

Abstract
Objective
This study delineates the clinical and molecular spectrum of ANKLE2‐related microcephaly (MIC), as well as highlights shared pathological mechanisms between ANKLE2 and the Zika virus.
Methods
We identified 12 individuals with MIC and variants in ANKLE2 with a broad range of features. Probands underwent thorough phenotypic evaluations, developmental assessments, and anthropometric measurements. Brain imaging studies were systematically reviewed for developmental abnormalities. We functionally interrogated a subset of identified ANKLE2 variants in Drosophila melanogaster.
Results
All individuals had MIC (z‐score ≤ −3), including nine with congenital MIC. We identified a broad range of brain abnormalities including simplified cortical gyral pattern, full or partial callosal agenesis, increased extra‐axial spaces, hypomyelination, cerebellar vermis hypoplasia, and enlarged cisterna magna. All probands had developmental delays in at least one domain, with speech and language delays being the most common. Six probands had skin findings characteristic of ANKLE2 including hyper‐ and hypopigmented macules. Only one individual had scalp rugae. Functional characterization in Drosophila recapitulated the human MIC phenotype. Of the four variants tested, p.Val229Gly, p.Arg236*, and p.Arg536Cys acted as partial‐loss‐of‐function variants, whereas the c.1421‐1G>C splicing variant demonstrated a strong loss‐of‐function effect.
Interpretation
Deleterious variants in the ANKLE2 gene cause a unique MIC syndrome characterized by congenital or postnatal MIC, a broad range of structural brain abnormalities, and skin pigmentary changes. Thorough functional characterization has identified shared pathogenic mechanisms between ANKLE2‐related MIC and congenital Zika virus infection. This study further highlights the importance of a thorough diagnostic evaluation including molecular diagnostic testing in individuals with MIC.
Introduction
Microcephaly (MIC) refers to a developmentally small head and is defined as an occipitofrontal circumference (OFC) that is two or more standard evaluations (SD) below the mean for age and sex, while clinically significant MIC is generally defined as three or more SD below the mean. 1 , 2 MIC at birth, or congenital MIC, affects an estimated 8.7 per 10,000 live births in the United States. 3 Both congenital and postnatal MIC are causally heterogeneous and can be due to a broad range of acquired and genetic factors. 1 Genetic variants in a rapidly growing number of genes and pathways have been identified in patients presenting with MIC and are known to play key roles in early brain development including cell cycle control, such as centrosome biogenesis and assembly, spindle pole organization, transcriptional regulation, and DNA damage repair. 4 , 5 , 6 These highly evolutionarily conserved molecular pathways in the developing human brain underlie neocortical expansion and overall brain size. 7 , 8 , 9 , 10 Recently, pathogenic variants in ANKLE2 (Ankyrin Repeat And LEM Domain Containing 2), a gene encoding a critical nuclear envelope protein, were identified as a cause of a rare and severe form of congenital MIC known as primary autosomal recessive MIC‐16 (MCPH16, MIM#616681). 6 , 11 , 12 ANKLE2 is a gene that encodes a critical endoplasmic reticulum and nuclear envelope protein that has been shown to interact with the kinase vaccina‐related kinase 1 (VRK1) to regulate cell division and is important for nuclear envelope formation after division by regulating barrier to autointegration factor phosphorylation through protein phosphatase 2A activity. 11 , 13 Mutations in Drosophila Ankle2 cause a small brain, while expression of reference human ANKLE2 rescues this phenotype. Variants in ANKLE2 found in MIC patients could not rescue loss of fly Ankle2, indicating that the function of human ANKLE2 is conserved, and variants are associated with loss‐of‐function phenotypes. 11 While phenotypes associated with loss of ANKLE2 are established, the mechanisms by which ANKLE2 interacts with and regulates proteins such as VRK1 are unknown. Interestingly, recent studies have also identified shared mechanistic links between ANKLE2 and viral causes of MIC in children, namely the Zika virus (ZIKV). 11 A specific ZIKV protein component, namely NS4A, physically interact with human ANKLE2 in human cell lines, and expression of ZIKV NS4A in Drosophila causes reduced brain volume. Wild type, but not patient variant, can rescue phenotypes associated with NS4A, showing that NS4A causes brain phenotypes by inhibiting ANKLE2 activity. 14 Despite the relatively recent identification of MCPH16, the clinical, molecular, and functional spectrum of ANKLE2‐related MIC remain poorly understood due to the small number of prior observations.
In this study, we describe the clinical, molecular, and neuroimaging spectrum of the largest cohort of children with ANKLE2‐related MIC identified to date. This cohort includes three previously unpublished children (probands 10–12) as well as updated and expanded phenotypic and molecular data on nine children who were previously published either as single observations in large series or with limited data (probands 1–9). 6 , 11 , 12 We also report on several novel pathogenic ANKLE2 genetic variants which recapitulate the human MIC phenotype in vivo. Overall, this study delineates a much broader spectrum of ANKLE2‐related MIC than previously recognized. It further highlights the need for thorough genetic evaluations in children with MIC, including in the setting of exposure to viral infections given the clinical overlap as well as the shared pathogenetic mechanisms between ANKLE2‐ and ZIKV‐related human MIC as a specific example.
Methods
Patient cohort
Affected individuals were identified and recruited through an international network of clinicians and investigators, facilitated by nodes of the MatchMaker Exchange (MME) including GeneMatcher and MyGene2. 15 , 16 Previously published patients were similarly recruited through collaboration with clinicians who evaluated affected families. Individuals or their legally authorized representatives either provided informed written consent for the use of their medical information for research or were included under an IRB‐approved waiver of consent for de‐identified data at participating institutions. Clinical and molecular data were analyzed retrospectively by the investigators. Brain imaging studies were acquired as part of standard diagnostic workup and clinical care. Available brain magnetic resonance (MR) images were reviewed by the investigators with specific attention paid to the type and severity of MIC, types and distribution of associated brain abnormalities involving the cerebral cortex, white matter, corpus callosum, basal ganglia, thalami, cerebellum, and brainstem. Z‐scores for growth parameters of all probands were computed based on the age provided in clinical records. Z‐scores for weight and height or length measurements were computed using the PediTools medical calculator using appropriate age‐ and sex‐matched scales. 17 Weight and length/height parameters from birth to 24 months were further determined using the WHO infant growth chart (birth–24 months) and measurements over 24 months were calculated using the CDC children and adolescent growth chart (2–20 years). Z‐scores for OFC values between the ages of birth to 24 months were computed using the WHO infant growth chart (birth–24 months) and z‐scores for patients over the age of 24 months were calculated using the Nellhaus Head Circumference Chart. 18 Variants in ANKLE2 were identified through molecular diagnostic testing, including either targeted multi‐gene panel testing or exome sequencing (ES), performed clinically at participating institutions.
