

Abstract
Objective
Spastic paraplegia‐12 (SPG12) is a subtype of hereditary spastic paraplegia caused by Reticulon‐2 (RTN2) mutations. We described the clinical and genetic features of three SPG12 patients, functionally explored the potential pathogenic mechanism of RTN2 mutations, and reviewed RTN2‐related cases worldwide.
Methods
The three patients were 31, 36, and 50 years old, respectively, with chronic progressive lower limb spasticity and walking difficulty. Physical examination showed elevated muscle tone, hyperreflexia and Babinski signs in the lower limbs. Patients 1 and 3 additionally had visual, urinary, and/or coordination dysfunctions. Patient 2 also had epileptic seizures. RTN2 mutations were identified by whole‐exome sequencing, followed by Sanger sequencing, segregation analysis, and phenotypic reevaluation. Functional examination of identified mutations was further explored.
Results
Three variants in RTN2 were identified in Patient 1 (c.103C>T, p.R35X), Patient 2 (c.230G>A, p.G77D), and Patient 3 (c.337C>A, p.P113T) with SPG, respectively. Western blotting revealed the p.R35X with smaller molecular weight than WT and other two missense mutants. Immunostaining showed the wild type colocalized with endoplasmic reticulum (ER) in vitro. p.R35X mutant diffusely distributes in the cytoplasm, losing colocalization with ER. p.G77D and p.P113T co‐localized with ER, which was abnormally aggregated in clumps.
Interpretation
In this study, we identified three cases with complicated SPG12 due to three novel RTN2 mutations, respectively, presenting various phenotypes: classic SPG symptoms with (1) visual abnormalities and sphincter disturbances or (2) seizures. The phenotypic heterogeneity might arise from the abnormal subcellular localization of mutant Reticulon‐2 and improper ER morphogenesis, revealing the RTN2‐related spectrum is still expanding.
Introduction
The reticulon family includes a class of proteins, which play a fundamental role in generating the correct tubular endoplasmic reticulum (ER) network. 1 , 2 As a single membrane bound organelle, ER plays a critical role in protein synthesis and transport, lipid biosynthesis, glucose homeostasis, carbohydrate metabolism, calcium ion store, protein folding, and contacting and regulating other organelles. 3 , 4 , 5 , 6 Several genes encoding the prototypic ER‐shaping proteins have been associated with hereditary spastic paraplegias (HSPs), which are mainly characterized by progressive spastic paralysis and weakness of the legs due to length‐dependent degeneration of the axons of the corticospinal tract. 7 , 8 Almost half of HSP patients carry mutations affecting the ER‐shaping proteins which are involved in maintaining proper morphology of this organelle. 9 , 10 Among these, Reticulon‐2 (RTN2) participates the ER shaping network involved, via interacting with Spastin and other HSP proteins. In 2012, the RTN2 gene was firstly identified to cause autosomal dominant (AD) HSP subtype. 11 RTN2‐related axonopathy is also known as spastic paraplegia‐12 (SPG12). While individual cases with genetic diagnosis of SPG12 have been reported to be characterized by pure form of HSP, with or without ataxia, in United Kingdom, Italy, North America and Germany. 11 , 12 , 13 , 14 , 15 SPG12 is still an extremely rare subtype of AD inherited SPG with incomplete penetrance, which is not fully interpreted both clinically and functionally.
Herein, we identified three patients manifesting as different phenotypes of complicated SPG due to three novel RTN2 mutations, respectively. On the basis of thorough clinical and genetic analysis, we further functionally investigate the effects of mutant RTN2 proteins on ER morphology.
Material and Methods
Participants
We identified three patients fulfilling the diagnosis of HSP according to progressive spasticity of lower limbs and walking difficulty. Three patients and their family members were clinically examined.
Ethical approval
The ethics committee of Shanghai Jiao Tong University Affiliated Sixth People's Hospital approved the study. All participants provided written informed consent.
