

Abstract
Many individuals with muscular dystrophies remain genetically undiagnosed despite clinical diagnostic testing, including exome sequencing. Some may harbor previously undetected structural variants (SVs) or cryptic splice sites. We enrolled 10 unrelated families: nine had muscular dystrophy but lacked complete genetic diagnoses and one had an asymptomatic DMD duplication. Nanopore genomic long‐read sequencing identified previously undetected pathogenic variants in four individuals: an SV in DMD, an SV in LAMA2, and two single nucleotide variants in DMD that alter splicing. The DMD duplication in the asymptomatic individual was in tandem. Nanopore sequencing may help streamline genetic diagnostic approaches for muscular dystrophy.
Introduction
Genomic advances enable us to pinpoint genetic diagnoses ever more thoroughly in Mendelian disorders. Yet a persistent subset of affected individuals remains genetically unsolved or partially solved. Numerous individuals with muscular dystrophy have either no pathogenic variants or one heterozygous pathogenic variant in genes that are primarily recessive. 1 , 2 , 3 , 4 In limb‐girdle muscular dystrophy, clinical genetic testing yields diagnoses in a quarter of affected individuals, 4 while exome sequencing identifies pathogenic variants in up to half the remainder. 1 , 2 , 3 , 5 Diagnostic genetic sequencing is currently based on short‐read sequencing (SRS) techniques (second‐generation sequencing), including targeted sequence panels 6 and whole‐exome sequencing (WES), 7 which may not reliably detect certain structural variant (SVs) or pathogenic intronic variants such as aberrant splice sites that lie outside the captured sequences. Long‐read sequencing (LRS) (third‐generation sequencing) may be more sensitive for detecting SVs and intronic variants than SRS. 8 Among LRS techniques, nanopore sequencing generates data from native nucleic acid strands and detects a broad range of variants, from single nucleotide variant (SNVs) to large SVs. 8 , 9
To determine whether LRS can detect SVs and intronic SNVs not detected via standard clinical genetic techniques, we used nanopore LRS to examine a cohort of unsolved or partially unsolved individuals suspected of having pathogenic variants in specific muscular dystrophy genes.
Methods
Under the auspices of institutional review board protocols at the University of Minnesota, the University of Florida, and Boston Children's Hospital, we enrolled 10 families without a definitive molecular diagnosis and performed LRS (Fig. 1). Genomic DNA (gDNA) was extracted from blood and saliva samples. Whole‐genome sequencing libraries were generated (ONT Ligation Sequencing Kit SQK_LSK109 or SQK_LSK110; Oxford Nanopore Technologies), then loaded on flow cells (R9.4.1) for sequencing on the Nanopore MinION or GridION. We implemented an analytic pipeline that includes basecalling and FASTQ file generation using guppy 5.0.11, mapping of reads to GRCh38 using minimap2 (v.2.22), and SV detection using sniffles (v.1.0.12). Nanopore data were visualized using integrative genomics viewer (igv). SVs supported by three or more reads were analyzed further. Assemblies were inspected for SNVs in genes of interest. Variants were confirmed using a combination of PCR and Sanger sequencing. To confirm altered DMD splicing in one proband, we performed a hybrid minigene assay (Data S1). 10 , 11 , 12 We classified confirmed variants in families 1441, 1462, 1480, 1466, 120, and 1443 according to ACMG criteria. 13
Figure 1.
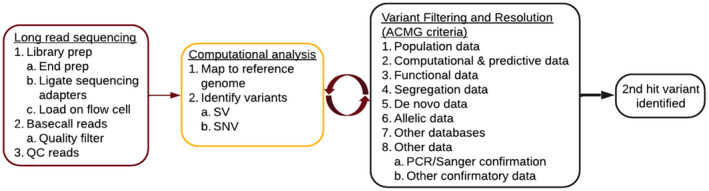
DNA to diagnosis. Families without a definitive molecular diagnosis were eligible for inclusion in the study. Long‐read nanopore sequencing was performed on genomic DNA from informative individuals. Computational analysis and variant filtering and resolution were performed in an iterative manner. QC, quality control; SV, structural variant; SNV, single nucleotide variant; ACMG, American College of Medical Genetics and Genomics; PCR, polymerase chain reaction. [Colour figure can be viewed at wileyonlinelibrary.com]
Results
We generated and analyzed whole‐genome LRS data in 12 individuals from 10 unrelated families with undiagnosed or incompletely diagnosed muscular dystrophy (Table 1 and Table S1). Depth of coverage was similar in blood and saliva samples and was greater than 10x in regions encompassing affected genes. The average read length N50 was 8.38 and 7.07 kb for blood and saliva samples, respectively.