Functional studies
Drosophila melanogaster studies
Several fly lines were generated and used to functionally validate identified ANKLE2 variants using previously published protocols, specifically: Ankle2 A , P{UASt‐ hANKLE2} VK37; P{UASt‐hANKLE2 p.Val229Gly}VK37; P{UASt‐hANKLE2 p.Arg236*}VK37; P{UASt‐hANKLE2 p.Arg536Cys}VK37; P{UASt‐hANKLE2 p.Gly510*}VK37, daughterless‐GAL4 (P{w [+mW.hs] = GAL4‐da.G32}UH1). 19 The p.Gly510* variant is predicted based on the splice site variant c.1421‐1G>C. All flies were maintained and crossed at 22°C and grown on standard cornmeal and molasses medium in plastic vials. Hemizygous males were analyzed as Ankle2 mutants (as the gene is located on the fly X chromosome). Drosophila brain volume measurements were performed in late third instar larvae (based on gut clearance and extruding spiracles), as previously described. 11 da‐GAL4 was used to express human ANKLE2 constructs ubiquitously.
Generation of human ANKLE2 constructs
Q5 Site‐directed mutagenesis was performed on P{UASt‐Flag‐hANKLE2} (New England Biosciences, Ipswich, MA). Each plasmid was sequence‐verified and injected into VK37 flies expressing phiC31 integrase for site‐specific integration in the fly genome. 20
Brain volume and immunostaining
Late third instar larval brains were dissected in PBS and fixed with 4% PFA/PBS/0.3%Triton for 20 min. For immunostaining, brains were blocked in PBS/0.3%Triton/1%BSA/5% normal goat serum and incubated in primary antibody in PBS/0.3%Triton/1%BSA overnight. Primary antibodies include rat anti‐Deadpan (Abcam Cat# ab195172, 1:250 or 1:500, Waltham, MA, USA) and mouse anti‐Prospero MR1A (Developmental Studies Hybridoma Bank, 1:1000, Iowa City, IA, USA). The donkey secondary antibodies were used at 1:500 (Jackson ImmunoResearch, West Grove, PA). Brains were mounted with double sided tape spacers and imaged on a Leica Sp8 confocal with 2.0 μm sections throughout the entire brain lobe using a 25× water immersion lens. Brain lobe volumes were quantified using Imaris (Bitplane) and the Volume function (units are μm3). Volume was then normalized to Ankle2 A ; hANKLE2 wt rescue animals. Rescue animals were set to 100%. Mutant and variant rescue animals are calculated as percent (%) of rescue volume for display purposes. Number of animals tested are as follows: hANKLE2 wt (6), Ankle2 A (8), Vall229Gly (10), Arg236* (5), Arg536Cys (8), and Gly510* (8). hANKLE2 p.Arg236* animals (n = 5) were difficult to obtain due to lower‐than‐expected Mendelian ratios. Note that Ankle2 A is a temperature sensitive mutation, and slight fluctuation in temperature can affect severity of brain volume defects.
Results
Clinical and molecular features
We identified a total of 12 individuals from 10 unrelated families with MIC who had variants in ANKLE2 including eight females and four males, ranging in age at the time of presentation from birth to 15 years. The clinical, neuroimaging, and molecular data for all affected probands are summarized in Table 1 and detailed in Table S1. Three families were known to be consanguineous. Pedigrees of all identified families and their variants are shown in Figure 1. All affected individuals presented with severe MIC (OFC z‐score ≤3 below the mean), including nine who had congenital MIC. OFC z‐scores at birth ranged from −7.07 to −1.00 (mean −4.25) below the mean, while postnatal OFC z‐scores ranged from −13.72 to −3.18 (mean −7.04). Of the nine children who met criteria for congenital MIC, five had severe MIC with OFC z‐scores ≤ −3 (range −7.07 to −6.11). Available OFC and stature measurements for all probands are graphically shown in Figure 2.
Table 1.
Summary of clinical and molecular features of individuals with ANKLE2‐related microcephaly, with comparison to ZIKV‐ and CMV‐related microcephaly.
| N | ANKLE2 variant(s) | Congenital MIC | Postnatal MIC | Brain MRI abnormalities | Seizures | Developmental delays/intellectual disability | Scalp rugae | Skin pigmentary abnormalities |
|---|---|---|---|---|---|---|---|---|
| 1 | c.1717C>G; p.Leu573Val | + | + | CTXD, WM, ACC, ↑ EAS | + | GM/FM/SLD | + | + |
| c.2344C>T; p.Gln782Ter | ||||||||
| 2 | c.1717C>G; p.Leu573Val | + | + | ND | NS | NS | − | + |
| c.2344C>T; p.Gln782Ter | ||||||||
| 3 | c.1754G>T; p.Gly585Val | − | + | ACC (CT scan) | − | GM/FM/SLD | − | − |
| 4 | c.601G>T; p.Gly201Trp | + | + | Normal (CT scan) | − | GM/FM/SLD | NS | NS |
| 5 | c.601G>T; p.Gly201Trp | + | + | CTXD, CBLH, pACC | − | GM/FM/SLD | − | − |
| 6 | c.686T>G; p.Val229Gly | + | + | CTXD, pACC | + | GM/FM/SLD | − | + |
| 7 | c.686T>G; p.Val229Gly | + | + | CTXD, pACC | + | GM/FM/SLD | − | + |
| 8 | c.325G>C; p.Ala109Pro | − | + | Mild CTXD, mildly thin CC, mild WM | − | SLD | − | + |
| c.1421‐1G>C; Splicing | ||||||||
| 9 | c.706C>T; p.Arg236Ter | + | + | CTXD, pACC, WM, ↑ EAS | − | GM | − | + |
| c.1606C>T; p.Arg536Cys | ||||||||
| 10 | c.1891+1701_2615+14delinsA; deletion exons 11/12 | + | + | CTXD, pACC | − | GM/FM/SLD | − | − |
| c.1606C>T; p.Arg536Cys | ||||||||
| 11 | c.940C>T; p.Arg314Trp | + | + | Mild CTXD, mildly thin CC | − | GM/FM/SLD | − | − |
| c.2505T>G; p.Asp835Glu | ||||||||
| 12 | c.1175T>C; p.Leu392Pro | − | + | CTXD | − | SLD | − | − |
| c.1352A>T; p.Asp451Val | ||||||||
| ZIKV | − | + | + | + | + | + (29%) | + | − |
| CMV | − | + | + | + (~70%) | + (5–10%) | + (50–75%) | − | − |
| LCMV | − | + | + | + | + | + | − | − |
ACC, agenesis of the corpus callosum; MIC, microcephaly; CBLH, cerebellar hypoplasia; CMV, Cytomegalovirus; CTXD, cortical dysplasia including simplification of the cortical gyral pattern; EAS, extra‐axial spaces; FM, fine motor; GM, gross motor; LCMV, lymphocytic choriomeningitis virus; ND, no data; NS, not specified; pACC, partial agenesis of the corpus callosum; SLD, speech and language development; WM, white matter; ZIKV, Zika‐virus related microcephaly.