Mutation analysis
Genomic DNA was extracted using a standard phenol/chloroform extraction protocol. Healthy individuals (n = 300) of matched geographic ancestry were included as normal controls. Exome sequencing was performed for the patients, using Agilent SureSelect v6 reagents for capturing exons and the Illumina HiSeq X Ten platform. Alignment to human genome assembly hg19 (GRCh37) was carried out followed by recalibration and variant calling. Population allele frequencies compiled from public databases of normal human variation (dbSNP, ESP6500, and 1000 g) were used to initially filter the data to exclude variants at >1‰ frequency in the population. The variants were further interpreted and classified according to the American College of Medical Genetics and Genomics (ACMG) Standards and Guidelines. 16 In this segment, two neurogeneticists analyzed the inheritance pattern, allele frequency (from 1000 g, ESP6500, dbSNP, Exome Aggregation Consortium [ExAC], and 300 in‐house ethnically matched healthy controls), amino acid conservation, pathogenicity prediction (Mutationtaster [http://www.mutationtaster.org]). Putative pathogenic variants were further confirmed by Sanger sequencing both forward and reverse strands.
Cell culture, transfection, and Western blotting
HEK 293T cells were obtained from the Cell Bank of Chinese Academy of Sciences (www.cellbank.org.cn) and maintained in Dulbecco's Modified Eagle Medium with 10% fetal bovine serum and 1% penicillin/streptomycin (PS) at 37°C in a humidified incubator with 5% CO2. One day before transfection, cells were plated at 150,000 cells per well in 6‐well culture dish. The next day, cells were transfected with 2.5 μg of EGFP control plasmid DNA or RTN2‐EGFP wild‐type (RTN2‐WT), mutant (c.103C>T, c.230G>A and c.337C>A) and one previously reported mutation (c.178insC, p.R60Pfs*11) 11 plasmid DNA using Lipofectamine 3000 transfection reagent (Invitrogen). Meanwhile, CALR‐mcherry construct (express a fluorescent protein located to ER), was also co‐transfected with the plasmid mentioned above. Forty‐eight hours later, the cells were split in radio immunoprecipitation assay buffer (Beyotime, Shanghai, China) to extract protein for western blot analysis. Cell lysates were diluted in 6X SDS‐PAGE Sample Loading Buffer (Beyotime) for protein denaturation. For cell lysates, equal volumes were run on 8% SDS polyacrylamide gels. Total RTN2 levels were detected using the anti‐GFP antibody (1:2500, GFP‐1010; AVES, Davis, CA, USA). GAPDH primary antibody (1:1000, 2118‐14C10; Cell Signaling Technology, Danvers, MA, USA) was used to ensure equal protein loading. Blots were then incubated with anti‐chicken and anti‐rabbit secondary HRP‐conjugated antibodies (1:5000; Beyotime) and bands were detected by enhanced chemiluminescence using Western Blot Enhancer reagents (Thermo Scientific, Waltham, MA, USA).
Immunofluorescence
HEK 293 T cells transfected with the respective expression constructs were washed in PBS and fixed using 4% paraformaldehyde for immunofluorescence test. Cells were blocked with 10% normal donkey serum and 0.3% Triton X‐100 in PBS for 60 min, incubated with primary antibody (GFP‐1010; AVES) in blocking solution at 4°C overnight and incubated with Alexa Fluor 488 secondary antibody (1:1000; Life, Waltham, MA, USA). Images were taken with a Zeiss 710 confocal microscope under 63× (oil) and 40× (dry) objectives.
Data availability statement
The original dataset used and analyzed for this study is available from the corresponding author on reasonable request.
Results
Clinical findings
Patient 1 (M0464) was a 31‐year‐old female, with progressively worsened stiffness in lower limbs for 13 years. Initially, weakness affected the bilateral lower limbs, leading to slow walking speed. In the past 5 years, she had occasional falls during walking, urinary incontinence, and diplopia when looking into the distance. Assessments of mental ability were comparable for her age. There was no evidence of multiple joint contractures and no history of seizures. Upon physical examination, she presented fluent and clear speech. She had normal strength in neck flexion, upper limbs and lower limbs (5/5), without muscle atrophy. Muscle tone in lower limbs was increased (2/4 on the modified Ashworth scale graded 0–4). Tendon reflexes were brisk in four limbs with marked bilateral ankle clonus and bilateral Babinski signs. Abnormal results were disclosed during heel–knee‐tibia test and Romberg test. The patient was able to walk alone slowly with a scissor gait without abnormal movements. She had normal serum lactate and creatine kinase. Magnetic resonance imaging of the brain and spinal cord and electromyography examinations showed no abnormalities. The similar symptoms of her mother during lifetime were reported, which was bedridden at the age of 55 years old, and then died of refractory pneumonia.
