

Abstract
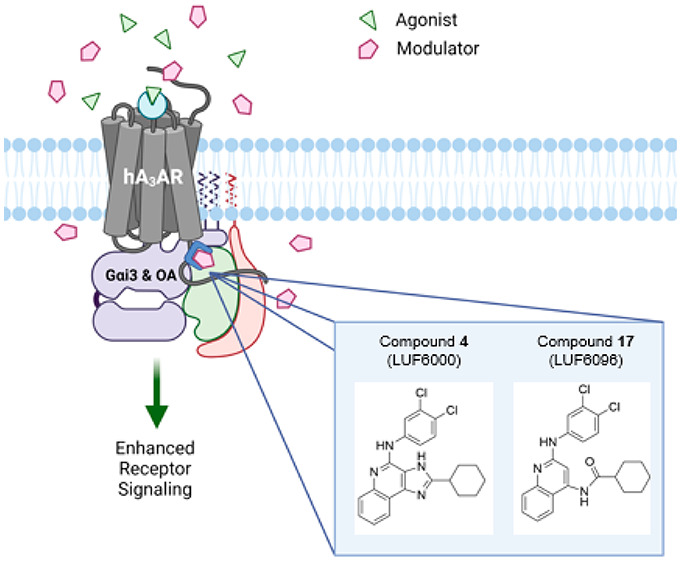
The A3 adenosine receptor (A3AR) is a promising therapeutic target for inflammatory diseases, cancer, and chronic neuropathic pain, with agonists already in advanced clinical trials. Here we report an in-depth comparison of the pharmacological properties and structure–activity relationships of existing and expanded compound libraries of 2-substituted 1H-imidazo[4,5-c]quinolin-4-amine and 4-amino-substituted quinoline derivatives that function as A3AR positive allosteric modulators (PAMs). We also show that our lead compound from each series enhances adenosine-induced A3AR signaling preferentially toward activation of Gαi3 and GαoA isoproteins, which are coexpressed with the A3AR in immune cells and spinal cord neurons. Finally, utilizing an extracellular/intracellular chimeric A3AR approach composed of sequences from a responding (human) and a nonresponding (mouse) species, we provide evidence in support of the idea that the imidazoquinolin-4-amine class of PAMs variably interacts dually with the orthosteric ligand binding site as well as with a separate allosteric site located within the inner/intracellular regions of the receptor. This study has advanced both structural and pharmacological understanding of these two classes of A3AR PAMs, which includes leads for future pharmaceutical development.
Keywords: adenosine receptor, positive allosteric modulator, G protein coupling, structure−activity relationship
Introduction
The biological effects of adenosine, a purine nucleoside produced locally during ischemia, hypoxia, and inflammation, are mediated by a family of four G protein-coupled receptors (GPCRs) designated A1, A2A, A2B, and A3.1 The A3 adenosine receptor (A3AR) is a Gi protein-coupled receptor that is abundantly expressed in granulocytic cells (neutrophils, eosinophils, basophils, and mast calls) and microglia, where it mediates the effects of adenosine to control immune cell chemotaxis and cellular activation.2−8 Several A3AR agonists have been developed that are being tested in phase 2 and 3 clinical trials for the treatment of psoriasis, rheumatoid arthritis, nonalcoholic steatohepatitis (NASH), and cancer.9−13 Due to combined anti-inflammatory and central nervous system effects, A3AR agonists are also being investigated for the treatment of neuropathic pain.14−18
To circumvent complications associated with the use of direct GPCR agonists, we have pursued the development of positive allosteric modulators (PAMs) for the A3AR. PAMs are ligands that increase the potency and/or efficacy of endogenous ligands by binding to a site (allosteric site) that is topographically distinct from the binding site for the endogenous agonist (orthosteric site).19−21 Mechanistically, binding of a PAM enhances potency by increasing the binding affinity of the orthosteric ligand, which is achieved by changing its association and/or dissociation binding rates. For efficacy modulation, the conformational change in the receptor produced by PAM binding increases signaling capacity, thereby enhancing coupling to downstream effectors. PAMs theoretically offer several pharmacological and therapeutic advantages over traditional orthosteric agonists including improved receptor subtype selectivity and spatiotemporal specificity.19−21
To discover novel A3AR PAMs, we conducted a small compound library screen and identified derivatives of the 1H-imidazo[4,5-c]quinoline-4-amine template (Figure 1) that slow the rate of orthosteric agonist dissociation via an allosteric mechanism,22 which predictably would increase binding affinity and therefore functional potency. In functional assays, it was noted that some members of this class, such as DU124183 (Figure 1), also increased the signaling efficacy of orthosteric agonists.22 However, rather than producing potency enhancement as predicted, they greatly decreased agonist potency.22 Given that certain imidazoquinoline-4-amines have been described in earlier studies to function as competitive AR antagonists,23 we speculated that these compounds have dual effects to compete for orthosteric ligand binding while acting simultaneously at a separate overlapping allosteric site. Structural modification of DU124183 and further structure-activity-relationship (SAR) exploration at the 2- and 4-amino positions of the imidazoquinolin-4-amine template, which aimed to separate orthosteric and allosteric effects, led to several novel compounds with A3AR modulatory activity including the current lead PAM from this class, LUF6000 (Figure 1).24−26 Additional modification, whereby the imidazole ring of LUF6000 was opened and replaced with an amide, produced the 2,4-disubstituted quinoline LUF6096 (Figure 1).27 Like DU124183, LUF6000 and LUF6096 increased the maximal efficacy of adenosine as well as the A3AR agonist 2-chloro-N6-(3-iodobenzyl)adenosine-5′-N-methylcarboxamide (Cl-IB-MECA) on the order of 2–3-fold in functional assays, but with less propensity to decrease their potency.24,26,27 Another key modification identified previously and maintained in the current series was the addition of a (3,4-dichlorophenyl)amino substitution that improved receptor subtype selectivity.25−27 Although LUF6000 and LUF6096 exhibit PAM activity at the human (h) A3AR (as well as rabbit and dog), they are strikingly less potent at rodent (mouse and rat) receptors, indicating species variability with this class of modulators.24
Figure 1.
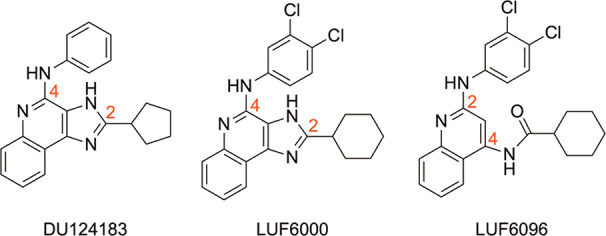
Structures of previously reported A3AR PAMs including the reference imidazoquinolin-4-amine LUF6000 (4) and the reference quinoline LUF6096 (17).
Presently, an A3AR structure remains to be solved experimentally, which limits our ability to utilize structure-based techniques to inform drug-design decisions. The purpose of this investigation was to re-examine our current and expanded compound library in more detail, with an emphasis on allosteric ligand potency and influences on orthosteric ligand binding potency and efficacy. Direct comparisons were made between the imidazoquinolin-4-amine and 2,4-disubstituted quinoline scaffolds containing equivalent substitutions. In addition, we expanded the assessment of our two current lead compounds (LUF6000 and LUF6096) to include a more comprehensive analysis of effects on specific G protein isoform coupling and β-arrestin-2 recruitment. Lastly, we utilized human/mouse A3AR chimeric receptors to investigate the molecular mechanism of action of our lead PAMs. These studies provide in depth characterization of two promising chemical classes of A3AR allosteric modulators and reveal that our lead PAMs preferentially enhance adenosine-induced G-protein signaling by binding to an allosteric site within intracellular regions of the A3AR.
Methods
Chemical Synthesis
Please refer to the synthetic methods in the Supporting Information (for 2,4-disubstituted quinoline derivatives) and in the submitted paper by Fallot et al.28 (1H-imidazo[4,5-c]quinoline-4-amine derivatives).
Creation of Stable HEK293 Cell Lines Expressing Wild-Type and Human/Mouse Chimeric A3ARs
The full-length mouse (as previously described29), human (purchased from the cDNA Resource Center), and human/mouse chimeric (custom synthesized by TOP Gene Technologies, St. Laurent, Quebec) A3AR cDNAs were subcloned into pcDNA3.1. The constructs were transfected into HEK293 cells (American Type Culture Collection, Manassas, VA) using Lipofectamine 2000 reagent (Invitrogen, Waltham, MA) and selected with 2 mg/mL of G418 in cell culture media (DMEM with 10% fetal bovine serum, 100 units/mL penicillin, and 100 μg/mL streptomycin). Cell lines derived from individual clones were maintained in cell culture media containing 0.6 mg/mL G418. The level of receptor expression in each of the cell lines was equivalent (∼1 500 fmol/mg) based on saturation radioligand binding analyses.