Table 1.
Summary of nanopore LRS findings.
| Individual | Clinically identified variant | Nanopore LRS results and (ACMG category) | Confirmation or supporting findings |
|---|---|---|---|
| 1441‐1 | No variants identified in DMD |
Identified novel maternally inherited 5.9 Mbp inversion that disrupts DMD exons 3–79 (ACMG: P/LP) |
PCR and Sanger sequencing confirmed 1441‐1 is hemizygous and 1441‐2 (mother) is heterozygous for inversion |
| 1441‐2 | Mother of 1441‐1; asymptomatic | Identified novel 5.9 Mbp inversion that disrupts DMD exons 3–79 (ACMG: P/LP) | PCR and Sanger sequencing confirmed 1441‐2 is heterozygous for inversion |
| 1462‐1 | No variants identified in DMD |
Identified maternally inherited intronic splice variant DMD c.5548+67A>G (ACMG: P) |
Sanger sequencing confirmed 1462‐1 is hemizygous and 1462‐2 (mother) is heterozygous for the DMD splice variant; PCR and Sanger sequencing of RNA from muscle specimen confirmed aberrant splicing |
| 1462‐2 | Mother of 1462–1; asymptomatic |
Identified intronic splice variant DMD c.5548+67A>G (ACMG: P) |
Sanger sequencing confirmed 1462‐2 is heterozygous for the DMD splice variant |
| 1480‐1 | No variants identified in DMD |
Identified intronic splice variant DMD c.5155‐16T>A (ACMG: P) |
Sanger sequencing confirmed 1480‐1 is hemizygous for DMD splice variant; aberrant splicing confirmed via minigene assay |
| 1466‐1 | Duplication of DMD exons 10–26, suspected to be nontandem | Determined duplication including DMD exons 10–26 was in tandem and identified breakpoints (ACMG: VUS) | PCR and Sanger sequencing confirmed 1466‐1 is hemizygous for tandem duplication |
| 120‐1 |
LAMA2 c.2962C>T; p.Gln988Ter (ACMG: P/LP) |
An identified novel heterozygous LAMA2 3463 bp duplication (chr6:129,339,012–129,342,475, hg38) (ACMG: P); confirmed clinical SNV | PCR and Sanger sequencing confirmed maternally inherited SV; LRS confirmed previously reported paternally inherited SNV |
| 1126‐1 |
LAMA2 c.2538‐1G>C; (splice variant) (ACMG: LP) |
Confirmed clinical SNV | NA |
| 1443‐1 | Decreased D4Z4 methylation; no FSHD1 or FSHD2 variants identified |
Confirmed SMCHD1 c.182_183 delGT heterozygous variant (ACMG: LP) |
NA |
| 110‐1 |
ANO5 c.692G>T; (p.Gly231Val) (ACMG: P/LP) |
Confirmed clinical SNV | NA |
| 122‐1 |
CAPN3 c.1505T>C; p.Ile502Thr (ACMG: LP) |
Confirmed clinical SNV | Sanger sequencing results suggest SNV is paternally inherited |
| 125‐1 |
CAPN3 c.640G>A; p.Gly214Arg (ACMG: P/LP) |
Confirmed clinical SNV | Sanger sequencing confirmed SNV is paternally inherited |
In 10 individuals, nanopore LRS identified four previously undetected pathogenic or likely pathogenic variants (shown in bold in families 1441, 1462, 1480, and 120), fully characterized a duplication noted on clinical testing, and confirmed all previously noted pathogenic SNVs. Variants identified in this study (in families 1441, 1462, 1480, 1466, 120, and 1443) were classified according to ACMG criteria; previously identified variants were classified by the reporting laboratory or according to their ClinVar designation. LRS, long‐read sequencing; FSHD, facioscapulohumeral muscular dystrophy; P, pathogenic; LP, likely pathogenic; VUS, variant of unknown significance; SNV, single nucleotide variant.
