

Abstract
Objective
ATXN1 encodes the polyglutamine protein ataxin‐1, which we have demonstrated exerting an immunomodulatory function in the context of central nervous system (CNS) autoimmunity, in addition to its classical role in the neurodegenerative disorder spinocerebellar ataxia type 1 (SCA1). In this study, we dissected the contribution of DNA methylation to the regulation of ATXN1 in multiple sclerosis (MS).
Methods
We interrogated a DNA methylation dataset previously generated via bisulfate DNA sequencing (BS‐seq) in sorted peripheral immune cytotypes (CD4+ and CD8+ T cells, CD19+ B cells, and CD14+ monocytes) isolated from untreated MS patients at symptoms onset.
Results
Here, we report that ATXN1 undergoes hypo‐methylation at four distinct regions upon MS, exclusively in B cells. We also highlight how these differentially methylated sites overlap with other regulatory epigenetic marks and MS risk variants. Lastly, we employ luciferase assays to assess the functionality of these regions, showing that the loss of methylation leads to an increase in ATXN1 expression.
Interpretation
Altogether, these findings provide biological insights into ataxin‐1 regulation in the immune system as well as into the molecular mechanisms underlying MS risk.
Introduction
The ATXN1 gene encodes the polyglutamine protein ataxin‐1, a transcription repressor that is primarily known for causing the neurodegenerative disorder spinocerebellar ataxia type 1 (SCA1) upon the pathological expansion of the polyglutamine domain, which in turn leads to toxic buildup of the protein within the nucleus. 1 Conversely, ATXN1 loss‐of‐function mutations have been linked to Alzheimer's disease (AD) pathogenesis and the etiology of various types of cancers. 2 , 3 A large genome‐wide association study (GWAS) designated ATXN1 as the most plausible gene associated with multiple sclerosis (MS) risk within a disease locus mapping at 6p22.3. 4 More recently, we have shown that ataxin‐1 exerts a protective function against autoimmune demyelination in the context of experimental autoimmune encephalomyelitis (EAE), 5 a murine disease model that shares several key pathogenic mechanisms with MS at the cellular and molecular levels. 6 Specifically, we documented a previously unknown immunomodulatory activity for ataxin‐1 in B cells mediated through the extracellular signal‐regulated kinase (ERK) and signal transducer and activator of transcription (STAT) pathways as well as B cell receptor (BCR) signaling. 5 , 7 However, the molecular mechanisms regulating ATXN1 expression and function in the immune system are yet to be defined.
Epigenetic modifications are heritable changes in the DNA molecule that affect the transcription process without modifications in its primary nucleotide sequence. 8 Among the different epigenetic marks, DNA methylation is the best‐characterized modification in humans and involves the covalent transfer of a methyl group onto the C5 position of a cytosine to form 5‐methylcytosine (typically in cytosine‐guanine rich DNA sequences, or CpG islands). 9 DNA methylation is strictly regulated in space and time, and aberrant methylation patterns have been connected to the etiology of numerous chronic disorders including neurodegenerative, cardiovascular, and autoimmune diseases. 10 , 11 In MS, distinct epigenetic signatures have been detected in multiple peripheral immune cell populations, including global methylation alterations in CD19+ B cells associated with clinical onset and initiation of cell differentiation programs, 12 , 13 , 14 , 15 fueling our rationale for investigating the epigenetic regulation of ATXN1 in response to an autoimmune attack against the central nervous system (CNS).
Here, we show that ATXN1 is hypomethylated at the time of clinical onset primarily in B cells, resulting in an increase in expression as confirmed by RNA‐seq data and luciferase assays. Moreover, we investigated the overlap between the methylation sites and other epigenetic signatures as well as MS risk variants, confirming that ATXN1 plays an important role in MS pathogenesis.
Materials and Methods
Subjects
Study participants, 29 multiple sclerosis (MS) patients and 24 healthy controls, were recruited at the UCSF MS Center as part of the UCSF multiple sclerosis EPIC and ORIGINS studies and have been previously described. 15 The UCSF‐EPIC cohort includes 637 participants with over 13 years of annual clinical follow‐up and biospecimens accrual at different clinical stages. 16 Patients included in this study were all treatment naive and met clinical and radiographic diagnostic criteria for relapsing–remitting MS (RR‐MS). The ORIGINS study was designed to recruit participants within the first few days of symptoms (symptomatic for fewer than 90 days). Study participants are evaluated within 24 h of the initial contact. All individuals were white, with western European ancestry, female, and had no history of smoking. Mean ages at sample collection were 38 (range from 21 to 56) for cases and 37 (range: 22–58) for controls. There was no significant difference in age between MS and control samples (Wilcoxon test, P = 0.47). Both studies were approved by the UCSF Institutional Review Board, and informed consent was obtained from all subjects.