Membrane Preparations
Transfected HEK293 cells were washed with PBS followed by homogenization in buffer A containing 10 mM Na+-HEPES, 10 mM EDTA, 1 mM benzamidine (pH 7.4) and centrifugation at 27 000 × g for 30 min at 4 °C. Cell pellets were subsequently rehomogenized in HE buffer consisting of 10 mM Na+-HEPES, 1 mM EDTA, 1 mM benzamidine (pH 7.4) and recentrifuged. The supernatant was discarded, and cell pellets were resuspended in HE buffer containing 10% sucrose and stored at −20 °C.
[35S]GTPγS Binding Assays
Cell membranes (5 μg of protein) isolated from transfected HEK293 cells were pretreated with modulators for 1 h in 100 μL of GTPγS binding buffer (50 mM Tris HCl [pH 7.4], 1 mM EGTA, 10 mM MgCl2, 100 mM NaCl, 0.004% CHAPS, and 0.5% BSA) in a 96-well large-volume polypropylene assay plate. In all assays, ZM-241385 and PSB-603 (each at a final concentration of 300 nM) were included to block A2BARs expressed endogenously in HEK293 cells.30 Adenosine deaminase (ADA) (1 unit/mL) was also included to degrade any endogenous adenosine that might have been produced during the assay, except when adenosine was used as the orthosteric agonist. The reactions were initiated by the addition of ∼0.2 nM [35S]GTPγS and agonist in 100 μL of GTPγS binding buffer and allowed to incubate for 2 h at room temperature. At the end of the 2 h incubation period, the membranes were harvested by rapid filtration through Whatman GF/B filters that had been presoaked for 2 h in GTPγS binding buffer containing 0.02% CHAPS using a 96-well cell harvester (Brandel, Gaithersburg, MD). Radioactivity trapped in the filters was measured by liquid scintillation counting. Nonspecific binding of [35S]GTPγS was determined in the presence of 10 μM unlabeled GTPγS.
Binding Assays with [125I]I-AB-MECA
Cell membranes (50 μg) isolated from transfected HEK293 cells as described earlier were incubated in 100 μL of binding buffer (50 mM Tris-HCl [pH 7.4], 10 mM MgCl2, 1 mM EDTA, and 1 unit/mL ADA) containing ∼0.3 nM [125I]I-AB-MECA and indicated concentrations of the A3AR allosteric modulator compounds. The reactions were incubated at room temperature for the times indicated, after which bound and free radioligands were separated by rapid filtration through GF/C glass fiber filters. Radioactivity trapped in the filters was measured using a gamma counter. For dissociation studies, [125I]I-AB-MECA (∼0.3 nM) was incubated with HEK293 cell membranes (50 μg) expressing ARs for 3 h at room temperature in binding buffer (100 μL), after which the assay was initiated by the addition of 100 μM adenosine-5′-N-ethylcarboxamide (NECA; a potent nonselective AR agonist) along with the enhancer compounds or equivalent vehicle. Specific [125I]I-AB-MECA binding was measured by rapid filtration at the indicated time intervals. For equilibration binding assays, membranes (50 μg) were incubated with [125I]I-AB-MECA (∼0.3 nM) and the modulator compounds for the indicated times before filtration. For all assays, nonspecific binding was determined by incubation in the presence of 100 μM NECA. [125I]I-AB-MECA was prepared by radioiodination of AB-MECA using the chloramine-T method and purified by high-performance liquid chromatography (HPLC).31
Bioluminescence Resonance Energy Transfer Assays
To measure A3AR-mediated β-arrestin-2 recruitment by BRET1, HEK293T cells were cotransfected (1:15) with two constructs (pcDNA3.1) encoding the human A3AR with C-terminally fused Renilla Luciferase (Rluc8) and N-terminally fused Venus-tagged β-arrestin-2 in DMEM supplemented with 10% fetal bovine serum, 100 units/mL penicillin, and 100 μg/mL streptomycin using TransIT-293 transfection reagent (Mirus Bio, Madison, WI). To measure A3AR G protein coupling via βγ subunit dissociation, as assessed by BRET2 (TRUPATH open-source biosensor platform32), HEK293T cells were cotransfected (1:1:1:1) in DMEM supplemented with 10% fetal bovine serum, 100 units/mL penicillin, and 100 μg/mL streptomycin with constructs encoding the indicated Rluc8-fused human Gα isoprotein, human Gβ1, C-terminally fused GFP2 fused to human Gγ2, and either the human or mouse A3AR using TransIT-293. After 24 h, transfected cells were seeded in poly-l-lysine-coated, white-walled, clear-bottom, 96-well cell culture plates at a density of 30 000 cells per well in DMEM with 1% dialyzed fetal bovine serum, 100 units/mL penicillin, and 100 μg/mL streptomycin and 25 mM HEPES (pH 7.4). The next day, the cell culture medium was aspirated, and cells were washed once with assay buffer (Hank’s buffered salt solution with Mg2+, Ca2+, and 20 mM HEPES [pH 7.4]) and allowed to incubate for 1 h at 37 °C in 60 μL of fresh assay buffer. Cells were subsequently treated with 30 μL of drugs (3×; agonist and modulator compounds) prepared in drug buffer (Hank’s buffered salt solution with Mg2+, Ca2+, 0.3% BSA, 0.03% ascorbic acid, and 20 mM HEPES [pH 7.4]), and bioluminescence signals were assessed at the times indicated using a Mithras 940 Multimode Plate reader. For β-arrestin-2 recruitment assays, 10 μL of the Rluc8 substrate coelenterazine h (Promega, Madison, WI; 5 μM final concentration) was added per well 15 min prior to measurements to allow for cell penetration and was read for bioluminescence at 480 nm and fluorescence eYFP emission at 515 nm. BRET1 ratios were calculated (emission at 515/emission at 480) first, and then the NET BRET1 was determined by subtracting the BRET ratio from control wells containing cells transfected with only the donor plasmid (human A3AR-Rluc8) from the BRET ratio of experimental wells. For BRET2 G isoprotein-coupling assays, 10 μL of the RLuc8 substrate coelenterazine 400a (Nanolight Technology, Pinetop, AZ; 5 μM final concentration) was added per well 15 min before measurement of bioluminescence at 385 nM and fluorescent GFP2 emission at 510 nm. NET BRET2 ratios were calculated in the same manner as described earlier for the BRET1 assays.
cAMP Production Assays
HEK293T cells were cotransfected (1:30) with pcDNA3.1 encoding either the human or mouse A3AR and the pGloSensor-22F reporter plasmid (Promega, Madison, WI) in DMEM supplemented with 10% fetal bovine serum, 100 units/mL penicillin, and 100 μg/mL streptomycin using TransIT-293 transfection reagent (Mirus Bio, Madison, WI). After 24 h, transfected cells were seeded in poly-l-lysine-coated, white-walled, clear-bottom, 96-well cell culture plates at a density of 30 000 cells per well in DMEM with 1% dialyzed fetal bovine serum, 100 units/mL penicillin, and 100 μg/mL streptomycin and 25 mM HEPES (pH 7.4). The next day, the cell culture medium was aspirated, and cells were washed once with assay buffer (Hank’s buffered salt solution with Mg2+, Ca2+, and 20 mM HEPES [pH 7.4]) and allowed to incubate for 2 h at 37 °C in 50 μL of fresh assay buffer containing d-luciferin (4 mM). ZM-241385 and PSB-603 (300 nM each) were also included to block A2BARs expressed endogenously in HEK293T cells. After incubation with the substrate, the plate was removed from the incubator and allowed to equilibrate at room temperature for 15 min, at which point a baseline chemiluminescence reading was acquired. Cells were subsequently treated with 5 μL of test compounds (12×; agonist and modulator compounds) prepared in assay buffer for 15 min followed by the addition of 5 μL of forskolin (12×; 1 μM final concentration) to stimulate production of cAMP. Chemiluminescence readings were obtained 30 min after the addition of forskolin.
Data Analysis
Emax and EC50 values were calculated from data obtained from agonist concentration–response curves according to E = (Emax × x)/(EC50 + x), in which x is the agonist concentration. All values were statistically compared using a one-way ANOVA followed by Bonferroni-adjusted t tests for post hoc comparison or a two-tailed Student’s t test, as indicated. For [125I]I-AB-MECA dissociation binding assays, data were fit to a one-phase exponential decay model: Y = (Y0 – NS)(−k×t), in which Y0 is the specific binding at time 0, k is the dissociation rate constant, and t is the elapsed time. Affinity estimation of the 2-cyclopropyl derivative for the orthosteric site at HumanOut/MouseIn chimeras was calculated from data obtained from agonist concentration–response curves according to Y = bottom + (top – bottom)/(1 + 10((log(EC–x))×Hillslope)), in which x is the agonist concentration with Hillslope and Schild slope values constrained to 1. All values are presented as the mean ± SEM. A p value <0.05 was considered statistically significant.