Figure 1.
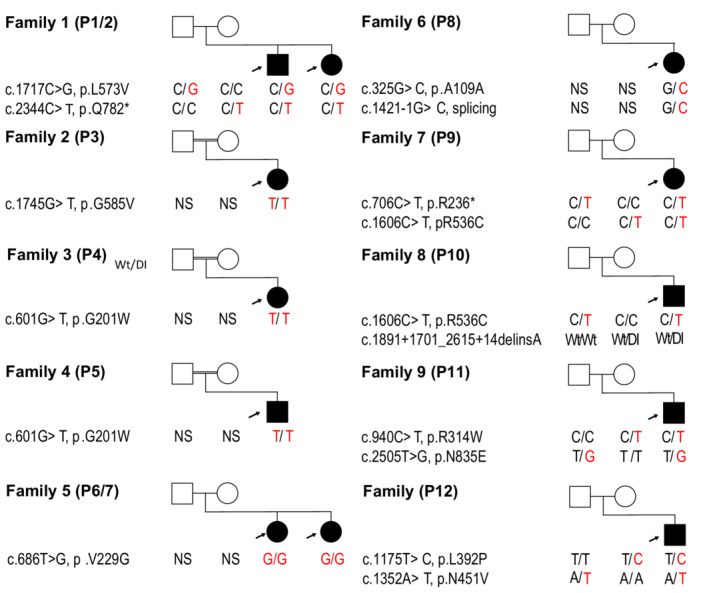
Pedigrees of all families with ANKLE2 variants. Family pedigrees and segregation of identified ANKLE2 variants shown in respective probands including parents. Shaded icons represent affected individuals. Gene name, nucleotide changes, protein changes, and respective variants (underlined) are indicated. [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 2.
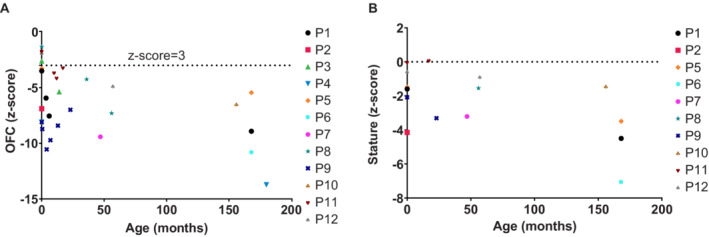
Graph plot of growth measurements of probands with ANKLE2 variants. Available OFC (A) and stature (B) measurements at birth and subsequent clinical evaluations are plotted with the age in months on the x‐axis and z‐scores on the y‐axis. Most affected probands had congenital microcephaly (OFC z‐scores ≤ −2) and all probands for whom postnatal OFC measurements were available had severe microcephaly (OFC z‐scores ≤ −3). Microcephaly was notably progressive in nature in all affected probands. OFC, occipitofrontal circumference. [Colour figure can be viewed at wileyonlinelibrary.com]
Of the 11 probands who underwent brain MR imaging, six probands had severe brain abnormalities including markedly reduced brain volume with diffuse, and often severe, simplification of the cortical gyral pattern with foreshortening of the frontal lobes. Other notable brain abnormalities include full or partial agenesis of the corpus callosum (ACC; n = 7), hypomyelination or other white matter abnormalities (n = 7), increased extra‐axial spaces (n = 3), cerebellar vermis hypoplasia, and enlarged cisterna magna (each seen in one proband). In contrast, four probands had mild brain involvement with only a mildly simplified cortical gyral pattern, with either mild or no ACC, and without other major brain abnormalities. Representative brain MR images are shown in Figure 3. Notably, no other cortical malformations, such as polymicrogyria or other types of cortical dysplasia, were identified in this series. Three individuals (probands 1, 6 and 7) had early onset focal epilepsy, two of which are affected siblings who both had neonatal‐onset focal seizures. Five probands had mild hypotonia while two had increased tone including one individual with spastic quadriparesis.
Figure 3.
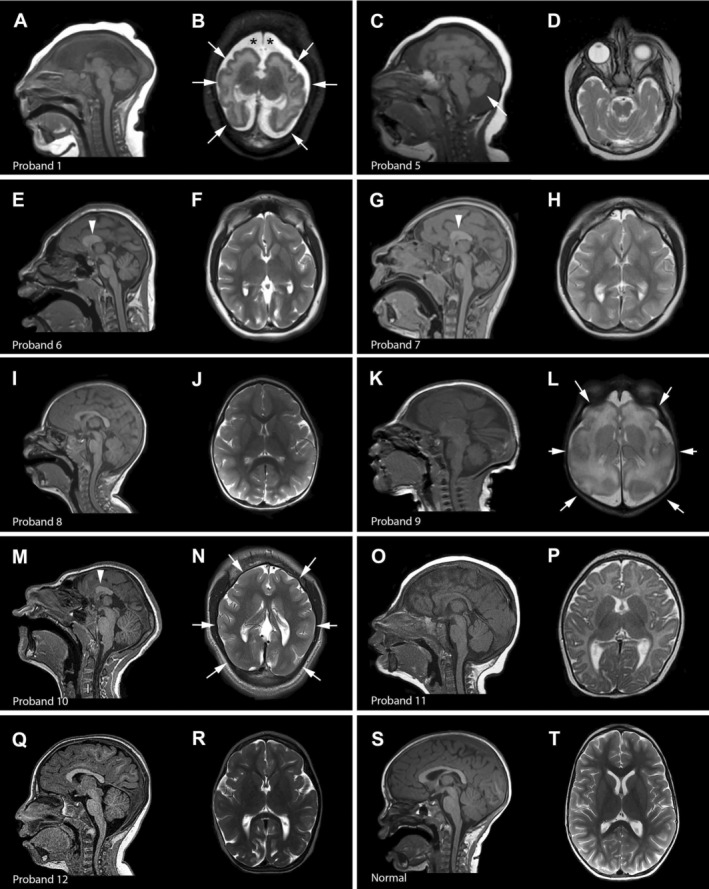
Brain imaging in ANKLE2‐related microcephaly. Select brain MR images of individuals with ANKLE2 variants are shown. (A and B) (proband 1), diffuse and severe undersulcation of the cortical gyral pattern with thin‐normal cortical thickness (arrows), increased extra‐axial space (asterisks), severe white matter involvement, dysplastic ventricles with complete agenesis of the corpus callosum with open communication between the trigones and medial extra‐axial space; (C and D) (proband 5), diffuse simplification and undersulcation of the cortical gyral pattern, partial agenesis of the corpus callosum (arrowhead), cerebellar vermis hypoplasia (arrow) (limited images available); (E and F) (proband 6), diffuse simplification of the cortical gyral pattern with severely foreshortened frontal lobes, very short and thick corpus callosum with partial agenesis (arrowhead), relatively preserved brainstem and cerebellum; (G and H) (proband 7), similar appearance as the sibling (proband 6) with diffuse simplification of the cortical gyral pattern, partial agenesis of the corpus callosum (arrowhead), and relatively preserved brainstem and cerebellum; (I and J) (proband 8), mild simplification of the cortical gyral pattern with mild foreshortening of the frontal lobes and mildly thin corpus callosum; (K and L) (proband 9), diffuse and severe simplification of the cortical gyral pattern (arrows), diffuse white matter abnormalities, mildly increased extra‐axial spaces; (M and N) (proband 10), diffuse severe simplification of the cortical gyral pattern (arrows), severe foreshortening of the frontal lobes, partial agenesis of the corpus callosum (arrowhead), relatively preserved brainstem and cerebellum; (O and P) (proband 11), mildly thin corpus callosum, with mild simplification of the cortical gyral pattern; (Q and R) (proband 12), diffusely simplified gyral pattern with paucity of the sulci in various areas, mild reduction in supratentorial white matter and a cisterna magna; (S and T) (normal), normal mid‐sagittal and axial images.
