

Abstract
Evaluation of arrhythmogenic drugs is required by regulatory agencies before any new compound can obtain market approval. Despite rigorous review, cardiac disorders remain the second most common cause for safety-related market withdrawal. On the other hand, false-positive preclinical findings prohibit potentially beneficial candidates from moving forward in the development pipeline. Complex in vitro models using cardiomyocytes derived from human-induced pluripotent stem cells (hiPSC-CM) have been identified as a useful tool that allows for rapid and cost-efficient screening of proarrhythmic drug risk. Currently available hiPSC-CM models employ simple two-dimensional (2D) culture formats with limited structural and functional relevance to the human heart muscle. Here, we present the use of our 3D cardiac microphysiological system (MPS), composed of a hiPSC-derived heart micromuscle, as a platform for arrhythmia risk assessment. We employed two different hiPSC lines and tested seven drugs with known ion channel effects and known clinical risk: dofetilide and bepridil (high risk); amiodarone and terfenadine (intermediate risk); and nifedipine, mexiletine, and lidocaine (low risk). The cardiac MPS successfully predicted drug cardiotoxicity risks based on changes in action potential duration, beat waveform (i.e., shape), and occurrence of proarrhythmic events of healthy patient hiPSC lines in the absence of risk cofactors. We showcase examples where the cardiac MPS outperformed existing hiPSC-CM 2D models.
Keywords: microphysiological system, hiPSC-CM, cardiac microtissue, arrhythmia, drug risk assessment, safety pharmacology
Before achieving market approval, all drug candidates are screened for cardiac side effects. Despite efforts to detect cardiac risk before a new pharmaceutical enters the clinic, a recent meta-analysis reported that cardiac disorders are the second most common cause for safety-related market withdrawal (18.8%) after hepatotoxicity (27.1%).1 The study, which investigated 133 drugs that have been withdrawn from the market for safety reasons between 1990 and 2010 in countries belonging to the World Health Organization (WHO), also found that the average time from the introduction of a drug to its withdrawal was 20.3 years. To prevent public health risks and costly late drug withdrawals, while keeping good drug candidates in the pipeline, there is a need to improve clinical prediction of new drug candidates for cardiac liabilities.
One important aspect of cardiac safety is the absence of arrhythmogenic properties including a drug’s potential to induce torsades de pointes (TdP), a rare but potentially fatal form of arrhythmia.2 Regulatory guidelines currently in place to prevent drugs with proarrhythmic properties from entering the market (ICH S7B and ICH E14) request screening for QT prolonging effects, which purportedly is a predictive marker of arrhythmic potential including TdP induction.3 However, in some cases, QT prolongation does not correlate with either arrhythmia or TdP, suggesting that screening simply on QT prolongation is not sufficient to identify cardiac liabilities4 and may eliminate promising drugs from the developmental pipeline.
A promising in vitro tool for screening for a drug’s arrhythmogenic properties are human-induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CM), which offer a human genetic background and therefore avoid issues associated with inter-species differences in drug response. Furthermore, cell-based assays are significantly cheaper than animal models and allow for much higher throughput. In recent years, several studies have investigated the utility of hiPSC-CMs for the predication of clinically relevant proarrhythmic drug properties.5−10 Since QT prolongation cannot be directly measured in vitro, testing typically includes evaluation of a drug’s ability to block the human ether-a-go-go-related (hERG) potassium channel (IKr), which is associated with clinical QT prolongation and/or observation of prolongation of QT-proxies such as action potential (APD) or field potential (FPD) duration.6,8 However, while screening for QT prolongation helps to improve drug safety, current methods can fail to detect arrhythmogenic drugs that do not necessarily cause QT prolongation and, on the other hand, also prohibit safe pharmaceuticals from entering the market since QT prolongation alone is not a definite predictor of increased arrhythmic potential.4
Several studies including the CiPA (comprehensive in vitro proarrhythmia assay) initiative,5,6,11 which is dedicated specifically to the in vitro evaluation of arrhythmia risk in drug compounds using hiPSC-CM models, have pointed out other parameters that should be considered in combination with QT prolongation for more accurate safety predictions. Important proarrhythmic markers can be derived from drug-induced changes in the beat waveform (i.e., shape as measured by triangulation12), observation of arrhythmia-like events such as early after depolarizations,6 and the beat pattern (such as irregular beating12 or drug-induced quiescence.6).
Models using hiPSC-CM have been used successfully to assess cardiac arrhythmic risk of various ion channel blockers. However, for some compounds, in vitro results are known to deviate from the official drug risk classification and clinical observations. Most cases of incorrect drug effect predication can be attributed to one or more of the following factors: (i) the clinical observations of arrhythmic events are associated with additional risk factors such as age, gender, underlying illness (e.g., liver disease, electrolyte imbalance, diabetes, and overweight), and drug–drug interactions.13−15 These co-factors typically are not recreated in in vitro assays, and consequently, the associated risk is not reported (e.g., bepridil and terfenadine). (ii) Risk classification of the drug is controversial (e.g., amiodarone).16 (iii) The drug has multi-ion channel activity with similar IC50 values but compensatory effects on the action potential (such as QT prolongation via IKr block and QT shortening via ICa block).5 This issue is enhanced if ion channels are differentially expressed in hiPSC-CM compared to adult human ventricular cardiomyocytes (e.g., lower density of late sodium current is suspected for the deviation of in vitro and in vivo effects of mexiletine).6
In the present study, we use a recently optimized version of our cardiac microphysiological system (MPS)17−19 to evaluate the proarrhythmic properties of seven drugs with well-known effects on cardiac ion channels. The compounds have classically been categorized into high-, intermediate-, and low-risk classes according to their clinical arrhythmia risk.11,20 We investigated two to three candidates from each category: dofetilide and bepridil (high risk); amiodarone and terfenadine (intermediate risk); and nifedipine, mexiletine, and lidocaine (low risk). Our drug selection comprises compounds with typically well-predicted effects in hiPSC-CM models (dofetilide, nifedipine, and lidocaine) as well as several examples of drugs that yielded false-negative or false-positive results in existing in vitro studies (bepridil, terfenadine, amiodarone, and mexiletine).6,11 Our compound selection included a mixture of drugs with predominant single-ion channel interaction and drugs with multi-channel activities (Tables S5–S7).
The cardiac MPS is a microfluidic tissue chip model comprising a three-dimensional (3D) heart micromuscle derived from hiPSC-CMs, which structurally and functionally closely resembles a human heart muscle fiber.19 Data stream readouts are based on high frame rate video acquisition of fluorescent markers that are specific for changes in the transmembrane potential (action potential) and intracellular calcium handling. Lack of maturity has repeatedly been reported as a critical limitation of hiPSC-CM models,21 and immature ion channel expression (such as lower densities of the late sodium current in hiPSC-CMs than the adult human ventricle) has been associated with limited predictive value for certain drugs.6 To address this, we recently optimized our in vitro model by implementing a metabolic maturation step using fatty-acid rich media.18 We demonstrated that this advancement helps to push the maturation characteristics of the cardiomyocytes within the MPS from a fetal to a more adult-like state with lower variance in drug response. Here, we demonstrated the predictive drug screening potential of the cardiac MPS using two hiPSC lines from healthy donors and compared our findings to published studies using 2D hiPSC-CM models.
Results
Characterization and Operation of the Cardiac MPS
Our microphysiological system is composed of elongated cell chambers of 60 μm height (Figure 1A and Figure S8A,B) that promote matrix-free self-assembly of 3D cardiac microtissues as previously reported.18,19 The tissues are formed from a single-cell suspension of 80% hiPSC-CM and 20% isogenic stromal cells (hiPSC-SCs) and start spontaneous beating activity within 24–48 h. In this study, we used hiPSC lines from two different healthy donors. Both cell lines allowed for efficient differentiation of hiPSC-CMs and hiPSC-SCs and successful formation of structurally and functionally stable micromuscles in the cardiac MPS (Figure 1B) that are typically 7–12 cell layers thick.18 A detailed comparative study of the two cell lines with regard to the maturation state and baseline characteristics such as the structure, ion channel expression, and electrophysiology was recently published.18Table S1 highlights the main ion channel activity differences between the two cell lines. WTC and SCVI20 MPS baseline (i.e., without drugs) characteristics show mean spontaneous beat rates ∼1 Hz, cAPD80 ∼500 ms (WTC) and 450 ms (SCVI20), and triangulation ∼0.3 (Table S2 and Figure S9). The drugs were perfused via the media channels that run parallel on either side of the cell chamber, and media exchange to the tissue via diffusion was facilitated by an array of small connection channels (i.e., a fenestration barrier) (Figure 1A and Figure S8B).19 The microfluidic devices either featured a single tissue or four individual tissues arranged in parallel (Figure S8A). Each drug was applied in an acute dose-escalation regimen where the same tissue was subjected to increasing doses of the compound, and electrophysiological effects were recorded after a 30 min incubation (Figure S8C). We derived action potential (AP) and calcium transients from high-speed (100 fps) videos (Figure S8D) and analyzed the traces for APD, AP shape (triangulation metric, see the Materials and Methods section; Figure 1C), and incidence of arrhythmia-like events in AP or calcium traces (Figure 1D). Importantly, the triangulation metric allows valuation of the later phase of the repolarization trajectory independent of the absolute change in APD.
Figure 1.

Design and function of the cardiac MPS. (A) Layout of a multiplexed cardiac MPS device comprising four parallel tissue chambers with individual cell loading ports and a common media inlet (in) and outlet (out). The media channels run parallel to each tissue chamber, and the fenestration barrier provides protection from fluid shear stress while allowing for exchange of media via diffusion. Anchor pillars located at both extremities of the tissue chamber provide attachment points to keep the cardiac muscle elongated and prevent collapsing. Scale bars represent 2000 μm in the full device (left panel) and 100 μm in the close up (right panel). (B) Phase contrast images of the MPS with cardiac tissues formed using WTC (top) and SCVI20 (bottom) cells. Scale bars represent 100 μm. (C) Fluorescent voltage traces (black and dashed red curves) are analyzed for action potential duration at 80 and 30% signal amplitude (APD80 and APD30, respectively). The beat shape metric triangulation is calculated as (APD80 – APD30)/APD80. A higher triangulation value (dashed red curve) is associated with increased arrhythmia risk. Importantly, normalizing to the APD80 decouples the triangulation value from other electrophysiological changes such as beat rate or peak width. (D) Classification of beat shape quality: representative voltage fluorescence traces of a normal beat shape, three types of arrhythmia-like events (early afterdepolarization (EAD); irregular beat pattern; fast spikes without discernable baseline), a weak and noisy trace for which the beat features could not be analyzed, and a trace of a non-beating tissue. The colored rectangles represent the color code used in the bar charts throughout the article.
Dofetilide
Dofetilide is classified as a category 1 drug,20 which means that prolonging repolarization is an intended, desirable effect of this drug. Dofetilide is labeled for QT and TdP risk by the FDA and accordingly has been placed in the high-risk category by the CiPA initiative.11 The cardiac MPS derived from both cell lines demonstrated an increase in cAPD80 with increasing doses of dofetilide (Figure 2A,D). The transition from long APDs to a fibrillation-like state caused a spread in the data set so that statistical significance was determined only at 0.1 nM for WTC and 10 nM for SCVI20. Starting at 10 nM (5× the maximum clinical free plasma concentration (Cmax)), more than half of the tested tissues exhibited arrhythmia-like events or stopped beating (Figure 2C,F). At this dose, IKr is blocked about 25% (Figure 2G). At higher doses, the prolonged beats developed EADs, which eventually split into narrow separate beats with a high degree of triangulation (see the representative traces in Figure S10A and triangulation in Figure 2B,E). Therefore, the increasing cAPD80 could only be observed and reported for doses up to 10 nM (WTC) or 100 nM (SCVI20) (Figure 2A,D). At high doses of 100 and 1000 nM (50–500× Cmax), an increasing number of MPS ceased spontaneous beating activity for both cell lines, indicating detrimental effects on the entire action potential machinery (Figure 2C,F). Using external stimulation, weak action potentials could still be induced in most, but not all of these MPS (data not shown).
Figure 2.
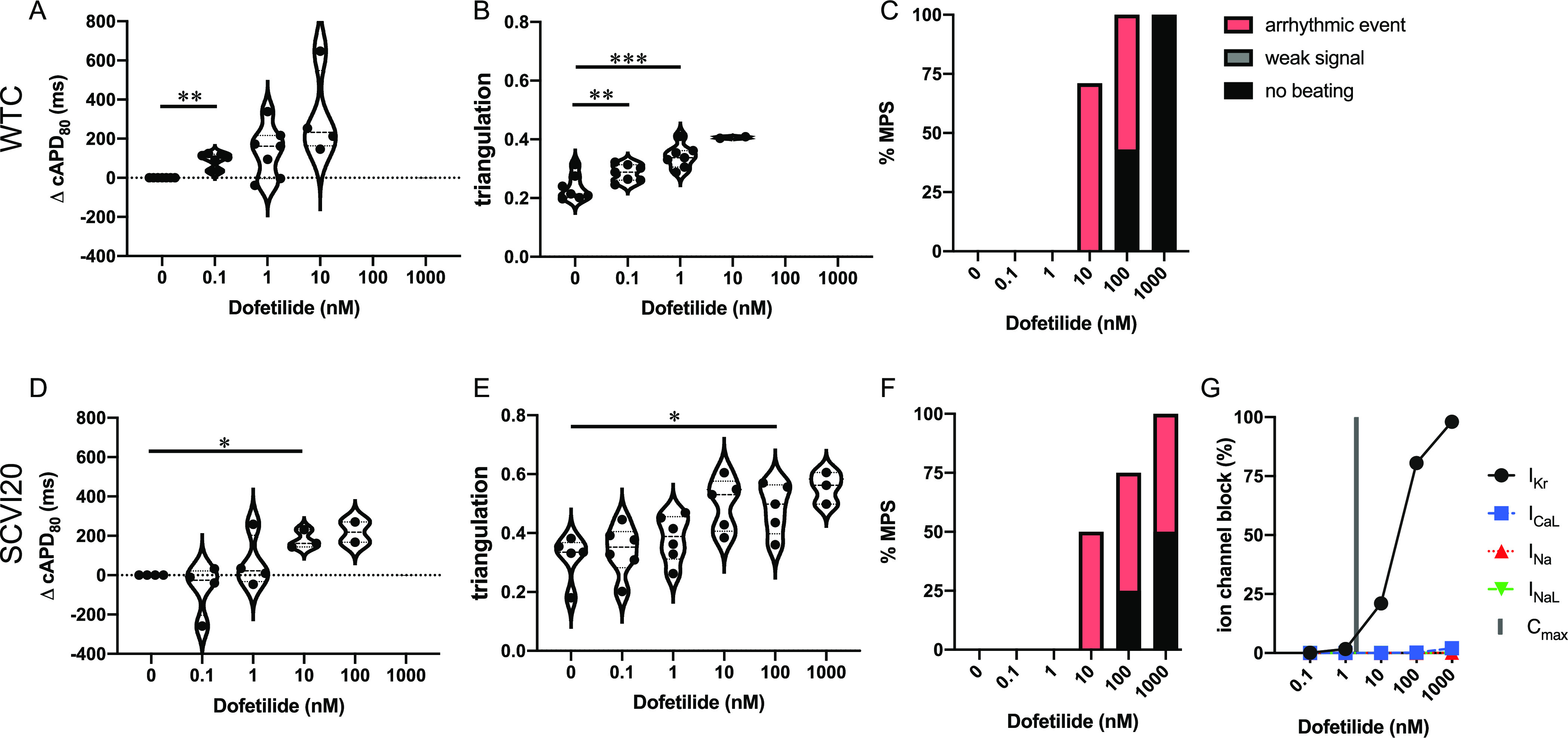
Evaluation of pro-arrhythmic effects of the high-risk drug dofetilide. Results are shown for the MPS generated from the WTC cell line (A–C) and the SCVI20 cell line (D–F). Panels (A, D) show the dose-dependent change in action potential duration (corrected to 1 Hz beat rate). Panels (B, E) show the beat shape metric triangulation calculated as (APD80 – APD30)/APD80 for voltage traces. Panels (C, F) show the percentage of the MPS exhibiting arrhythmia-like event (red), weak signals (gray), or non-beating tissues (black), as defined in Figure 1. Spontaneous voltage and calcium traces were considered for this analysis. (G) Expected percent ion channel block determined from tested drug doses from the literature. Statistical analysis: mixed-effects analysis with the Geisser–Greenhouse correction followed by Dunnett’s multiple comparison test to dose 0. Replicates are from independent tissues. Reported significance levels are p < 0.05 (*), 0.01 (**), 0.001 (***), and 0.0001 (****).