Results
Pharmacological Characterization of Novel A3AR PAMs
Equilibrium binding assays using an orthosteric agonist radioligand at a nonsaturating concentration gauge the net effect of modulator compounds on orthosteric agonist binding due to combined allosterism and potential competitive antagonism, which should inform effects on agonist potency. In prior work,22,24−27 we have not succeeded in identifying any derivatives that increase orthosteric agonist radioligand equilibrium binding (in fact, nearly all reduce it). However, these initial studies using [125I]I-AB-MECA were performed using conventional incubation times (≤2 h), which we have since learned may not be sufficient to achieve equilibrium conditions because the modulators can slow the binding kinetics.24 In preliminary studies, we retested a previously synthesized series of imidazoquinolin-4-amine derivatives25,26 with hydrophobic mono- or bicycloalkyl substitutions at the 2- position and the 3,4-dichlorophenyl group at the 4-amino position (2–5, 11, and 12; Table 1), including our current lead compound 4 (LUF6000) containing a 2-cyclohexyl substituent, where the incubation time was extended to 18 h. Under these conditions, it was revealed that several derivatives at a concentration of 10 μM increased binding by as much as ∼30%. Because binding tended to increase as the 2-cycloalkyl substituent increased in size, we expanded this series to include ring systems ranging from 3 to 12 carbons, as well as a flexible hydrophobic heptan-4-yl derivative (1, 6–10, and 13; Table 1). To allow a direct comparison, we also expanded our existing library of quinoline derivatives (15–17, 21, and 22; Table 1)27 containing a 2-(3,4-dichlorophenyl)amine substituent with matching substitutions (up to C-9) at the 4-amide position (18–20 and 23; Table 1). Within this series, compound 17 (LUF6096) is the cyclohexyl derivative analogous to reference compound 4 in the imidazoquinolin-4-amine series. Synthesis of the new quinoline derivatives is included in the Supporting Information. Detailed synthetic methods for the imidazoquinolin-4-amine derivates will be reported elsewhere.28
Table 1. 2-Substituted Imidazoquinolin-4-amine and 4-Substituted Quinoline Derivatives.

A full comparison of the two series of modulator compounds on [125I]I-AB-MECA equilibrium binding using membranes prepared from HEK293 cells expressing the human (h) A3AR is presented in Figure 2A (values reported in Table S1). For the imidazoquinolin-4-amine series, it was remarkable that derivatives with larger 2-cycloalkyl substitutions, beginning with the 2-cycloheptyl derivative (5), began to increase [125I]I-AB-MECA equilibrium binding. However, this effect was lost as the ring size increased beyond C-9 (7). The more flexible heptan-4-yl derivative (13) produced the greatest increase in binding (70%), and the tricyclic 2-adamantanyl derivative (12) increased binding modestly (20%). Notably, the C-3 derivative (1) reduced binding by up to 70%. In contrast, most of the quinoline derivatives reduced [125I]I-AB-MECA equilibrium binding, but the direction of this effect upon changes of the 2-position substitution followed a similar SAR as the imidazoquinolin-4-amine series. Thus, the cyclohexyl (17) and cycloheptanyl (18) groups in the quinoline series had the least-negative values for the [125I]I-AB-MECA equilibrium binding. The exception to this pattern was the 4-heptan-4-yl derivative in the quinoline series (23), which surprisingly produced the greatest decrease in binding. Thus, the SARs of the PAMs derived from the two chemical scaffolds are highly divergent.
Figure 2.

(A) Equilibrium binding assays with membranes prepared from HEK293 cells expressing the hA3AR. Membranes were incubated with 0.3 nM [125I]I-AB-MECA and 10 μM modulator compounds for 18 h. Data are presented as the percent change in specific binding compared to vehicle. (B) Dissociation binding assays with membranes prepared from HEK293 cells expressing the hA3AR. Membranes were incubated with ∼0.3 nM [125I]I-AB-MECA and 10 μM modulator compounds for 3 h when the assay was initiated by the addition of 100 μM NECA. After 60 min, the amount of specific binding remaining was determined. Data are presented as mean ± SEM. *p < 0.05 vs vehicle by Student’s t tests. Percent change from vehicle (A) and % [125I]I-AB-MECA remaining after 60 min (B) values are reported in Tables S1 and S2.
To evaluate pure allosteric actions, we assessed the effects of the modulators in a [125I]I-AB-MECA dissociation binding assay using transfected HEK293 cell membranes (Figure 2B and Table S2). Cell membranes were incubated with [125I]I-AB-MECA for 3 h, and then binding was determined after the addition of test compounds and excess NECA (100 μM), which prevented [125I]I-AB-MECA from rebinding after it had dissociated. In the absence of modulator (vehicle), ∼25% of the radioligand remained bound 1 h after the addition of NECA (dotted line in Figure 2B). An increase in binding by a modulator compound indicates an allosteric effect to slow [125I]I-AB-MECA dissociation. Using this assay, it was observed that several of the derivatives from both series at a concentration of 10 μM slowed [125I]I-AB-MECA dissociation with a SAR similar to that observed in equilibrium binding assays, whereby increasingly bulky cycloalkyl substitutions from C-4 to C-8/9 (2–7 and 15–19) produced maximal effects. The bicyclic (except for the adamantanyl-substituted quinoline derivative, 22) and especially the heptan-4-yl derivatives (13 and 23) also slowed [125I]I-AB-MECA dissociation. Maximal slowing was produced by the 2-heptan-4-yl imidazoquinolin-4-amine modulator (13).
Several interesting observations arise when the equilibrium binding data in Figure 2A and the dissociation binding data in Figure 2B are considered together. Neither compound with the cyclopropyl substitution (1 and 14) affected the [125I]I-AB-MECA dissociation binding rate and therefore lacked allosteric actions. We hypothesized that the reduction in equilibrium binding produced in both compound series presumably was caused by competitive antagonism rather than negative allosteric modulation. This hypothesis was later supported using chimeric receptor constructs (see below). For other compounds that slowed [125I]I-AB-MECA dissociation without increasing equilibrium binding, which occurred with several of the quinoline derivatives (16, 19, 21, and 23), prominent competitive antagonism was also assumed. All compounds that increased equilibrium binding, including compounds 5, 6, 12, and 13 within the imidazoquinolin-4-amine series containing C-7/8, cycloalkyl, bicyclic, or the heptan-4-yl substitutions, produced prominent effects to slow [125I]I-AB-MECA dissociation, implicating prominent allosterism with diminished competitive antagonism.
We next evaluated the two series of modulators in a [35S]GTPγS binding assay using isolated HEK293 cell membranes expressing the hA3AR, which detects receptor-induced G protein activation. For these studies, concentration–response curves with the orthosteric agonist Cl-IB-MECA were assessed in the presence of vehicle or 0.1, 1, or 10 μM modulator compound. As shown in Figures 3 (imidazoquinolin-4-amine series) and 4 (quinoline series), a clear SAR emerged with each series of compounds that correlated well with the single-point radioligand binding assays (EC50 and Emax for Cl-IB-MECA are reported in Table S3). Indeed, the cyclopropyl derivatives from both chemical series (1 and 14) right-shifted the Cl-IB-MECA concentration–response curve in a concentration-dependent manner without altering the efficacy, indicative of competitive antagonism. As the ring size increased, efficacy enhancement became apparent, beginning with the C-4 derivatives (2 and 15), while potency reduction diminished, correlating with the effect of the modulators in radioligand binding assays to slow [125I]I-AB-MECA dissociation while tending to improve [125I]I-AB-MECA equilibrium binding. The greatest increase in efficacy enhancement within the imidazoquinolin-4-amine series occurred with the C-6 (4), C-9 (7), norbornanyl (11), and adamantanyl (12) derivatives; among these, the C-9 (7) and adamantanyl (12) derivatives tended to increase the potency of Cl-IB-MECA, whereas the C-6 (4) and norbornyl (11) derivatives tended to reduce it. While the magnitude of the efficacy-enhancing action of the heptan-4-yl derivative (13) was not as prominent, it is notable that it appeared to be the most potent among this series given that it produced a maximal degree of efficacy enhancement at a concentration of only 100 nM. In comparison, the lowest concentration to produce maximal activity for the other compounds exhibiting substantial PAM activity, including compound 4, was 1 μM. It is necessary to mention that efforts to fit the [35S]GTPγS binding data to operational models33,34 to calculate affinity constants and cooperativity factors were not successful, presumably because the compounds have additional pharmacological actions (i.e., competitive antagonism) or because of the limited amount of data that was collected. Within the quinoline series (Figure 4), our lead compound 17 containing the cyclohexyl substitution produced the greatest increase in efficacy without reducing the potency of Cl-IB-MECA. Correlating with the [125I]I-AB-MECA equilibrium binding data, this series of derivatives in general produced a greater reduction in agonist potency.
Figure 3.