Sample preparation and BS‐seq analysis
Peripheral blood mononuclear cells (PBMCs) were isolated immediately after blood collection using a standard Ficoll (GE Healthcare) gradient procedure. CD4+ and CD8+ T cells, CD14+ monocytes, and CD19+ B cells were purified from PBMCs using a MoFlo Astrios cell sorter (Beckman Coulter) and the following antibodies: CD3 APC (UCHT1, Beckman Coulter), CD4 Brilliant Violet 421(OKT4, BioLegend), CD8 APC AlexaFluor750 (B9.11, Beckman Coulter), CD19 PerCP Cy5.5 (J3‐119, Beckman Coulter), CD14 PE (61D3, eBioscience). Genomic DNA from sorted cells was extracted using the Quick DNA Kit (Zymo Research) and processed for BS‐seq library construction as described. 15 Briefly, genomic DNA was fragmented, end‐repaired, A‐tailed, and ligated to methylated adaptors. Ligated fragments were subsequently bisulfite converted using the EZ DNA Methylation Gold Kit (Zymo Research) and amplified using the HiFi HotStart Uracil+ ReadyMix PCR Kit (KAPA). Libraries were finally sequenced on a NovaSeq platform (Illumina). The generated reads were analyzed following an established bioinformatics pipeline. After filtering out low‐quality reads, the remaining paired reads were uniquely mapped to the reference human genome using the Bismark software. 17 The number of “C” bases from the sequencing reads were counted as methylated (denoted as NC) and the number of “T” bases as unmodified (denoted as NT). The methylation levels were then estimated as NC/(NC + NT).
Identification of differentially methylated regions (DMRs)
Three different algorithms (metilene, SMART, and Defiant) for DMR identification were applied. The pipeline using the metilene package (version 0.2–8) to call the DMRs has been previously described. 15 SMART is an entropy‐based algorithm to calculate the methylation similarity between neighboring CpG sites. 18 By continuous scanning the methylation similarity on each chromosome, the CpG sites with similar methylation patterns across all samples are merged into genome segments. After genome segmentation, the methylation level of each genome segment is calculated as mean methylation level of the CpG sites in it. In this study, the genome segments with differences in methylation ≥10% between MS and HC groups (Student's t‐test p ≤ 0.05) and a minimum of 3 CpGs were defined as DMRs. Defiant employs a weighted welch expansion (WWE) algorithm to detect the DMRs. 19 We applied the Defiant software to identify DMRs with a 10% minimum absolute mean methylation difference and a minimum of 3 CpGs per DMR (parameter ‐c 3 ‐CpN 3 ‐P 10).
mRNA‐seq analysis
Transcriptomic analysis was performed on the B cells from 23 MS patients and 14 healthy controls selected within our study cohort. The poly(A)‐selected paired‐end sequencing libraries were constructed using the mRNA Hyper Prep Kit (KAPA), following the manufacturer's instructions. The sequencing was performed on a HiSeq X Ten platform (Illumina), generating ~30 million pair‐end 150 bp reads per sample. For the data analysis, low‐quality and adapter sequences were trimmed using the Trimmomatic tool. 20 The remaining reads were then aligned to the human reference using hisat2. 21 The htseq‐count tool and human gtf file from Ensembl were used to count aligned reads for each gene. 22 Differentially expressed genes (DEGs) between RR‐MS and HC individuals were finally identified using the edgeR package. 23
Bioinformatics analyses
The methylation of each DMR was calculated as the mean methylation of all CpG sites in the region for each sample. The methylation differences of each DMR between MS and HC groups in each cell type were assessed with Mann–Whitney U test. DNase‐seq and ATAC‐seq data for T cells, B cells, and monocytes were obtained from the ENCODE database (Accession numbers: ENCFF508FIN, ENCFF531CKE, ENCFF160SGQ, and ENCFF273DGN). 24 H3K27ac peak data for four immune cell types (CD4+ and CD8+ T cells, CD19+ B cells, and CD14+ monocytes) were also downloaded from the ENCODE portal (Accession numbers: ENCFF969HEX, ENCFF471QGU, ENCFF053LHH, and ENCFF823JZP). DNAse‐seq and ATAC‐seq identify open chromatin regions along the genome. 25 H3K27ac ChIP‐seq instead maps those loci enriched in histone H3 acetylated at the lysine residue K27, which represents a robust marker for active enhancers. 26 The r2 measurements of linkage disequilibrium (LD) were calculated with PLINK, using the European data from the 1000 Genomes Project. 27 The summary statistics from the last MS meta‐analysis (14,802 subjects with MS and 26,703 controls) were used to identify SNPs associated with MS susceptibility with nominal significance (p < 0.05). 4 The tracks of open chromatin peaks, H3K27ac peaks, DMR positions, SNP significance, and LD matrix were visualized using WashU Epigenome Browser. 28
Cell lines
The HeLa cell line was obtained from the ATCC biobank. The cells were cultured in Dulbecco's Modified Eagle's Medium (GIBCO) supplemented with 10% v/v fetal bovine serum (GIBCO) and maintained at 37°C in a humidified atmosphere with 5% CO2.
