al. : Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149:1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu T, Robotham James L, Yoon Y: Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proceedings of the National Academy of Sciences 2006, 103:2653–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, Rossen J, Joesch-Cohen L, Humeidi R, Spangler Ryan D, et al. : Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375:1254–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kerr JFR, Wyllie AH, Currie AR: Apoptosis: A Basic Biological Phenomenon with Wideranging Implications in Tissue Kinetics. British Journal of Cancer 1972, 26:239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fricker M, Tolkovsky AM, Borutaite V, Coleman M, Brown GC: Neuronal Cell Death. Physiological Reviews 2018, 98:813–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shimohama S: Apoptosis in Alzheimer’s disease--an update. Apoptosis 2000, 5:9–16. [DOI] [PubMed] [Google Scholar]
- 10.Cotman CW, Anderson AJ: A potential role for apoptosis in neurodegeneration and Alzheimer’s disease. Mol Neurobiol 1995, 10:19–45. [DOI] [PubMed] [Google Scholar]
- 11.Söllvander S, Nikitidou E, Brolin R, Söderberg L, Sehlin D, Lannfelt L, Erlandsson A: Accumulation of amyloid-β by astrocytes result in enlarged endosomes and microvesicle-induced apoptosis of neurons. Molecular Neurodegeneration 2016, 11:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yao M, Nguyen T-VV, Pike CJ: β-Amyloid-Induced Neuronal Apoptosis Involves c-Jun N-Terminal Kinase-Dependent Downregulation of Bcl-w. The Journal of Neuroscience 2005, 25:1149–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW: Apoptosis is induced by beta-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci U S A 1993, 90:7951–7955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ivins KJ, Thornton PL, Rohn TT, Cotman CW: Neuronal Apoptosis Induced by β-Amyloid Is Mediated by Caspase-8. Neurobiology of Disease 1999, 6:440–449. [DOI] [PubMed] [Google Scholar]
- 15.Robbins JP, Perfect L, Ribe EM, Maresca M, Dangla-Valls A, Foster EM, Killick R, Nowosiad P, Reid MJ, Polit LD, et al. : Clusterin Is Required for β-Amyloid Toxicity in Human iPSC-Derived Neurons. Frontiers in Neuroscience 2018, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kudo W, Lee HP, Smith MA, Zhu X, Matsuyama S, Lee Hg: Inhibition of Bax protects neuronal cells from oligomeric Aβ neurotoxicity. Cell Death & Disease 2012, 3:e309–e309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stadelmann C, Bruck W, Bancher C, Jellinger K, Lassmann H: Alzheimer Disease: DNA Fragmentation Indicates Increased Neuronal Vulnerability, but not Apoptosis. Journal of Neuropathology & Experimental Neurology 1998, 57:456–464. [DOI] [PubMed] [Google Scholar]
- 18.Hollville E, Romero SE, Deshmukh M: Apoptotic cell death regulation in neurons. Febs j 2019, 286:3276–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou W, Yuan J: Necroptosis in health and diseases. Seminars in Cell & Developmental Biology 2014, 35:14–23. [DOI] [PubMed] [Google Scholar]
- 20.Dhuriya YK, Sharma D: Necroptosis: a regulated inflammatory mode of cell death. Journal of Neuroinflammation 2018, 15:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Behl C, Davis JB, Klier FG, Schubert D: Amyloid β peptide induces necrosis rather than apoptosis. Brain Research 1994, 645:253–264. [DOI] [PubMed] [Google Scholar]
- 22.Tanaka H, Homma H, Fujita K, Kondo K, Yamada S, Jin X, Waragai M, Ohtomo G, Iwata A, Tagawa K, et al. : YAP-dependent necrosis occurs in early stages of Alzheimer’s disease and regulates mouse model pathology. Nature Communications 2020, 11:507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang S-H, Shin J, Shin NN, Hwang J-H, Hong S-C, Park K, Lee JW, Lee S, Baek S, Kim K, et al. : A small molecule Nec-1 directly induces amyloid clearance in the brains of aged APP/PS1 mice. Scientific Reports 2019, 9:4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caccamo A, Branca C, Piras IS, Ferreira E, Huentelman MJ, Liang WS, Readhead B, Dudley JT, Spangenberg EE, Green KN, et al. : Necroptosis activation in Alzheimer’s disease. Nature Neuroscience 2017, 20:1236–1246. [DOI] [PubMed] [Google Scholar]