[35S]GTPγS binding assays with the imidazoquinolin-4-amine series of 2-positioned derivatives and membranes prepared from HEK293 cells expressing the human A3AR. Concentration–response curves with Cl-IB-MECA were conducted in the presence of vehicle or 0.1, 1.0, or 10 μM modulator compounds. Results were normalized to the Emax value obtained in the presence of vehicle. Data are presented as mean + SEM; n = 3. * p < 0.05 vs vehicle by one-way ANOVA followed by Bonferroni-adjusted t tests for post hoc comparison. EC50 and Emax values are reported in Table S3.
Figure 4.

[35S]GTPγS binding assays with the quinoline series of derivatives and membranes prepared from HEK293 cells expressing the human A3AR. Concentration–response curves with Cl-IB-MECA were conducted in the presence of vehicle or 0.1, 1.0, or 10 μM modulator compounds. Results were normalized to the Emax value obtained in the presence of vehicle. Data are presented as mean ± SEM; n = 3. * p < 0.05 vs vehicle by one-way ANOVA followed by Bonferroni-adjusted t tests for post hoc comparison. EC50 and Emax values are reported in Table S3.
Results of Agonist Activity at the mA3AR for the Newly Synthesized Derivatives
We assessed activity of the two series of modulators in assays using HEK293 cells expressing the mouse (m) A3AR. Both of our current leads (4 and 17) display minimal allosteric activity with rodent A3AR’s,24 and we are eager to identify pan-species derivatives that will facilitate efforts to characterize their biological activity in disease models. Unfortunately, none of the newly synthesized compounds showed improved activity. Figure 5 presents results of the 2-substituted cyclopropyl (1), cyclohexyl (4), cyclononanyl (7), and heptan-4-yl (13) derivatives from the imidazoquinolin-4-amine series that performed best in assays with the hA3AR. In the [35S]GTPγS binding assay using mA3AR membranes (Figure 5A), 4 and the heptan-4-yl derivative (13) increased the efficacy of Cl-IB-MECA by only ∼35 and ∼60% at the highest concentration tested, respectively (10 μM; EC50 and Emax values for Cl-IB-MECA are presented in Table S4). In comparison, in assays using hA3AR membranes, compound 4 and the heptan-4-yl derivative at a concentration of 10 μM increased the efficacy of Cl-IB-MECA by ∼250% and ∼220%, respectively. In [125I]I-AB-MECA binding assays, none of the derivatives from either series enhanced [125I]I-AB-MECA equilibrium binding (Figure 5B; Table S4), nor did they slow the rate of [125I]I-AB-MECA dissociation (Figure 5C; Table S4) in assays with the mA3AR.
Figure 5.

Characterization of select imidazoquinolin-4-amine derivatives at the mouse A3AR. (A) [35S]GTPγS binding assays, (B) [125I]I-AB-MECA dissociation binding assays, and (C) [125I]I-AB-MECA equilibrium binding assays. Assays were conducted using HEK293 cells stably expressing the mouse A3AR. Data are presented as mean ± SEM; n = 3. EC50 and Emax values from the [35S]GTPγS binding assays and values for the [125I]I-AB-MECA binding assays are presented in Table S4.
Evidence That the Imidazoquinolin-4-amine PAMs Act at Two Distinct Sites on the hA3AR
Taking advantage of species differences,24 we generated stable HEK293 cell lines expressing human/mouse chimeric A3 receptors in efforts to begin to localize the allosteric binding site. Two chimeric A3ARs were prepared in which the lower transmembrane regions of the human sequence were changed to the mouse sequence (HOut/MIn) and vice versa (MOut/HIn). Division between the upper and lower segments of the transmembrane regions were at residue x.50 defined by the Ballesteros–Weinstein numbering scheme, where x.50 is the most-conserved residue in that helix. Figure 6 illustrates differences in amino acid sequences in the species chimeras, which were most prominent in the extracellular loops and extracellular portions of the transmembrane helices, but differences were also notable in the intracellular loop segments and helix 8. The residues previously identified by mutagenesis and modeling to coordinate the orthosteric nucleoside agonists are all contained in the upper segments.16,17 In [35S]GTPγS binding assays with the MOut/HIn chimera, strikingly compound 4 retained the ability to increase the efficacy of Cl-IB-MECA to a similar extent as in assays with the wild-type hA3AR, whereas efficacy enhancement was minimal in assays with the HOut/MIn chimera, similar to that observed with the wild-type mA3AR (Figure 7A; EC50 and Emax values for Cl-IB-MECA are reported in Table S5). An identical profile was obtained in [125I]I-AB-MECA dissociation binding assays, wherein compound 4 slowed dissociation from the MOut/HIn chimera but not from the HOut/MIn chimera (Figure 7B; Table S5). This finding that the allosteric activity of compound 4 is retained when intracellular portions of the A3AR are composed of the human amino acid sequence suggests that the imidazoquinolin-4-amine allosteric binding site is distinct from the orthosteric site and that it resides within the inner cytoplasmic portions of the receptor.
Figure 6.

Snake diagram of the human A3AR amino acid sequence. Residues highlighted in green indicate outside residues, while those in white indicate inside residues of the human/mouse chimeric receptors. Amino acid residues that differ between the human and mouse sequences are highlighted in red. This diagram was prepared using the GPCRdb database (http://www.gpcrdb.org).
Figure 7.

Characterization of human/mouse chimeric A3ARs. (A, C) [35S]GTPγS binding assays with Cl-IB-MECA (agonist) and either compound 4 (A) or compound 1 (C). (B) [125I]I-AB-MECA dissociation binding assays in the presence of vehicle or 10 μM compound 4. (D, E) [125I]I-AB- MECA equilibrium binding assays in the presence of increasing concentrations of compound 1 (D) or compound 4 (E). EC50 and Emax values from the [35S]GTPγS binding assays and values for the [125l]l-AB-MECA dissociation binding assays are presented in Table S5.
We noticed in the chimeric receptor studies that compound 4 had less propensity to reduce the potency of Cl-IB-MECA in [35S]GTPγS binding assays with the MOut/HIn chimera (Figure 7A; Table S5). In fact, the EC50 of Cl-IB-MECA in the presence of 10 μM compound 4 was significantly reduced 5-fold from 10.5 to 2.24 nM (p < 0.05), while in assays with the wild-type hA3AR the EC50 was increased 5-fold by 10 μM compound 4 (15.1 vs 72.5 nM; p < 0.05). Because the binding affinity of non-nucleoside typical non-nucleoside hA3AR antagonist ligands in the mA3AR is greatly reduced as compared to the hA3AR,35 we predicted that the enhanced potency of Cl-IB-MECA observed with the MOut/HIn chimera is explained by reduced affinity of compound 4 for the orthosteric binding site composed of the mouse sequence. To further test this idea, we examined the 2-cyclopropyl imidazoquinolin-4-amine derivative (1) in [35S]GTPγS binding assays with human/mouse chimeric receptors, which lacks efficacy-enhancing allosteric activity but greatly right-shifts the Cl-IB-MECA concentration–response relationship in assays with the hA3AR in a manner suggestive of competitive antagonism. As shown in Figure 7C (EC50 and Emax values for Cl-IB-MECA are reported in Table S5), compound 1 produced a similar rightward shift in assays with the HOut/MIn chimera but had little effect in assays with the MOut/HIn chimera, confirming competitive antagonism. Cl-IB-MECA concentration–response curves treated with vehicle, 0.1, 1.0, or 10 μM of the 2-cyclopropyl derivative (1) at the HOut/MIn chimera were used to estimate the affinity of this derivative for the orthosteric site by Schild analysis, where a purely competitive antagonist effect was observed and determined to be ∼135 nM (pA2 = 6.87 ± 0.19). Lastly, we compared the effects of compounds 1 and 4 in [125I]I-AB-MECA equilibrium binding assays using the human/mouse A3AR chimeras. As shown in Figure 7D, the results support the idea that compound 1 functions as a competitive antagonist with greater potency for the hA3AR as compared to the mA3AR, and data in Figure 7E support a dual mechanism of action of compound 4, evidenced by a reduction in binding in assays with the HOut/MIn chimera (binds with high affinity to the human orthosteric binding site but has low activity at the mouse allosteric site) and an increase in binding with the MOut/HIn chimera (binds with low affinity to the mouse orthosteric binding site while fully activating the human allosteric site). Thus, the chimeric receptor studies not only aided in localizing the allosteric binding site to cytosolic regions of the receptor but also provided further evidence that this family of PAMs can also act dually as competitive antagonists for the orthosteric binding site and allowed us to derive modulator affinity estimates for the orthosteric site.
Preferential Enhancement of Adenosine-Induced hA3AR Gα Isoprotein Coupling by Compounds 4 and 17
We investigated the activity of our two lead PAM compounds to modulate A3AR-induced activation of specific G protein isoforms and β-arrestin-2 translocation using BRET technology to explore the possibility that they may preferentially enhance select signaling pathways. The G protein-coupling assays involved measurement of BRET2 fluorescence after expressing various Gα-Rluc8 fusion proteins and the Gγ2-GFP2 fusion protein in HEK293T cells (TRUPATH32). With this assay, loss of BRET2 signal, reflecting dissociation of the Gα and Gβγ subunits, indicates receptor-mediated activation of each specific G protein isoform. β-arrestin-2 recruitment was assessed by expressing both the hA3AR-Rluc8 and β-arrestin-2-YFP fusion proteins in HEK293T cells and measuring the attainment of BRET1 fluorescence.