Patient 2 (M1543) was a 36‐year‐old male, with progressive walking difficulty for 19 years. Since the age of 17, he had developed weakness with stiffness, swelling and soreness of muscles in both lower limbs. Gradually, she had to take a break every few minutes when walking. During the past 8 years, the symptoms were slowly worsened with occasional falls during walking. Then, stiffness and swelling of the upper limb muscles also appeared. He had seizures every 5–6 months between the ages of 6 and 9 years old. He used to be treated with oxcarbazepine showing improvement. After the age of 10 years old, seizures were relieved without medication. There was no evidence of dysphagia and dysarthria. Upon physical examination, he presented fluent and clear speech, normal mental ability without nystagmus. He had normal strength in neck flexion, upper limbs and lower limbs (5/5), without muscle atrophy. Muscle tone in lower limbs was mildly increased. Tendon reflexes were brisk in four limbs with right ankle clonus and bilateral Babinski signs. He had no abnormalities during coordination evaluation. The patient was able to walk alone slowly with a scissors gait without abnormal movements.
Patient 3 (T4157) was a 50‐year‐old female, with progressive walking difficulty for more than 26 years. At the age of 24, the abnormal walking posture was first noticed, with stiffness of the right lower limb. Gradually, she developed weakness in both lower limbs and difficulty in lifting the legs. Then the symptoms were progressively aggravated. She eventually used a cane, then a walker, during walking. Since the age of 39, she had constipation and dysuria with frequent urinary tract infections. The history of poor vision since the childhood was also reported. She developed cataracts in both eyes at the age of 8 years old. Upon physical examination, she presented fluent and clear speech, normal mental ability with bilateral visual acuity <0.8. without nystagmus. She had normal strength in neck flexion and four limbs (5/5), without muscle atrophy. Muscle tone in lower limbs was increased. Tendon reflexes were brisk in lower limbs with bilateral ankle clonus and bilateral Babinski signs. She had no abnormalities during coordination evaluation. The patient was able to walk slowly with the help of walker with a scissor gait. Magnetic resonance imaging of the brain and spinal cord showed no abnormalities. She also reported the similar illness history of her mother during lifetime, which became bedridden and then died of uncertain reason at the age of 47.
The detailed clinical features of the three cases are all summarized in Table 1.
Table 1.
Clinical features of patients with RTN2 variants.
| Ethnicity | AAO (years) | Inheritance | Phenotype | Hypertonia | Hyperreflexia | Weakness | Ankle clonus | Babinski signs | Additional features | Electrophysiology | SPRS | Variant | ACMG | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UL | LL | UL | LL | UL | LL | |||||||||||
| UK 11 | 5 | AD | Pure | − | + | − | + | − | + | / | + | BD | / | / |
c.178dupC p.R60Pfs*11 |
P |
| Italy 11 | 7 | AD | Pure | − | + | − | + | − | + | / | + | BD, DVS, foot deformity, spinal cord atrophy | − | / |
c.178dupC p.R60Pfs*11 |
P |
| USA 11 | 36 | S | Pure | − | + | − | + | − | + | / | + | BD, DVS | − | / | RTN2‐del | P |
| Germany 11 | 39 | S | Pure | + | + | − | + | − | − | / | + | Brain white matter hyperintensities in T2 | − | / |
c.1100C>T p.S367F |
P |
| Norway 12 | 50 | AD | Pure | / | / | / | / | / | / | / | / | / | / | / |
c.916G>T p.V306F |
LP |
| Canada 17 | / | AD | Autism | / | / | / | / | / | / | / | / | / | / | / |
c.939delT p.T314Lfs*8 |
LP |
| China‐P1 | 18 | AD | C | − | + | + | + | − | − | + | + | Diplopia, coordination dysfunction, BD | / | 35 |
c.103C>T p.R35X |
P |
| China‐P2 | 17 | S | C | − | + | + | + | − | + | +R | + | Seizures | − | 32 |
c.230G>A p.G77D |
LP |
| China‐P3 | 24 | AD | C | − | + | − | + | − | + | + | + | Constipation, BD, cataract | − | 31 |
c.337C>A p.P113T |
LP |
| Total | / | / | / | 1/7 | 7/7 | 2/7 | 7/7 | 0/7 | 5/7 | 3/3 | 7/7 | / | 0/5 | / | / | / |
AAO, age at onset; AD, autosomal dominant; S, sporadic; C, complicated form; UL, upper limbs; LL, lower limbs; +, positive; −, negative; /, not available; R, right side; BD, bladder disturbance; DVS, decreased vibration sensation; SPRS, Spastic Paraplegia Rating Scale; ACMG, American College of Medical Genetics and Genomics; P, pathogenic; LP, likely pathogenic.