- 25.Jayaraman A, Htike TT, James R, Picon C, Reynolds R: TNF-mediated neuroinflammation is linked to neuronal necroptosis in Alzheimer’s disease hippocampus. Acta Neuropathologica Communications 2021, 9:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koper MJ, Van Schoor E, Ospitalieri S, Vandenberghe R, Vandenbulcke M, von Arnim CAF, Tousseyn T, Balusu S, De Strooper B, Thal DR: Necrosome complex detected in granulovacuolar degeneration is associated with neuronal loss in Alzheimer’s disease. Acta Neuropathologica 2020, 139:463–484. [DOI] [PubMed] [Google Scholar]
- 27.Broz P, Dixit VM: Inflammasomes: mechanism of assembly, regulation and signalling. Nature Reviews Immunology 2016, 16:407–420. [DOI] [PubMed] [Google Scholar]
- 28.Man SM, Karki R, Kanneganti T-D: Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunological Reviews 2017, 277:61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zengeler KE, Lukens JR: Innate immunity at the crossroads of healthy brain maturation and neurodevelopmental disorders. Nature Reviews Immunology 2021, 21:454–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lammert CR, Frost EL, Bellinger CE, Bolte AC, McKee CA, Hurt ME, Paysour MJ, Ennerfelt HE, Lukens JR: AIM2 inflammasome surveillance of DNA damage shapes neurodevelopment. Nature 2020, 580:647–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yogarajah T, Ong KC, Perera D, Wong KT: AIM2 Inflammasome-Mediated Pyroptosis in Enterovirus A71-Infected Neuronal Cells Restricts Viral Replication. Scientific Reports 2017, 7:5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yan J, Xu W, Lenahan C, Huang L, Wen J, Li G, Hu X, Zheng W, Zhang JH, Tang J: CCR5 Activation Promotes NLRP1-Dependent Neuronal Pyroptosis via CCR5/PKA/CREB Pathway After Intracerebral Hemorrhage. Stroke 2021, 52:4021–4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paldino E, D’Angelo V, Sancesario G, Fusco FR: Pyroptotic cell death in the R6/2 mouse model of Huntington’s disease: new insight on the inflammasome. Cell Death Discovery 2020, 6:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT: The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nature Immunology 2008, 9:857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng T-C, et al. : NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493:674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, Schwartz S, Albasset S, McManus RM, Tejera D, et al. : NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575:669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tan MS, Tan L, Jiang T, Zhu XC, Wang HF, Jia CD, Yu JT: Amyloid-β induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer’s disease. Cell Death & Disease 2014, 5:e1382–e1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pontillo A, Catamo E, Arosio B, Mari D, Crovella S: NALP1/NLRP1 genetic variants are associated with Alzheimer disease. Alzheimer Dis Assoc Disord 2012, 26:277–281. [DOI] [PubMed] [Google Scholar]
- 39.Golzari-Sorkheh M, Brown CE, Weaver DF, Reed MA: The NLRP3 Inflammasome in the Pathogenesis and Treatment of Alzheimer’s Disease. Journal of Alzheimer’s Disease 2021, 84:579–598. [DOI] [PubMed] [Google Scholar]
- 40.Stancu I-C, Cremers N, Vanrusselt H, Couturier J, Vanoosthuyse A, Kessels S, Lodder C, Brône B, Huaux F, Octave J-N, et al. : Aggregated Tau activates NLRP3–ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathologica 2019, 137:599–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK, Kagan VE, et al. : Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171:273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yao Y, Chen Z, Zhang H, Chen C, Zeng M, Yunis J, Wei Y, Wan Y, Wang N, Zhou M, et al. : Selenium-GPX4 axis protects follicular helper T cells from ferroptosis. Nat Immunol 2021, 22:1127–1139. [DOI] [PubMed] [Google Scholar]