We initially identified the specific G proteins that are activated by the hA3AR in response to adenosine. As expected, adenosine induced activation of all three Gαi isoforms (Gαi1,2,3) with EC50 values in the range of 1–10 μM (Figure 8A; Table S6). Coupling to GαoA and GαoB was also detectable, with the latter being highly variable. No coupling was observed to the GαZ, GαSS, GαSL, Gαq, or Gα12/13 isoforms. In these assays, bioluminescence emission values for the various Gα-Rluc constructs were similar (Table S7). To address the potential for probe-dependence, we further assessed G protein activation profiles of 2-chloroadenosine (2-CADO) and Cl-IB-MECA, which were similar to adenosine, except that Cl-IB-MECA was ∼35% more efficacious at activating Gαi3 and 2-CADO was unable to activate GαoA up to a concentration of 100 μM (Figure 8C and Table S8). In BRET1 assays for the assessment of β-arrestin-2 signaling, all three of the agonists also stimulated β-arrestin-2 recruitment (Figure 8B and C). However, Cl-IB-MECA and 2-CADO were, respectively, ∼40% and 20% less efficacious as compared to adenosine and thus appeared to function as partial agonists in this assay. A heat map comparing relative potencies and efficacies of the three agonists to stimulate G isoprotein activation and β-arrestin-2 recruitment is displayed in Figure 8D.
Figure 8.

Agonist-induced G isoprotein activation and β-arrestin-2 recruitment by the human A3AR. (A) BRET2 G protein profiling assays using adenosine as the orthosteric agonist. Data for each G isoprotein are presented relative to vehicle. (B) BRET1 assays assessing β-arrestin-2 recruitment. (C) BRET2 and BRET1 assays assessing effects of adenosine, 2-CADO, and Cl-IB-MECA on activation of G isoproteins or β-arrestin-2 as indicated. Results were normalized to the Emax value obtained in the presence of adenosine. (D) Heat maps relating potency and efficacy of the agonists to activate the indicated G isoproteins and β-arrestin-2 recruitment. BRET assays were conducted with HEK293T cells transfected with the human A3AR and constructs for BRET assessment as described in the Methods section. Colors indicate log EC50 and efficacy values. Data are presented as mean ± SEM; n = 3. EC50 and Emax values are presented in Table S8.
We next assessed the actions of compounds 4 and 17 on hA3AR-induced G isoprotein activation and β-arrestin-2 recruitment in response to adenosine. HEK293T cells transfected with the hA3AR were simultaneously stimulated with increasing concentrations of adenosine and 10 μM of either 4 or 17, and BRET measurements were assessed for up to 2 h. As shown in Figure 9 (EC50 and Emax values for adenosine are reported in Table S9), both compounds enhanced the efficacy of adenosine to activate both Gαi3 and GαoA by ∼35 and 60%, respectively, whereas neither compound affected adenosine-induced activation of Gαi1 or Gαi2, although compound 17 exhibited agonism in the Gαi1 and Gαi2 protein-coupling assays, an effect that was also observed in a cAMP production assay (Figure S1). An efficacy-enhancing effect of both compounds was observed as early as 15 min after the addition of adenosine in assays of GαoA, whereas efficacy enhancement was only observed at the 60 min time point in assays with Gαi3. At the 60 min time point, compounds 4 and 17 were found to not only increase the efficacy of adenosine but also increase their potencies by ∼7- and 40-fold, respectively. Neither compound influenced adenosine-induced β-arrestin-2 recruitment. Thus, the modulators preferentially enhanced adenosine-induced activation of the Gαi3 and GαoA isoforms. Figure 10 (Table S10) displays concentration–response curves with compounds 4 and 17 on Gαi3 and GαoA isoprotein coupling, in comparison with β-arrestin-2 recruitment. Similar data for compounds 7 and 13 are reported in the Supporting Information (Figure S2).
Figure 9.

Effects of compound 4 and compound 17 on agonist-induced G isoprotein activation and β- arrestin-2 recruitment by the hA3AR over time. BRET2 assays to assess G protein coupling and BRET1 assays to assess β-arrestin-2 recruitment were conducted with adenosine in the presence of vehicle, 10 μM compound 4, or 10 μM compound 17 as described in the Methods section. Results were normalized to the Emax value obtained in the presence of vehicle. Data represent mean + SEM; n = 3–5. EC50 and Emax values are presented in Table S9.
Figure 10.

In-depth analysis of specific G isoproteins identified to be potentiated by compounds 4 and 17 at select time points. BRET2 and BRET1 assays to assess (A) GaαoA protein coupling and β-arrestin- 2 recruitment at 30 min and (B) Gaαl3 protein coupling and β-arrestin-2 recruitment at 60 min were conducted with adenosine in the presence of vehicle, compound 4, or compound 17 as described in the Methods section. Results were normalized to the Emax value obtained in the presence of vehicle. Data represent mean ± SEM; n = 3. EC50 and Emax values are presented in Table S9.
Discussion
Because adenosine is produced locally at sites of tissue injury, hypoxia, and inflammation,1 an allosteric tactic is likely an optimal approach to therapeutically target each of the four adenosine receptor subtypes. In the present study, we have compared and expanded knowledge of the pharmacological properties of two previously identified classes of compounds that function as A3AR PAMs, namely, 1H-imidazo[4,5-c]-imidazoquinolin-4-amines represented by our lead compound LUF6000 (4)24−26 and 2,4-disubstituted quinolines represented by LUF6096 (17).24,27 Salient findings from this work are as follows. First, expanded SAR studies revealed that bulkier cycloalkyl substitutions at the 2-position of the imidazoquinolin-4-amine and the 4-amide position of the quinoline scaffold, each containing a 3,4-dichlorophenyl substituent at the 4- and 2-amino positions, respectively, produced substantial improvements in allosteric actions of the derivatives to promote agonist efficacy enhancement with lessened propensity to reduce agonist potency. Maximal responses were obtained with ring systems containing 6–9 carbons yielding EC50 values estimated to be in the range of 0.1–1 μM. Second, more in-depth evaluation of our lead compounds using cell-based BRET assays with transfected HEK293T cells determined that, among the four G isoproteins coupled with the A3AR (αi1, αi2, αi3, and αoA), both compounds 4 and 17 enhanced adenosine-induced activation of the Gαi3 and GαoA isoproteins; neither compound influenced the potency or efficacy of adenosine to elicit β-arrestin-2 recruitment. Thus, we provide evidence that compounds 4 and 17 potentiate adenosine-mediated A3AR signaling preferentially to specific Gα isoproteins. Two newly synthesized imidazoquinolin-4-amine modulators described in this investigation, compounds 7 and 13, exhibited similar pharmacological profiles. Finally, utilizing an extracellular/intracellular chimeric A3AR approach composed of sequences from a responding (human) and a nonresponding (mouse) species, we have confirmed our prior prediction that the imidazoquinolin-4-amine class of PAMs retains variable competitive orthosteric antagonist activity, which likely explains the propensity of these compounds to reduce agonist potency. Loss of PAM activity when intracellular portions of the A3AR are composed of the mouse sequence (nonresponding) provided evidence that the allosteric binding pocket is located distal to the orthosteric site within the inner regions of the receptor.