In 1441‐1 (DMD without a pathogenic variant), nanopore sequencing identified a 5.9 Mb inversion (chrX:26,911,024–32,855,668, hg38) that disrupts DMD exons 3–79 (NM_004006.3) (Fig. 2A and B) and is predicted to result in a truncated DMD transcript that is degraded by nonsense‐mediated decay (NMD). 14 The breakpoint at chrX:26,911,024 is within an LTR (THE1B‐int); the other breakpoint is in a LINE (LIMA3). PCR and Sanger sequencing confirmed that the proband is hemizygous and that the unaffected mother is heterozygous for this inversion (Fig. 2C–E). This SV was absent from the database of genomic variants (DGV), ClinVar, and gnomAD‐SVs (v2.1). This copy‐neutral variant is consistent with the absent dystrophin on muscle biopsy and would not be expected to be detected consistently on clinical SRS or multiplex ligation‐dependent probe amplification (MLPA).
Figure 2.
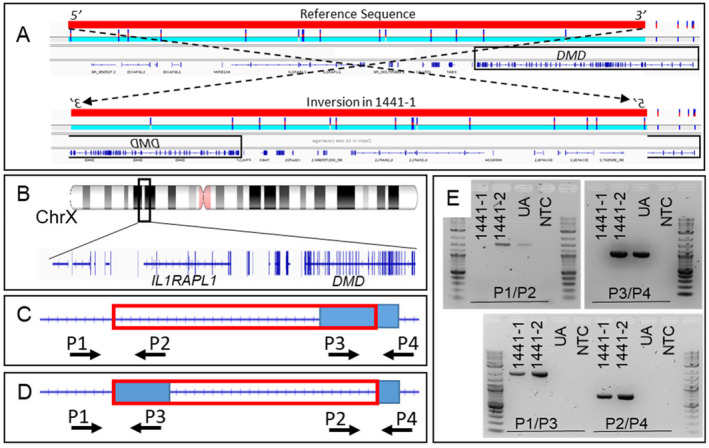
Detection and confirmation of inversion for family 1441 that includes exons 3–79 of DMD. (A) Nanopore LRS data for family 1441 shows inversion on the X chromosome that includes DMD exons 3–79. The solid red bar represents the inverted region. A black box highlights the location of the DMD gene. The top schematic shows the reference sequence, the bottom schematic shows the effect of the inversion found in 1441‐1. The region in the schematic is 6730 kb from chrX:26,750,000–33,480,000 (hg38). (B) The location of the DMD inversion. (C) gDNA positions are targeted by primer pairs P1/P2 and P3/P4, which straddle the predicted breakpoints when the inversion is not present. The red box outlines the predicted inversion and the blue box outlines the DMD gene. (D) gDNA positions are targeted by primer pairs P1/P3 and P2/P4 when the inversion is present. The red box outlines the predicted inversion and the blue box outlines the DMD gene. (E) PCR reactions are performed using primer pairs P1/P2, P3/P4, P1/P3, and P2/P4 to amplify gDNA extracted from 1441‐1 (proband), 1441‐2 (mother), and an unaffected individual (UA). A no template control (NTC) is also included in all reactions. In 1441‐1 and 1441‐2, primers P1/P3 amplify an ~2000 bp amplicon and primers P2/P4 amplify an ~800 bp amplicon, indicating that they carry the inversion, while the unaffected individual does not. Primers P1/P2 and P3/P4 do not produce an amplicon in 1441‐1 but do for 1441‐2 and the unaffected individual, indicating that 1441‐1 is hemizygous for the inversion and 1441‐2 is a heterozygous carrier. [Colour figure can be viewed at wileyonlinelibrary.com]
In 1462‐1 (DMD without a pathogenic variant), nanopore sequencing identified a hemizygous pathogenic SNV (DMD NM_004006.3, chrX:32,348,339T>C; c.5548+67A>G; (p.Met1816_Asn1817insValAsnTrpLeu*); rs72468626). 15 Sanger sequencing confirmed that this variant was present in the hemizygous state in 1462‐1 and in the heterozygous state in the mother, 1462–2. This SNV is predicted to activate a cryptic donor site in intron 38 (SpliceAI donor loss 0.19, donor gain 0.83; HSF3 variation score 38%; regSNP‐intron probability 0.83). An altered splicing pattern was confirmed via PCR, and via Sanger sequencing on RNA isolated from 1462‐1's muscle biopsy.