Bepridil
Bepridil is classified as a category 3 drug,20 meaning that there have been numerous case reports of TdP in humans, and has been labeled by the FDA for QT prolongation14 and TdP risk. Due to its torsadogenic potential, bepridil has been assigned to the high-risk category by the CiPA initiative.11 Significant cAPD80 prolongation was observed from 3.2 to 3200 nM bepridil (0.1–100× Cmax) in WTC (Figure 3A). The beat shape as measured by triangulation remained stable until 3200 nM (100× Cmax) where it started to increase (Figure 3B). For SCVI20, the same trend was observed but to a much lesser extent, which did not yield significant changes at any dose (Figure 3D,E). Interestingly, at extremely high doses of 1000× Cmax (32,000 nM), cAPD80 dropped in both cell lines while triangulation increased further. At this concentration, 75% of the SCVI20 MPS stopped spontaneous beating activity. We did not observe arrhythmia-like events in any dose or cell line (Figure 3C,F). Around its clinically relevant dose (Cmax = 32 nM; Table 1), bepridil predominantly blocks IKr while it targets multiple ion channels at higher doses. At 32,000 nM (1000× Cmax), ICaL, INaL, and INa are all blocked almost 100% while IKr is blocked about 85% (Figure 3G).
Figure 3.
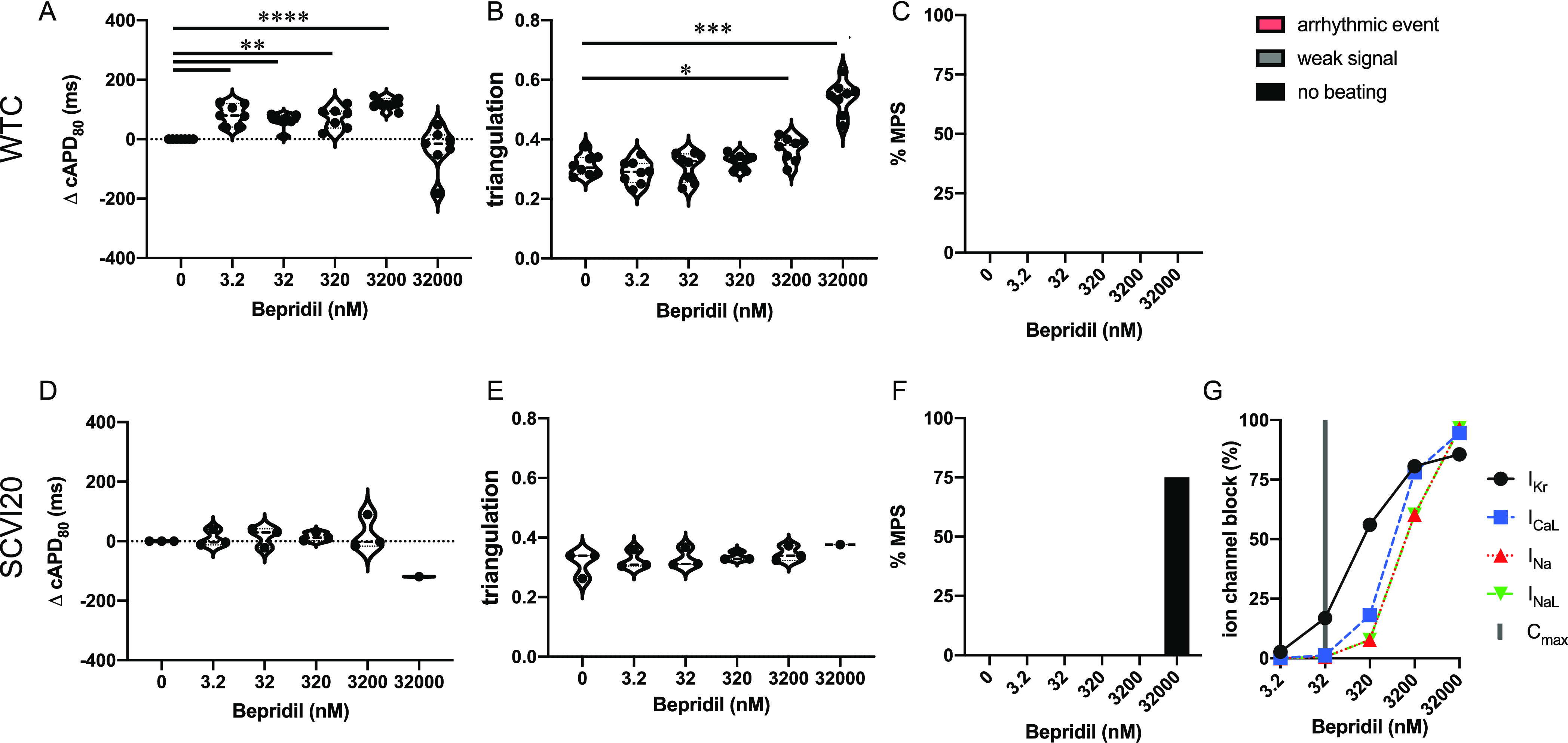
Evaluation of pro-arrhythmic effects of the high-risk drug bepridil. Results are shown for the MPS generated from the WTC cell line (A–C) and the SCVI20 cell line (D–F). Panels (A, D) show the dose-dependent change in action potential duration (corrected to 1 Hz beat rate). Panels (B, E) show the beat shape metric triangulation calculated as (APD80 – APD30)/APD80 for voltage traces. Panels (C, F) show the percentage of the MPS exhibiting any sort of arrhythmia-like event (red), weak signals (gray), or non-beating tissues (black). Spontaneous voltage and calcium traces were considered for this analysis. (G) Expected percent ion channel block at tested drug doses based on literature values. Statistical analysis: mixed-effects analysis with the Geisser–Greenhouse correction followed by Dunnett’s multiple comparison test to dose 0. Replicates are from independent tissues. Reported significance levels are p < 0.05 (*), 0.01 (**), 0.001 (***), and 0.0001 (****).
Table 1. Summary of Observed Drug Effects in the Cardiac MPS for Two Cell Lines and Literature Values from Other 3D hiPSC CM Tissue Formats.
| drug | studies | TdP risk | Cmax (μM) | prediction | cAPD80 | triangulation | arrhythmia or no beating | matches prediction? |
|---|---|---|---|---|---|---|---|---|
| dofetilide | MPS WTC | high | 0.002 | ↑ QT, ↑TdP20 | ↑ 0.0001 μM | ↑ 0.0001–0.001 μM | ↑ 0.01–1 μM | yes |
| MPS SCVI20 | ↑ 0.01 μM | ↑ 0.1 μM | ↑ 0.01–1 μM | yes | ||||
| Biowire31 | ↑ 10–1000 nM | yes | ||||||
| tissue rings32 | ↑25a nM32 | yes | ||||||
| bepridil | MPS WTC | high | 0.032 | ↑ QT, ↑TdP20 | ↑ 0.0032–3.2 μM | ↑ 3.2–32 μM | N/A | partly (QT) |
| MPS SCVI20 | N/A | N/A | ↑ 32 μM | partly (TdP) | ||||
| amiodarone | MPS WTC | intermed | 0.0008 | ↑ QT, ↑TdP5 | ↑ 0.01–1 μM | N/A | N/A | partly (QT) |
| MPS SCVI20 | N/A | N/A | ↑ 0.01–1 μM | partly (TdP) | ||||
| terfenadine | MPS WTC | intermed | 0.00028 | ↑ QT, ↑TdP5,20 | N/A | N/A | ↑ 0.3–3 μM | yes |
| MPS SCVI20 | ↓ Dose5 | N/A | ↑ 0.003–3 μM | yes | ||||
| anisotropy strip33 | ↑ reentry events 300 nMa | yes | ||||||
| nifedipine | MPS WTC | low | 0.0077 | ↑TdP20 | ↓ 0.1–1 μM | ↑ 1 μM | N/A | yes |
| MPS SCVI20 | N/A | ↑ 10 μM | ↑ 0.1–10 μM | yes | ||||
| tissue rings30 | ↑ 30 nM–1 μMa30 | yes | ||||||
| mexiletine | MPS WTC | low | 2.5 | ↓ QT,11INaL block | N/A | ↑ 100 μM | ↑ 100–1000 μM | yes |
| MPS SCVI20 | ↓ 100 μM | ↑ 100 μM | ↑ 100–1000 μM | yes | ||||
| lidocaine | MPS WTC | low | 2.5 | N/A,5INaL block | N/A | N/A | ↑ 100–1000 μM | yes |
| MPS SCVI20 | N/A | N/A | ↑ 1000 μM | yes | ||||
| tissue rings34 | no arrhythmia events 100 μMa | yes |
Parameter available in the literature not identical to the parameter used in this publication but comparable.
Amiodarone
Amiodarone has risks of QT prolongation and TdP risk on its FDA label.5 It has not been classified by the CiPA initiative but can be considered intermediate risk based on its similarities to other drugs in this category.5,6,20 We detected a weak cAPD80 increase (in WTC only) and no change in triangulation (Figure 4A,B,D,E). Similarly, we observed no (WTC) or few (SCVI20) arrhythmia-like events at any of the tested doses ranging up to 1250× Cmax (Figure 4C,F). The arrhythmia-like events were classified as fibrillations, meaning that no clear baseline could be established between consecutive beats. Amiodarone has no to little ion channel block activity at concentrations under 1000× Cmax (Cmax = 0.8 nM; Table 1) and targets mainly IKr and ICaL at high doses (Figure 4G).
Figure 4.
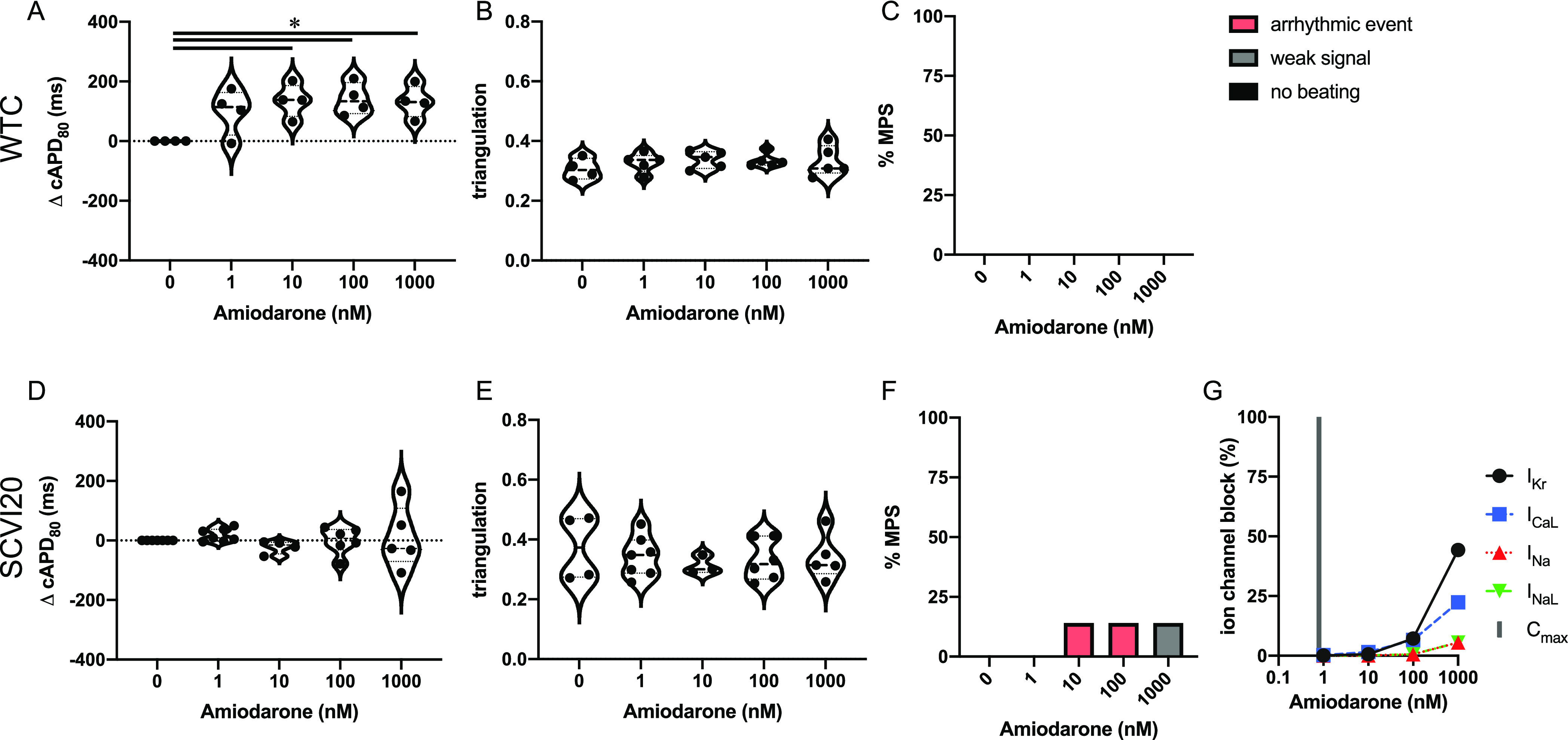
Evaluation of pro-arrhythmic effects of the intermediate-risk drug amiodarone. Results are shown for the MPS generated from the WTC cell line (A–C) and the SCVI20 cell line (D–F). Panels (A, D) show the dose-dependent change in action potential duration (corrected to 1 Hz beat rate). Panels (B, E) show the beat shape metric triangulation calculated as (APD80 – APD30)/APD80 for voltage traces. Panels (C, F) show the percentage of the MPS exhibiting any sort of arrhythmia-like event (red), weak signals (gray), or non-beating tissues (black). Spontaneous voltage and calcium traces were considered for this analysis. (G) Expected percent ion channel block at tested drug doses based on literature values. Statistical analysis: mixed-effects analysis with the Geisser–Greenhouse correction followed by Dunnett’s multiple comparison test to dose 0. Replicates are from independent tissues. Reported significance levels are p < 0.05 (*), 0.01 (**), 0.001 (***), and 0.0001 (****).