Plasmids
The CpG‐free firefly luciferase reporter vector pCpGL‐basic was a kind gift from Dr. Michael Rehli (University Hospital, Regensburg, Germany). The Renilla luciferase reporter vector pRL‐TK was purchased from Promega. The four DMRs identified within the ATXN1 gene by BS‐seq were amplified from genomic DNA with Phusion High‐Fidelity DNA Polymerase (New England Biolabs) using the primers listed in Table S1, and the PCR products were then cloned into the pCpGL‐basic vector using the restriction enzymes SpeI and NcoI (New England Biolabs). Individual clones were confirmed by Sanger sequencing. The different pCpGL constructs were methylated in vitro by incubating 6 μg of each plasmid with 12 U of DNA methyltransferase SssI (New England Biolabs) for 4 h at 37°C, in the presence of 320 μM S‐Adenosylmethionine (SAM; New England Biolabs) with additional 320 μM SAM supplemented after the first 2 h. Mock reactions were carried out in the same conditions but without the enzyme. All the plasmids were finally precipitated overnight at −20°C with ethanol and resuspended in sterile water.
Luciferase assays
HeLa cells were grown in 12‐well plates at 90% confluency and co‐transfected with 700 ng of the different pCpGL constructs, either methylated or non‐methylated, and 70 ng of the pRL‐TK vector using Lipofectamine 2000 (Invitrogen). After 24 h, cells were washed once with PBS and lysed in 250 mL of Passive Lysis Buffer (Promega), for 15 min at room temperature with agitation. Twenty microliters of each cell lysate were loaded in duplicate in black 96‐well plates and the activities of both firefly and Renilla luciferases were measured with the Dual‐Luciferase Reporter Assay System (Promega), using a Veritas Microplate Luminometer (Turner BioSystems). The firefly luciferase activity of each construct was first normalized against Renilla luciferase activity, and then the normalized signals of the methylated constructs were expressed as fold‐changes of the corresponding non‐methylated vectors. Data from three biologically independent experiments were expressed as mean ± SEM. Differences were assessed with two‐tailed Student's t test, and p values equal to 0.05 or less were considered significant.
Results
ATXN1 is differentially methylated upon multiple sclerosis
We have recently reported the results of a comprehensive BS‐seq screening on four peripheral blood immune cell types (CD4+ and CD8+ T cells, CD19+ B cells, and CD14+ monocytes) isolated from 29 female non‐smokers MS patients at disease clinical onset and 24 healthy controls matched in sex, age, smoking history, and ancestry. 15 Here, we extracted from the dataset the DNA methylation profiles for the ATXN1 locus (hg19, chr6:16350981–16598133). Four differentially methylated regions (DMRs) were identified using the metilene package within the third, sixth, and seventh introns of the ATXN1 sequence, named hereafter DMR1 to DMR4 (Fig. S1). They cover, respectively, 12, 7, 18, and 26 CpG sites. We re‐analyzed the raw sequencing data with two alternative packages (defiant and SMART) to call the DMRs, and the same four methylated regions in ATXN1 were identified by at least two of the algorithms, thus confirming the robustness of the results (Fig. S1). All four DMRs were significantly hypo‐methylated in the B cells of MS patients as compared to controls (p DMR1 = 4.141 × 10−14; p DMR2 = 2.814 × 10−12; p DMR3 = 7.049 × 10−13; p DMR4 = 4.041 × 10−14), whereas DMR4 was also hypo‐methylated in monocytes (p = 0.042) (Fig. 1). We extended the analysis to the genomic regions flanking the ATXN1 gene (±100 kb from the transcriptional start site) but no additional DMRs were detected. In summary, these results are consistent with the B cell‐specific epigenetic regulation of ATXN1 expression in response to autoimmune demyelination.
Figure 1.