The involvement of specific Gα isoproteins in mediating the biological actions of the A3AR remain unknown; thus, we can only speculate at the present time on the potential therapeutic advantages that may be provided by the PAMs described in this investigation. The Gαi3 isoprotein is the least ubiquitously expressed of the Gαi/o isoforms and is enriched in immune cell populations including granulocytic cells and microglia where expression of the A3AR is also prominent.36−38 This has been elegantly demonstrated by single-cell mRNA sequencing of mouse and human tissues.36,38 The Gαi3 isoprotein has been implicated in the regulation of leukocyte chemotaxis,39 and our prior work suggests that increased A3AR signaling may indirectly influence oxidant production and chemotaxis through sequestration of cellular components involved in cellular activation, including rac1.7,40 Taken together, preferential enhancement of adenosine-induced Gαi3 activation by the A3AR in leukocytes might be an effective and more tolerable means to treat inflammatory diseases. On the other hand, the GαoA isoprotein is known to be found in neurons in the brain as well as in the spinal cord where the A3AR is also reported to be abundantly expressed.36,38 Preclinical work has demonstrated efficacy of A3AR agonists in a number of different models of neuropathic pain due to mechanical injury and chemotherapeutics that provide a benefit not only by reducing inflammatory responses but also by suppressing neuronal circuits in the spinal cord involved in pain sensation.15,41−44 Thus, the current families of PAMs described herein might be highly effective therapies for the treatment of neuropathic pain due to selective enhancement of two analgesic mechanisms. It is unfortunate that we are currently limited in our ability to investigate the biological actions of the PAMs we have characterized thus far in animal models in detail because we have yet to identify any derivatives that display meaningful activity with rodent A3ARs.24 In a recent study, however, we reported that compound 17 was effective at limiting infarct size in a dog model of ischemia/reperfusion injury.45 Because it produced a reduction in infarct size when it was given at the onset of reperfusion, we predict that it was effective in this model in part by suppressing inflammatory responses.45
As part of the G isoprotein BRET studies, we have reported the G protein coupling profile of the A3AR in a live cell system for the first time. The endogenous agonist adenosine elicited activation of Gαi1, αi2, αi3, and αoA. Two synthetic agonists commonly used in adenosinergic research, specifically 2-CADO and the selective A3AR agonist Cl-IB-MECA, produced similar responses, although Cl-IB-MECA was more efficacious at activating Gαi3 and 2-CADO was not able to activate GαoA. In comparison to adenosine and 2-CADO, Cl-IB-MECA was ∼50% less effective at activating β-arrestin-2 recruitment, and thus it appears to favor G protein activation of the A3AR when compared to adenosine. This property of Cl-IB-MECA may explain its broad efficacy in preclinical models of inflammatory diseases.4,14,43,46,47
We were surprised by our findings from the species chimera experiments that the imidazoquinolin-4-amine PAMs appear to compete for orthosteric ligand binding while acting separately within an inner region of the receptor to produce allosteric enhancement. This is supported by (1) our findings that PAM activity is only present when intracellular regions of the receptor comprise the human sequence and (2) findings with the HOut/MIn chimera where the efficacy-enhancing activity of compound 4 is absent, yet competitive antagonistic activity of this compound as well as compound 1, which lacks PAM activity, remains. We speculate that the imidazoquinolin-4-amine PAMs, and presumably the quinoline PAMs, may interact with the orthosteric site, or perhaps with an extracellular vestibule described in other GPCRs that controls the entry and efflux of agonist ligands into the orthosteric site.48,49 In an early guided molecular dynamics simulation and molecular modeling study, compound 4 was predicted to interact at both of these sites.50 At the present time, we do not know the location of the imidazoquinolin-4-amine allosteric site, and we are pursuing additional mutagenesis studies in combination with molecular modeling and structural approaches to gain further insights. It is notable that there are a number of structure-based reports of small-molecule PAMs binding to sites located within inner regions of other GPCRs.48,49 For example, compound 6FA, a PAM of the β2 adrenergic receptor, binds to the inner surface of the receptor in a pocket created by intracellular loop 1 and transmembrane helices 3 and 4.51 Compound 6FA binding is thought to stabilize the loop in an α-helical formation that maintains an opening for insertion of the terminal α-helix of the G protein. An ago-PAM for the class B1 human glucagon-like peptide-1 (GLP-1) receptor, termed compound 1, binds covalently to the membrane-facing surface of TMD6, causing its outward movement along with conformation changes in the extracellular domains that facilitate insertion of the amino terminus into the GLP-1 binding site.52 Finally, a cryo-electromicroscopic structure of the A1 adenosine receptor complexed with a Gαi2 heterotrimer, adenosine, and the 2-amino-3-benzoylthiophene PAM MIPS521 revealed an extrahelical lipid-facing allosteric binding pocket involving transmembrane helices 1, 6, and 7.53 MIPS521 binding is hypothesized to stabilize the adenosine–receptor–G protein complex.
In conclusion, we present expanded SAR information for two promising families of A3AR PAMs that selectively enhance G isoprotein-dependent signaling. Mechanistic studies provide support for a dual mechanism of action and the presence of an allosteric binding site within the inner/intracellular regions of the receptor. This study has advanced both structural and pharmacological understanding of these two classes of A3AR PAMs. Our findings will guide future computational and structure-based approaches to develop improved A3AR PAMs that have potential as anti-inflammatory and analgesic therapeutics.
Acknowledgments
This research was supported in part by the National Institutes of Health (Grant R01HL133589 to J.A.A. and R35GM128840 to B.C.S.), the National Intramural Research Program (Grant ZIADK031117 to K.A.J.), the American Heart Association (Grant 898217 to C.L.F.), and the Medical College of Wisconsin Therapeutic Accelerator Program (J.A.A.). The Graphical Abstract was created using BioRender (BioRender.com).
Glossary
Abbreviations
- 2-CADO
2-Chloroadenosine
- ADA
Adenosine deaminase
- AR
Adenosine receptor
- BSA
Bovine serum albumin
- BRET
Bioluminescence resonance energy transfer
- cAMP
Cyclic adenosine monophosphate
- CHAPS
3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate
- Cl-IB-MECA
2-Chloro-N6-(3-iodobenzyl)adenosine-5′-N-methylcarboxamide
- DMEM
Dulbecco’s modified Eagle medium
- EDTA
Ethylenediaminetetraacetic acid
- EGTA
Triethylene glycol diamine tetraacetic acid
- GPCR
G protein-coupled receptor
- [35S]GTPγS
Guanosine 5′-[γ[35S]thio]triphosphate
- HEK293
Human embryonic kidney 293
- HEPES
2-[4-(2-Hydroxyethyl)piperazin-1-yl]ethanesulfonic acid
- [125I]I-AB-MECA
N6-(4-amino-3-[125I]iodobenzyl)adenosine-5′-N-methycarboxamide
- NASH
Nonalcoholic steatohepatitis
- NECA
Adenosine-5′-N-ethylcarboxamide
- RLuc8
Renilla luciferase 8
- PAM
Positive allosteric modulator
- PBS
Phosphate-buffered saline
- SAR
Structure–activity relationship
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.2c00076.
Synthetic methods of quinoline derivatives; values from [125I]I-AB-MECA and [35S]GTPγS binding assays with human, mouse, and human/mouse chimeric A3ARs; and values from BRET G isoprotein coupling, β-arrestin-2 recruitment, and cAMP accumulation assays (PDF)
The authors declare no competing financial interest.
Special Issue
Published as part of the ACS Pharmacology & Translational Science virtual special issue “GPCR Signaling”.
Supplementary Material
References