Terfenadine
Terfenadine has been categorized as a class 2 agent,20 indicating market withdrawal due to an unacceptable risk of TdP for the conditions being treated. It is labeled as QT prolonging and TdP risk by the FDA,5 consistent with its primary mechanism as an hERG blocker (Figure 5G). We found variable effects on cAPD80 with no significant prolongation at any dose but a clear cAPD80 decrease at the highest dose (significant for SCVI20) (Figure 5A,D). We observed very little impact on the beat shape measured by triangulation (Figure 5B,E) and no arrhythmia-like events up to 300 nM (1000× Cmax) followed by a few observations of irregular beat patterns at 3000 nM (10,000× Cmax) in WTC cells (Figure 5C,F). Quiescence was detected in a few MPS of each cell line, reaching up to 33% in SCVI20 at 300 nM. At high concentrations, terfenadine shows considerable multi-ion channel activity (Figure 5G).
Figure 5.
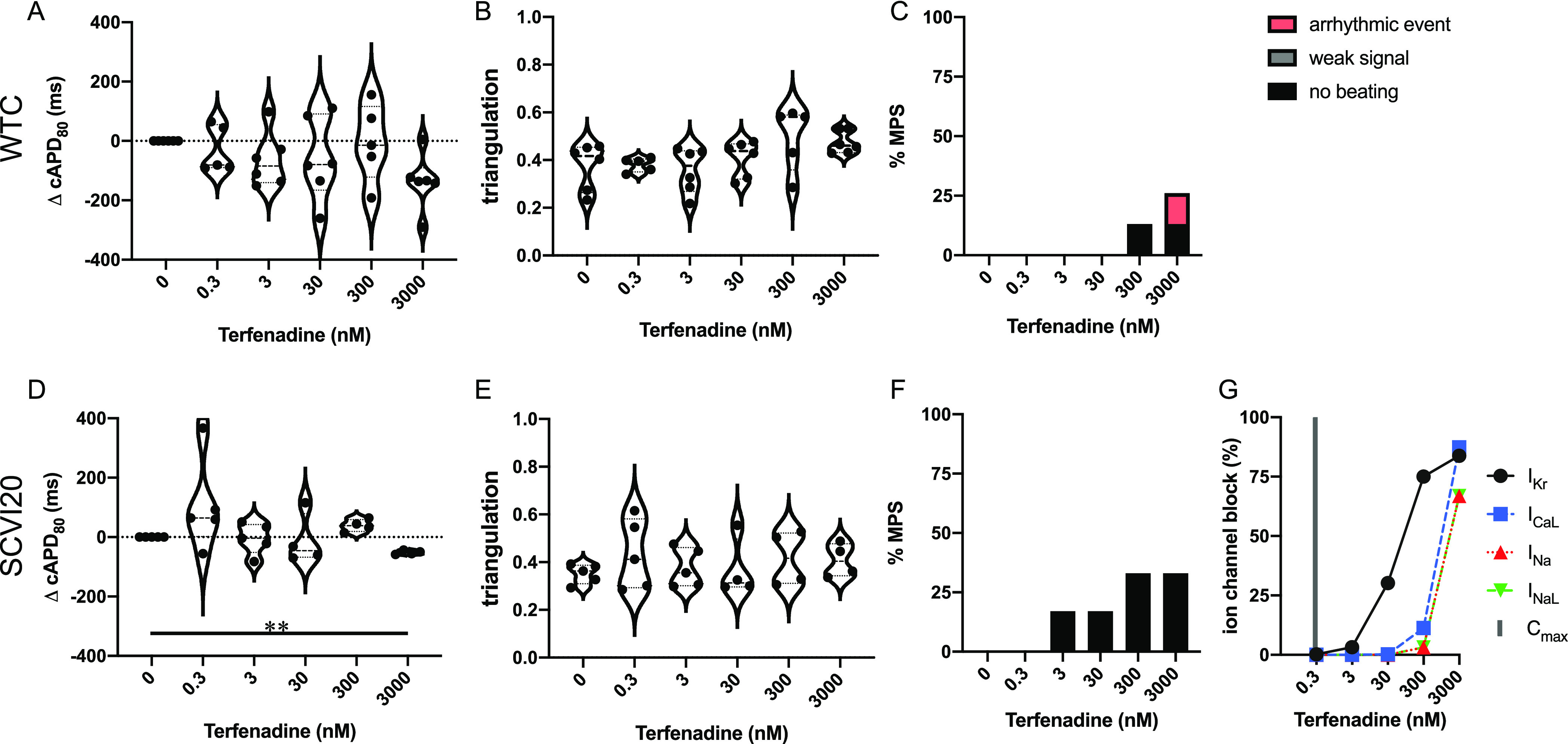
Evaluation of pro-arrhythmic effects of the intermediate-risk drug terfenadine. Results are shown for the MPS generated from the WTC cell line (A–C) and the SCVI20 cell line (D–F). Panels (A, D) show the dose-dependent change in action potential duration (corrected to 1 Hz beat rate). Panels (B, E) show the beat shape metric triangulation calculated as (APD80 – APD30)/APD80 for voltage traces. Panels (C, F) show the percentage of the MPS exhibiting any sort of arrhythmia-like event (red), weak signals (gray), or non-beating tissues (black). Spontaneous voltage and calcium traces were considered for this analysis. (G) Expected percent ion channel block at tested drug doses based on literature values. Statistical analysis: mixed-effects analysis with the Geisser–Greenhouse correction followed by Dunnett’s multiple comparison test to dose 0. Replicates are from independent tissues. Reported significance levels are p < 0.05 (*), 0.01 (**), 0.001 (***), and 0.0001 (****).
Nifedipine
Nifedipine only has isolated reports of TdP in humans (Redfern class 4).20 This ICaL blocker (Figure 6G) has been placed in the very low-risk group by the CiPA initiative.11 We observed APD shortening in both cell lines (significant in WTC and non-significant in SCVI20) (Figure 6A,D). At the highest dose, beat shape changes were detected as triangulation increases in both cell lines (Figure 6B,E). No arrhythmia-like events were detected (Figure 6C,F). A low incidence of non-beating tissues was observed in SCVI20 at the two highest doses of 1 and 10 μM (125 and 1250× Cmax, respectively). However, action potentials could still be induced in all these tissues by external electrical stimulation (data not shown).
Figure 6.
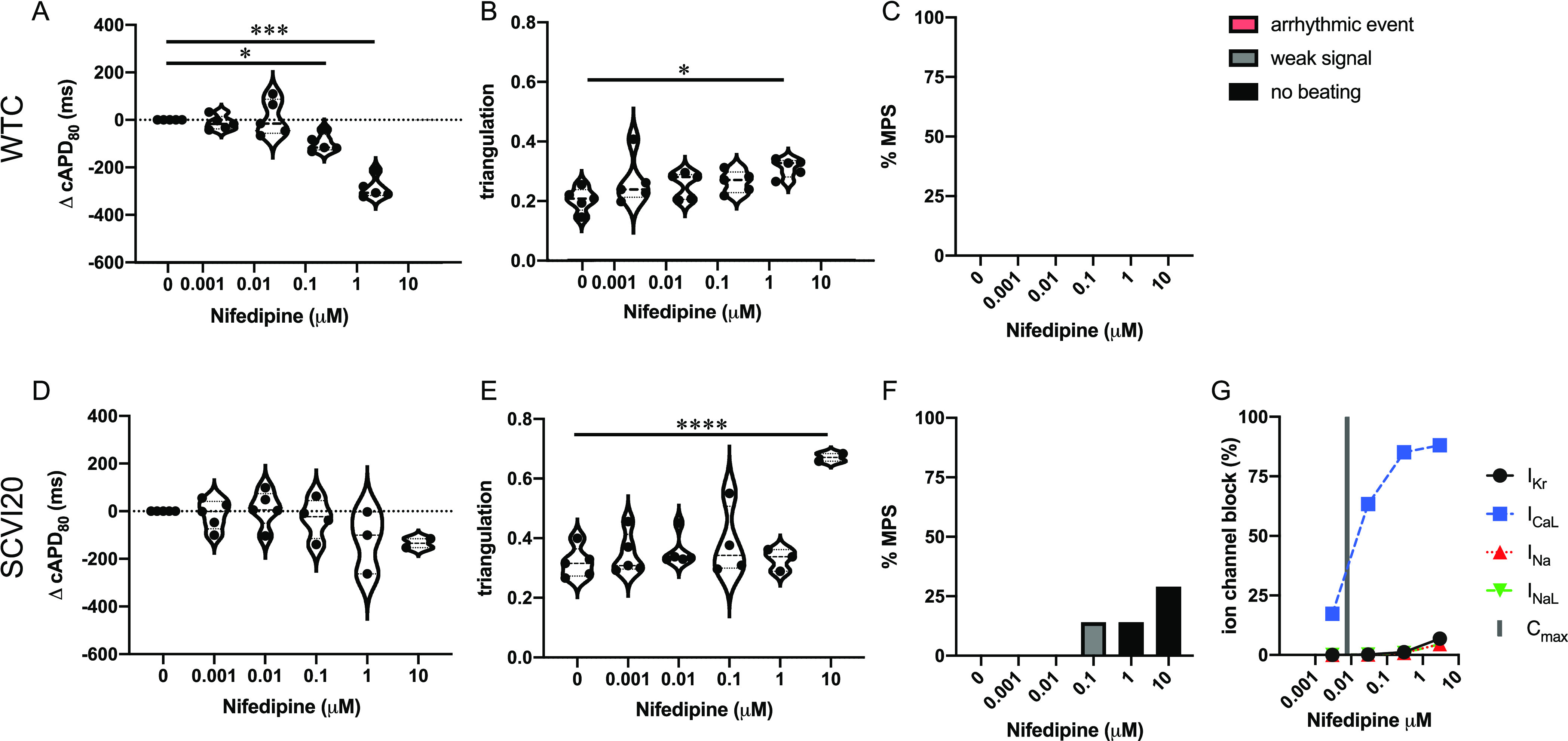
Evaluation of pro-arrhythmic effects of the low-risk drug nifedipine. Results are shown for the MPS generated from the WTC cell line (A–C) and the SCVI20 cell line (D–F). Panels (A, D) show the dose-dependent change in action potential duration (corrected to 1 Hz beat rate). Panels (B, E) show the beat shape metric triangulation calculated as (APD80 – APD30)/APD80 for voltage traces. Panels (C, F) show the percentage of the MPS exhibiting any sort of arrhythmia-like event (red), weak signals (gray), or non-beating tissues (black). Spontaneous voltage and calcium traces were considered for this analysis. (G) Expected percent ion channel block at tested drug doses based on literature values. Statistical analysis: mixed-effects analysis with the Geisser–Greenhouse correction followed by Dunnett’s multiple comparison test to dose 0. Replicates are from independent tissues. Reported significance levels are p < 0.05 (*), 0.01 (**), 0.001 (***), and 0.0001 (****).
Mexiletine
Mexiletine is a low-risk drug11 and does not have FDA labels for QT prolongation or TdP risk.5 It not only has predominant INaL blocking action but also affects IKR, ICaL, and INa at higher doses (Figure 7G). We report cAPD80 shortening (significant for SCVI20 at 40× Cmax (Figure 7A,D)) and triangulation increase in both cell lines (Figure 7B,E). No arrhythmic events were observed, and most tissues stopped beating between 100 and 1000 μM mexiletine (Figure 7C,F). Our observation is in line with the drug blocking INaL, IKR, ICaL, and INa 50–100% at 100 μM and >95% at 1000 μM (Figure 7G).
Figure 7.