- 43.Angeli JPF, Shah R, Pratt DA, Conrad M: Ferroptosis Inhibition: Mechanisms and Opportunities. Trends Pharmacol Sci 2017, 38:489–498. [DOI] [PubMed] [Google Scholar]
- 44.Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, Roveri A, Peng X, Porto Freitas F, Seibt T, et al. : Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172:409–422.e421. [DOI] [PubMed] [Google Scholar]
- 45.Tian R, Abarientos A, Hong J, Hashemi SH, Yan R, Dräger N, Leng K, Nalls MA, Singleton AB, Xu K, et al. : Genome-wide CRISPRi/a screens in human neurons link lysosomal failure to ferroptosis. Nat Neurosci 2021, 24:1020–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen L, Hambright WS, Na R, Ran Q: Ablation of the Ferroptosis Inhibitor Glutathione Peroxidase 4 in Neurons Results in Rapid Motor Neuron Degeneration and Paralysis*. Journal of Biological Chemistry 2015, 290:28097–28106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hambright WS, Fonseca RS, Chen L, Na R, Ran Q: Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biology 2017, 12:8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jhelum P, Santos-Nogueira E, Teo W, Haumont A, Lenoël I, Stys PK, David S: Ferroptosis Mediates Cuprizone-Induced Loss of Oligodendrocytes and Demyelination. J Neurosci 2020, 40:9327–9341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E, Seravalli J, Ai Y, Sansing LH, Ste.Marie EJ, et al. : Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell 2019, 177:1262–1279.e1225. [DOI] [PubMed] [Google Scholar]
- 50.Wang T, Tomas D, Perera ND, Cuic B, Luikinga S, Viden A, Barton SK, McLean CA, Samson AL, Southon A, et al. : Ferroptosis mediates selective motor neuron death in amyotrophic lateral sclerosis. Cell Death & Differentiation 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen L, Na R, Danae McLane K, Thompson CS, Gao J, Wang X, Ran Q: Overexpression of ferroptosis defense enzyme Gpx4 retards motor neuron disease of SOD1G93A mice. Scientific Reports 2021, 11:12890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Do Van B, Gouel F, Jonneaux A, Timmerman K, Gelé P, Pétrault M, Bastide M, Laloux C, Moreau C, Bordet R, et al. : Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol Dis 2016, 94:169–178. [DOI] [PubMed] [Google Scholar]
- 53.Angelova PR, Choi ML, Berezhnov AV, Horrocks MH, Hughes CD, De S, Rodrigues M, Yapom R, Little D, Dolt KS, et al. : Alpha synuclein aggregation drives ferroptosis: an interplay of iron, calcium and lipid peroxidation. Cell death and differentiation 2020, 27:2781–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen L, Dar NJ, Na R, McLane KD, Yoo K, Han X, Ran Q: Enhanced defense against ferroptosis ameliorates cognitive impairment and reduces neurodegeneration in 5xFAD mice. Free Radical Biology and Medicine 2022, 180:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Feng H, Schorpp K, Jin J, Yozwiak CE, Hoffstrom BG, Decker AM, Rajbhandari P, Stokes ME, Bender HG, Csuka JM, et al. : Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Reports 2020, 30:3411–3423.e3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cregan SP, Dawson VL, Slack RS: Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene 2004, 23:2785–2796. [DOI] [PubMed] [Google Scholar]
- 57.David KK, Andrabi SA, Dawson TM, Dawson VL: Parthanatos, a messenger of death. FBL 2009, 14:1116–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fatokun AA, Dawson VL, Dawson TM: Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. British Journal of Pharmacology 2014, 171:2000–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shanbhag NM, Evans MD, Mao W, Nana AL, Seeley WW, Adame A, Rissman RA, Masliah E, Mucke L: Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease. Acta Neuropathologica Communications 2019, 7:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mullaart E, Boerrigter ME, Ravid R, Swaab DF, Vijg J: Increased levels of DNA breaks in cerebral cortex of Alzheimer’s disease patients. Neurobiol Aging 1990, 11:169–173. [DOI] [PubMed] [Google Scholar]