- IJzerman A. P.; Jacobson K. A.; Muller C. E.; Cronstein B. N.; Cunha R. A. International Union of Basic and Clinical Pharmacology. CXII: Adenosine Receptors: A Further Update. Pharmacol Rev. 2022, 74 (2), 340–372. 10.1124/pharmrev.121.000445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonioli L.; Pacher P.; Hasko G. Adenosine and inflammation: it’s time to (re)solve the problem. Trends Pharmacol. Sci. 2022, 43 (1), 43–55. 10.1016/j.tips.2021.10.010. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Corriden R.; Inoue Y.; Yip L.; Hashiguchi N.; Zinkernagel A.; Nizet V.; Insel P. A.; Junger W. G. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science 2006, 314 (5806), 1792–1795. 10.1126/science.1132559. [DOI] [PubMed] [Google Scholar]
- Ge Z. D.; van der Hoeven D.; Maas J. E.; Wan T. C.; Auchampach J. A. A3 adenosine receptor activation during reperfusion reduces infarct size through actions on bone marrow-derived cells. J. Mol. Cell Cardiol 2010, 49 (2), 280–286. 10.1016/j.yjmcc.2010.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasko G.; Linden J.; Cronstein B.; Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug Discov 2008, 7 (9), 759–770. 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan J. E.; Thourani V. H.; Auchampach J. A.; Robinson J. A.; Wang N. P.; Vinten-Johansen J. A3 adenosine receptor activation attenuates neutrophil function and neutrophil-mediated reperfusion injury. Am. J. Physiol. 1999, 277 (5), H1895–H1905. 10.1152/ajpheart.1999.277.5.H1895. [DOI] [PubMed] [Google Scholar]
- van der Hoeven D.; Wan T. C.; Auchampach J. A. Activation of the A3 adenosine receptor suppresses superoxide production and chemotaxis of mouse bone marrow neutrophils. Mol. Pharmacol. 2008, 74 (3), 685–696. 10.1124/mol.108.048066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker B. A.; Jacobson M. A.; Knight D. A.; Salvatore C. A.; Weir T.; Zhou D.; Bai T. R. Adenosine A3 receptor expression and function in eosinophils. Am. J. Respir. Cell Mol. Biol. 1997, 16 (5), 531–537. 10.1165/ajrcmb.16.5.9160835. [DOI] [PubMed] [Google Scholar]
- David M.; Gospodinov D. K.; Gheorghe N.; Mateev G. S.; Rusinova M. V.; Hristakieva E.; Solovastru L. G.; Patel R. V.; Giurcaneanu C.; Hitova M. C.; Purcaru A. I.; Horia B.; Tsingov I. I.; Yankova R. K.; Kadurina M. I.; Ramon M.; Rotaru M.; Simionescu O.; Benea V.; Demerdjieva Z. V.; Cosgarea M. R.; Morariu H. S.; Michael Z.; Cristodor P.; Nica C.; Silverman M. H.; Bristol D. R.; Harpaz Z.; Farbstein M.; Cohen S.; Fishman P. Treatment of Plaque-Type Psoriasis With Oral CF101: Data from a Phase II/III Multicenter, Randomized, Controlled Trial. J. Drugs Dermatol. 2016, 15 (8), 931–938. [PubMed] [Google Scholar]
- Fishman P.; Cohen S.; Itzhak I.; Amer J.; Salhab A.; Barer F.; Safadi R. The A3 adenosine receptor agonist, namodenoson, ameliorates nonalcoholic steatohepatitis in mice. Int. J. Mol. Med. 2019, 44 (6), 2256–2264. 10.3892/ijmm.2019.4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman M. H.; Strand V.; Markovits D.; Nahir M.; Reitblat T.; Molad Y.; Rosner I.; Rozenbaum M.; Mader R.; Adawi M.; Caspi D.; Tishler M.; Langevitz P.; Rubinow A.; Friedman J.; Green L.; Tanay A.; Ochaion A.; Cohen S.; Kerns W. D.; Cohn I.; Fishman-Furman S.; Farbstein M.; Yehuda S. B.; Fishman P. Clinical evidence for utilization of the A3 adenosine receptor as a target to treat rheumatoid arthritis: data from a phase II clinical trial. J. Rheumatol. 2008, 35 (1), 41–48. [PubMed] [Google Scholar]
- Stemmer S. M.; Benjaminov O.; Medalia G.; Ciuraru N. B.; Silverman M. H.; Bar-Yehuda S.; Fishman S.; Harpaz Z.; Farbstein M.; Cohen S.; Patoka R.; Singer B.; Kerns W. D.; Fishman P. CF102 for the treatment of hepatocellular carcinoma: a phase I/II, open-label, dose-escalation study. Oncologist 2013, 18 (1), 25–26. 10.1634/theoncologist.2012-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmer S. M.; Manojlovic N. S.; Marinca M. V.; Petrov P.; Cherciu N.; Ganea D.; Ciuleanu T. E.; Pusca I. A.; Beg M. S.; Purcell W. T.; Croitoru A. E.; Ilieva R. N.; Natosevic S.; Nita A. L.; Kalev D. N.; Harpaz Z.; Farbstein M.; Silverman M. H.; Bristol D.; Itzhak I.; Fishman P. Namodenoson in Advanced Hepatocellular Carcinoma and Child-Pugh B Cirrhosis: Randomized Placebo-Controlled Clinical Trial. Cancers (Basel) 2021, 13 (2), 187. 10.3390/cancers13020187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes K.; Symons-Liguori A. M.; Jacobson K. A.; Salvemini D. Identification of A3 adenosine receptor agonists as novel non-narcotic analgesics. Br. J. Pharmacol. 2016, 173 (8), 1253–1267. 10.1111/bph.13446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little J. W.; Ford A.; Symons-Liguori A. M.; Chen Z.; Janes K.; Doyle T.; Xie J.; Luongo L.; Tosh D. K.; Maione S.; Bannister K.; Dickenson A. H.; Vanderah T. W.; Porreca F.; Jacobson K. A.; Salvemini D. Endogenous adenosine A3 receptor activation selectively alleviates persistent pain states. Brain 2015, 138 (1), 28–35. 10.1093/brain/awu330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletta S.; Tosh D. K.; Finley A.; Gizewski E. T.; Moss S. M.; Gao Z. G.; Auchampach J. A.; Salvemini D.; Jacobson K. A. Rational design of sulfonated A3 adenosine receptor-selective nucleosides as pharmacological tools to study chronic neuropathic pain. J. Med. Chem. 2013, 56 (14), 5949–5963. 10.1021/jm4007966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh D. K.; Finley A.; Paoletta S.; Moss S. M.; Gao Z. G.; Gizewski E. T.; Auchampach J. A.; Salvemini D.; Jacobson K. A. In vivo phenotypic screening for treating chronic neuropathic pain: modification of C2-arylethynyl group of conformationally constrained A3 adenosine receptor agonists. J. Med. Chem. 2014, 57 (23), 9901–9914. 10.1021/jm501021n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh D. K.; Paoletta S.; Chen Z.; Crane S.; Lloyd J.; Gao Z. G.; Gizewski E. T.; Auchampach J. A.; Salvemini D.; Jacobson K. A. Structure-Based Design, Synthesis by Click Chemistry and in Vivo Activity of Highly Selective A3 Adenosine Receptor Agonists. Medchemcomm 2015, 6, 555–563. 10.1039/C4MD00571F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin Q.; Hopper A. T.; Blanco M. J.; Tirunagaru V.; Robichaud A. J.; Doller D. Allosteric Modalities for Membrane-Bound Receptors: Insights from Drug Hunting for Brain Diseases. J. Med. Chem. 2019, 62 (13), 5979–6002. 10.1021/acs.jmedchem.8b01651. [DOI] [PubMed] [Google Scholar]
- Slosky L. M.; Caron M. G.; Barak L. S. Biased Allosteric Modulators: New Frontiers in GPCR Drug Discovery. Trends Pharmacol. Sci. 2021, 42 (4), 283–299. 10.1016/j.tips.2020.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Yu Z.; Xiao W.; Lu S.; Zhang J. Allosteric binding sites at the receptor-lipid bilayer interface: novel targets for GPCR drug discovery. Drug Discov Today 2021, 26 (3), 690–703. 10.1016/j.drudis.2020.12.001. [DOI] [PubMed] [Google Scholar]
- Gao Z. G.; Kim S. G.; Soltysiak K. A.; Melman N.; Ijzerman A. P.; Jacobson K. A. Selective allosteric enhancement of agonist binding and function at human A3 adenosine receptors by a series of imidazoquinoline derivatives. Mol. Pharmacol. 2002, 62 (1), 81–89. 10.1124/mol.62.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Galen P. J. M.; Nissen P.; van Wijngaarden I.; IJzerman A. P.; Soudijn W. 1H-imidazo[4,5-c]quinolin-4-amines: novel non-xanthine adenosine antagonists. J. Med. Chem. 1991, 34 (3), 1202–1206. 10.1021/jm00107a046. [DOI] [PubMed] [Google Scholar]
- Du L.; Gao Z. G.; Paoletta S.; Wan T. C.; Gizewski E. T.; Barbour S.; van Veldhoven J. P. D.; IJzerman A. P.; Jacobson K. A.; Auchampach J. A. Species differences and mechanism of action of A3 adenosine receptor allosteric modulators. Purinergic Signalling 2018, 14 (1), 59–71. 10.1007/s11302-017-9592-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göblyös A.; Gao Z. G.; Brussee J.; Connestari R.; Santiago S. N.; Ye K.; Ijzerman A. P.; Jacobson K. A. Structure-activity relationships of new 1H-imidazo[4,5-c]quinolin-4-amine derivatives as allosteric enhancers of the A3 adenosine receptor. J. Med. Chem. 2006, 49 (11), 3354–6331. 10.1021/jm060086s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.; de Castro S.; Gao Z. G.; Ijzerman A. P.; Jacobson K. A. Novel 2- and 4-substituted 1H-imidazo[4,5-c]quinolin-4-amine derivatives as allosteric modulators of the A3 adenosine receptor. J. Med. Chem. 2009, 52 (7), 2098–2108. 10.1021/jm801659w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitman L. H.; Goblyos A.; Zweemer A. M.; Bakker R.; Mulder-Krieger T.; van Veldhoven J. P.; de Vries H.; Brussee J.; Ijzerman A. P. A series of 2,4-disubstituted quinolines as a new class of allosteric enhancers of the adenosine A3 receptor. J. Med. Chem. 2009, 52 (4), 926–931. 10.1021/jm8014052. [DOI] [PubMed] [Google Scholar]