All affected probands had developmental delays in at least one domain with speech‐language delays (SLD) being the most common, affecting 10 probands. Eight individuals had developmental delays in all domains namely gross motor (GM), fine motor (FM), and SLD. Proband 9 had only mild GM delay, whereas probands 8 and 12 had isolated SLD. Subtle facial dysmorphisms associated with MIC were identified in most individuals. Four probands also had micrognathia. Skin findings including characteristic hypo‐ and hyper‐pigmented macules were observed in six affected individuals. Only proband 1 had scalp rugae. Representative clinical photographs of selected probands (1, 9, and 11) are shown in Figure 4.
Figure 4.
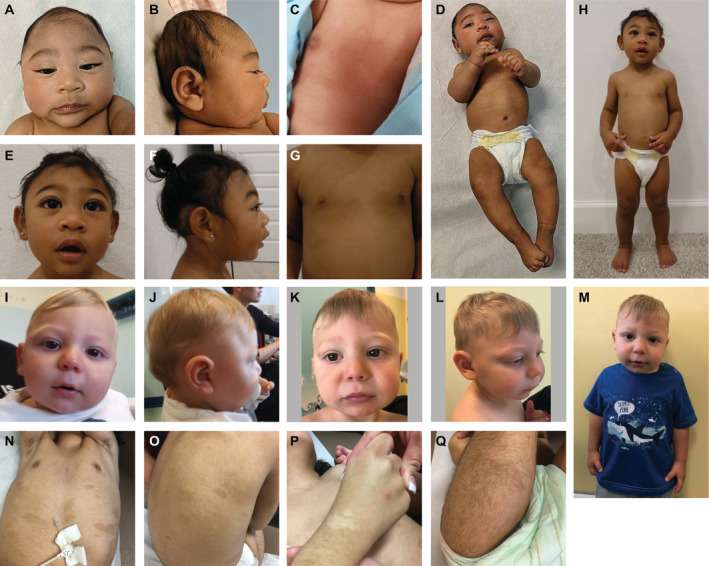
Clinical photographs of individuals with ANKLE2 variants. Proband 9 at 4 months (A–D) and 23 months of age (E–H). (A and B) frontal and lateral profile of the head at 4 months demonstrating apparent microcephaly with a low‐sloping forehead. (C) anterior view of the chest demonstrating hypopigmented and hyperpigmented macules. (D and H) full length photographs at 4 and 23 months further demonstrating skin pigmentary abnormalities and apparent microcephaly. (E and F) frontal and lateral profile of the head at 23 months. (G) anterior view of the chest demonstrating hypopigmented and hyperpigmented macules. Proband 11 at 10 months (I and J) and 20 months of age (K–M). (I and J) frontal and lateral profile of the head at 10 months demonstrating a narrow skull shape with prominent supraorbital ridges. (K and L) frontal and oblique profile of the head at 24 months demonstrating a mildly sloping forehead. (M) mid‐length body photograph demonstrating apparent microcephaly. Proband 1 (N–Q). Multiple hypopigmented and hyperpigmented macules located on (N) anterior chest, (O) back, (P) dorsal hand, (Q) posterior thigh. [Colour figure can be viewed at wileyonlinelibrary.com]
Most of the ANKLE2 variants identified in this series were detected through diagnostic ES, whereas two were identified via a multi‐gene panel (i.e., an exome slice). The pathogenic ANKLE2 variants include single‐nucleotide variants (SNVs) altering the coding regions of the gene (n = 10 probands), as well as a splice site variant (n = 1) and a multi‐exon deletion (n = 1) both predicted to result in premature protein termination. Seven children had compound heterozygous variants, and five had homozygous variants, four of whom were known to be from consanguineous (i.e., first cousin) unions. Two variants were seen recurrently in unrelated families including the homozygous p.Gly201Trp variant and the heterozygous p.Arg536Cys variant (proband 9 of Indian‐Filipino ancestry and proband 10 of Egyptian ancestry). The pathogenic ANKLE2 variants identified in our cohort spanned the length of the protein and were distributed across several functional domains as schematically shown in Figure 5. Of note, proband 9 had confirmed prenatal exposure to ZIKV from parental travel to an endemic country prior to conception. 21 Extensive testing for ZIKV and other congenital infections was performed on this child and all testing was negative (Data S1).
Figure 5.