In 1480‐1 (DMD without a pathogenic variant), nanopore sequencing identified a hemizygous SNV (DMD NM_004006.3, chrX:32,362,974A>T; c.5155‐16T>A; (p.Lys1718_Arg1719insLeuMetGluTyrSerVal*)) with unclear pathogenicity (rs72468631). 16 Sanger sequencing confirmed this variant in 1480‐1. This SNV is predicted to activate a cryptic acceptor site in intron 36 that is expected to outcompete the canonical acceptor site (SpliceAI acceptor loss 0.81, acceptor gain 0.70; HSF3 variation score 51%; regSNP‐intron probability 0.66). An altered splicing pattern was confirmed via PCR, and Sanger sequencing of cDNA was obtained from a minigene assay.
Individual 1466‐1 was found to have an in‐frame duplication of DMD exons 10–26 during a genetic evaluation for an unrelated issue. He is a 40‐year‐old construction worker with no weakness on physical examination and a normal serum creatine kinase level, thus it was suspected that the duplication may be nontandem. Nanopore LRS determined that the duplication was indeed in tandem, suggesting that this SV is benign. We confirmed the breakpoints of this duplication (chrX:32,444,637 and chrX:32,649,432; hg38) in 1466‐1 using PCR and Sanger sequencing. This SV was absent from DGV, ClinVar, and gnomAD‐SVs (v2.1).
Individual 120‐1 presented with a sporadic congenital muscular dystrophy phenotype suggestive of merosin deficiency. Clinical WES identified a paternally inherited, heterozygous LAMA2 c.2962C>T, p.Gln988Ter pathogenic variant. 17 Nanopore LRS identified a heterozygous 3463 bp duplication (chr6:129,339,012–129,342,475, hg38) that includes all of exon 30 and 4 bp of intron 30 in LAMA2 (Fig. 3A and B), predicted to result in a frameshift and premature termination. This SV was absent from DGV, ClinVar, and gnomAD‐SVs (v2.1). This heterozygous duplication was confirmed via PCR in 120‐1 (proband) and 120‐2 (mother) (Fig. 3C–E). Reanalysis of the clinical SRS data provided additional confirmation of the duplication (data not shown).
Figure 3.
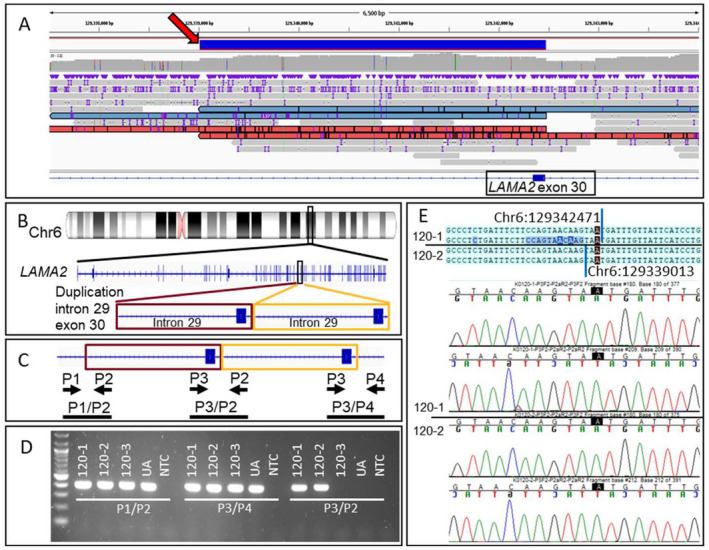
Detection and confirmation of duplication in LAMA2 for family 120. (A) View of nanopore LRS reads in the region of the 3463 bp duplication. A red arrow points to the solid blue bar that represents the duplicated region. Reads supporting the duplication are shown in red or blue. The IGV view shows ~6500 bp from chr6:129,337,495–129,343,993 (hg38). (B) Diagram indicates the location of the chromosome 6 duplication found on LRS that falls within LAMA2. The IGV view shows 27 bp from chr6:129,297,778–129,297,804 (hg38). (C) Diagram indicates relative positions of primers P1, P2, P3, and P4 in duplicated gDNA. Primer pair P3/P2 should only produce an amplicon if the duplication is present. Maroon and gold boxes outline the duplicated region and the black bars show expected amplicons. (D) PCR reactions are performed using primer sets P1/P2, P3/P4, and P3/P2 to amplify gDNA extracted from 120‐1 (proband), 120‐2 (mother), 120‐3 (father), and a control UA. A NTC is also included in all reactions. Primers P3/P2 yield an ~400 bp amplicon in 120‐1 and 120‐2, indicating that these two individuals carry the duplication, whereas 120‐3 and the unaffected individual do not. (E) Sanger sequencing of the P3/P2 amplicon from 120‐1 and 120‐2 confirms the tandem duplication and refines the breakpoint positions as chr6:129,339,013 and chr6:129,342,471 (hg38). Sequence data from proband and mother are separated by a black line. LRS, long‐read sequencing; IGC, integrative genomics viewer; ua, unaffected individual; NTC, no template control. [Colour figure can be viewed at wileyonlinelibrary.com]