Genetic findings
Patient 1 was identified with a heterozygous nonsense variant c.103C>T (p.R35X) in RTN2 gene (Fig. 1A), which was negative in the unaffected father (I:1) and brother (II:2). Patient 2 was detected with a de novo missense variant c.230G>A (p.G77D) (Fig. 1B), which was not identified in his parents (I:1 and I:2). Patient 3 was disclosed with a heterozygous missense variant c.337C>A (p.P113T) in the RTN2 gene (Fig. 1C), which was negative in the unaffected siblings (II:2 and II:3). The amino acid sites affected are all highly conserved among different species. All of the three variants were not identified in 300 healthy controls, dbSNP (http://www.ncbi.nlm.nih.gov/snp), 1000 Genome Project (http://browser.1000genomes.org), NHLBI Exome Sequencing Project (ESP) Exome Variant Server (http://evs.gs.washington.edu/EVS) or ExAC. p.R35X was predicted to be disease causing by Mutationtaster (probability score 1.000). p.G77D was predicted to be probably damaging by PolyPhen2 (probability score 0.99), damaging by SIFT (SIFT score: 0.0000). p.P113T was predicted to be probably damaging by PolyPhen2 (probability score 0.96), damaging by SIFT (SIFT score: 0.0000).
Figure 1.
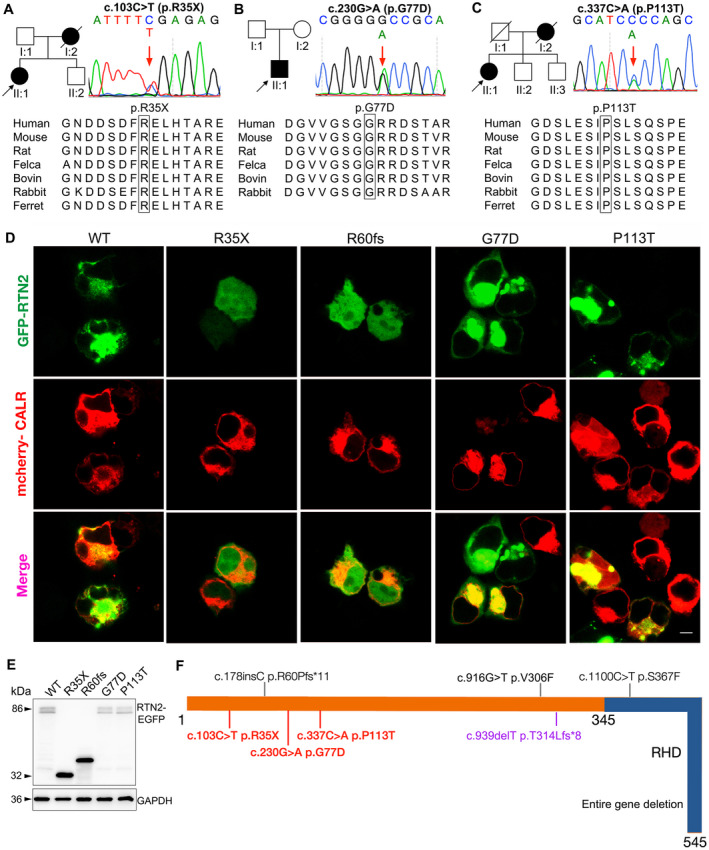
(A) Pedigree of Patient 1’s family. Sequence chromatograms of RTN2 gene displays one heterozygous nonsense mutation of c.103C>T p.R35X (arrow) in the proband (II:1), which is negative in unaffected father (I:1) and brother (II:2). (B) Pedigree of Patient 2’s family. Sequence chromatograms of RTN2 gene displays one heterozygous missense mutation of c.230G>A p.G77D (arrow) in the proband (II:1), which is negative in the parents (I:1 and I:2). (C) Pedigree of Patient 3’s family. Sequence chromatograms of RTN2 gene displays one heterozygous missense mutation of c.337C>A p.P113T (arrow) in the proband (II:1), which is negative in unaffected bothers (II:2 and II:3). (A‐C) The mutations located in the highly conserved region of proteins are shown in the bottom half. (D) RTN2‐WT/Mut‐EGFP transfected HEK 293 T cells showing the presence of WT in cytoplasmic distribution colocalizing with CALR‐mcherry (ER), but R35X and R60fs are expressed in diffuse distribution in both nuclear and cytoplasm without specific colocalization with ER. G77D and P113T still colocalize with CALR‐mcherry (ER), with ER tending to form punctate aggregates of ER. The scale bar represents 10 μm. (E) Western blotting showed the signals at ~86 kDa from expressed HEK 293 T cells expressing RTN2‐WT‐EGFP, RTN2‐G77D‐EGFP, or RTN2‐P113T‐EGFP. The signals with relative smaller molecular weight were detected in R35X group (~32 kDa) and the pathogenic control R60fs group (~40 kDa). (F) The schematic diagram of RTN2 structure with all mutations documented. Full length of RTN2 (NM_005619) consists of 545 amino acids. RHD, reticulon homology domain (aa 345–545). Mutations identified with pure SPG12, complicated SPG12 and autism are in black, red, and purple, respectively. Mutations firstly identified in this paper are in bold font, c.103C>T (p.R35X), c.230G>A (p.G77D) and c.337C>A (p.P113T). [Colour figure can be viewed at wileyonlinelibrary.com]