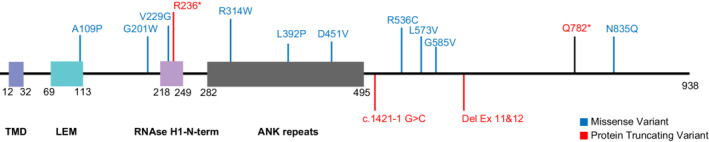
Diagram of the ANKLE2 protein and identified variants. Diagram of the ANKLE2 protein with the most important functional domains and the identified ANKLE2 variants in this manuscript. ANK, ankyrin repeats; Cauli‐VI, Caulimovirus viroplasmin VI domain; LEM, Lap2, Emerin, MAN1 domain; TMD, transmembrane domain. [Colour figure can be viewed at wileyonlinelibrary.com]
Modeling Ankle2 function in vivo
To investigate the mechanisms by which ANKLE2 variants cause MIC, we used an established Drosophila melanogaster model to assess variant function in vivo. 12 This scalable system is well‐suited for testing genetic variants in human disorders as it can be used to create sophisticated and specific genetic manipulations with rapid variant generation and high homology to the human genome, with 23% identity and 35% similarity between the Drosophila Ankle2 and human ANKLE2 genes. 22 , 23 Previous studies demonstrated that genetic variants in Drosophila Ankle2 lead to reduced brain volume in third instar larvae, recapitulating the human phenotype. 12 Ubiquitous expression of the reference, but not variant, human ANKLE2 rescued lethality and MIC of Ankle2 mutants. To investigate whether novel ANKLE2 variants in our series also affect gene function, we ubiquitously expressed each variant and tested their rescue ability compared with the reference cDNA. Analogous to previous studies, expression of wild type human ANKLE2 rescued phenotypes associated with loss of fly Ankle2 (Fig. 6A, B, and G). 11 , 12 However, when compared with controls, variants did not fully rescue Ankle2‐induced MIC, suggesting that the variants act as hypomorphic loss‐of‐function alleles (Fig. 6C–G). Furthermore, none of the tested variants rescued the lethality associated with loss of Ankle2. Taken together, these data further support the link between identified ANKLE2 variants and MIC.
Figure 6.
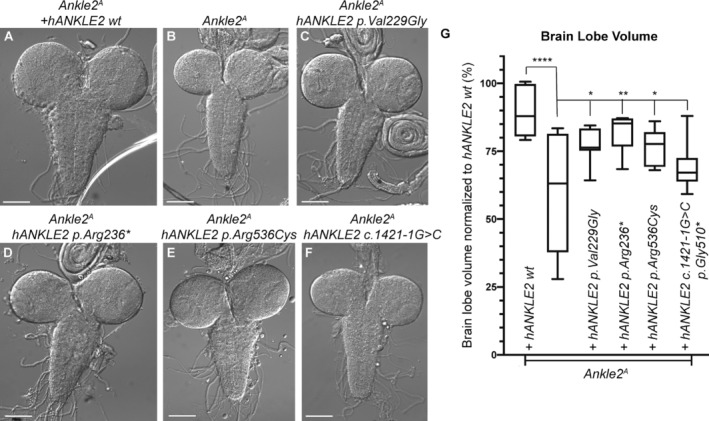
Functional modeling of ANKLE2 variants recapitulates microcephaly phenotypes in Drosophila melanogaster. (A–F) Bright field images of third instar larval brains from (A) Ankle2 A , da‐hANKLE2 wt, (B) Ankle2 A , (C) Ankle2 A , da‐hANKLE2 p.Val229Gly, (D) Ankle2 A , da‐hANKLE2 p.Arg236*, (E) Ankle2 A , da‐hANKLE2 p.Arg536Cys, (F) Ankle2 A , da‐hANKLE2 c.1421‐1G>C. (G) Brain volume quantification of animals from (A–F). One‐way ANOVA with post hoc test. *p < 0.05, **p < 0.01, ****p < 0.0001.
Discussion
MIC refers to an abnormally small head and is a common feature seen in many pediatric neurodevelopmental disorders. Clinically significant congenital MIC (OFC z‐score ≤3) has an incidence of 0.54% among newborns. 1 At least 28 genetic loci have been identified in association with congenital or primary MIC (Online Mendelian Inheritance in Man; OMIM). 5 Within this large and diverse group of disorders, variants within the ANKLE2 gene have been recently identified as an ultra‐rare cause of a congenital MIC syndrome (MIM#616681). 6 , 11 , 12 In this study, we report the largest series to date of individuals with ANKLE2‐related MIC. This series illustrates a broader clinical and molecular spectrum than previously reported single observations yielding a severe MIC phenotype with a markedly reduced brain volume, severe cortical dysgenesis, and other brain MRI abnormalities. In this study, we also report expanded clinical and molecular data on the first identified family of two affected siblings who had severe neurological involvement with early lethality (Family 1). 12 Importantly, we have identified several individuals with a milder neurological phenotype presenting with mild MIC with only mild simplification of the cortical gyral pattern and lacking other syndromic features such as scalp rugae, cutaneous pigmentary abnormalities, and other brain MRI abnormalities. Further, while most affected individuals had congenital MIC, three children (probands 3, 8, and 12) were normocephalic at birth but became progressively microcephalic postnatally, suggesting that ANKLE2 variants are associated with a much broader neurological syndrome than previously appreciated and can be associated with postnatal as well as congenital MIC. As expected, more severe congenital MIC with additional brain MRI abnormalities (other than ACC) were associated with more severe neurodevelopmental features including more profound tone abnormalities and developmental delays, whereas individuals with milder MIC or normal OFC at birth without cortical or subcortical abnormalities (e.g., white matter abnormalities) had better neurodevelopmental outcomes. Interestingly, linear growth appeared to be impacted in all individuals, albeit to a lesser degree than head size, with stature z‐scores ranging from −7.08 to +0.1 (mean −2.67), suggesting that ANKLE2 variants may have more severe effects on growth in general but with preferential involvement of the brain (Fig. 2).
Given this broad clinical and molecular spectrum, the findings in our study support the inclusion of ANKLE2‐related MIC in the differential diagnosis of individuals presenting with both mild and severe congenital MIC, as well as in children who present with postnatal MIC. The co‐occurrence of characteristic hyper‐ and hypopigmented cutaneous markings at birth in conjunction with congenital MIC may constitute a useful physical diagnostic aid for clinicians to consider ANKLE2‐related MIC specifically, although these skin findings were not seen in all affected children in our series. While we were unable to derive conclusive genotype–phenotype correlations from this series, we observed that the three missense variants may confer a milder MIC phenotype (probands 11 and 12). Notably, two of the most severely affected individuals (probands 1 and 2 in family 1) were compound heterozygous for a missense and a protein‐truncating variant (PTV). Similarly, proband 10, who was also more severely affected, had a variant that caused deletion of two exons predicted to be a PTV. Interestingly, the proband with a splice site variant had a more severe MIC (OFC −5.45) with only a simplified cortical gyral pattern; however, the Drosophila studies supported a more significant loss‐of‐function for this variant. The combined with the expression data of the Drosophila Ankle2 system suggest that pathogenic ANKLE2 variants may act in a hypomorphic loss‐of‐function manner.