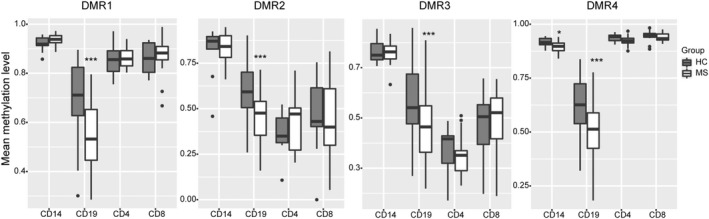
ATXN1 is hypo‐methylated in B cells upon MS. The boxplots represent mean methylation levels at four differentially methylated regions within ATXN1 genomic sequence (DMR1, chr6:16640696–16641053; DMR2, chr6:16473394–16473577; DMR3, chr6:16420309–16422015; DMR4, chr6:16306158–16306782) in four relevant immune peripheral cytotypes (CD4+ and CD8+ T cells, CD19+ B cells, and CD14+ monocytes) between untreated MS patients (N = 17 for T cells, N = 29 for B cells, N = 12 for monocytes) and healthy controls (N = 12 for T cells, N = 24 for B cells, N = 10 for monocytes). Differences between MS patients and controls in each cell type were assessed by Mann–Whitney U test. *p < 0.05, ***p < 0.001.
ATXN1 methylated sites overlap with euchromatin regions
We compared different epigenetic signatures in the ATXN1 locus deriving publicly available data from ENCODE DNAse‐seq, ATAC‐seq, and H3K27ac ChIP‐seq analyses in T cells, B cells, and monocytes (Fig. 2). Interestingly, DMR1 does not overlap with any other epigenetic mark in any cell type analyzed, likely mapping to a heterochromatin region in both lymphocytes and monocytes. DMR2 and MDR3 show similar patterns as both overlap with DNAse/ATAC‐seq peaks and ChIP‐seq peaks in B cells and T cells, but not in monocytes. Finally, DMR4 overlaps with a DNAse‐sensitive region only in B cells. Altogether, these data indicate that DMR2 and DMR3 likely function as enhancer regions for the lymphocyte lineage. On the contrary, DMR4 marks a B cell‐specific transcriptionally active region.
Figure 2.
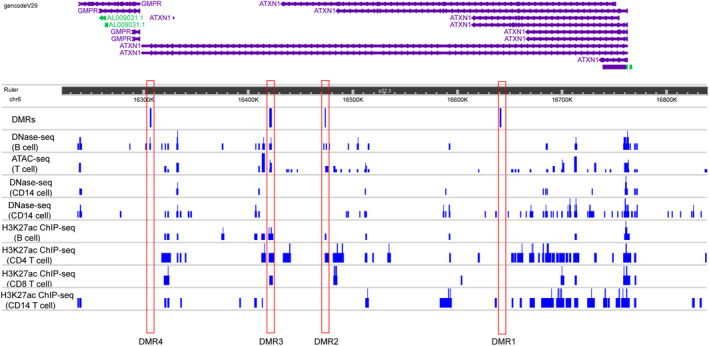
ATXN1 DMRs overlap with other epigenetic marks. The plot depicts the epigenetic annotation at the ATXN1 locus in four different immune cell types (CD4+ and CD8+ T cells, CD19+ B cells, and CD14+ monocytes), based on publicly available ENCODE data. DNAse‐seq and ATAC‐seq peaks highlight genomic regions of open chromatin, while H3K27ac ChIP‐seq peaks indicate sites functioning as active enhancers. [Colour figure can be viewed at wileyonlinelibrary.com]
ATXN1 methylated sites are close to MS‐risk variants
The single nucleotide polymorphism (SNP) rs719316 in the third intron of ATXN1 has been found as the strongest MS risk variant in the 6p22.3 locus (p = 1.62 × 10−13, OR = 1.072) in the largest GWAS and meta‐analysis from the International Multiple Sclerosis Genetics Consortium (IMSGC). 4 As DRM1 also maps to the third intron, we were keen to explore the spatial relationship between DNA methylation patterns and MS susceptibility hits within the ATXN1 gene. To do so, we first plotted the linkage disequilibrium (LD) across the region using the European data from the 1000 Genomes Project as a reference panel. Subsequently, we superimposed all the SNPs associated with MS susceptibility with nominal significance (p < 0.05) from the IMSGC study (Fig. 3). As expected, both rs719316 and DMR1 co‐localize in a large LD block which shows the most significant association with MS risk. Also, DMR2 and DMR3 map to two adjacent LD blocks containing several MS risk markers. In contrast, DMR4 is located in an LD block that failed to show any association with MS susceptibility.
Figure 3.