- 61.Shackelford DA: DNA end joining activity is reduced in Alzheimer’s disease. Neurobiology of Aging 2006, 27:596–605. [DOI] [PubMed] [Google Scholar]
- 62.Tanaka H, Kondo K, Fujita K, Homma H, Tagawa K, Jin X, Jin M, Yoshioka Y, Takayama S, Masuda H, et al. : HMGB1 signaling phosphorylates Ku70 and impairs DNA damage repair in Alzheimer’s disease pathology. Communications Biology 2021, 4:1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Love S, Barber R, Wilcock GK: Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer’s disease. Brain 1999, 122:247–253. [DOI] [PubMed] [Google Scholar]
- 64.Park H, Kam T-I, Dawson TM, Dawson VL: Chapter One - Poly (ADP-ribose) (PAR)-dependent cell death in neurodegenerative diseases. In International Review of Cell and Molecular Biology. Edited by Spetz JKE, Galluzzi L: Academic Press; 2020:1–29. vol 353.] [DOI] [PubMed] [Google Scholar]
- 65.James SA, Volitakis I, Adlard PA, Duce JA, Masters CL, Cherny RA, Bush AI: Elevated labile Cu is associated with oxidative pathology in Alzheimer disease. Free Radic Biol Med 2012, 52:298–302. [DOI] [PubMed] [Google Scholar]
- 66.Morioka S, Perry JSA, Raymond MH, Medina CB, Zhu Y, Zhao L, Serbulea V, Onengut-Gumuscu S, Leitinger N, Kucenas S, et al. : Efferocytosis induces a novel SLC program to promote glucose uptake and lactate release. Nature 2018, 563:714–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’Loughlin E, Xu Y, Fanek Z, et al. : The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47:566–581.e569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sarhan M, Land WG, Tonnus W, Hugo CP, Linkermann A: Origin and Consequences of Necroinflammation. Physiological Reviews 2018, 98:727–780. [DOI] [PubMed] [Google Scholar]
- 69.Banjara M, Ghosh C: Sterile Neuroinflammation and Strategies for Therapeutic Intervention. International Journal of Inflammation 2017, 2017:8385961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Adamczak SE, de Rivero Vaccari JP, Dale G, Brand FJ, Nonner D, Bullock M, Dahl GP, Dietrich WD, Keane RW: Pyroptotic Neuronal Cell Death Mediated by the AIM2 Inflammasome. Journal of Cerebral Blood Flow & Metabolism 2014, 34:621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Leng F, Edison P: Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nature Reviews Neurology 2021, 17:157–172. [DOI] [PubMed] [Google Scholar]
- 72.Conos Stephanie A, Chen Kaiwen W, De Nardo D, Hara H, Whitehead L, Núñez G, Masters Seth L, Murphy James M, Schroder K, Vaux David L, et al. : Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proceedings of the National Academy of Sciences 2017, 114:E961–E969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Riegman M, Sagie L, Galed C, Levin T, Steinberg N, Dixon SJ, Wiesner U, Bradbury MS, Niethammer P, Zaritsky A, et al. : Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nature Cell Biology 2020, 22:1042–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Koehler RC, Dawson VL, Dawson TM: Targeting Parthanatos in Ischemic Stroke. Frontiers in Neurology 2021, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McQuade A, Blurton-Jones M: Microglia in Alzheimer’s Disease: Exploring How Genetics and Phenotype Influence Risk. Journal of Molecular Biology 2019, 431:1805–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rohn TT, Vyas V, Hernandez-Estrada T, Nichol KE, Christie LA, Head E: Lack of pathology in a triple transgenic mouse model of Alzheimer’s disease after overexpression of the anti-apoptotic protein Bcl-2. J Neurosci 2008, 28:3051–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang L, Qian Y, Li J, Zhou X, Xu H, Yan J, Xiang J, Yuan X, Sun B, Sisodia SS, et al. : BAD-mediated neuronal apoptosis and neuroinflammation contribute to Alzheimer’s disease pathology. iScience 2021, 24:102942. [DOI] [PMC free article] [PubMed] [Google Scholar]