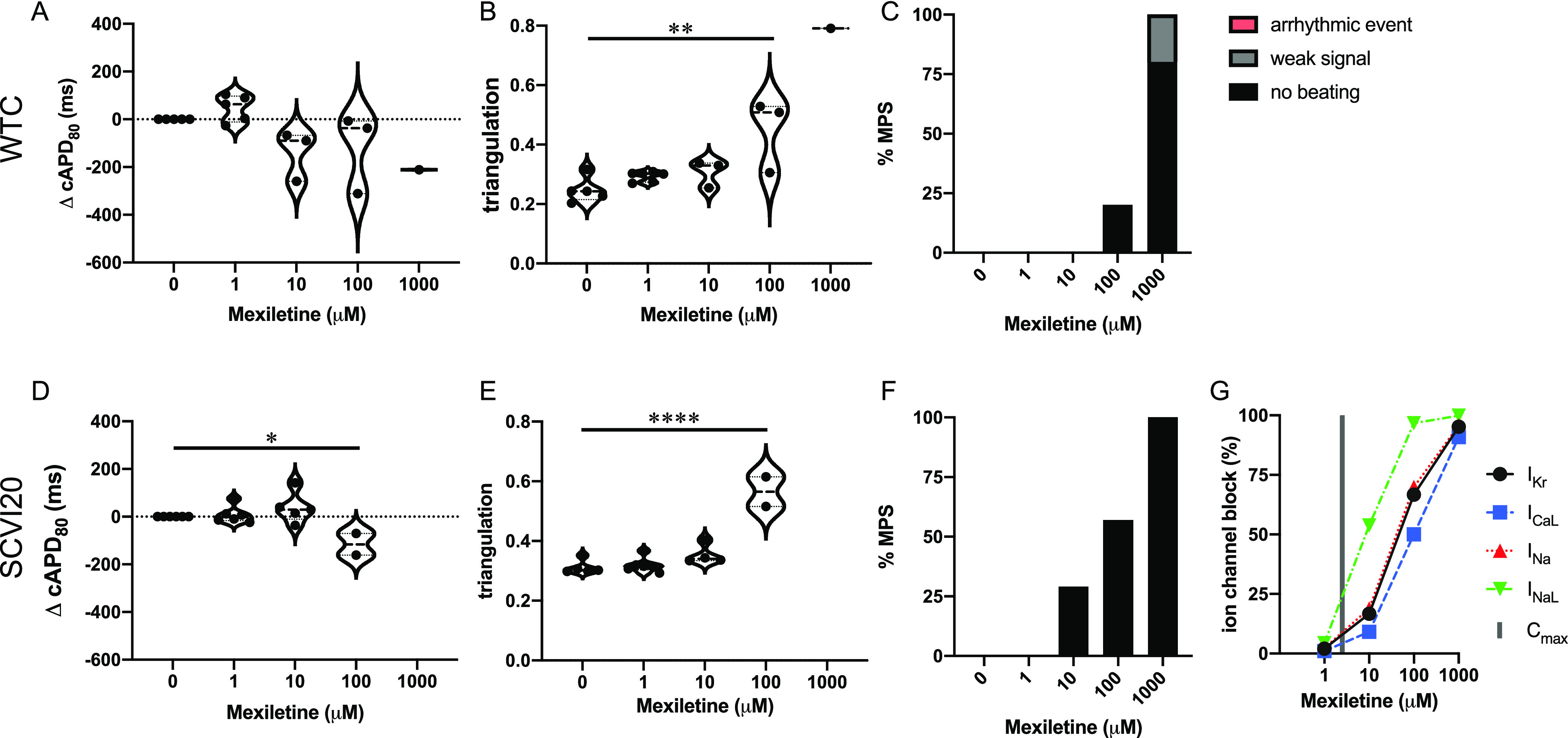
Evaluation of pro-arrhythmic effects of the low-risk drug mexiletine. Results are shown for the MPS generated from the WTC cell line (A–C) and the SCVI20 cell line (D–F). Panels (A, D) show the dose-dependent change in action potential duration (corrected to 1 Hz beat rate). Panels (B, E) show the beat shape metric triangulation calculated as (APD80 – APD30)/APD80 for voltage traces. Panels (C, F) show the percentage of the MPS exhibiting any sort of arrhythmia-like event (red), weak signals (gray), or non-beating tissues (black). Spontaneous voltage and calcium traces were considered for this analysis. (G) Expected percent ion channel block at tested drug doses based on literature values. Statistical analysis: mixed-effects analysis with the Geisser–Greenhouse correction followed by Dunnett’s multiple comparison test to dose 0. Replicates are from independent tissues. Reported significance levels are p < 0.05 (*), 0.01 (**), 0.001 (***), and 0.0001 (****).
Lidocaine
Lidocaine is an exclusive INaL blocker (class 1B sodium channel blocker) (Figure 8G) with no FDA label for QT prolongation or TdP risk5 and can therefore be considered low TdP risk. Expected changes in the APD are a mild delay in the upstroke and slightly shortened repolarization. We found a slight average decrease in cAPD80 (not significant) and no statistically significant effect on the triangulation metric (Figure 8A,B,D,E). We did not observe any occurrence of arrhythmia-like events but recorded a stop in spontaneous beating activity between 100 and 1000 μM (400× Cmax) in almost all tissues (Figure 8C,F). At that dose, INaL is blocked nearly 100% (Figure 8G), which will induce beating quiescence. Blockage of the sodium channels abolishes the raising phase of the action potential, so an AP cannot be generated.
Figure 8.
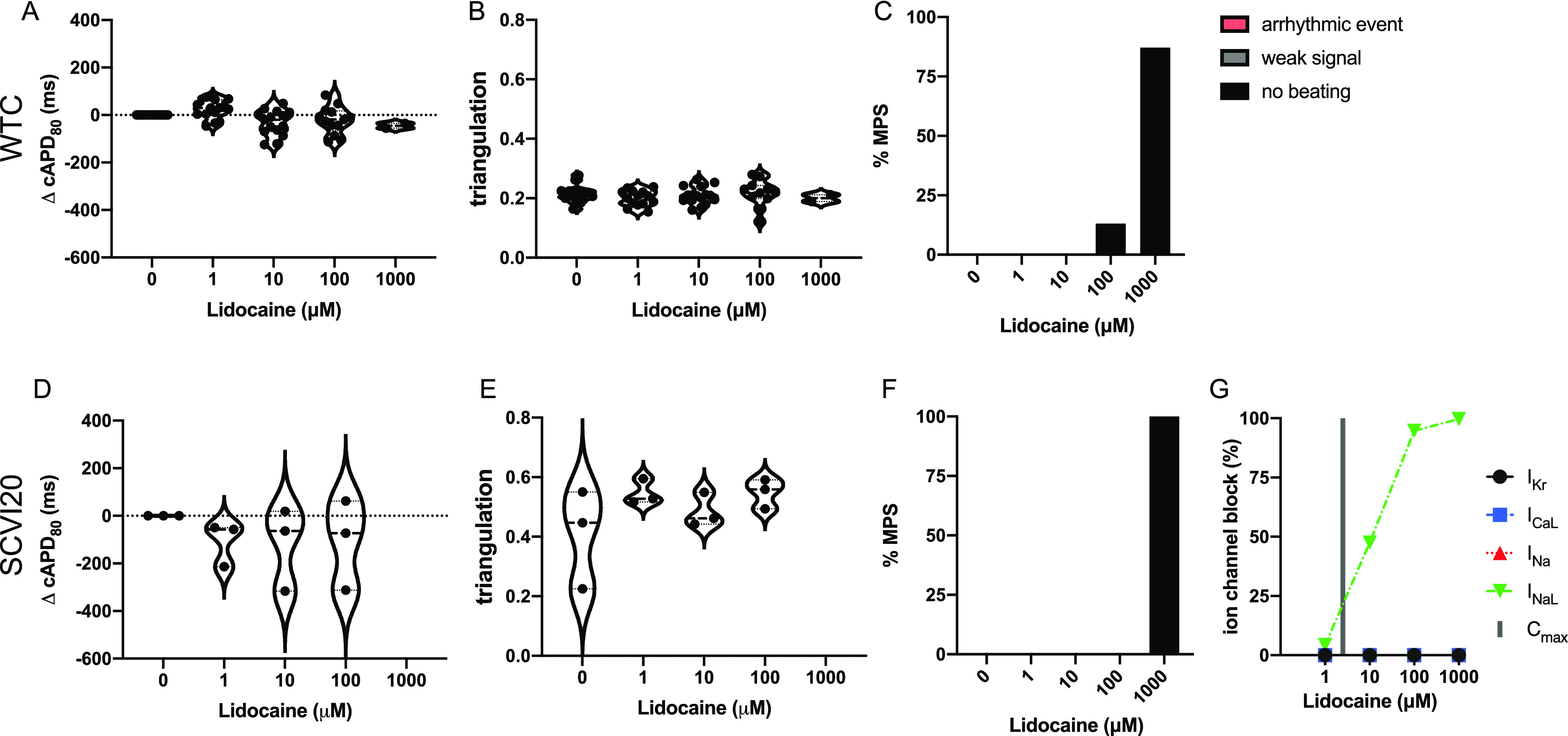
Evaluation of pro-arrhythmic effects of the low-risk drug lidocaine. Results are shown for the MPS generated from the WTC cell line (A–C) and the SCVI20 cell line (D–F). Panels (A, D) show the dose-dependent change in action potential duration (corrected to 1 Hz beat rate). Panels (B, E) show the beat shape metric triangulation calculated as (APD80 – APD30)/APD80 for voltage traces. Panels (C, F) show the percentage of the MPS exhibiting any sort of arrhythmia-like event (red), weak signals (gray), or non-beating tissues (black). Spontaneous voltage and calcium traces were considered for this analysis. (G) Expected percent ion channel block at tested drug doses based on literature values. Statistical analysis: mixed-effects analysis with the Geisser–Greenhouse correction followed by Dunnett’s multiple comparison test to dose 0. Replicates are from independent tissues. Reported significance levels are p < 0.05 (*), 0.01 (**), 0.001 (***), and 0.0001 (****).
Discussion
Evaluation of Seven Drugs in the MPS
In this study, we evaluated the arrhythmogenic drug risk with a novel micro heart muscle tissue combined with a microfluidic platform. The cardiac MPS features a structural and functional mimic of a human cardiac muscle fiber derived from iPSC-CM in combination with support cells. The geometric confinement of the microfabricated cell chamber encourages self-assembly into a uniaxial beating microtissue, and combined with fatty-acid rich media, the maturity of the micro heart muscle was enhanced.18
We investigated seven drugs with known ion channel inhibition and clinical effects. The drugs were selected to include examples with high (dofetilide and bepridil), intermediate (amiodarone and terfenadine), and low (nifedipine, mexiletine, and lidocaine) arrhythmia risk.
Dofetilide
The desired arrhythmia-free APD prolongation of dofetilide was successfully predicted by our system up to 5× Cmax. The observed cAPD80 increase around a Cmax of 2 nM correctly indicates a small safety margin for this drug. Based on the AP prolongation accompanied by beat shape changes and occurrences of arrhythmia-like events, the cardiac MPS system successfully predicted the arrhythmogenic risk associated with dofetilide in both cell lines (Figure 2). Due to its relatively high TdP risk, the clinical use of dofetilide is subjected to extensive contraindications, strict dosing guidelines, and FDA-mandated inpatient monitoring during drug loading.22
Bepridil
The observed cAPD80 prolongation around 0.1–100× Cmax is in concordance with clinical QT prolongation and effects observed in other hiPSC-CM-based in vitro studies.5,6 APD prolongation together with the increasing triangulation at higher doses points toward bepridil’s arrhythmic risk. The effects at very high bepridil doses can be attributed to multiple ion channel blockage, which will shorten and eventually completely abolish all beating activity (Figure 3F,G).
Early clinical studies have shown instances of TdP associated with bepridil treatment, albeit at low rates (0.01–1%).14,23 Even though bepridil is classified as a drug with TdP risk, we did not find any arrhythmia-like events (Figure 3C,F), similar to other state-of-the-art hiPSC-CM studies. This can be explained by three main factors: (i) the almost exclusive occurrence of TdP correlated with old age (especially women over 70 years); (ii) electrolyte imbalance (especially hypokalemia) as an additional risk factor; and (iii) more hiPSC lines are needed to predict TdP with low clinical incidence.14,15 Several other studies also reported QT prolongation with high interindividual variation but reported a low incidence of cardiovascular side effects and no episodes of TdP, especially when accounting for the established risk factors.15,24 Taken together, the low rate of clinical TdP events and their association with additional risk factors (hypokalemia and older age) suggest that current complex tissue in vitro screening tools are unlikely to predict proarrhythmic properties for bepridil but can reveal drug-induced QT increase.
Amiodarone
Amiodarone blocks multiple ion channels around similar concentrations (Figure 4G),5 which makes placement in any specific risk and antiarrhythmic class controversial:16 this atypical class III antiarrhythmic also exhibits class I, II, and IV activities. The observed low prevalence of pro-arrhythmic events as well their type (lack of stable baseline between consecutive beats) can be explained by a concurrent block of multiple ion channels at high drug concentrations (ICaL and INaL; Figure 4G) that prevents hERG-related arrhythmias. The outcome of our study matches what has been previously reported in comparable in vitro studies. Despite the drug’s label for QT increase, literature shows a hERG block of 50% and higher only for concentrations around 100–1000× Cmax (Figure 4G). Similarly, Redfern et al. noted that, out of the 11 category 1 drugs (i.e., compounds that have repolarization prolongation as a desired effect), amiodarone was the only compound that did not show in vitro APD prolongation around the estimated therapeutic plasma concentration (ETPC) but did at significantly higher doses, reaching a safety margin of 1400-fold.20 Therefore, it can be expected that in vitro studies will only reveal an impact on APD, beat shape, or arrhythmic events only at very high amiodarone concentrations above Cmax. While amiodarone was not one of the 28 drugs of the 2018 multi-center CiPA study,6 it was included in an earlier trial where no arrhythmic events were found even at concentrations that caused a strong hERG block.5 Furthermore, in the clinical setting, chronic amiodarone treatment is necessary to achieve its therapeutic effects.16 Therefore, we suggest that a chronic in vitro exposure scenario is necessary to fully reveal QT changes and the torsadogenic potential of this drug.9
Terfenadine
Terfenadine effects have historically been difficult to detect using hiPSC-CM in vitro test systems. Literature reports that the clinically observed TdP risk of terfenadine could not be predicted in hiPSC-based testing, and outcomes for APD changes were variable.5,6,20 At doses up to 100 nM, mostly APD increases were reported followed by a decrease and eventually quiescence around 1000 nM.6 These effects are in line with the drug’s multi-ion channel blockage activity at high doses (Figure 5G). While terfenadine is labeled as QT prolonging and TdP risk by the FDA,5 in the CiPA initiative, it was only placed in the intermediate-risk category for torsadogenic potential based on published data and expert opinion.11 Clinical cases of arrhythmia in patients without additional risk factors have rarely been reported.25 Thus, the cardiac MPS’ prediction is accurate for exposure to healthy patients. The observation of irregular beat patterns at the highest tested dose (3000 nM) in the WTC cell line is in-line with the drug’s reported arrhythmia risk. However, the low prevalence and occurrence only at a very high dose make it a weak predictor for clinical arrhythmias. Terfenadine has been associated with QT increase and risk of TdP, for which it was removed from the market in 1998.26 Increased risk for these cardiotoxic effects was connected to terfenadine accumulation in the patient due to inhibition of its metabolic breakdown via CYP3A4 by a second drug, ketoconazole treatment,13 and other predisposing factors. To better capture the clinical risk of terfenadine, further in-depth studies tailored to the specific pharmacokinetics of this drug are required. Such follow-up tests may include assessing the risk of the terfenadine metabolite fexofenadine.
Nifedipine
Our MPS system correctly predicted the QT shortening effect of this ICaL blocker (Figure 6G). In combination with the APD decrease, the observed triangulation increase at high doses (Figure 6B,E) does not present a proarrhythmic risk factor and is in concordance with the expected effects of reduced calcium currents, which leads to a reduction of the AP plateau phase. Our results are in line with similar studies on hiPSC-CM models that reported a decrease in QT, no occurrence of arrhythmia-like events, and few cases of quiescence at >10× Cmax (>0.1 nM).11
Mexiletine
Clinically, mexiletine shortens the QT interval in different forms of long QT syndrome.27 In the cardiac MPS, these effects were accurately predicted by APD shortening, especially in the presence of long baseline APDs (Figure 7A,D and Figure S12B). Together with the absence of proarrhythmic events, the observed behavior was in line with the expected effects for this low-risk drug. Our findings for mexiletine are in better concordance to the clinical effects than the large multi-center CiPA study with 2D hiPSC-CM models,6 where a mild repolarization prolongation (no effect in some test centers) was reported together with occasional reports of arrhythmias at ≥4-fold Cmax. The authors indicated lower densities of the late sodium current in hiPSC-CMs than human ventricular cardiomyocytes as a possible explanation for the unexpected arrhythmia-like events since late INa is necessary to balance the proarrhythmic hERG block effects. The absence of such non-physiological events in our data suggests that our MPS provides a more physiologically relevant model than simple 2D hiPSC-CM formats.