Mutant protein detection
The expression of RTN2‐WT/Mut‐EGFP was examined by western blotting after transfecting HEK 293T cells with RTN2 constructs, showing the R35X (~32 kDa) and R60fs (~40 kDa) with smaller molecular weight than WT and another two missense mutants (~86 kDa) (Fig. 1E). In immunofluorescence staining (Fig. 1D), the cytoplasmic colocalization of RTN2‐WT and CALR‐cherry (ER) was confirmed. Both truncated mutants, R35X and R60fs were diffusely present in the cytoplasm as well as the nucleus and showed no specific colocalization with ER markers (Fig. 1D). Though G77D and P113T still colocalized with ER, the punctate structures of ER formed (Fig. 1D). According to the ACMG evaluation and grading guidelines, 16 all the mutants identified in this paper can be rated as “pathogenic” or “likely pathogenic” (Table 1).
Discussion
We described three patients with complicated HSP due to three novel mutations in RTN2 (c.103C>T, c.230G>A, and c.337C>A), respectively. In addition to the typical features of HSP, Patient 2 also manifested with epilepsy seizures. Both Patient 1 and Patient 3 had visual abnormality and sphincter disturbance. So far, a total of 8 mutations (including this paper) in RTN2 have been associated with diseases, including 4 missense, 1 nonsense, 2 frameshift, and 1 entire gene deletion variants (summarized in Fig. 1D). Previously, three AD‐HSP families and two sporadic HSP with different RTN2 mutations have been reported. 11 , 12 Interestingly, all these patients reported were characterized by pure form of HSP with or without bladder disturbances. As it is shown in Table 1 and Figure 2, the most common features include hypertonia (7/7) and hyperreflexia (7/7) in lower limbs, ankle clonus (3/3), positive Babinski sign (7/7), bladder disturbance (5/7), and lower limb weakness (5/7). However, the relatively uncommon characteristics are also reported in several cases, such as hypertonia (1/7) and hyperreflexia in upper limbs (2/7), constipation (1/7), coordination dysfunction (1/7), seizures (1/7), diplopia (1/7), and cataract (1/7). Out of the 5 patients with MRI examinations, there are 2 cases with atrophied spinal cord and white matter hyperintensities in brain, respectively. However, the electrophysiological examination seems to be normal for all the 5 patients. Moreover, only one patient manifested as AD‐autism spectrum disorder due to c.939delT (p.T314Lfs*8). 17
Figure 2.