Pathogenic variants previously detected on clinical genetic testing were confirmed on nanopore LRS for families 110, 122, 125, and 1126 (Table 1). Individual 1443‐1 presented with an facioscapulohumeral muscular dystrophy (FSHD) phenotype and nondiagnostic clinical genetic testing. Research studies showed decreased D4Z4 methylation and a heterozygous SMCHD1 c.182_183delGT variant, 18 confirmed on nanopore LRS.
Discussion
In our cohort, nanopore LRS identified four pathogenic SVs and SNV splice variants that were undetected on clinical testing and confirmed all pathogenic SNVs that were found on clinical testing. Current SV analytic protocols for SRS in clinical diagnostic laboratories rely on statistical analyses of read‐depth variations and discordant paired‐read mapping, which detect some SVs but are less accurate in highly repetitive regions. LRS detects SVs that are not reliably found by such clinical SRS protocols 19 ; however, prior studies did not focus on muscular dystrophy. In addition to screening for SVs in known disease genes, potential clinical applications of LRS in muscular dystrophy include the quantification of macrosatellite repeats in FSHD 20 , 21 , 22 and the quantification of microsatellite repeats in myotonic dystrophy (DM1). 23 , 24
Previously, LRS technology had high error rates for identifying smaller variants such as SNVs compared with SRS. Recent iterations of nanopore LRS have shown improved SNV detection. 25 We found that a mean read depth of 10–20X was generally sufficient to ascertain SVs. SNVs were usually identifiable at 10X coverage, but we found that 20–30X was more reliable. A nanopore library preparation from recently extracted gDNA typically yields 10–20 Gbp of basecalled data. We estimate that in our laboratory, the cost of detecting SVs for a gene of interest is $1800–3600 per gDNA sample, whereas the cost of detecting both SNVs and SVs is $3600–5400 per sample. We anticipate that these costs will continue to diminish.
A key clinical diagnostic question that arises from this study is whether it is better to optimize bioinformatic analyses of SRS data or to develop LRS further. Our finding in LAMA2 for family 120 would support the former, as more sensitive computational pipelines may have detected the second variant in the SRS data. However, it is difficult to pinpoint the diagnostic gaps in current clinical testing without more sensitive techniques such as LRS for comparison. Furthermore, whole‐genome LRS detects variant types that would not have been detected by other techniques, even when optimized. For example, exome sequences do not capture deep intronic splice variants. The DMD inversion in 1441‐1 is copy‐neutral, and thus would not be expected to be detected even via optimized analysis of SRS genome sequencing. Other genomic techniques such as transcriptome sequencing (RNAseq) rely on the availability of a tissue that expresses the genes under investigation; a consistent substitute for skeletal muscle samples has not been found to date. 26
A limitation of this study is the small cohort. Our depth of coverage is lower than in comparable SRS projects, though our average read length (N50) is much larger, facilitating the identification of SVs. To offset a potential bias toward false positives, the previously undetected pathogenic variants in the current study were confirmed by PCR and/or Sanger sequencing.
Nanopore LRS detects pathogenic variants ranging from SNVs to SVs in muscular dystrophy. Currently, LRS may be most appropriate for cases with specific candidate genes. As analytic pipelines mature, potential clinical applications will expand. We anticipate that future studies will build a foundation for clinical applications of LRS.
Author Contributions
C. C. B., L. M. K., and P. B. K. contributed to the conception and design of the study; all authors contributed to the acquisition and analysis of data; C. C. B. and P. B. K. contributed to drafting the manuscript and figures.