Lidocaine
Our findings predict expected effects of the non-cardiac risk lidocaine. We observed a mildly shortened APD and no arrhythmia-like events. Beat shape changes caused by this class 1B sodium channel blocker did not result in significant changes in the triangulation metric.
Cardiac MPS Model for Drug Risk Prediction
In this study, we focused on a screening protocol employing a complex in vitro tissue model where drugs were acutely exposed to healthy patient hiPSC lines in the absence of risk cofactors. Under these circumstances, the cardiac MPS correctly predicted clinically relevant changes in the APD (a QT-proxy), proarrhythmic changes in beat waveform (increased triangulation), and arrhythmia-like events (e.g., EADs and irregular beat patterns). The improved predictive outcome for mexiletine compared to 2D hiPSC-CM studies highlights the need for cardiac tissue models beyond simple 2D formats for in vitro drug risk screening. We hypothesize that the higher physiological accuracy of our 3D tissue geometry and uniaxial alignment combined with a fatty acid rich media supports tissue maturation and more relevant ion channel expression.18
Data retrieved from the cardiac MPS without additional experiments can be used to investigate a range of research questions.17−19 For example, the fluorescence video traces can be examined for additional properties such as upstroke velocity (to characterize INaL blockers such as lidocaine), intensity amplitude, and inter-beat irregularities (Poincaré plots9,12). Data from externally paced tissues can be probed for the relationship of voltage to calcium traces using phase plots and to measure conduction velocity within a tissue. The microfluidic system further allows for precise control over the cellular microenvironment and collection of risk biomarkers from the media channels and chronic drug exposure studies.10 Finally, brightfield videos can be examined for tissue stress and strain as well as contractile forces.17,19
To improve prediction of drug-induced cardiac liabilities, our findings suggest that screening should be conducted with broader representation in patient lines to model clinical reality with respect to the genetic, racial, physiological, and pathological makeup of the population. Although it has been demonstrated that hiPSC-CM generated from different sources appears superficially similar,28 patient line-dependent variations in drug response should be expected.29 To begin to account for possible donor variability, we tested all drugs on the MPS generated from two hiPSC lines (WTC and SCVI20). Overall, the two cell lines showed good concordance in drug response; however, in several cases, drug effects were significant in only one patient line, while a similar but non-significant trend was observed in the other patient line. Similarly, the exact dose at which each patient line responded was different for most drugs. Our observations for cell line variations and similarities are comparable to recent findings in other 3D cardiac tissue formats.30,31 Taken together, our results not only further confirm the usefulness of hiPSC-CM models in general and our 3D MPS heart micromuscle model in particular for cardiac drug risk assessment but also underline the importance of using multiple patient lines for new molecule discovery.
Recently, enormous progress has been made in 3D cardiac tissue cultivation and new culture formats as application notes are published frequently.35 Several studies have demonstrated the benefits of 3D culture formats over simple 2D cell layers. 3D cultures of various formats such as tissue strips, tissue rings, and chambers yielded more mature phenotypes regarding gene expression, electrophysiology, and tissue mechanics.18,36 To date, only a few studies provide a direct comparison of drug effects in 2D vs 3D cultures. The ones that exist clearly point to increased accuracy of drug responses in 3D platforms.18,37 These early successes highlight the importance of 3D platforms in the drug risk assessment pipeline. Where applicable, we compared our own findings to results obtained in other 3D cardiac tissue culture systems (Table 1). However, few systems were directly comparable to our data since they either used different drug compounds,38 different metrics (such as structural metrics39 or mechanical analysis34,36,40), or both.41 We did not find 3D studies with comparable metrics for amiodarone and bepridil, and 3D cardiac tissue testing for mexiletine has not been previously published.
While our MPS model demonstrated high value for fast and inexpensive prediction of clinical proarrhythmia risk, some limitations need to be noted. First, even though we successfully implemented engineering (confined chambers that promote self-assembly into uniaxially beating tissues) and biological (fatty acid rich maturation media) strategies to improve tissue maturation, our MPS differ from adult human ventricular cardiomyocytes. One prominent indicator if incomplete maturation is the fact that our tissues retained spontaneous beating capabilities, while adult human cardiomyocytes require external stimulation to elicit an action potential. Our current efforts toward further maturation include ongoing optimization of the microdevice geometry, chronic pacing, and the combination of our in vitro data with mathematical models that extrapolate the expected clinical effects from experimental results.18,42,43In vitro or in silico maturation is an especially attractive approach for drugs with multichannel effects (i.e., IC50 values for multiple ion channels are within the same order of magnitude) such as bepridil, amiodarone, and mexiletine (Tables S5–S7). For these drugs, the exact clinical effect depends on a fine balance between the various ion channels and small differences in ion channel ratios between patient lines, and in vitro models and in vivo can result in contradicting outcomes.
For some compounds with known clinical side effects, our MPS did not show AP prolongation or arrhythmia-like events. These cases of “false negative” risk assessment occurred with drugs (bepridil, terfenadine, and amiodarone) that are associated with clinical cofactor or complex situation. Closer examination of clinical data often revealed that the cardiotoxic side effects were largely associated with the presence of additional risk factors such as pre-existing cardiac issues (congenital or acquired), electrolyte imbalance, or drug–drug interaction.13,14 To correctly predict the risk for these drugs, the in vitro models need to be adapted to mimic relevant risk factors. Accordingly, our MPS is well suited to recapitulate several conditions that could lead to increased prediction of arrhythmic potential. Patient-derived iPSC-CM can be used to mimic congenital heart disease such as long QT syndrome and for personalized risk assessment even in the absence of known genetic predispositions. A range of conditions such as electrolyte imbalance or high fat diet can be readily mimicked by dynamically altering the media composition with automated fluid handling. The cardiac MPS is also capable of replicating chronic treatment scenarios,10 which is critical to assess drugs that require long-term exposure to predict their clinical side effects (e.g., amiodarone). Furthermore, MPS technology facilitates the combination of different tissues44 to specifically probe multiorgan effects. In a recent study, we use our MPS to demonstrate the effect of the drug–drug interaction on liver metabolism and subsequently cardiac toxicity of cisapride.45 The combination of liver and heart MPS is essential in cases where unknown drug metabolites are suspected to confer cardiotoxicity.
To allow for screening of larger compound libraries, to increase technical replicates, and to cover broader dose ranges, platforms that allow for high throughput are desirable. In this work, we used four-plex devices containing four parallel tissue chambers (see Figure 1A) as opposed to the single-tissue devices used in our previous publications.18,19 Technically, there is no limitation in further multiplexing of our technology (e.g., 8, 16, 32, or 64 parallel chambers per device) and fitting approximately 200 tissues within the footprint of a standard multi-well plate. Regarding cell loading, handling, and maintenance, the effort is comparable to 2D cultures. From a tissue generation and maintenance perspective, our platform is amenable to achieve similar throughput as systems using a 96- or 384-well format.46 The main limitation for throughput in our current system is the data acquisition. Recording and saving three videos (brightfield and two fluorescence recordings) plus time for moving the objective between samples take about 1 min per tissue. In our current setup, all tissues within one experiment are exposed to the drug at the same time. To keep drug exposure timing concise, we limit acquisition to about 10 tissues per experiment. By adding more fluidic pumps that deliver drugs independently to different MPS, this number could be increased to about 35 tissues while keeping up with the 30 min drug exposure duration for each tissue. To further improve the throughput, multiple imaging systems could be used or alterations in the multiplex form factor to accommodate imaging multiple tissue chambers at the same time. In screening scenarios where throughput is of prime importance, content can be sacrificed in favor of throughput (e.g., limit acquisition to voltage traces or reduce frame rates).
Conclusions
Predicting arrhythmogenic risk is an important step in the drug development pipeline. Although hiPSC-CM models are gaining increasing popularity due to their low cost and suitability for high throughput analysis, most of the currently used hiPSC-CM test systems are set up as simple 2D culture formats, which inherently have limitations regarding their ability to recapitulate in vivo tissue structure and function, and hence drug response. We demonstrated that our advanced cardiac MPS (3D hiPSC-CM micromuscle model) allows for accurate prediction of drug-induced cardiac liabilities and improves on those predictions from 2D hiPSC-CM models. The specific biological and technological advantages of our microfluidic system, which we extensively reported on in several previous publications,17−19,43,45 allow the design of future studies to investigate cardiac drug side effects that occur only in combination with additional risk factors (e.g., genetic variations, drug–drug interaction, and co-morbidities) and have not been sufficiently addressed using existing hiPSC-CM models.
Materials and Methods
Microfluidic Device Design and Fabrication
The microfluidic design featured elongated cell chambers of 800 or 1550 μm length and 150 μm width connected to media channels (50 μm width) via an array of fenestrations (2 μm × 2 μm height/width; 40 μm length), which served to protect the tissue from fluid mechanical forces while allowing rapid media change via diffusion (Figure 1A and Figure S8B).19 Each MPS either contained a single cell chamber or four cell chambers multiplexed in parallel and fed via a single media inlet and outlet port (Figure S8A,B).
The microfluidic MPS devices were fabricated from polydimethylsiloxane (PDMS) using a traditional replica molding technique. The process has been described in detail elsewhere.9,18,19 In brief, an SU-8 master wafer was fabricated in a two-step process. First, 2 μm tall fenestration features were patterned on a silicon wafer followed by 60 μm tall structures for the fluidic channels and cell chamber. Replica molding of polydimethylsiloxane (PDMS; Dow Chemical, Sylgard 184) was performed to pattern the final microfluidic devices. After curing, the PDMS was cut to size and holes of 0.75 μm diameter were punched for the media inlet and outlet as well as the cell loading ports. To make the MPS, the replica-molded PDMS was treated with an oxygen plasma (21 W, 24 s, 600 mTorr) and then permanently bonded to a glass slide. Fluidic connections were made via the media ports (Instech, SC20/15) and tubing (IDEX, 1527 and 1568; Saint Gobain, AAD04103) that was connected using a pressure-driven pump (Fluigent, Flow EZ 345 mbar unit with M-switch) to precisely control media flow rates.
iPSC Cell Lines
All experiments were performed on cells derived from two different healthy donor human iPSC lines: Wild Type C (WTC; #GM25256, Coriell Institute) edited to express the fluorescent calcium reporter GCaMP6f17 and the Stanford University Cardiovascular Biobank Line 20 (SCVI20). Cell handling and differentiation were performed similar to published protocols.18 We have provided an extensive characterization of the MPS created from both cell sources in a recent publication using experimental data supplemented with computational modeling to deduce quantitative information on ion channel activity and calcium handling.18Table S1 summarizes the main differences in ion channel activity between the two cell lines.
Human hiPSC-CM Differentiation (hiPSC-CMs) and Purification
Human iPSCs were thawed and cultured in mTeSR-1 media (Stem Cell Technologies, 85851) on Matrigel (Corning, 354277)-coated tissue culture polystyrene (TCPS) for several passages until differentiation was initiated. Cells were split using Accutase (Millipore, SCR005) and plated at a density necessary for the colonies to reach full confluency after 3 days prior to induction of the Wnt/β-catenin signaling pathway. Using 8 μM glycogen synthase kinase 3 small molecule inhibitor CHIR 99021 (Peprotech, 2520691-10MG) in RPMI 1640 media (Gibco, 11875-093) supplemented with 2% of B-27 Minus Insulin (B27-I, Gibco, A18956-01), hiPSCs were induced for a period of 24 h followed by a media exchange to only RPMI + B27-I without any small molecules for another 24 h. Next, the Wnt/β-catenin pathway was blocked using 5 μM inhibitor of WNT Production-4 (IWP-4, Peprotech, 6861787-10MG) for 48 h followed by another media exchange to only RPMI + B27-I without small molecules for another 48 h. After this step, the differentiation into hiPSC-derived cardiomyocytes (hiPSC-CMs) was completed with subsequent media exchanges every other day with an RPMI + 2% B27 supplement that contained insulin (B27 + C, Gibco, 17504-044) until the cells began to beat spontaneously. Cells were then harvested for purification using collagenase type 2 (Worthington Biochemical Corporation, LS004176) for 45–60 min in a 5% CO2 controlled, 37 °C incubator with humidity. Once the cells were singularized, they were gently collected in EB20 media (KnockOut DMEM (Gibco, 10829-018) with 20% FBS (Gibco, 16000-044), 1% MEM non-essential amino acids (Gibco, 11140-050), 1% Glutamax (Gibco, 35050-061), and 400 nM 2-mercaptoethanol (Gibco, 21985-023)) freshly supplemented with 10 μM ROCK-inhibitor (RI, Y-27632 dihydrochloride, Peprotech, 1293823-10MG) and centrifuged for 5 min. The pellet was resuspended in EB20 + RI, and cells were plated onto Matrigel-coated TCPS plates at 150,000 cells/cm2 for the first 24 h. Media were exchanged the following day to RPMI + B27 + C to allow the cells to recover for 48 h; then, to purify the population for cardiomyocytes, the cells were exposed to glucose-free RPMI 1640 media (Sigma, R1383-10X1L) supplemented with 4 mM sodium l-lactate (Sigma, L7022-10G) for no more than 5 days.47 Once purification was completed, cells were fed with RPMI + B27 + C until cells were harvested for use in the cardiac MPS. To confirm cardiomyocyte purity, a portion of the purified iPSC-CMs was harvested, washed, fixed, permeabilized, and stained for cardiac troponin T (cTnT). The lactate purified hiPSC-CMs were quantified via flow cytometry using the Attune NxT (Life Technologies) and were consistently >95% cTnT+ upon loading.