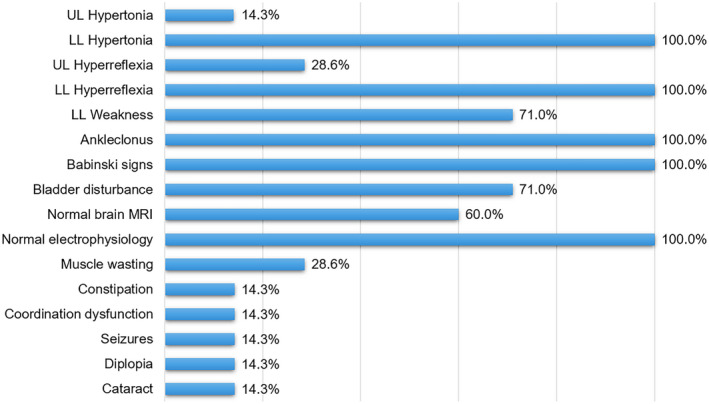
The clinical features of SPG12 patients with RTN2 mutations. For each clinical manifestation, the proportion of patients is indicated. [Colour figure can be viewed at wileyonlinelibrary.com]
The RTN2 gene encodes Reticulon‐2, belonging to the family of the reticulons, which are a group of integral membrane proteins (RTN1‐4) sharing a conserved C‐terminal reticulon homology domain (RHD). 18 By interacting with M1 Spastin, Reticulon‐2 participates in the network of hairpin loop‐containing ER morphogens that includes REEP1, atlastin‐1, and Spastin. Reticulon‐2, which includes two transmembrane domains (aa 368–388, 463–483) and 1 RHD (aa 345–545), is crucial to stabilizing the membrane curvature of ER and generating tubular ER. 11 p.R35X, located in the N‐terminal of Reticulon‐2, introduces a premature stop codon leading to 511 missing amino acids (93.8% of the whole length). This nonsense mutation is most likely to cause haploinsufficiency due to nonsense‐mediated mRNA decay, which is a surveillance pathway in eukaryotes to reduce errors in gene expression. All four truncated mutants (p.R35X, p.R60Pfs*11, p.T314Lfs*8, and entire‐RTN2 deletion) directly disturb the expression of RHD, which plays a fundamental role in regulating ER quality control by LC3 binding and lysosome delivery. 19 , 20 In this study, punctate ER structures were observed in the two missense mutants expressed cells. Similar phenotypes have also been observed in another missense mutant p.S367F reported before, indicating the related base substitution may lead to disrupted stability of ER. 11 Indeed, almost half HSP subtypes are associated with mutations affecting ER‐shaping proteins, leading to ER morphology maintenance dysfunction, including Atlastin (SPG3A), Spastin (SPG4), Reticulon 2 (SPG12), REEP1 (SPG31), Protrudin (SPG33), TGF (SPG57), ARL6IP1 (SPG61), RAB3GAP2 (SPG69) and REEP2 (SPG72). 8 , 9 , 10 , 21
Conclusion
This work reported new phenotypic features of RTN2‐related spectrum and identified three novel RTN2 mutations causing classic manifestations of HSP, with visual and coordination dysfunctions or seizures. The mutations lead to either abnormal Reticulon‐2 distribution or abnormal ER morphology. This study provides the evidence that Reticulon‐2 deficiency can be related with complicated SPG12. Thus, we suggest that RTN2‐related spectrum is still expanding.
Author Contributions
Wotu Tian, Haoran Zheng, and Zeyu Zhu contributed to data collection and analysis and drafted the manuscript. Chao Zhang contributed to data acquisition. Xinghua Luan and Li Cao contributed to study design and conceptualization, data acquisition, analysis and interpretation of data, and manuscript revision.
Conflict of Interest
The authors declare that they have no conflict of interest.
Acknowledgements
The authors appreciate all the participants for their contributions.
Annals of Clinical and Translational Neurology 2022;9(8): 1108–1115
Data Analysis: Wotu Tian, Li Cao, Department of Neurology, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai 200233, China.
Funding Information
This work is supported by grants from the National Natural Science Foundation of China (No. 81870889 and 82071258).
Funding Statement
This work was funded by National Natural Science Foundation of China grants 81870889 and 82071258.
