

Summary
A functional electron transport chain (ETC) is crucial for supporting bioenergetics and biosynthesis. Accordingly, ETC inhibition decreases proliferation in cancer cells but does not seem to impair stem cell proliferation. However, it remains unclear how stem cells metabolically adapt. In this study, we show that pharmacological inhibition of complex III of the ETC in skeletal stem and progenitor cells induces glycolysis side pathways and reroutes the tricarboxylic acid (TCA) cycle to regenerate NAD+ and preserve cell proliferation. These metabolic changes also culminate in increased succinate and 2-hydroxyglutarate levels that inhibit Ten-eleven translocation (TET) DNA demethylase activity, thereby preserving self-renewal and multilineage potential. Mechanistically, mitochondrial malate dehydrogenase and reverse succinate dehydrogenase activity proved to be essential for the metabolic rewiring in response to ETC inhibition. Together, these data show that the metabolic plasticity of skeletal stem and progenitor cells allows them to bypass ETC blockade and preserve their self-renewal.
Keywords: skeletal stem cells, proliferation, self-renewal, metabolic plasticity, electron transport chain, reverse succinate dehydrogenase, NAD regeneration, TCA rerouting, TET activity, cell-based regenerative medicine
Graphical abstract
Highlights
-
•
Skeletal stem/progenitor cells can proliferate upon electron transport chain blockade
-
•
Succinate dehydrogenase is reversed with fumarate functioning as electron acceptor
-
•
Pyruvate and aspartate are critical for NAD+ regeneration and proliferation
-
•
Metabolic changes prevent DNA demethylation and preserve self-renewal
Blocking mitochondrial respiration decreases proliferation in tumor cells but not in stem cells, although mechanistic insight is lacking. Tournaire et al. report that skeletal progenitors bypass this inhibition by reversing succinate dehydrogenase activity and using alternative NAD+ regenerating pathways. This metabolic plasticity maintains proliferation and self-renewal, improving bone regeneration.
Introduction
By transporting electrons from different sources to the final electron acceptor oxygen, the mitochondrial electron transport chain (ETC) is involved not only in bioenergetics but also in biosynthetic processes. Indeed, by accepting electrons from NADH, complex I of the ETC supports the regeneration of NAD+, a co-factor used as electron acceptor in several metabolic pathways such as nucleotide and amino acid synthesis (Birsoy et al., 2015; Lunt and Vander Heiden, 2011; Sullivan et al., 2015; Vasan et al., 2020). Similarly, complex II transfers electrons from FADH2 via ubiquinone to oxygen, but it is also directly linked to the tricarboxylic acid (TCA) cycle by converting succinate to fumarate via its succinate dehydrogenase (SDH) subunit. In addition, the ETC supports a crucial step in pyrimidine synthesis, as ubiquinone accepts electrons from dihydroorotate dehydrogenase (DHODH), when dihydroorotate is converted to orotate. Ubiquinone thus receives electrons from complex I, complex II, and DHODH, resulting in the generation of ubiquinol, which is oxidized back to ubiquinone by complex III. These pleiotropic functions of the ETC likely explain the proliferation defect observed in cancer and endothelial cells upon ETC inhibition (Birsoy et al., 2015; Diebold et al., 2019; Diehl et al., 2019; Sullivan et al., 2015). In contrast, blocking ETC complex III in stem cells, such as embryonic or hematopoietic stem cells, does not reduce proliferation but changes their differentiation potential (Anso et al., 2017; Pereira et al., 2013; Varum et al., 2009). These findings suggest a greater metabolic plasticity of stem cells compared with cancer cells, although it remains unknown how stem cells bypass ETC inhibition to ensure proliferation.
Skeletal stem and progenitor cells (SSPCs) are self-renewing cells found in bone marrow, growth plate, and periosteum, and can give rise to chondrocytes, osteoblasts, adipocytes, and hematopoiesis-supportive stromal cells (Bianco et al., 2013; Chan et al., 2015; Sacchetti et al., 2007). Periosteal SSPCs are crucial for bone repair (Colnot, 2009; Zhang et al., 2005) and start to proliferate and differentiate in an avascular milieu, suggesting that SSPCs, similar to other stem cell types, exhibit metabolic plasticity to adapt and proliferate in a challenging environment.