Human iPSC-Stromal Cell Differentiation (hiPSC-SC) and Purification
The same hiPSCs that were cultured and expanded for the cardiomyocyte differentiation were split into separate TCPS plates for endothelial cell differentiations as described previously.18,48 Once cells were confluent, after 3 days, they were treated with 8 μM CHIR 99021 in LaSR media (Advanced DMEM (Gibco, 12491-015), 2.5 mM Glutamax (Gibco, 35050-061), and 60 μg/mL l-ascorbic acid (Thermo Fisher Scientific, BP351-500)) for 48 h. Media were then changed to LaSR supplemented with 50 ng/mL Human VEGF 165 (Peprotech, 100-20-2UG) for two subsequent feedings lasting for 48 h each. After this step, cells were fed with endothelial cell growth media 2 (EGM2, PromoCell, C-22011) to complete the differentiation for 24 h. Cells were harvested no more than 48 h after completion of the differentiation and were singularized using 0.125% trypsin (Corning, 25-053-CI) for 5 min. Cells were collected, centrifuged, and washed with PBS in preparation for magnetic sorting. Using the anti-human CD31 primary antibody (eBioscience, 14-0319-82) 1:100 in PBS + 2% FBS, cells were incubated for 30 min on a rocker at room temperature to allow for binding. Afterward, cells were centrifuged and washed to remove unbound primaries and were then incubated with magnetic Dynabeads (Invitrogen, 11201D) 1:100 in PBS + 2% FBS for another 30 min at room temperature to allow for binding to the primary CD31 antibodies. CD31+ cells were then magnetically sorted out of the suspension, and the remaining CD31– cells were isolated. These stromal cells (hiPSC-SCs) have been characterized previously18 and were resuspended in EGM2 and plated in T75 flasks. hiPSC-SCs were split once and allowed to reach full confluency before being mixed with our pure iPSC-CMs at a ratio of 80% hiPSC-CM and 20% hiPSC-SC.
Cardiac MPS Loading and Maintenance
A cell mixture of 80% purified hiPSC-CM and 20% isogenic hiPSC-SC was prepared and loaded into the cardiac MPS. Both cell types were dissociated using trypsin (Gibco, 25200056) at 0.125% for no more than 5–8 min and resuspended in EB20 media (KnockOut DMEM (Gibco, 10829-01820%) with 20% FBS (Gibco, 16000-0), 1% MEM non-essential amino acids (Gibco, 11140-050), 1% Glutamax (Gibco, 35050-061), and 400 nM β-mercaptoethanol (Gibco, 21985023)) freshly supplemented with 10 μM RI. Cell viability was 83–90% for hiPSC-CM and 85–95% for hiPSC-SC. For each cell chamber, 3 μL of cell suspension containing 20,000 viable cells (∼6600 cells/μL) was injected into the cell loading port of the cardiac MPS using a pipette tip (Rainin, LTS ultrafine 20 μL). The tip was cut to approximately 1.5 cm length and remained in the chip during two subsequent centrifugation steps (first in the horizontal and then in the vertical position; each 300g for 3 min) that transferred the cells from the pipette tip into the cell chamber of the cardiac MPS (Figure 1A and Figure S8A). The pipette tip end was then sealed and after 1 h of recovery in the incubator, and additional pipette tips were inserted into the media inlet and outlet. Media (100 μL, EB20 + RI) were added to the media inlet tip and gently aspirated through the outlet tip to remove air bubbles. After this priming step, the media perfused gravimetrically from the inlet to the outlet tip. After 24 h, the media were replaced with RI-free media. A quality control step was performed, and any failed samples (such as incomplete loading or air bubbles in the device) were discarded (10–30%). After the onset of beating (typically 24–48 h after loading), the media were changed to fatty acid rich maturation media (MM) to metabolically promote cardiomyocyte maturation.18 The MM base media were prepared from RPMI 1640 (Sigma, R1383-10X1L) and supplemented with 0.5 g/L d-glucose (Thermo Fisher Scientific, BP350-1), 10 mM d-galactose (Sigma, G5388), and 2 g/L sodium bicarbonate (Thermo Fisher Scientific, S233-500). A fatty-acid cocktail was prepared from MM base with 9% bovine serum albumin (BSA; Thermo Fisher Scientific, BP1605-100), 10.23 μg/mL palmitic acid (Sigma, P0500-10G), and 0.8 mM oleic acid (Sigma, O1383-5G). For the final MM, 1 part FA solution was mixed with 3 parts MM base media yielding 2.5575 μg/mL palmitic acid and 0.2 mM oleic acid and supplemented with 2% B27 (Gibco, 17504-044) and 150 μg/mL ascorbic acid (Thermo Fisher Scientific, AC105021000). Fresh MM were applied to the MPS three times per week. The MPS subjected to at least 10 days of metabolic maturation in MM were used for the drug experiments. Only MPS with completely filled cell chambers featuring spontaneously and uniaxial beating tissues with 0.6–1.4 Hz baseline beat rate were included in the drug evaluation experiments (65–90% of tissues). Typically, beat rates were relatively consistent and the majority of tissue failures were attributed to morphological defects such as incomplete fiber formation.
Tissue Staining
Tissues were stained to allow for fluorescence-based recording of changes in the action potential and calcium transients. A calcium dye (Oregon Green) was only used on the SCVI20 cell line since WTC cells have a genetically encoded calcium reporter (GCaMP). The voltage sensitive dye BeRST-1 was obtained from Professor Evan Miller’s lab.49 Stocks of 1 mM BeRST-1 in DMSO were stored at room temperature in the dark for up to 3 months. A 500 nM staining solution in MM was prepared fresh, and MPS were incubated with the dye overnight. For the calcium-sensitive dye Oregon Green 488 BAPTA-1 AM (OGB-1-AM; Thermo Fisher Scientific, O6807), a 4 mM stock was prepared in DMSO and stored at −20 °C for up to 1 month. For staining, OGB-1-AM was diluted to 2 μM in MM and MPS were incubated for 2 h. Both dyes were washed out as the first drug dose was applied.
Drug Preparation
Drug doses were prepared in a concentration range starting around the maximum clinical free plasma concentration (Cmax) up to about 1000× Cmax at 10-fold increments. Stock solutions prepared in cell culture media were sterile-filtered using 0.2 μm pore size. Table S3 details the drug preparation, and Table S4 lists the doses applied for each compound. Drug doses were always prepared fresh on the day of the experiment by performing a serial dilution from the highest to lowest dose in MM. All drug doses including dose 0 were adjusted to the same solvent concentration (Table S4). To maintain a stable fluorescence signal intensity over the entire experiment, voltage and calcium dyes were spiked into all drug doses at 10% of the staining concentration: 50 nM BeRST-1 and 200 nM OGB-1-AM. OGB-1-AM was only added to SCVI20 cells. Before use, the final drug solutions were equilibrated to a 5% CO2 atmosphere until pH 7.5 was reached.
Drug Exposure
The MPS were subjected to increasing drug doses in a “dose escalation” format (Figure S8C): A fresh drug dose was applied and incubated for 30 min to allow for drug effect manifestation before the tissues were imaged. The first dose was always the drug-free control media followed by the doses outlined in Table S4. Drug application was either performed by manually changing the media or using a pressure-driven pump. In the manual scenario, the pipette tips in the media inlet and outlet were used as reservoirs and perfusion occurred gravimetrically as the levels on the inlet and outlet side equilibrated. For a media change, the media were aspirated from the outlet and inlet tip. Without introducing air bubbles, 200 μL of the next drug solution was added to the inlet tip. A similar drug exchange pattern was achieved by using a pressure-driven pump (Fluigent with M-switch). All vials containing increasing concentrations of a particular drug were connected to the pump at the beginning of the experiment, and the system was primed to get rid of any air bubbles. Each drug dose was perfused at 15 μL/min for the first 10 min to ensure that the drug reaches the tissue in a timely manner (2.3 min for 35 μL of tubing dead volume). To minimize fluid mechanical stress, a flow rate of 5 μL/min was applied for the remaining incubation time as well as during acquisition. The pump was programmed to automatically switch to the next dose. Occasionally, two devices were connected to the pump outlet in parallel using a flow splitter (IDEX, P-512). In that case, needle valves (IDEX, P-445NF) were used to ensure equal flow into both sides and volume flow rates were adjusted accordingly. Replicates for each drug acquisition were obtained from independent tissues derived from one or two independent differentiations.
Data Acquisition
All images and videos were obtained on a Nikon TE-300 inverted microscope base equipped with a motorized stage, z-focus, an LED light engine (Lumencor SpectraX), and a digital CMOS camera (HAMAMATSU, C11440/ORCA-Flash 4.0). A heated platform (Tokai Hit, TPi-SQX) was used to keep the cardiac microtissue and media in the MPS at 37 °C. A pulse generator (ION OPTIX Myopacer Field Simulator) was used to generate pacing pulses (20 ms, biphasic). All hardware was controlled via the JOBS module of Nikon NIS Elements software (version 5.02.00). For each drug dose and tissue, one or several of the following acquisitions were performed: brightfield videos (40 fps, no binning, 5–8 s), BeRST-1 voltage videos (red LED 100% power, 100 fps, 4 × 4 binning, 5–8 s), and GCaMP or OGB-1-AM calcium videos (cyan LED 100% power, 100 fps, 4 × 4 binning, 5–8 s). After the spontaneous beating activity had been recorded for all MPS, the samples were electrically stimulated at 1 Hz (20 V for manual drug applications via 1.5” long blunt stainless-steel needles (OD 0.032, “ID 0.023”; McMaster-Carr, 75165A757 (Vita Needle M937)) inserted into the media inlet and outlet pipette tips; 3 V for pump perfusion via hollow stainless-steel couplers (Instech, SC20/15) bent to 90° and directly inserted into the PDMS inlet and outlet punch holes) for at least 15 s before videos of the paced activity were recorded.
Data Analysis
An in-house library of python scripts was developed and used to process voltage and calcium recordings. Fluorescence intensity profiles were derived from the videos followed by background drift correction and extraction of individual beats. Examples of time-intensity traces for each drug are provided in Figures S10–S12. All peaks from each video were then overlaid, and any peaks with properties deviating more than one standard deviation from the average were excluded. Figure S8D shows an example of the relative time-stamped final AP and calcium waveforms extracted from videos of paced tissue.
Characteristic parameters such as beat rate and beat duration (at 80 and 30% of the total amplitude) were calculated. Action potential duration values (APD80 and APD30) were derived from the red voltage traces (Figure 1C). Similarly, duration of the calcium transient (CaD80 and CaD30) was derived from the cyan calcium traces (Figure S8D). However, this data was largely omitted for further analysis as we focused on the action potential traces. To make data comparable, all values were either obtained from tissues paced at 1 Hz or the beat duration was scaled to 1 Hz using the Fridericia method:50 cAPD80 = APD80/BR–1/3 with BR being the beat rate in Hz and cAPD80 being the action potential duration at 80% peak height corrected to a beat rate of 1 Hz. The change in cAPD upon drug exposure (ΔcAPD80) was calculated as the difference in cAPD80 at a certain drug dose compared to cAPD80 of the same tissue at drug-free baseline. It is worth noting that, while many other studies look at APD90 values to determine the peak width, we chose APD80 since this parameter was more robust in our dataset and could be reliably measured using our automated scripts even in the presence of background drift or low signal amplitude.
While elongation of the action potential duration is an important predictor for proarrhythmic drug effects, it is not sufficient to predict drug-induced arrythmia.51 Therefore, we used triangulation, a parameter describing changes in the beat waveform shape, as introduced by Hondeghem et al.12 We computed triangulation as (APD80 – APD30)/APD80 (Figure 1C). This formula calculates the difference in peak width at the base (APD80) compared to the top (APD30). Since the parameter is normalized to the base peak width, a beat rate correction is not necessary. The triangulation metric quantifies changes in the later phase of the repolarization trajectory independent of the absolute APD change. Lower triangulation values indicate a more rectangular beat shape, while higher values indicate stronger triangulation, which is associated with increased proarrhythmic potential.
Arrhythmia-like events were categorized in a non-parametric fashion, as outlined in Figure 1D. Each trace was manually classified as normal, displaying any sort of arrhythmia-like event, very weak (beats were noticeable but too weak and noisy for any more detailed analysis), or non-beating. For every drug dose, the number of MPS falling into each category was counted and plotted as % of total MPS. Both calcium and voltage traces were considered for this analysis.
Data was preprocessed in Microsoft Excel for Mac (version 16.16.27). All graphs were plotted in GraphPad Prism version 8.4.1 for macOS. Figures were assembled using Adobe Illustrator (version 25.0.1).
Statistical analysis to compare dose effects was performed by an initial mixed-effects analysis with the Geisser–Greenhouse correction for paired samples with missing values, which uses the maximum likelihood method. This analysis was followed by Dunnett’s multiple comparison test to dose 0, with individual variances computed for each comparison. Effects were reported for the significance levels of p < 0.05 (*), 0.01 (**), 0.001 (***), and 0.0001 (****).
Channel Block Percentages Based on Literature Values
We computed ion channel block percentages for specific drug doses using the following formula:
| 1 |
where D is the drug dose, p(D) is the corresponding ion channel block percentage, pmax is the maximal block percentage, IC50 is the half maximal inhibitory concentration, and H is the Hill coefficient. Table S5 provides an overview of the main ion channel targets for each drug. Table S6 reports theoretical block percentages for the specified drugs and doses. Drug characteristics (pmax, IC50, and H) were obtained by Kramer et al.(52) and used to compute the block percentages in Table S6. Kramer et al.(52) provide data of the drug effects on the hERG (IKr) current, the Cav1.2 (ICaL) current, and the Nav1.5 (INa) current. In Table S6, we have assumed that the block percentage for Nav1.5 applies to both the fast and the late component of the sodium current (INa and INaL). For the drugs not considered in the study by Kramer et al. (mexiletine and lidocaine), we have used IC50 and H values from Crumb et al. and Mirams et al.(53) and assumed that pmax = 100%. Note that, in some cases, there are quite large variations in the drug characteristics reported in the literature. To illustrate this, Table S7 compares various drug IC50 values from the literature.
Acknowledgments
We thank Bruce Conklin, Ph.D. (Gladstone Institutes, San Francisco, USA) and Joseph C. Wu, MD, Ph.D. (Stanford Cardiovascular Institute funded by NIH 75N92020D00019) for providing the WTC and SCVI20 iPSC lines, respectively, and for providing technical advice regarding each cell line. We thank Tafadzwa Amani, Nicholas C. Jeffreys, Nicole Lee, Gabriel Neimann, and Elizabeth Gill (Department of Bioengineering and California Institute for Quantitative Biosciences (QB3), University of California at Berkeley, USA) for their help with data processing.
Glossary
Abbreviations
- APD
action potential duration
- cAPD
beat rate corrected APD
- Cmax
maximum clinical free plasma concentration
- DMSO
dimeythyl sulfoxide
- ETPC
estimated therapeutic plasma concentration
- hiPSC
human-induced pluripotent stem cell
- IC50
half maximum inhibitory concentration
- MPS
microphysiological system
- n.s.
non-significant
- PDMS
polydimethylsiloxane
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.2c00088.