Conflicts of Interest
A. O’. L. has received consulting fees from Tome Biosciences as a paid clinical consultant and from Congenica as a scientific advisory board member; both companies are in the field of genomic medicine and may develop therapies for muscular dystrophy. Y. W. and C. C. are employees of PerkinElmer Genomics, a fee‐for‐service clinical testing laboratory; C. C. holds stock or stock options in PerkinElmer Genomics. B. T. D. has received grant support from Ionis Pharmaceuticals, Sarepta Pharmaceuticals, and FibroGen and has served as an ad hoc scientific advisory board member for Vertex, Genentech, and Roche; all of which produce or are developing therapies for muscular dystrophy or neuromuscular disorders. L. M. K. has received consulting fees from Dyne Therapeutics, Peter Bio, and Kate Therapeutics and holds stock or stock options in Dyne Therapeutics; all of which produce or are developing therapies for muscular dystrophy. P. B. K. has served on advisory boards for Sarepta Therapeutics and NS Pharma and receives grant support from Sarepta Therapeutics and ML Bio; all of which produce or are developing therapies for muscular dystrophy.
Supporting information
Data S1. Supplemental Methods. Minigene assay to assess splicing effect in DMD in 1480‐1.
Table S1. Summary of individuals and clinical findings.
Acknowledgments
We thank the patients and their families who contributed to the samples used in this study and helped to make this research possible. This project was funded in part by the NIH U01HG011755 (A. O’. D. L. and L. P.), T32HG010464 (V. S. G.), R01AR064300 (L. M. K.), the Bernard F. and Alva B. Gimbel Foundation (L. M. K.), and NIH R01NS080929 (P. B. K.). Short‐read sequencing and analysis support were provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG), funded by the National Human Genome Research Institute (NHGRI) grant numbers UM1HG008900 and R01HG009141. The authors thank the Minnesota Supercomputing Institute (MSI) at the University of Minnesota (http://www.msi.umn.edu) and Research Computing at the University of Florida (UFRC). The authors are grateful to Abby Schmitt, Dinesha Walek, and Kenneth Beckman at the University of Minnesota Genomics Center (UMGC) for providing platforms for Sanger sequencing and long‐read nanopore sequencing.
Annals of Clinical and Translational Neurology 2022;9(8): 1302–1309
Funding Information
This project was funded in part by the NIH U01HG011755 (A. O’. L. and L. P.), T32HG010464 (V. S. G.), R01AR064300 (L. M. K.), the Bernard F. and Alva B. Gimbel Foundation (L. M. K.), and NIH R01NS080929 (P. B. K.). Short‐read sequencing and analysis support were provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG), funded by the National Human Genome Research Institute (NHGRI) grant numbers UM1HG008900 and R01HG009141.
Funding Statement
This work was funded by Bernard F. and Alva B. Gimbel Foundation; National Human Genome Research Institute grants R01HG009141 and UM1HG008900; National Institutes of Health grants R01AR064300, R01NS080929, T32HG010464, and U01HG011755.