In this study, we used SSPCs as a model to investigate the metabolic flexibility of stem cells upon ETC inhibition. We show that when ETC complex III is inhibited, extensive metabolic rewiring at the level of the TCA cycle, primarily through reverse SDH activity, in combination with NAD+ regenerating pathways, allows SSPCs to expand and preserve their multilineage potential. These findings provide mechanistic insight into the metabolic plasticity of adult stem cell populations and support the development of metabolic strategies to improve cell-based regenerative medicine.
Results
SSPCs preserve proliferation, self-renewal, and trilineage potential upon ETC dysfunction
To confirm our hypothesis that cancer and stem cells respond differently to ETC inhibition, we analyzed the effect of the ETC complex III inhibitor antimycin A (AMA) on the proliferation of four cancer cell types versus mouse periosteal-derived cells (mPDCs), a cell source rich in SSPCs. Of note, all cells were cultured in the same medium, containing glucose (5.5 mM), pyruvate (1 mM), glutamine (2 mM), and aspartate (0.22 mM). AMA treatment decreased the proliferation of tumor cells (Figures S1A and S1B) but did not affect proliferation nor survival of mPDCs (Figures 1A and 1B). However, mPDCs showed a marked decrease in oxygen consumption upon AMA treatment (Figure S1C), indicating that these cells have a functional ETC and can metabolically adapt to ETC blockade. The ability to sustain proliferation was specific for SSPCs, as AMA treatment reduced proliferation of lineage-committed skeletal cells, such as mouse growth plate chondrocytes and mature osteoblasts, differentiated from calvarial osteoprogenitors (Figures S1D–S1J). Thus, the capacity to ensure proliferation upon mitochondrial complex III blockade is specific for SSPCs and absent in lineage-committed skeletal cells.
Figure 1.

SSPCs preserve proliferation, self-renewal, and trilineage potential upon ETC dysfunction
(A) Flow-cytometry analysis of BrdU-positive (BrdU+) mouse periosteal cells (mPDCs) treated for 24 h or 3 weeks with vehicle (veh) or 10 μM antimycin A (AMA) (n = 3).
(B) Cell viability of veh- and AMA-treated mPDCs, analyzed by annexin V-propidium iodide flow cytometry (n = 5).
(C and D) Visualization (methylene blue staining) and quantification of primary colonies, formed by mPDCs treated with veh or AMA for 1 week (n = 8).
(E) Flow-cytometry analysis of the percentage of skeletal stem cells (SSCs) in freshly isolated mPDCs (fresh) or after treatment with veh or AMA for 1 week (n = 6).
(F) Flow-cytometry analysis of the percentage of pre-bone, cartilage, and stromal progenitors (pre-BCSP) in freshly isolated mPDCs or after treatment with veh or AMA for 1 week (n = 6).
(G–J) Visualization (methylene blue staining) and quantification of primary (G and H; n = 10) and secondary (I and J; n = 4) colonies, formed by mPDCs treated with veh or AMA for 3 weeks.
(K and L) Flow-cytometry analysis of the percentage of SSCs (K) and pre-BCSPs (L) in mPDCs treated with veh or AMA for 3 weeks (n = 8).
(M and N) Chondrogenic differentiation of mPDCs treated with veh or AMA for 3 weeks and assessed by visualization and quantification of chondrogenic matrix (Alcian blue staining; M, n = 6) and Col2a1 and Acan mRNA levels (N, n = 5).
(O and P) Osteogenic differentiation of mPDCs treated with veh or AMA for 3 weeks and assessed by staining for alkaline phosphatase activity (ALP) and mineralized matrix (Alizarin red staining; O, n = 5) and quantification of Runx2, Tnap, and Col1a1 mRNA levels (P, n = 5–6).