- Fallot L. B.; Suresh R. R.; Fisher C. L.; Kaufman N.; Gao Z.-G.; Auchampach J. A.; Jacobson K. A.. Structure activity studies of 1H-Imidazo[4,5-c]quinolin-4-amine derivatives as A3 adenosine receptor positive allosteric modulators. J. Med. Chem. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreckler L. M.; Wan T. C.; Ge Z. D.; Auchampach J. A. Adenosine inhibits tumor necrosis factor-alpha release from mouse peritoneal macrophages via A2A and A2B but not the A3 adenosine receptor. J. Pharmacol Exp Ther 2006, 317 (1), 172–180. 10.1124/jpet.105.096016. [DOI] [PubMed] [Google Scholar]
- Gao Z.; Chen T.; Weber M. J.; Linden J. A2B adenosine and P2Y2 receptors stimulate mitogen-activated protein kinase in human embryonic kidney-293 cells. cross-talk between cyclic AMP and protein kinase c pathways. J. Biol. Chem. 1999, 274 (9), 5972–5980. 10.1074/jbc.274.9.5972. [DOI] [PubMed] [Google Scholar]
- Auchampach J. A.; Jin X.; Wan T. C.; Caughey G. H.; Linden J. Canine mast cell adenosine receptors: cloning and expression of the A3 receptor and evidence that degranulation is mediated by the A2B receptor. Mol. Pharmacol. 1997, 52 (5), 846–860. 10.1124/mol.52.5.846. [DOI] [PubMed] [Google Scholar]
- Olsen R. H. J.; DiBerto J. F.; English J. G.; Glaudin A. M.; Krumm B. E.; Slocum S. T.; Che T.; Gavin A. C.; McCorvy J. D.; Roth B. L.; Strachan R. T. TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. Nat. Chem. Biol. 2020, 16 (8), 841–849. 10.1038/s41589-020-0535-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai A. J.; Mechin I.; Nagarajan K.; Valant C.; Wootten D.; Lam P. C. H.; Orry A.; Abagyan R.; Nair A.; Sexton P. M.; Christopoulos A.; Miller L. J. Molecular Basis of Action of a Small-Molecule Positive Allosteric Modulator Agonist at the Type 1 Cholecystokinin Holoreceptor. Mol. Pharmacol. 2019, 95 (3), 245–259. 10.1124/mol.118.114082. [DOI] [PubMed] [Google Scholar]
- Jakubik J.; Randakova A.; Chetverikov N.; El-Fakahany E. E.; Dolezal V. The operational model of allosteric modulation of pharmacological agonism. Sci. Rep 2020, 10 (1), 14421. 10.1038/s41598-020-71228-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z. G.; Suresh R. R.; Jacobson K. A. Pharmacological characterization of DPTN and other selective A3 adenosine receptor antagonists. Purinergic Signal 2021, 17 (4), 737–746. 10.1007/s11302-021-09823-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson M.; Zhang C.; Mear L.; Zhong W.; Digre A.; Katona B.; Sjostedt E.; Butler L.; Odeberg J.; Dusart P.; Edfors F.; Oksvold P.; von Feilitzen K.; Zwahlen M.; Arif M.; Altay O.; Li X.; Ozcan M.; Mardinoglu A.; Fagerberg L.; Mulder J.; Luo Y.; Ponten F.; Uhlen M.; Lindskog C. A single-cell type transcriptomics map of human tissues. Sci. Adv. 2021, 7 (31), eabh2169 10.1126/sciadv.abh2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiss V.; Reisinger E.; Speidel A.; Beer-Hammer S.; Nurnberg B. Analyses of Gnai3-iresGFP reporter mice reveal unknown Gαi3 expression sites. Sci. Rep 2021, 11 (1), 14271. 10.1038/s41598-021-93591-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018, 562 ( (7727), ), 367–372. 10.1038/s41586-018-0590-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwano Y.; Adler M.; Zhang H.; Groisman A.; Ley K. Gαi2 and Gαi3 Differentially Regulate Arrest from Flow and Chemotaxis in Mouse Neutrophils. J. Immunol 2016, 196 (9), 3828–3833. 10.4049/jimmunol.1500532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hoeven D.; Gizewski E. T.; Auchampach J. A. Activation of the A3 adenosine receptor inhibits fMLP-induced Rac activation in mouse bone marrow neutrophils. Biochem. Pharmacol. 2010, 79 (11), 1667–1673. 10.1016/j.bcp.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppi E.; Cherchi F.; Fusco I.; Failli P.; Vona A.; Dettori I.; Gaviano L.; Lucarini E.; Jacobson K. A.; Tosh D. K.; Salvemini D.; Ghelardini C.; Pedata F.; Di Cesare Mannelli L.; Pugliese A. M. Adenosine A3 receptor activation inhibits pronociceptive N-type Ca2+ currents and cell excitability in dorsal root ganglion neurons. Pain 2019, 160 (5), 1103–1118. 10.1097/j.pain.0000000000001488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durante M.; Squillace S.; Lauro F.; Giancotti L. A.; Coppi E.; Cherchi F.; Di Cesare Mannelli L.; Ghelardini C.; Kolar G.; Wahlman C.; Opejin A.; Xiao C.; Reitman M. L.; Tosh D. K.; Hawiger D.; Jacobson K. A.; Salvemini D. Adenosine A3 agonists reverse neuropathic pain via T cell-mediated production of IL-10. J. Clin Invest 2021, 131 (7), e139299 10.1172/JCI139299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvemini D.; Jacobson K. A. Highly selective A3 adenosine receptor agonists relieve chronic neuropathic pain. Expert Opin Ther Pat 2017, 27 (8), 967. 10.1080/13543776.2017.1341018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A. K.; Mahalingam R.; Squillace S.; Jacobson K. A.; Tosh D. K.; Dharmaraj S.; Farr S. A.; Kavelaars A.; Salvemini D.; Heijnen C. J. Targeting the A3 adenosine receptor to prevent and reverse chemotherapy-induced neurotoxicities in mice. Acta Neuropathol Commun. 2022, 10 (1), 11. 10.1186/s40478-022-01315-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L.; Gao Z.-G.; Nithipatikom K.; IJzerman A. P.; van Veldhoven J. P. D.; Jacobson K. A.; Gross G. J.; Auchampach J. A. Protection from myocardial ischemia/reperfusion injury by a positive allosteric modulator of the A3 adenosine receptor. J. Pharmacol Exp Ther 2012, 340 (1), 210–217. 10.1124/jpet.111.187559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Z. D.; Peart J. N.; Kreckler L. M.; Wan T. C.; Jacobson M. A.; Gross G. J.; Auchampach J. A. Cl-IB-MECA [2-chloro-N6-(3-iodobenzyl)adenosine-5′-N-methylcarboxamide] reduces ischemia/reperfusion injury in mice by activating the A3 adenosine receptor. J. Pharmacol Exp Ther 2006, 319 (3), 1200–1210. 10.1124/jpet.106.111351. [DOI] [PubMed] [Google Scholar]
- Mulloy D. P.; Sharma A. K.; Fernandez L. G.; Zhao Y.; Lau C. L.; Kron I. L.; Laubach V. E. Adenosine A3 receptor activation attenuates lung ischemia-reperfusion injury. Ann. Thorac Surg 2013, 95 (5), 1762–1767. 10.1016/j.athoracsur.2013.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan H. C. S.; Li Y.; Dahoun T.; Vogel H.; Yuan S. New Binding Sites, New Opportunities for GPCR Drug Discovery. Trends Biochem. Sci. 2019, 44 (4), 312–330. 10.1016/j.tibs.2018.11.011. [DOI] [PubMed] [Google Scholar]
- Lu S.; Zhang J. Small Molecule Allosteric Modulators of G-Protein-Coupled Receptors: Drug-Target Interactions. J. Med. Chem. 2019, 62 (1), 24–45. 10.1021/acs.jmedchem.7b01844. [DOI] [PubMed] [Google Scholar]
- Deganutti G.; Cuzzolin A.; Ciancetta A.; Moro S. Understanding allosteric interactions in G protein-coupled receptors using Supervised Molecular Dynamics: A prototype study analysing the human A3 adenosine receptor positive allosteric modulator LUF6000. Bioorg. Med. Chem. 2015, 23 (14), 4065–4071. 10.1016/j.bmc.2015.03.039. [DOI] [PubMed] [Google Scholar]
- Liu X.; Masoudi A.; Kahsai A. W.; Huang L. Y.; Pani B.; Staus D. P.; Shim P. J.; Hirata K.; Simhal R. K.; Schwalb A. M.; Rambarat P. K.; Ahn S.; Lefkowitz R. J.; Kobilka B. Mechanism of β2AR regulation by an intracellular positive allosteric modulator. Science 2019, 364 (6447), 1283–1287. 10.1126/science.aaw8981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Z.; Chen L. N.; Ma H.; Zhou Q.; Zou X.; Ye C.; Dai A.; Liu Q.; Huang W.; Sun X.; Wang X.; Xu P.; Zhao L.; Xia T.; Zhong W.; Yang D.; Eric Xu H.; Zhang Y.; Wang M. W. Molecular insights into ago-allosteric modulation of the human glucagon-like peptide-1 receptor. Nat. Commun. 2021, 12 (1), 3763. 10.1038/s41467-021-24058-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper-Joyce C. J.; Bhola R.; Wang J.; Bhattarai A.; Nguyen A. T. N.; Cowie-Kent I.; O’Sullivan K.; Chia L. Y.; Venugopal H.; Valant C.; Thal D. M.; Wootten D.; Panel N.; Carlsson J.; Christie M. J.; White P. J.; Scammells P.; May L. T.; Sexton P. M.; Danev R.; Miao Y.; Glukhova A.; Imlach W. L.; Christopoulos A. Positive allosteric mechanisms of adenosine A1 receptor-mediated analgesia. Nature 2021, 597 (7877), 571–576. 10.1038/s41586-021-03897-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.