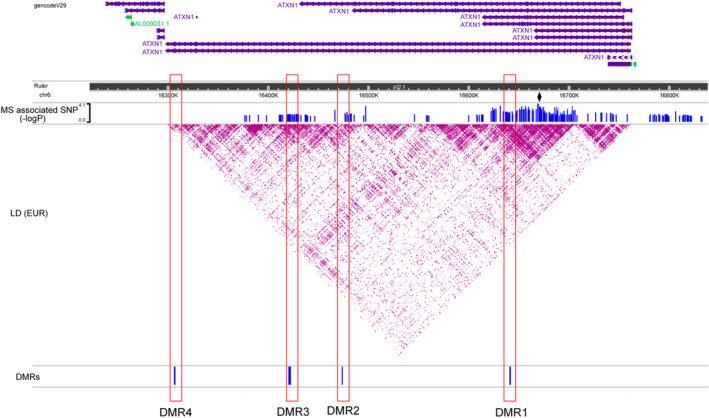
ATXN1 DMRs overlap with MS‐risk variants. The plot displays the linkage‐disequilibrium (LD) across the ATXN1 locus according to r 2 values calculated from the European dataset in the 1000 Genomes Project. Variants associated with MS susceptibility with nominal significance (p < 0.05) in the most recent MS meta‐analysis from the International Multiple Sclerosis Genetics Consortium (IMSGC) are also depicted. The diamond highlights the strongest association in the meta‐analysis (rs719316) mapping to the third intron of ATXN1. The DMRs are shown below the LD plot. [Colour figure can be viewed at wileyonlinelibrary.com]
DNA methylation affects ATXN1 expression
DNA methylation typically results in the repression of gene transcription. 29 To experimentally address whether this epigenetic mark negatively affects ATXN1 expression, we established a luciferase assay to test the functionality of the four DMRs we identified. 30 We cloned each DMR into a CpG‐free luciferase reporter vector, and the different constructs were transfected in HeLa cells after being methylated in vitro. The luciferase activity for each construct was then measured and compared to that of the non‐methylated counterpart. Consistent with our expectation, the constructs carrying the methylated DMR1, DMR3, and DMR4 expressed significantly lower levels of luciferase than their non‐methylated versions (Fig. 4A). Methylation of DRM2 failed to exert any detectable effect on luciferase transcription, possibly due to the limited length of this site (183 bp) compared to the other three (357 bp for DMR1, 1706 bp for DMR3, and 624 bp for DMR4) (Fig. 4A).
Figure 4.
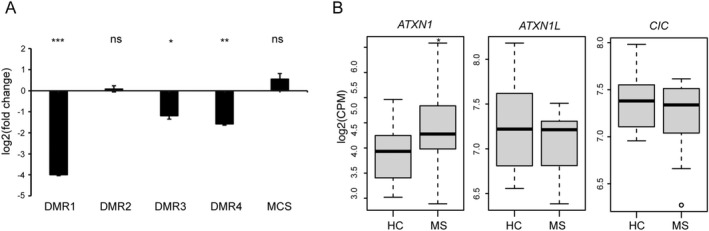
DNA methylation modulates ATXN1 expression. (A) Luciferase reporter vectors bearing the four DMRs were methylated in vitro and transfected in HeLa cells. The luciferase activity for each construct was tested 24 h after transfection and compared to the non‐methylated counterpart. A luciferase vector lacking the promoter region (MCS) was used as additional control. The data were expressed as log2 transformed fold change (log2FC) and plotted as means ± SE. Differences between experimental conditions were assessed by two‐tailed Student's t test. (B) The boxplots represent the RNA levels for ATXN1, ATXN1L, and CIC gene in B cells from MS patients (N = 23) and healthy controls (N = 14). The data were expressed as log2 transformed counts per million (log2CPM) and derived from RNA‐seq. Differential expression analysis was performed using the exact test implemented in the edgeR package. *p < 0.05, **p < 0.01, ***p < 0.001.
To further confirm the results of the luciferase assays, we extracted the expression data for ATXN1 from a transcriptomic dataset that we generated using the B cells isolated from the same cohort of the initial BS‐seq screening. Consistent with the in vitro results, ATXN1 expression was significantly higher in MS patients (Fig. 4B). We also analyzed the mRNA levels of two genes encoding key ataxin‐1 interactors, namely Capicua (CIC) and ataxin‐1‐like (ATXN1L), but no differences were found between cases and controls (Fig. 4B). Likewise, no DMRs were detected in their DNA coding sequences (data not shown).