Table S1: Differences in ion channel activity and other elements of the cellular electrophysiological machinery in WTC vs SCVI20 determined by mathematical modeling; Table S2: inter- and intra-batch variation of key electrophysiological parameters in the WTC and SCVI20 cell line; Table S3: drug properties; Table S4: drug dose preparation; Table S5: ion channel targets; Table S6: block percentages for drug doses based on drug characteristics from the literature; Table S7: IC50 values reported in different studies; Figure S8: cardiac MPS and experimental plan; Figure S9: baseline characterization of WTC and SCVI20 MPS; Figure S10: representative voltage traces for high-risk drugs; Figure S11: representative voltage traces for intermediate-risk drugs; Figure S12: representative voltage traces for low-risk drugs (PDF)
Author Contributions
⊥ V.C. and B.C. contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. V.C. and B.C. conducted the main experiments; B.A.S. prepared the cells and helped with MPS loading and MPS maintenance; N.H. helped with the drug testing experiments; H.F. and S.W. developed the code to analyze action potential and calcium waveforms; A.T. and K.H.J. provided the percentage channel block values; A.G.E., S.W., and N.H. helped with the interpretation of the electrophysiological data; E.M. provided BeRST-1 and staining protocols; V.C., B.C., and B.A.S. analyzed the data. V.C. prepared the figures. V.C., B.C., and K.E.H. wrote the manuscript. All authors participated in the study design and review of the manuscript; K.E.H. funded the work.
This work was funded by the California Institute for Regenerative Medicine (DISC2/10090 and DISC2P/10090), the National Heart, Lung, and Blood Institute of the National Institutes of Health (NIH-NHLBI/HL130417), and the Research Council of Norway (309871/E50).
The authors declare the following competing financial interest(s): KEH, VC, BAS, HF, KHJ, AGE, NH, SW and AT have a financial relationship with Organos Inc., and they and the company may benefit from commercialization of the results of this research.
Supplementary Material
References
- Craveiro N. S.; Lopes B. S.; Tomás L.; Almeida S. F. Drug Withdrawal Due to Safety: A Review of the Data Supporting Withdrawal Decision. Curr. Drug Saf. 2020, 15, 4–12. 10.2174/1574886314666191004092520. [DOI] [PubMed] [Google Scholar]
- Stockbridge N.; Morganroth J.; Shah R. R.; Garnett C. Dealing with global safety issues : was the response to QT-liability of non-cardiac drugs well coordinated?. Drug. Saf. 2013, 36, 167–182. 10.1007/s40264-013-0016-z. [DOI] [PubMed] [Google Scholar]
- a Ahmad K.; Dorian P. Drug-induced QT prolongation and proarrhythmia: an inevitable link?. Europace 2007, 9 Suppl 4, iv16-22. 10.1093/europace/eum167. [DOI] [PubMed] [Google Scholar]; b Antoniou C. K.; Dilaveris P.; Manolakou P.; Galanakos S.; Magkas N.; Gatzoulis K.; Tousoulis D. QT Prolongation and Malignant Arrhythmia: How Serious a Problem?. Eur Cardiol. 2017, 12, 112–120. 10.15420/ecr.2017:16:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondeghem L. M. QT prolongation is an unreliable predictor of ventricular arrhythmia. Heart Rhythm. 2008, 5, 1210–1212. 10.1016/j.hrthm.2008.05.006. [DOI] [PubMed] [Google Scholar]
- Blinova K.; Stohlman J.; Vicente J.; Chan D.; Johannesen L.; Hortigon-Vinagre M. P.; Zamora V.; Smith G.; Crumb W. J.; Pang L.; Lyn-Cook B.; Ross J.; Brock M.; Chvatal S.; Millard D.; Galeotti L.; Stockbridge N.; Strauss D. G. Comprehensive Translational Assessment of Human-Induced Pluripotent Stem Cell Derived Cardiomyocytes for Evaluating Drug-Induced Arrhythmias. Toxicol. Sci. 2017, 155, 234–247. 10.1093/toxsci/kfw200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blinova K.; Dang Q.; Millard D.; Smith G.; Pierson J.; Guo L.; Brock M.; Lu H. R.; Kraushaar U.; Zeng H.; Shi H.; Zhang X.; Sawada K.; Osada T.; Kanda Y.; Sekino Y.; Pang L.; Feaster T. K.; Kettenhofen R.; Stockbridge N.; Strauss D. G.; Gintant G. International Multisite Study of Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes for Drug Proarrhythmic Potential Assessment. Cell Rep. 2018, 24, 3582–3592. 10.1016/j.celrep.2018.08.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Johannesen L.; Vicente J.; Mason J. W.; Erato C.; Sanabria C.; Waite-Labott K.; Hong M.; Lin J.; Guo P.; Mutlib A.; Wang J.; Crumb W. J.; Blinova K.; Chan D.; Stohlman J.; Florian J.; Ugander M.; Stockbridge N.; Strauss D. G. Late sodium current block for drug-induced long QT syndrome: Results from a prospective clinical trial. Clin. Pharmacol. Ther. 2016, 99, 214–223. 10.1002/cpt.205. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Clements M.; Thomas N. High-throughput multi-parameter profiling of electrophysiological drug effects in human embryonic stem cell derived cardiomyocytes using multi-electrode arrays. Toxicol. Sci. 2014, 140, 445–461. 10.1093/toxsci/kfu084. [DOI] [PubMed] [Google Scholar]
- a Yamamoto W.; Asakura K.; Ando H.; Taniguchi T.; Ojima A.; Uda T.; Osada T.; Hayashi S.; Kasai C.; Miyamoto N.; Tashibu H.; Yoshinaga T.; Yamazaki D.; Sugiyama A.; Kanda Y.; Sawada K.; Sekino Y. Electrophysiological Characteristics of Human iPSC-Derived Cardiomyocytes for the Assessment of Drug-Induced Proarrhythmic Potential. PLoS One 2016, 11, e0167348 10.1371/journal.pone.0167348. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bot C. T.; Juhasz K.; Haeusermann F.; Polonchuk L.; Traebert M.; Stoelzle-Feix S. Cross - site comparison of excitation-contraction coupling using impedance and field potential recordings in hiPSC cardiomyocytes. J. Pharmacol. Toxicol. Methods 2018, 93, 46–58. 10.1016/j.vascn.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrez B.; Charwat V.; Siemons B. A.; Goswami I.; Sakolish C.; Luo Y. S.; Finsberg H.; Edwards A. G.; Miller E. W.; Rusyn I.; Healy K. E. Heart Muscle Microphysiological System for Cardiac Liability Prediction of Repurposed COVID-19 Therapeutics. Front Pharmacol. 2021, 12, 684252 10.3389/fphar.2021.684252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrez B.; Charwat V.; Siemons B.; Finsberg H.; Miller E. W.; Edwards A. G.; Healy K. E. In vitro safety ″clinical trial″ of the cardiac liability of drug polytherapy. Clin. Transl. Sci. 2021, 14, 1155–1165. 10.1111/cts.13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fermini B.; Hancox J. C.; Abi-Gerges N.; Bridgland-Taylor M.; Chaudhary K. W.; Colatsky T.; Correll K.; Crumb W.; Damiano B.; Erdemli G.; Gintant G.; Imredy J.; Koerner J.; Kramer J.; Levesque P.; Li Z.; Lindqvist A.; Obejero-Paz C. A.; Rampe D.; Sawada K.; Strauss D. G.; Vandenberg J. I. A New Perspective in the Field of Cardiac Safety Testing through the Comprehensive In Vitro Proarrhythmia Assay Paradigm. J. Biomol. Screen. 2016, 21, 1–11. 10.1177/1087057115594589. [DOI] [PubMed] [Google Scholar]
- Hondeghem L. M.; Carlsson L.; Duker G. Instability and Triangulation of the Action Potential Predict Serious Proarrhythmia, but Action Potential Duration Prolongation Is Antiarrhythmic. Circulation 2001, 103, 2004–2013. 10.1161/01.cir.103.15.2004. [DOI] [PubMed] [Google Scholar]
- Aronson J. K.Meyler’s side effects of drugs: the international encyclopedia of adverse drug reactions and interactions. Elsevier 2016, 10.1016/B978-0-444-53717-1.09974-1. [DOI]
- Singh B. N. Safety profile of bepridil determined from clinical trials in chronic stable angina in the United States. Am. J. Cardiol. 1992, 69, 68D–74D. 10.1016/0002-9149(92)90962-x. [DOI] [PubMed] [Google Scholar]
- Coumel P. Safety of bepridil: from review of the European data. Am. J. Cardiol. 1992, 69, 75D–78D. 10.1016/0002-9149(92)90963-y. [DOI] [PubMed] [Google Scholar]
- Kodama I.; Kamiya K.; Toyama J. Cellular electropharmacology of amiodarone. Cardiovasc. Res. 1997, 35, 13–29. 10.1016/s0008-6363(97)00114-4. [DOI] [PubMed] [Google Scholar]
- Huebsch N.; Loskill P.; Mandegar M. A.; Marks N. C.; Sheehan A. S.; Ma Z.; Mathur A.; Nguyen T. N.; Yoo J. C.; Judge L. M.; Spencer C. I.; Chukka A. C.; Russell C. R.; So P. L.; Conklin B. R.; Healy K. E. Automated Video-Based Analysis of Contractility and Calcium Flux in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes Cultured over Different Spatial Scales. Tissue Eng., Part C 2015, 21, 467–479. 10.1089/ten.TEC.2014.0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebsch N.; Charrez B.; Neiman G.; Siemons B.; Boggess S. C.; Wall S.; Charwat V.; Jæger K. H.; Cleres D.; Telle Å.; Lee-Montiel F. T.; Jeffreys N. C.; Deveshwar N.; Edwards A. G.; Serrano J.; Snuderl M.; Stahl A.; Tveito A.; Miller E. W.; Healy K. E. Metabolically driven maturation of human-induced-pluripotent-stem-cell-derived cardiac microtissues on microfluidic chips. Nat. Biomed. Eng. 2022, 6, 372–388. 10.1038/s41551-022-00884-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur A.; Loskill P.; Shao K.; Huebsch N.; Hong S.; Marcus S. G.; Marks N.; Mandegar M.; Conklin B. R.; Lee L. P.; Healy K. E. Human iPSC-based cardiac microphysiological system for drug screening applications. Sci. Rep. 2015, 5, 8883. 10.1038/srep08883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redfern W. S.; Carlsson L.; Davis A. S.; Lynch W. G.; MacKenzie I.; Palethorpe S.; Siegl P. K.; Strang I.; Sullivan A. T.; Wallis R.; Camm A. J.; Hammond T. G. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc. Res. 2003, 58, 32–45. 10.1016/s0008-6363(02)00846-5. [DOI] [PubMed] [Google Scholar]
- a Vunjak Novakovic G.; Eschenhagen T.; Mummery C. Myocardial tissue engineering: in vitro models. Cold Spring Harbor Perspect. Med. 2014, 4, a014076. 10.1101/cshperspect.a014076. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ogle B. M.; Bursac N.; Domian I.; Huang N. F.; Menasché P.; Murry C. E.; Pruitt B.; Radisic M.; Wu J. C.; Wu S. M.; Zhang J.; Zimmermann W. H.; Vunjak-Novakovic G. Distilling complexity to advance cardiac tissue engineering. Sci. Transl. Med. 2016, 8, 342ps313. 10.1126/scitranslmed.aad2304. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Laflamme M. A.; Murry C. E. Heart regeneration. Nature 2011, 473, 326–335. 10.1038/nature10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham J. M.; Saliba W. I.; Vekstein C.; Lawrence D.; Bhargava M.; Bassiouny M.; Janiszewski D.; Lindsay B.; Militello M.; Nissen S. E.; Poe S.; Tanaka-Esposito C.; Wolski K.; Wilkoff B. L. Safety of oral dofetilide for rhythm control of atrial fibrillation and atrial flutter. Circ.: Arrhythmia Electrophysiol. 2015, 8, 772–776. 10.1161/CIRCEP.114.002339. [DOI] [PubMed] [Google Scholar]
- Prystowsky E. N. Effects of bepridil on cardiac electrophysiologic properties. Am. J. Cardiol. 1992, 69, 63D–67D. 10.1016/0002-9149(92)90961-w. [DOI] [PubMed] [Google Scholar]
- Egami Y.; Nishino M.; Taniike M.; Makino N.; Kato H.; Shutta R.; Tanouchi J.; Yamada Y. Efficacy and Safety of Bepridil for Patients with Persistent Atrial Fibrillation after Failed Electrical Cardioversion. J. Arrhythmia 2011, 27, 131–136. 10.1016/S1880-4276(11)80021-9. [DOI] [Google Scholar]
- a June R. A.; Nasr I. Torsades de pointes with terfenadine ingestion. Am. J. Emerg. Med. 1997, 15, 542–543. 10.1016/s0735-6757(97)90206-0. [DOI] [PubMed] [Google Scholar]; b Yap Y. G.; Camm A. J. Arrhythmogenic mechanisms of non-sedating antihistamines. Clin. Exp. Allergy 1999, 29, 174–181. 10.1046/j.1365-2222.1999.0290s3174.x. [DOI] [PubMed] [Google Scholar]
- Pawankar R.; Holgate S. T.; Rosenwasser L. J.. Allergy Frontiers: Therapy and Prevention; Springer, 2010, 10.1007/978-4-431-99362-9. [DOI] [Google Scholar]
- a Bos J. M.; Crotti L.; Rohatgi R. K.; Castelletti S.; Dagradi F.; Schwartz P. J.; Ackerman M. J. Mexiletine Shortens the QT Interval in Patients With Potassium Channel-Mediated Type 2 Long QT Syndrome. Circ.: Arrhythmia Electrophysiol. 2019, 12, e007280 10.1161/CIRCEP.118.007280. [DOI] [PubMed] [Google Scholar]; b Mazzanti A.; Maragna R.; Faragli A.; Monteforte N.; Bloise R.; Memmi M.; Novelli V.; Baiardi P.; Bagnardi V.; Etheridge S. P.; Napolitano C.; Priori S. G. Gene-Specific Therapy With Mexiletine Reduces Arrhythmic Events in Patients With Long QT Syndrome Type 3. J. Am. Coll Cardiol. 2016, 67, 1053–1058. 10.1016/j.jacc.2015.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sicouri S.; Antzelevitch D.; Heilmann C.; Antzelevitch C. Effects of sodium channel block with mexiletine to reverse action potential prolongation in in vitro models of the long term QT syndrome. J Cardiovasc. Electrophysiol. 1997, 8, 1280–1290. 10.1111/j.1540-8167.1997.tb01019.x. [DOI] [PubMed] [Google Scholar]