Contributor Information
Xinghua Luan, Email: green_lxh@hotmail.com.
Li Cao, Email: caoli2000@yeah.net.
References
- 1. Collins RN. How the ER stays in shape. Cell. 2006;124(3):464‐466. [DOI] [PubMed] [Google Scholar]
- 2. Voeltz GK, Prinz WA, Shibata Y, Rist JM, Rapoport TA. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell. 2006;124(3):573‐586. [DOI] [PubMed] [Google Scholar]
- 3. English AR, Zurek N, Voeltz GK. Peripheral ER structure and function. Curr Opin Cell Biol. 2009;21(4):596‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wu Y, Whiteus C, Xu CS, et al. Contacts between the endoplasmic reticulum and other membranes in neurons. Proc Natl Acad Sci USA. 2017;114(24):E4859‐E4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Phillips MJ, Voeltz GK. Structure and function of ER membrane contact sites with other organelles. Nat Rev Mol Cell Biol. 2016;17(2):69‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Öztürk Z, O'Kane CJ, Pérez‐Moreno JJ. Axonal endoplasmic reticulum dynamics and its roles in neurodegeneration. Front Neurosci. 2020;14:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blackstone C, O'Kane CJ, Reid E. Hereditary spastic paraplegias: membrane traffic and the motor pathway. Nat Rev Neurosci. 2011;12(1):31‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meyyazhagan A, Orlacchio A. Hereditary spastic paraplegia: an update. Int J Mol Sci. 2022;23(3):1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Allison R, Edgar JR, Pearson G, et al. Defects in ER‐endosome contacts impact lysosome function in hereditary spastic paraplegia. J Cell Biol. 2017;216(5):1337‐1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gumeni S, Vantaggiato C, Montopoli M, Orso G. Hereditary spastic paraplegia and future therapeutic directions: beneficial effects of small compounds acting on cellular stress. Front Neurosci. 2021;15:660714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Montenegro G, Rebelo AP, Connell J, et al. Mutations in the ER‐shaping protein reticulon 2 cause the axon‐degenerative disorder hereditary spastic paraplegia type 12. J Clin Invest. 2012;122(2):538‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Iqbal Z, Rydning SL, Wedding IM, et al. Targeted high throughput sequencing in hereditary ataxia and spastic paraplegia. PLoS One. 2017;12(3):e0174667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reid E, Dearlove AM, Osborn O, Rogers MT, Rubinsztein DC. A locus for autosomal dominant “pure” hereditary spastic paraplegia maps to chromosome 19q13. Am J Hum Genet. 2000;66(2):728‐732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Orlacchio A, Kawarai T, Rogaeva E, et al. Clinical and genetic study of a large Italian family linked to SPG12 locus. Neurology. 2002;59(9):1395‐1401. [DOI] [PubMed] [Google Scholar]
- 15. Reid E, Grayson C, Rogers MT, et al. Locus‐phenotype correlations in autosomal dominant pure hereditary spastic paraplegia. A clinical and molecular genetic study of 28 United Kingdom families. Brain. 1999;122(Pt 9):1741‐1755. [DOI] [PubMed] [Google Scholar]
- 16. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yuen RK, Thiruvahindrapuram B, Merico D, et al. Whole‐genome sequencing of quartet families with autism spectrum disorder. Nat Med. 2015;21(2):185‐191. [DOI] [PubMed] [Google Scholar]
- 18. Diekmann H, Klinger M, Oertle T, et al. Analysis of the reticulon gene family demonstrates the absence of the neurite growth inhibitor Nogo‐A in fish. Mol Biol Evol. 2005;22(8):1635‐1648. [DOI] [PubMed] [Google Scholar]
- 19. D'Eletto M, Oliverio S, Di Sano F. Reticulon homology domain‐containing proteins and ER‐Phagy. Front Cell Dev Biol. 2020;21(8):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tian X, Teng J, Chen J. New insights regarding SNARE proteins in autophagosome‐lysosome fusion. Autophagy. 2021;17(10):2680‐2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roda RH, Schindler AB, Blackstone C. De novo REEP2 missense mutation in pure hereditary spastic paraplegia. Ann Clin Transl Neurol. 2017;4(5):347‐350. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The original dataset used and analyzed for this study is available from the corresponding author on reasonable request.