Discussion
We have recently provided experimental evidence supporting a modulatory role for ATXN1 on B cell function, a key component of MS pathogenesis. 5 Here, we expand its functional characterization by showing that ATXN1 expression in B cells is epigenetically regulated in the early stages of disease expression. Specifically, we identified within the ATXN1 genomic sequence four sites hypo‐methylated in B cells at clinical onset. Furthermore, we demonstrated that the loss of methylation mechanistically leads to increased levels of ATXN1 messenger RNA. These results are in line with the immunoregulatory function we have previously documented for ataxin‐1 in the EAE model. In fact, the EAE data indicate that the two mouse Atxn1 splicing variants are similarly upregulated in the spleen at disease peak, as part of a novel homeostatic mechanism to control the aberrant immune response. 5
Our current knowledge of the epigenetic regulation of ATXN1 in health and disease is limited, mostly coming from studies in the SCA1 field. The RNA‐binding protein PUMILIO1 (PUM1) was found to regulate ataxin‐1 levels in the mouse brain by affecting the Atxn1 transcript stability via direct interactions with the 3'UTR. 31 The microRNAs miR‐19, miR‐101, and miR‐130 also target the 3′UTR of ATXN1 transcript and cooperatively inhibit its translation. 32 More recently, the 5'UTR has been demonstrated to participate in ATXN1 regulation as well. The microRNA miR‐760 was shown to bind a conserved site in this region to induce RNA degradation and translational inhibition. 33 Also, 12 out‐of‐frame upstream AUGs (uAUGs) were detected in the ATXN1 5′UTR with the ability to impair the translational efficiency of the transcript. 34 Our study is the first to report an additional mechanism of ATXN1 regulation outside the central nervous system, with relevance for B cell biology and autoimmunity. It would be noteworthy to investigate whether, and to what extent, the other known regulatory circuits take part in the control of ATXN1 expression in the peripheral immune system upon MS.
The overall methylation status of the DNA molecule is a dynamic process that results from the concerted activity of DNA methyltransferases such as DNMT1, and enzymes that can oxidize 5‐methylcytosines to unmodified cytosines such as the 10 to 11 translocation (TET) family. 35 In addition, passive dilution of methylated cytosines during the S phase can also lead to a change in global DNA methylation levels. 36 Although we still ignore which of the two mechanisms is the main contributor for ATXN1 hypo‐methylation, the fact that our previous RNA‐seq screening found TET1 upregulated with nominal significance in patient B cells supports the hypothesis of an active demethylation process acting on the ATXN1 locus. 15
From a molecular standpoint, ataxin‐1 shuttles between the cytosol and the nucleus where it binds the promoters of specific target genes as part of a multimeric complex and inhibits their transcription. 37 We have previously tested two key ataxin‐1 interactors, ATXN1L, and CIC, for their role in autoimmune demyelination and at least CIC showed a modifying activity in the EAE paradigm. 5 Interestingly, we did not detect any differences in DNA methylation and mRNA levels for ATXN1L and CIC genes, suggesting that their regulation upon disease might take place post‐transcriptionally, at the protein level. In fact, it has been shown that the levels of CIC protein are controlled by the cooperative binding of both ATXN1 and ATXN1L. 38 On the other hand, we cannot rule out the possibility that other ataxin‐1 binding partners within the complex are subjected to epigenetic regulatory events in B cells in a pro‐inflammatory milieu.
In summary, we provide insights into the regulatory mechanisms of a novel MS risk gene in the peripheral immune system at the time of diagnosis. The mapping of three of the four DMRs nearby susceptibility‐associated SNPs is suggestive of the direct involvement of MS risk genetic factors in modulating ATXN1 methylation. The correlation between allelic carriage and DNA methylation levels has been shown at several MS risk loci including HLA‐DRB1*15:01, TNFSF14, and CYP24A1, 12 , 39 and it will be important to formally test the ATXN1 locus in larger cohorts for cis methylation effects associated with the susceptibility variant rs719316. Altogether, our findings support a working model in which positive feedback loops epigenetically drive the expression of ataxin‐1 in the immune system in response to encephalitogenic challenges in order to limit the aberrant activation and proliferation of B cells and eventually control the magnitude of the autoimmune attack. Among others, ataxin‐1 negatively regulates the B cell‐mediated expression of interleukin 18 (IL‐18), a critical factor mediating autoreactive Th1 and humoral responses. 5 , 40 A consistent body of evidence highlights an active role of B cells in MS pathogenesis through antigen presentation, pro‐inflammatory cytokine production, and perhaps autoantibody secretion. 41 Additional studies will be required to address the heterogeneity of the B cell compartment. In this light, we have already documented that ataxin‐1 ablation drives the expansion of specific B cell subsets such as B‐1a and B‐1b. 5 Single‐cell approaches will be instrumental to verify whether ATXN1 undergoes the same regulation in different B cell subtypes.