- Hwang H. S.; Kryshtal D. O.; Feaster T. K.; Sánchez-Freire V.; Zhang J.; Kamp T. J.; Hong C. C.; Wu J. C.; Knollmann B. C. Comparable calcium handling of human iPSC-derived cardiomyocytes generated by multiple laboratories. J. Mol. Cell. Cardiol. 2015, 85, 79–88. 10.1016/j.yjmcc.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hortigon-Vinagre M. P.; Zamora V.; Burton F. L.; Green J.; Gintant G. A.; Smith G. L. The Use of Ratiometric Fluorescence Measurements of the Voltage Sensitive Dye Di-4-ANEPPS to Examine Action Potential Characteristics and Drug Effects on Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Toxicol. Sci. 2016, 154, 320–331. 10.1093/toxsci/kfw171. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Huo J.; Kamalakar A.; Yang X.; Word B.; Stockbridge N.; Lyn-Cook B.; Pang L. Evaluation of Batch Variations in Induced Pluripotent Stem Cell-Derived Human Cardiomyocytes from 2 Major Suppliers. Toxicol. Sci. 2017, 156, 25–38. 10.1093/toxsci/kfw235. [DOI] [PubMed] [Google Scholar]; c Liang P.; Lan F.; Lee A. S.; Gong T.; Sanchez-Freire V.; Wang Y.; Diecke S.; Sallam K.; Knowles J. W.; Wang P. J.; Nguyen P. K.; Bers D. M.; Robbins R. C.; Wu J. C. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation 2013, 127, 1677–1691. 10.1161/CIRCULATIONAHA.113.001883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannhardt I.; Saleem U.; Mosqueira D.; Loos M. F.; Ulmer B. M.; Lemoine M. D.; Larsson C.; Améen C.; de Korte T.; Vlaming M. L. H.; Harris K.; Clements P.; Denning C.; Hansen A.; Eschenhagen T. Comparison of 10 Control hPSC Lines for Drug Screening in an Engineered Heart Tissue Format. Stem Cell Rep. 2020, 15, 983–998. 10.1016/j.stemcr.2020.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Rafatian N.; Feric N. T.; Cox B. J.; Aschar-Sobbi R.; Wang E. Y.; Aggarwal P.; Zhang B.; Conant G.; Ronaldson-Bouchard K.; Pahnke A.; Protze S.; Lee J. H.; Davenport Huyer L.; Jekic D.; Wickeler A.; Naguib H. E.; Keller G. M.; Vunjak-Novakovic G.; Broeckel U.; Backx P. H.; Radisic M. A Platform for Generation of Chamber-Specific Cardiac Tissues and Disease Modeling. Cell 2019, 176, 913–927.e918. 10.1016/j.cell.2018.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfracht I.; Efraim Y.; Shinnawi R.; Kovalev E.; Huber I.; Gepstein A.; Arbel G.; Shaheen N.; Tiburcy M.; Zimmermann W. H.; Machluf M.; Gepstein L. Engineered heart tissue models from hiPSC-derived cardiomyocytes and cardiac ECM for disease modeling and drug testing applications. Acta Biomater. 2019, 92, 145–159. 10.1016/j.actbio.2019.05.016. [DOI] [PubMed] [Google Scholar]
- Shum A. M.; Che H.; Wong A. O.; Zhang C.; Wu H.; Chan C. W.; Costa K.; Khine M.; Kong C. W.; Li R. A. A Micropatterned Human Pluripotent Stem Cell-Based Ventricular Cardiac Anisotropic Sheet for Visualizing Drug-Induced Arrhythmogenicity. Adv. Mater. 2017, 29, 1602448. 10.1002/adma.201602448. [DOI] [PubMed] [Google Scholar]
- Goldfracht I.; Protze S.; Shiti A.; Setter N.; Gruber A.; Shaheen N.; Nartiss Y.; Keller G.; Gepstein L. Generating ring-shaped engineered heart tissues from ventricular and atrial human pluripotent stem cell-derived cardiomyocytes. Nat. Commun. 2020, 11, 75. 10.1038/s41467-019-13868-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremner S. B.; Gaffney K. S.; Sniadecki N. J.; Mack D. L. A Change of Heart: Human Cardiac Tissue Engineering as a Platform for Drug Development. Curr. Cardiol. Rep. 2022, 24, 473–486. 10.1007/s11886-022-01668-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R. A.; Keung W.; Cashman T. J.; Backeris P. C.; Johnson B. V.; Bardot E. S.; Wong A. O. T.; Chan P. K. W.; Chan C. W. Y.; Costa K. D. Bioengineering an electro-mechanically functional miniature ventricular heart chamber from human pluripotent stem cells. Biomaterials 2018, 163, 116–127. 10.1016/j.biomaterials.2018.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Saleem U.; van Meer B. J.; Katili P. A.; Mohd Yusof N. A. N.; Mannhardt I.; Garcia A. K.; Tertoolen L.; de Korte T.; Vlaming M. L. H.; McGlynn K.; Nebel J.; Bahinski A.; Harris K.; Rossman E.; Xu X.; Burton F. L.; Smith G. L.; Clements P.; Mummery C. L.; Eschenhagen T.; Hansen A.; Denning C. Blinded, Multicenter Evaluation of Drug-induced Changes in Contractility Using Human-induced Pluripotent Stem Cell-derived Cardiomyocytes. Toxicol. Sci. 2020, 176, 103–123. 10.1093/toxsci/kfaa058. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lu H. F.; Leong M. F.; Lim T. C.; Chua Y. P.; Lim J. K.; Du C.; Wan A. C. A. Engineering a functional three-dimensional human cardiac tissue model for drug toxicity screening. Biofabrication 2017, 9, 025011 10.1088/1758-5090/aa6c3a. [DOI] [PubMed] [Google Scholar]
- a Kofron C. M.; Kim T. Y.; Munarin F.; Soepriatna A. H.; Kant R. J.; Mende U.; Choi B. R.; Coulombe K. L. K. A predictive in vitro risk assessment platform for pro-arrhythmic toxicity using human 3D cardiac microtissues. Sci. Rep. 2021, 11, 10228. 10.1038/s41598-021-89478-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lemme M.; Ulmer B. M.; Lemoine M. D.; Zech A. T. L.; Flenner F.; Ravens U.; Reichenspurner H.; Rol-Garcia M.; Smith G.; Hansen A.; Christ T.; Eschenhagen T. Atrial-like Engineered Heart Tissue: An In Vitro Model of the Human Atrium. Stem Cell Rep. 2018, 11, 1378–1390. 10.1016/j.stemcr.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer C. R.; Sargeant R.; Basak J.; Pilling J.; Barnes J. R.; Pointon A. Characterization and Validation of a Human 3D Cardiac Microtissue for the Assessment of Changes in Cardiac Pathology. Sci. Rep. 2018, 8, 10160. 10.1038/s41598-018-28393-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Thavandiran N.; Hale C.; Blit P.; Sandberg M. L.; McElvain M. E.; Gagliardi M.; Sun B.; Witty A.; Graham G.; Do V. T. H.; Bakooshli M. A.; Le H.; Ostblom J.; McEwen S.; Chau E.; Prowse A.; Fernandes I.; Norman A.; Gilbert P. M.; Keller G.; Tagari P.; Xu H.; Radisic M.; Zandstra P. W. Functional arrays of human pluripotent stem cell-derived cardiac microtissues. Sci. Rep. 2020, 10, 6919. 10.1038/s41598-020-62955-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Feric N. T.; Pallotta I.; Singh R.; Bogdanowicz D. R.; Gustilo M. M.; Chaudhary K. W.; Willette R. N.; Chendrimada T. P.; Xu X.; Graziano M. P. Engineered cardiac tissues generated in the biowire II: a platform for human-based drug discovery. Toxicol. Sci. 2019, 172, 89–97. 10.1093/toxsci/kfz168. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Mannhardt I.; Eder A.; Dumotier B.; Prondzynski M.; Krämer E.; Traebert M.; Söhren K. D.; Flenner F.; Stathopoulou K.; Lemoine M. D.; Carrier L.; Christ T.; Eschenhagen T.; Hansen A. Blinded Contractility Analysis in hiPSC-Cardiomyocytes in Engineered Heart Tissue Format: Comparison With Human Atrial Trabeculae. Toxicol. Sci. 2017, 158, 164–175. 10.1093/toxsci/kfx081. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Keung W.; Chan P. K. W.; Backeris P. C.; Lee E. K.; Wong N.; Wong A. O. T.; Wong G. K. Y.; Chan C. W. Y.; Fermini B.; Costa K. D.; Li R. A. Human Cardiac Ventricular-Like Organoid Chambers and Tissue Strips From Pluripotent Stem Cells as a Two-Tiered Assay for Inotropic Responses. Clin. Pharmacol. Ther. 2019, 106, 402–414. 10.1002/cpt.1385. [DOI] [PubMed] [Google Scholar]
- a Mills R. J.; Parker B. L.; Quaife-Ryan G. A.; Voges H. K.; Needham E. J.; Bornot A.; Ding M.; Andersson H.; Polla M.; Elliott D. A.; Drowley L.; Clausen M.; Plowright A. T.; Barrett I. P.; Wang Q. D.; James D. E.; Porrello E. R.; Hudson J. E. Drug Screening in Human PSC-Cardiac Organoids Identifies Pro-proliferative Compounds Acting via the Mevalonate Pathway. Cell Stem Cell 2019, 24, 895–907.e896. 10.1016/j.stem.2019.03.009. [DOI] [PubMed] [Google Scholar]; b Arai K.; Murata D.; Takao S.; Nakamura A.; Itoh M.; Kitsuka T.; Nakayama K. Drug response analysis for scaffold-free cardiac constructs fabricated using bio-3D printer. Sci. Rep. 2020, 10, 8972. 10.1038/s41598-020-65681-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Tadano K.; Miyagawa S.; Takeda M.; Tsukamoto Y.; Kazusa K.; Takamatsu K.; Akashi M.; Sawa Y. Cardiotoxicity assessment using 3D vascularized cardiac tissue consisting of human iPSC-derived cardiomyocytes and fibroblasts. Mol. Ther. Methods Clin. Dev. 2021, 22, 338–349. 10.1016/j.omtm.2021.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jæger K. H.; Charwat V.; Charrez B.; Finsberg H.; Maleckar M. M.; Wall S.; Healy K. E.; Tveito A. Improved Computational Identification of Drug Response Using Optical Measurements of Human Stem Cell Derived Cardiomyocytes in Microphysiological Systems. Front Pharmacol 2020, 10, 1648. 10.3389/fphar.2019.01648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tveito A.; Jaeger K. H.; Huebsch N.; Charrez B.; Edwards A. G.; Wall S.; Healy K. E. Inversion and computational maturation of drug response using human stem cell derived cardiomyocytes in microphysiological systems. Sci. Rep. 2018, 8, 17626. 10.1038/s41598-018-35858-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loskill P.; Marcus S. G.; Mathur A.; Reese W. M.; Healy K. E. μOrgano: A Lego®-Like Plug & Play System for Modular Multi-Organ-Chips. PLoS One 2015, 10, e0139587 10.1371/journal.pone.0139587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Montiel F. T.; Laemmle A.; Charwat V.; Dumont L.; Lee C. S.; Huebsch N.; Okochi H.; Hancock M. J.; Siemons B.; Boggess S. C.; Goswami I.; Miller E. W.; Willenbring H.; Healy K. E. Integrated Isogenic Human Induced Pluripotent Stem Cell-Based Liver and Heart Microphysiological Systems Predict Unsafe Drug-Drug Interaction. Front Pharmacol. 2021, 12, 667010 10.3389/fphar.2021.667010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feyen D. A. M.; McKeithan W. L.; Bruyneel A. A. N.; Spiering S.; Hörmann L.; Ulmer B.; Zhang H.; Briganti F.; Schweizer M.; Hegyi B.; Liao Z.; Pölönen R. P.; Ginsburg K. S.; Lam C. K.; Serrano R.; Wahlquist C.; Kreymerman A.; Vu M.; Amatya P. L.; Behrens C. S.; Ranjbarvaziri S.; Maas R. G. C.; Greenhaw M.; Bernstein D.; Wu J. C.; Bers D. M.; Eschenhagen T.; Metallo C. M.; Mercola M. Metabolic Maturation Media Improve Physiological Function of Human iPSC-Derived Cardiomyocytes. Cell Rep. 2020, 32, 107925 10.1016/j.celrep.2020.107925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z.; Koo S.; Finnegan M. A.; Loskill P.; Huebsch N.; Marks N. C.; Conklin B. R.; Grigoropoulos C. P.; Healy K. E. Three-dimensional filamentous human diseased cardiac tissue model. Biomaterials 2014, 35, 1367–1377. 10.1016/j.biomaterials.2013.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne S.; Hossainy S.; Healy K. Hyaluronic Acid Macromer Molecular Weight Dictates the Biophysical Properties and in Vitro Cellular Response to Semisynthetic Hydrogels. ACS Biomater. Sci. Eng. 2020, 6, 1135–1143. 10.1021/acsbiomaterials.9b01419. [DOI] [PubMed] [Google Scholar]
- Huang Y. L.; Walker A. S.; Miller E. W. A Photostable Silicon Rhodamine Platform for Optical Voltage Sensing. J. Am. Chem. Soc. 2015, 137, 10767–10776. 10.1021/jacs.5b06644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberk B.; Vandael E.; Robyns T.; Vandenberghe J.; Garweg C.; Foulon V.; Ector J.; Willems R. Which QT Correction Formulae to Use for QT Monitoring?. J. Am. Heart Assoc. 2016, 5, e003264 10.1161/jaha.116.003264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondeghem L. M. QT and TdP. QT: an unreliable predictor of proarrhythmia. Acta Cardiol. 2008, 63, 1–7. 10.2143/AC.63.1.2025324. [DOI] [PubMed] [Google Scholar]
- Kramer J.; Obejero-Paz C. A.; Myatt G.; Kuryshev Y. A.; Bruening-Wright A.; Verducci J. S.; Brown A. M. MICE models: superior to the HERG model in predicting Torsade de Pointes. Sci. Rep. 2013, 3, 2100. 10.1038/srep02100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Crumb W. J.; Vicente J.; Johannesen L.; Strauss D. G. An evaluation of 30 clinical drugs against the comprehensive in vitro proarrhythmia assay (CiPA) proposed ion channel panel. J. Pharmacol. Toxicol. Methods 2016, 81, 251–262. 10.1016/j.vascn.2016.03.009. [DOI] [PubMed] [Google Scholar]; b Mirams G. R.; Cui Y.; Sher A.; Fink M.; Cooper J.; Heath B. M.; McMahon N. C.; Gavaghan D. J.; Noble D. Simulation of multiple ion channel block provides improved early prediction of compounds’ clinical torsadogenic risk. Cardiovasc. Res. 2011, 91, 53–61. 10.1093/cvr/cvr044. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.