Despite increased clinical utilization of next generation sequencing methods to identify genetic causes in children presenting with MIC, acquired insults or injuries (notably infection during early fetal development) remain among the most common causes of MIC overall with interesting emerging convergence in the cellular pathways linking genetic and acquired forms of MIC. In utero infections linked to congenital MIC as well as malformations of the developing human brain include the classic TORCH (Toxoplasma gondii, rubella virus, cytomegalovirus [CMV] and herpes simplex virus) infections, lymphocytic choriomeningitis virus and congenital Zika syndrome (CZS). 24 , 25 , 26 , 27 , 28 There are marked similarities in phenotype between these congenital viral infections when the central nervous system is involved, as they may all cause MIC, ventriculomegaly, callosal abnormalities, intracranial calcifications, and in the most severe cases, fetal brain disruption sequence. However, these congenital viral infections also vary considerably in their manifestations and clinical severity in newborns ranging from asymptomatic to several neurological impairments with poor outcomes. 29
Among viral infections involving the brain, ZIKV has gained the most notoriety to date. The first documented ZIKV outbreak in humans was reported on the Yap Islands in Micronesia in 2007, where up to 73% of the population tested positive for ZIKV and, of those, 80% were asymptomatic. 30 , 31 Subsequently, severe outbreaks in French Polynesia, Brazil, and the United States from 2013 to 2016 resulted in a dramatically increased prevalence of congenital MIC due to in utero ZIKV infection. 32 Retrospective analysis of all congenital MIC cases in French Polynesia subsequently established the first causal link between ZIKV and MIC. 33 The disorder, termed CZS, is now known to be associated with multiple and often severe abnormalities of the developing central nervous system in children and adults including diffuse cortical dysgenesis, increased extra‐axial spaces, intracranial calcifications, hypoplasia or absence of corpus callosum, hippocampal and optic nerve anomalies. Other clinical features including severe MIC (birth OFC z‐score ≤ −3), partially collapsed skull, overriding sutures, scalp rugae, subcortical calcifications, macular scarring, focal pigmentary retinal mottling, joint contractures, severe hypertonia and extrapyramidal symptoms such as tremors and posturing have also been reported and proposed to represent the most severe end of the CZS spectrum. 34 Increasing numbers of children and adults with milder phenotypes due to ZIKV were subsequently reported including mild MIC and even normocephalic head size, and mild skeletal abnormalities such as club foot, which are seen in up to 15% of infants with CZS. 34
ANKLE2 encodes a key protein that regulates the reassembly of the nuclear envelope at the onset of anaphase prior to mitotic cell exit. 11 , 12 A recent study of ZIKV in non‐primate models identified ANKLE2 as a downstream target of the ZIKV and that genetic variants of ANKLE2 can disrupt shared cellular processes, namely asymmetric cell division in the developing brain, thereby recapitulating ZIKV‐related MIC. 12 We also previously demonstrated that ANKLE2 interacts with NS4A, a ZIKV protein that when expressed in the larval Drosophila brain causes MIC with decreased proliferation and increased apoptosis. 12 Further, MIC in hypomorphic Drosophila Ankle2 A mutants is rescued by the expression of human ANKLE2. Moreover, overexpression of ZIKV NS4A in D. melanogaster causes a significant decrease in larval brain volume, while expression of human ANKLE2 or Drosophila Ankle2 rescue this phenotype. 11 , 12 , 14 Our functional experiments modeling additional human ANKLE2 mutants in D. melanogaster in this study further recapitulated the human MIC phenotype. Taken together, these data demonstrate a robust link in pathogenic mechanisms between ZIKV‐ and ANKLE2‐related MIC, with growing evidence that ZIKV and ANKLE2 act on the same highly conserved molecular pathways crucial for normal human brain development. 11 , 12 , 14 However, additional functional experiments are necessary to further elucidate the mechanisms by which these ANKLE2 variants cause human MIC, as well as identify potential modifiers that may explain the wide variability in the clinical severity of ANKLE2‐related MIC. These emerging links between ANKLE2‐related MIC and CZS are further supported by the clinical and neuroimaging data within this study as we observed several phenotypic similarities between these two disorders (Table 1). Shared clinical features among both entities include congenital MIC of variable severity, and, in the most severely affected individuals, collapsed skull, increased extra‐axial spaces, scalp rugae, overriding sutures, spastic quadriparesis, and prominent occiput. 26 , 27 , 34 Interestingly, the pigmentary cutaneous findings in our series have not been reported in children with ZIKV‐related MIC to our knowledge, and may suggest involvement of other pathways which could be further explored in the Drosophila model system.
Since ZIKV and ANKLE2 mechanistically converge on the same neurodevelopmental pathways, we hypothesize that the severity of CZS in infected children could be potentiated by genetic variants in relevant genes in these pathways making carriers of heterozygous pathogenic variants more susceptible to congenital MIC after ZIKV infection in utero. This could explain why CZS causes congenital MIC in only about 6% of exposed fetuses. 35 , 36 , 37 If this hypothesis proves to be valid, we expect that more individuals who are heterozygous carriers of ANKLE2 variants would be identified among ZIKV‐exposed infants with MIC, and fewer among those without MIC. This hypothesis merits further investigation by genomic studies of ZIKV‐exposed infants with and without MIC, specifically searching for heterozygous variants in ANKLE2 and other MIC‐associated genes.
While the CZS has gained much public attention, it is important to remember that congenital cytomegalovirus (cCMV) continues to be the most common congenital viral infection globally and is a common cause of neurodevelopmental disabilities, sensorineural hearing loss, and vision loss in children. 38 cCMV infection can also perturb developmental processes in the fetal and neonatal brain resulting in abnormalities that overlap many congenital MIC syndromes including cortical dysgenesis, intracranial (predominantly periventricular) calcifications, dysgenesis of the corpus callosum, cerebellar hypoplasia with additional features as well such as ventriculomegaly, lenticulostriate vasculopathy, periventricular cystic malformations, and fetal brain sequence disruption or arrest syndrome. 39 , 40 , 41 Of note, intracranial calcifications, as seen with cCMV, CZS, and other in utero infections, may be underreported in our series as most children underwent brain imaging by MR which has limited sensitivity for their detection. All in all, the phenotypic features of children with cCMV are similar to, or at times indistinguishable from, CZS, and now ANKLE2‐related MIC. 26 , 29 This phenotypic overlap in brain involvement between genetic and acquired forms of MIC underscores the broad differential diagnosis of MIC in children and strongly supports that a systematic and comprehensive diagnostic workup examining both genetic and non‐genetic causes is often warranted. 1
In summary, this study reports on the clinical, neuroimaging, and molecular features of ANKLE2‐related MIC in children revealing a much broader spectrum than previously identified. Affected children have variable MIC, a range of brain abnormalities including variable degrees of cortical dysplasia or dysgenesis, white matter abnormalities, partial or complete callosal agenesis, increased extra‐axial spaces, and characteristic hyper‐ and hypo‐pigmented macules. This study further supports that bi‐allelic hypomorphic or loss‐of‐function variants in ANKLE2 cause MIC by perturbing several key neural pathways that overlap with congenital ZIKV infection. This study overall strongly supports the evolving paradigm that environmental (or acquired) and genetic factors underlying congenital MIC have highly overlapping mechanisms and many converge on the same key cellular pathways in the developing human brain.