Author Contribution
QM, AD, and JRO conceived and supervised the study. QM and AD carried out all the experiments. QM and AD analyzed the data and wrote the paper. All authors read and approved the final manuscript.
Conflict of Interest
The authors declare no competing interests.
Supporting information
Figure S1 DMR calling at the ATXN1 locus. The results of three different software (metilene, defiant, and SMART) used to identify differentially methylated regions (DMRs) from bisulfate DNA sequencing (BS‐seq) data are shown in the figure. The thickness of the bars is proportional to the extension of the different DMRs.
Table S1 Primers sequences were used to clone the four DMRs into the pCpGL‐basic vector.
Acknowledgments
This study was supported by a grant from the National Multiple Sclerosis Society (RG‐1901‐33219) to JRO and AD. QM is supported by a postdoctoral fellowship from the National Multiple Sclerosis Society (FG‐2108‐38348). The UCSF DNA biorepository is supported by a grant from the National Multiple Sclerosis Society (SI‐2001‐35701). The authors thank the UCSF MS‐EPIC Study Group for the recruitment of study participants, sample acquisition, and data management.
Annals of Clinical and Translational Neurology 2022;9(8): 1186–1194
Funding Information
This study was supported by a grant from the National Multiple Sclerosis Society (RG‐1901‐33219) to JRO and AD. QM is supported by a postdoctoral fellowship from the National Multiple Sclerosis Society (FG‐2108‐38348). The UCSF DNA biorepository is supported by a grant from the National Multiple Sclerosis Society (SI‐2001‐35701).
Funding Statement
This work was funded by National Multiple Sclerosis Society grants FG‐2108‐38348, RG‐1901‐33219, and SI‐2001‐35701.
References
- 1. Orr HT, Chung MY, Banfi S, et al. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet. 1993;4(3):221‐226. [DOI] [PubMed] [Google Scholar]
- 2. Suh J, Romano DM, Nitschke L, et al. Loss of Ataxin‐1 potentiates Alzheimer's pathogenesis by elevating cerebral BACE1 transcription. Cell. 2019;178(5):1159‐1175 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kang AR, An HT, Ko J, Kang S. Ataxin‐1 regulates epithelial‐mesenchymal transition of cervical cancer cells. Oncotarget. 2017;8(11):18248‐18259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. International Multiple Sclerosis Genetics Consortium . Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science. 2019;365(6460):eaav7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Didonna A, Canto Puig E, Ma Q, et al. Ataxin‐1 regulates B cell function and the severity of autoimmune experimental encephalomyelitis. Proc Natl Acad Sci USA. 2020;117(38):23742‐23750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Constantinescu CS, Farooqi N, O'Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol. 2011;164(4):1079‐1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ma Q, Didonna A. The novel multiple sclerosis susceptibility gene ATXN1 regulates B cell receptor signaling in B‐1a cells. Mol Brain. 2021;14(1):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28(10):1057‐1068. [DOI] [PubMed] [Google Scholar]
- 9. Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38(1):23‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Martinez‐Iglesias O, Carrera I, Carril JC, Fernandez‐Novoa L, Cacabelos N, Cacabelos R. DNA methylation in neurodegenerative and cerebrovascular disorders. Int J Mol Sci. 2020;21(6):2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mazzone R, Zwergel C, Artico M, et al. The emerging role of epigenetics in human autoimmune disorders. Clin Epigenetics. 2019;11(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kular L, Liu Y, Ruhrmann S, et al. DNA methylation as a mediator of HLA‐DRB1*15:01 and a protective variant in multiple sclerosis. Nat Commun. 2018;9(1):2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Graves MC, Benton M, Lea RA, et al. Methylation differences at the HLA‐DRB1 locus in CD4+ T‐cells are associated with multiple sclerosis. Mult Scler. 2014;20(8):1033‐1041. [DOI] [PubMed] [Google Scholar]
- 14. Maltby VE, Graves MC, Lea RA, et al. Genome‐wide DNA methylation profiling of CD8+ T cells shows a distinct epigenetic signature to CD4+ T cells in multiple sclerosis patients. Clin Epigenetics. 2015;7:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ma Q, Caillier SJ, Muzic S, et al. Specific hypomethylation programs underpin B cell activation in early multiple sclerosis. Proc Natl Acad Sci USA. 2021;118(51):e2111920118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. University of California San Francisco MS‐EPIC Team , Cree BA, Gourraud PA, Oksenberg JR, et al. Long‐term evolution of multiple sclerosis disability in the treatment era. Ann Neurol 2016;80(4):499–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for bisulfite‐seq applications. Bioinformatics. 