References
- 1. Ghaoui R, Cooper ST, Lek M, et al. Use of whole‐exome sequencing for diagnosis of limb‐girdle muscular dystrophy: outcomes and lessons learned. JAMA Neurol. 2015;72(12):1424‐1432. [DOI] [PubMed] [Google Scholar]
- 2. Reddy HM, Cho K‐A, Lek M, et al. The sensitivity of exome sequencing in identifying pathogenic mutations for LGMD in the United States. J Hum Genet. 2017;62(2):243‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harris E, Topf A, Barresi R, et al. Exome sequences versus sequential gene testing in the UK highly specialised service for limb girdle muscular dystrophy. Orphanet J Rare Dis. 2017;12(1):151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nallamilli BRR, Chakravorty S, Kesari A, et al. Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann Clin Transl Neurol. 2018;5(12):1574‐1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Saha M, Reddy HM, Salih MA, et al. Impact of PYROXD1 deficiency on cellular respiration and correlations with genetic analyses of limb‐girdle muscular dystrophy in Saudi Arabia and Sudan. Physiol Genomics. 2018;50(11):929‐939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Valencia CA, Rhodenizer D, Bhide S, et al. Assessment of target enrichment platforms using massively parallel sequencing for the mutation detection for congenital muscular dystrophy. J Mol Diagn. 2012;14(3):233‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ng SB, Turner EH, Robertson PD, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461(7261):272‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cretu Stancu M, van Roosmalen MJ, Renkens I, et al. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat Commun. 2017;8(1):1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Manrao EA, Derrington IM, Laszlo AH, et al. Reading DNA at single‐nucleotide resolution with a mutant MspA nanopore and phi29 DNA polymerase. Nat Biotechnol. 2012;30(4):349‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang Z, Lin Y, Qiu L, et al. Hybrid minigene splicing assay verified the pathogenicity of a novel splice site variant in the dystrophin gene of a Chinese patient with typical Duchenne muscular dystrophy phenotype. Clin Chem Lab Med. 2016;54(9):1435‐1440. [DOI] [PubMed] [Google Scholar]
- 11. van der Klift HM, Jansen AML, van der Steenstraten N, et al. Splicing analysis for exonic and intronic mismatch repair gene variants associated with lynch syndrome confirms high concordance between minigene assays and patient RNA analyses. Mol Genet Genomic Med. 2015;3(4):327‐345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang Y, Yan K, Liu B, et al. Comprehensive genetic diagnosis of patients with Duchenne/Becker muscular dystrophy (DMD/BMD) and pathogenicity analysis of splice site variants in the DMD gene. J Zhejiang Univ Sci B. 2019;20(9):753‐765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Maquat LE. When cells stop making sense: effects of nonsense codons on RNA metabolism in vertebrate cells. RNA. 1995;1(5):453‐465. [PMC free article] [PubMed] [Google Scholar]
- 15. Waldrop MA, Moore SA, Mathews KD, et al. Intron mutations and early transcription termination in Duchenne and Becker muscular dystrophy. Hum Mutat. 2022;43:511‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Flanigan KM, Dunn DM, von Niederhausern A, et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat. 2009;30(12):1657‐1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Løkken N, Born AP, Duno M, Vissing J. LAMA2‐related myopathy: frequency among congenital and limb‐girdle muscular dystrophies. Muscle Nerve. 2015;52(4):547‐553. [DOI] [PubMed] [Google Scholar]
- 18. Lemmers RJLF, van der Vliet PJ, Vreijling JP, et al. Cis D4Z4 repeat duplications associated with facioscapulohumeral muscular dystrophy type 2. Hum Mol Genet. 2018;27(20):3488‐3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miller DE, Sulovari A, Wang T, et al. Targeted long‐read sequencing identifies missing disease‐causing variation. Am J Hum Genet. 2021;108:1436‐1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mitsuhashi S, Nakagawa S, Takahashi Ueda M, Imanishi T, Frith MC, Mitsuhashi H. Nanopore‐based single molecule sequencing of the D4Z4 array responsible for facioscapulohumeral muscular dystrophy. Sci Rep. 2017;7(1):14789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mitsuhashi S, Nakagawa S, Sasaki‐Honda M, Sakurai H, Frith MC, Mitsuhashi H. Nanopore direct RNA sequencing detects DUX4‐activated repeats and isoforms in human muscle cells. Hum Mol Genet. 2021;30(7):552‐563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stence AA, Thomason JG, Pruessner JA, et al. Validation of optical genome mapping for the molecular diagnosis of facioscapulohumeral muscular dystrophy. J Mol Diagn. 2021;23(11):1506‐1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McGinty RJ, Rubinstein RG, Neil AJ, et al. Nanopore sequencing of complex genomic rearrangements in yeast reveals mechanisms of repeat‐mediated double‐strand break repair. Genome Res. 2017;27(12):2072‐2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chintalaphani SR, Pineda SS, Deveson IW, Kumar KR. An update on the neurological short tandem repeat expansion disorders and the emergence of long‐read sequencing diagnostics. Acta Neuropathol Commun. 2021;9(1):98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Feng Z, Clemente JC, Wong B, Schadt EE. Detecting and phasing minor single‐nucleotide variants from long‐read sequencing data. Nat Commun. 2021;12(1):3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cummings BB, Marshall JL, Tukiainen T, et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med. 2017;9(386):eaal5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental Methods. Minigene assay to assess splicing effect in DMD in 1480‐1.
Table S1. Summary of individuals and clinical findings.