Author Contributions
A. X. T., R. D. C., and G. M. M. conceived and designed the study, acquired and analyzed the data, and drafted the manuscript and Figs. N. L., G. D.‐H., L. A. R., E. C. P., A. E. S., S. M., J. S. C., A. C., P. P., A. V. d. P., G. D. C, L. G., S. G. C., C. F., R. Z., K. C. P., E. H. S., M. O. H., S. M., F. S. A., I. A. F. C., J. E. N., C. A. W., H. J. B., H.‐T. C. contributed to the acquisition and analysis of the data.
Conflicts of Interest
The authors do not have any real or apparent conflicts of interest.
Supporting information
Data S1. Full clinical and molecular data of probands with pathogenic ANKLE2 variants.
Table S1. Clinical and molecular features of individuals with ANKLE2‐related microcephaly.
Acknowledgments
We thank the patients, their families, and collaborators for their contribution to this study. Research reported in this publication was supported by Jordan's Guardian Angels, the Brotman‐Baty Institute, and the Sunderland Foundation (to G. M. M.). C. A. W. is supported in part by NINDS grant R01NS035129 and is an investigator of the Howard Hughes Medical Institute. H. J. B is an investigator of the Howard Hughes Medical Institute. H.‐T. C. is supported in part by the Child Neurology Society and Foundation, the Mark A. Wallace Endowment, Burroughs Wellcome Fund, and the McNair Medical Institute at The Robert and Janice McNair Foundation. Dr. Garavelli and Dr. Carlo Fusco are members of the European Reference Network on Rare Congenital Malformations and Rare Intellectual Disability ERN‐ITHACA (EU Framework Partnership Agreement ID: 3HP‐HP‐FPA ERN‐01‐2016/739516).
Annals of Clinical and Translational Neurology 2022;9(8): 1276–1288
Funding Information
Research reported in this publication was supported by Jordan's Guardian Angels, the Brotman‐Baty Institute, and the Sunderland Foundation (to G. M. M.). C. A. W. is supported in part by NINDS grant R01NS035129 and is an investigator of the Howard Hughes Medical Institute. H. J. B is an investigator of the Howard Hughes Medical Institute. H.‐T. C. is supported in part by the Child Neurology Society and Foundation, the Mark A. Wallace Endowment, Burroughs Wellcome Fund, and the McNair Medical Institute at The Robert and Janice McNair Foundation. Dr. Garavelli and Dr. Carlo Fusco are members of the European Reference Network on Rare Congenital Malformations and Rare Intellectual Disability ERN‐ITHACA (EU Framework Partnership Agreement ID: 3HP‐HP‐FPA ERN‐01‐2016/739516).
Funding Statement
This work was funded by Brotman‐Baty Institute; Burroughs Wellcome Fund ; EU Framework Partnership grant ERN‐01‐2016 / 739516; Howard Hughes Medical Institute ; McNair Medical Institute at The Robert and Janice McNair Foundation; NINDS grant R01NS035129; Sunderland Foundation; Mark A. Wallace Endowment; Child Neurology Society and Foundation; Jordan's Guardian Angels.
Contributor Information
Ajay X. Thomas, Email: ajay.thomas@bcm.edu.
Ghayda M. Mirzaa, Email: ghayda.mirzaa@seattlechildrens.org.
References
- 1. Ashwal S, Michelson D, Plawner L, Dobyns WB; Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society . Practice parameter: evaluation of the child with microcephaly (an evidence‐based review): report of the Quality Standards Subcommittee of the American Academy of neurology and the Practice Committee of the Child Neurology Society. Neurology. 2009;73(11):887‐897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Villar J, Cheikh Ismail L, Victora CG, et al. International standards for newborn weight, length, and head circumference by gestational age and sex: the Newborn Cross‐Sectional Study of the INTERGROWTH‐21st Project. Lancet. 2014;384(9946):857‐868. [DOI] [PubMed] [Google Scholar]
- 3. Cragan JD, Isenburg JL, Parker SE, et al. Population‐based microcephaly surveillance in the United States, 2009 to 2013: an analysis of potential sources of variation. Birth Defects Res A Clin Mol Teratol. 2016;106(11):972‐982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Morris‐Rosendahl DJ, Kaindl AM. What next‐generation sequencing (NGS) technology has enabled us to learn about primary autosomal recessive microcephaly (MCPH). Mol Cell Probes. 2015;29(5):271‐281. [DOI] [PubMed] [Google Scholar]
- 5. Pirozzi F, Nelson B, Mirzaa G. From microcephaly to megalencephaly: determinants of brain size. Dialogues Clin Neurosci. 2018;20(4):267‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shaheen R, Maddirevula S, Ewida N, et al. Genomic and phenotypic delineation of congenital microcephaly. Genet Med. 2019;21(3):545‐552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Florio M, Huttner WB. Neural progenitors, neurogenesis and the evolution of the neocortex. Development. 2014;141(11):2182‐2194. [DOI] [PubMed] [Google Scholar]
- 8. Montgomery SH, Capellini I, Venditti C, et al. Adaptive evolution of four microcephaly genes and the evolution of brain size in anthropoid primates. Mol Biol Evol. 2011;28(1):625‐638. [DOI] [PubMed] [Google Scholar]
- 9. Murray JE, Jackson AP. Exploring microcephaly and human brain evolution. Dev Med Child Neurol. 2012;54(7):580‐581. [DOI] [PubMed] [Google Scholar]
- 10. Northcutt RG, Kaas JH. The emergence and evolution of mammalian neocortex. Trends Neurosci. 1995;18(9):373‐379. [DOI] [PubMed] [Google Scholar]
- 11. Link N, Chung H, Jolly A, et al. Mutations in ANKLE2, a ZIKA virus target, disrupt an asymmetric cell division pathway in Drosophila neuroblasts to cause microcephaly. Dev Cell. 2019;51(6):713‐729.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yamamoto S, Jaiswal M, Charng W‐L, et al. A Drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases. Cell. 2014;159(1):200‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Asencio C, Davidson IF, Santarella‐Mellwig R, et al. Coordination of kinase and phosphatase activities by Lem4 enables nuclear envelope reassembly during mitosis. Cell. 2012;150(1):122‐135. [DOI] [PubMed] [Google Scholar]