2011;27(11):1571‐1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu H, Liu X, Zhang S, et al. Systematic identification and annotation of human methylation marks based on bisulfite sequencing methylomes reveals distinct roles of cell type‐specific hypomethylation in the regulation of cell identity genes. Nucleic Acids Res. 2016;44(1):75‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Condon DE, Tran PV, Lien YC, et al. Defiant: (DMRs: easy, fast, identification and ANnoTation) identifies differentially methylated regions from iron‐deficient rat hippocampus. BMC Bioinformatics. 2018;19(1):31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114‐2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph‐based genome alignment and genotyping with HISAT2 and HISAT‐genotype. Nat Biotechnol. 2019;37(8):907‐915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Anders S, Pyl PT, Huber W. HTSeq–a python framework to work with high‐throughput sequencing data. Bioinformatics. 2015;31(2):166‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. ENCODE Project Consortium . An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Karabacak Calviello A, Hirsekorn A, Wurmus R, Yusuf D, Ohler U. Reproducible inference of transcription factor footprints in ATAC‐seq and DNase‐seq datasets using protocol‐specific bias modeling. Genome Biol. 2019;20(1):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hah N, Murakami S, Nagari A, Danko CG, Kraus WL. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res. 2013;23(8):1210‐1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. 1000 Genomes Project Consortium , Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li D, Hsu S, Purushotham D, Sears RL, Wang T. WashU epigenome browser update 2019. Nucleic Acids Res. 2019;47(W1):W158‐W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Newell‐Price J, Clark AJ, King P. DNA methylation and silencing of gene expression. Trends Endocrinol Metab. 2000;11(4):142‐148. [DOI] [PubMed] [Google Scholar]
- 30. Klug M, Rehli M. Functional analysis of promoter CpG methylation using a CpG‐free luciferase reporter vector. Epigenetics. 2006;1(3):127‐130. [DOI] [PubMed] [Google Scholar]
- 31. Gennarino VA, Singh RK, White JJ, et al. Pumilio1 haploinsufficiency leads to SCA1‐like neurodegeneration by increasing wild‐type Ataxin1 levels. Cell. 2015;160(6):1087‐1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee Y, Samaco RC, Gatchel JR, Thaller C, Orr HT, Zoghbi HY. miR‐19, miR‐101 and miR‐130 co‐regulate ATXN1 levels to potentially modulate SCA1 pathogenesis. Nat Neurosci. 2008;11(10):1137‐1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nitschke L, Tewari A, Coffin SL, et al. miR760 regulates ATXN1 levels via interaction with its 5′ untranslated region. Genes Dev. 2020;34(17–18):1147‐1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Manek R, Nelson T, Tseng E, Rodriguez‐Lebron E. 5'UTR‐mediated regulation of Ataxin‐1 expression. Neurobiol Dis. 2020;134:104564. [DOI] [PubMed] [Google Scholar]
- 35. Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502(7472):472‐479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. He S, Sun H, Lin L, et al. Passive DNA demethylation preferentially up‐regulates pluripotency‐related genes and facilitates the generation of induced pluripotent stem cells. J Biol Chem. 2017;292(45):18542‐18555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lam YC, Bowman AB, Jafar‐Nejad P, et al. ATAXIN‐1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell. 2006;127(7):1335‐1347. [DOI] [PubMed] [Google Scholar]
- 38. Lee Y, Fryer JD, Kang H, et al. ATXN1 protein family and CIC regulate extracellular matrix remodeling and lung alveolarization. Dev Cell. 2011;21(4):746‐757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Roostaei T, Klein HU, Ma Y, et al. Proximal and distal effects of genetic susceptibility to multiple sclerosis on the T cell epigenome. Nat Commun. 2021;12(1):7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shi FD, Takeda K, Akira S, Sarvetnick N, Ljunggren HG. IL‐18 directs autoreactive T cells and promotes autodestruction in the central nervous system via induction of IFN‐gamma by NK cells. J Immunol. 2000;165(6):3099‐3104. [DOI] [PubMed] [Google Scholar]
- 41. Cencioni MT, Mattoscio M, Magliozzi R, Bar‐Or A, Muraro PA. B cells in multiple sclerosis ‐ from targeted depletion to immune reconstitution therapies. Nat Rev Neurol. 2021;17(7):399‐414. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 DMR calling at the ATXN1 locus. The results of three different software (metilene, defiant, and SMART) used to identify differentially methylated regions (DMRs) from bisulfate DNA sequencing (BS‐seq) data are shown in the figure. The thickness of the bars is proportional to the extension of the different DMRs.
Table S1 Primers sequences were used to clone the four DMRs into the pCpGL‐basic vector.