- 14. Shah PS, Link N, Jang GM, et al. Comparative flavivirus‐host protein interaction mapping reveals mechanisms of dengue and Zika virus pathogenesis. Cell. 2018;175(7):1931‐1945.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de Macena Sobreira NL, Hamosh A. Next‐generation sequencing and the evolution of data sharing. Am J Med Genet A. 2021;185:2633‐2635. [DOI] [PubMed] [Google Scholar]
- 16. Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36(10):928‐930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chou JH, Roumiantsev S, Singh R. PediTools electronic growth chart calculators: applications in clinical care, research, and quality improvement. J Med Internet Res. 2020;22(1):e16204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nellhaus G. Head circumference from birth to eighteen years. Practical composite international and interracial graphs. Pediatrics. 1968;41(1):106‐114. [PubMed] [Google Scholar]
- 19. Wodarz A, Hinz U, Engelbert M, Knust E. Expression of crumbs confers apical character on plasma membrane domains of ectodermal epithelia of Drosophila. Cell. 1995;82(1):67‐76. [DOI] [PubMed] [Google Scholar]
- 20. Venken KJT, He Y, Hoskins RA, Bellen HJ. P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster . Science. 2006;314(5806):1747‐1751. [DOI] [PubMed] [Google Scholar]
- 21. Honein MA, Cetron MS, Meaney‐Delman D. Endemic Zika virus transmission: implications for travellers. Lancet Infect Dis. 2019;19(4):349‐351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hu Y, Flockhart I, Vinayagam A, et al. An integrative approach to ortholog prediction for disease‐focused and other functional studies. BMC Bioinformatics. 2011;12:357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Link N, Bellen HJ. Using Drosophila to drive the diagnosis and understand the mechanisms of rare human diseases. Development. 2020;147(21):dev191411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bonthius DJ. Lymphocytic choriomeningitis virus: an underrecognized cause of neurologic disease in the fetus, child, and adult. Semin Pediatr Neurol. 2012;19(3):89‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bonthius DJ, Wright R, Tseng B, et al. Congenital lymphocytic choriomeningitis virus infection: spectrum of disease. Ann Neurol. 2007;62(4):347‐355. doi: 10.1002/ana.21161 [DOI] [PubMed] [Google Scholar]
- 26. Del Campo M, Feitosa IML, Ribeiro EM, et al. The phenotypic spectrum of congenital Zika syndrome. Am J Med Genet A. 2017;173(4):841‐857. [DOI] [PubMed] [Google Scholar]
- 27. Freitas DA, Souza‐Santos R, Carvalho LMA, et al. Congenital Zika syndrome: a systematic review. PLoS One. 2020;15(12):e0242367. doi: 10.1371/journal.pone.0242367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mitsikas D, Gabrani C, Giannakou K, Lamnisos D. Intrauterine exposure to Zika virus and hearing loss within the first few years of life: a systematic literature review. Int J Pediatr Otorhinolaryngol. 2021;147:110801. [DOI] [PubMed] [Google Scholar]
- 29. Moodley A, Payton KSE. The term newborn: congenital infections. Clin Perinatol. 2021;48(3):485‐511. [DOI] [PubMed] [Google Scholar]
- 30. Lanciotti RS, Kosoy OL, Laven JJ, et al. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg Infect Dis. 2008;14(8):1232‐1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Duffy MR, Chen T‐H, Hancock WT, et al. Zika virus outbreak on Yap Island, Federated States of Micronesia. N Engl J Med. 2009;360(24):2536‐2543. doi: 10.1056/NEJMoa0805715 [DOI] [PubMed] [Google Scholar]
- 32. Schuler‐Faccini L, Ribeiro EM, Feitosa IML, et al. Possible association between Zika virus infection and microcephaly – Brazil, 2015. MMWR Morb Mortal Wkly Rep. 2016;65(3):59‐62. [DOI] [PubMed] [Google Scholar]
- 33. Cauchemez S, Besnard M, Bompard P, et al. Association between Zika virus and microcephaly in French Polynesia, 2013–15: a retrospective study. Lancet. 2016;387(10033):2125‐2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moore CA, Staples JE, Dobyns WB, et al. Characterizing the pattern of anomalies in congenital Zika syndrome for pediatric clinicians. JAMA Pediatr. 2017;171(3):288‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eppes C, Rac M, Dunn J, et al. Testing for Zika virus infection in pregnancy: key concepts to deal with an emerging epidemic. Am J Obstet Gynecol. 2017;216(3):209‐225. [DOI] [PubMed] [Google Scholar]
- 36. Petersen EE, Staples JE, Meaney‐Delman D, et al. Interim guidelines for pregnant women during a Zika virus outbreak—United States, 2016. MMWR Morb Mortal Wkly Rep. 2016;65(2):30‐33. [DOI] [PubMed] [Google Scholar]
- 37. Honein MA, Dawson AL, Petersen EE, et al. Birth defects among fetuses and infants of US women with evidence of possible Zika virus infection during pregnancy. JAMA. 2017;317(1):59‐68. [DOI] [PubMed] [Google Scholar]
- 38. Jenks CM, Hoff SR, Mithal LB. Congenital cytomegalovirus infection: epidemiology, timely diagnosis, and management. Neoreviews. 2021;22(9):e606‐e613. [DOI] [PubMed] [Google Scholar]
- 39. Cheeran MC‐J, Lokensgard JR, Schleiss MR. Neuropathogenesis of congenital cytomegalovirus infection: disease mechanisms and prospects for intervention. Clin Microbiol Rev. 2009;22(1):99‐126. doi: 10.1128/CMR.00023-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kachramanoglou C, Jan W, Jones B, et al. Diagnostic analysis of baseline brain MRI features in infants with congenital cytomegalovirus infection: a simplified scoring system. Clin Radiol. 2021;76(12):942.e7‐942.e14. [DOI] [PubMed] [Google Scholar]
- 41. Zhou Y‐P, Mei M‐J, Wang X‐Z, et al. A congenital CMV infection model for follow‐up studies of neurodevelopmental disorders, neuroimaging abnormalities, and treatment. JCI Insight. 2022;7(1):e152551. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Full clinical and molecular data of probands with pathogenic ANKLE2 variants.
Table S1. Clinical and molecular features of individuals with ANKLE2‐related microcephaly.