(Q and R) Adipogenic differentiation of mPDCs treated with veh or AMA for 3 weeks and assessed by visualization of lipid deposits (oil red O staining; Q, n = 10) and quantification of Pparγ mRNA levels (R, n = 6).
Data are means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 versus vehicle (paired two-tailed Student’s t test). Scale bars, 1 cm (C, G, I, M, O) and 200 μm (Q). See also Figures S1 and S2.
To verify that the positive effect on proliferation did not exhaust SSPC properties, we used three approaches: colony formation, flow-cytometry-based analysis of SSPC markers (Chan et al., 2015), and trilineage differentiation potential (Figure S1K, short protocol). First, AMA-treated cells generated more colonies than vehicle-treated cells (Figures 1C and 1D). Second, using described stem and progenitor markers, we observed that the stromal fraction of freshly isolated mPDCs contained 2.04% ± 0.31% skeletal stem cells (SSCs; CD31−/CD45−/Ter119−/CD51+/CD90−/CD105−/BP1−/CD200+; n = 3; mean ± SD), 28.79% ± 0.93% pre-bone, cartilage, and stromal progenitors (pre-BCSPs; CD31−/CD45−/Ter119−/CD51+/CD90−/CD105−/BP1−/CD200−; n = 3; mean ± SD), and the rest were more lineage-restricted skeletal progenitors. After 1 week of culture, 75% of SSCs and 20% of pre-BCSPs were lost in vehicle-treated mPDCs, whereas AMA treatment preserved these populations (Figures 1E and 1F). Third, treating mPDCs seeded at colony-forming density with AMA for 10 days resulted in a higher number of colonies (Figures S1K, S2A, and S2B). Switching to differentiation medium, in the absence of AMA, induced a higher proportion of colonies to differentiate to the chondrogenic (Alcian blue staining), osteogenic (alkaline phosphatase [ALP] activity), and adipogenic (oil red O staining; Figures S2C and S2D) lineage, indicating that AMA treatment preserves the trilineage potential of SSPCs and prevents their exhaustion after cell expansion.
Similar to the short regimen, long-term treatment of mPDCs with AMA preserved proliferation (Figure S1K [long protocol] and Figure 1A) and SSPC characteristics, as evidenced by the increase in primary and secondary colonies (Figures 1G–1J), the higher percentage of SSCs (1.1% ± 0.52% versus 0.46% ± 0.27%, AMA versus vehicle) and pre-BCSPs (29.63% ± 10.11% versus 21.07% ± 9.06%, AMA versus vehicle; Figures 1K and 1L) and increased trilineage potential (Figures 1M–1R). Of note, while SSPCs expanded with AMA maintained their multipotency, AMA treatment during differentiation decreased the trilineage potential (Figures S2E–S2J), suggesting that ETC function is required for SSPC differentiation. To exclude that these findings are AMA specific, we confirmed that another inhibitor of mitochondrial complex III, myxothiazol (Myx), had no effect on proliferation or survival but increased SSPC properties (Figures S2K–S2O).
Thus, SSPCs preserve proliferation and self-renewal when ETC complex III is blocked.
SSPCs increase the NAD(H) pool to preserve proliferation when ETC is inhibited
The normal proliferation of SSPCs upon AMA treatment (Figure 1A) suggests that SSPCs have a high metabolic plasticity to sustain proliferation upon ETC blockade. Indeed, AMA treatment reduced proliferation during the first 12 h, but it was restored again after 24 h (Figure 2A).
Figure 2.

SSPCs increase NAD(H) pool and NAD+ regeneration by glycolysis side pathway and TCA cycle rerouting
(A) Flow-cytometry analysis of BrdU+ SSPCs treated with vehicle (veh) or 10 μM AMA for 12 h or 24 h (n = 3).
(B) NAD+ levels after treatment for indicated times (n = 4).
(C) Nampt mRNA levels after treatment for indicated times (n = 4).
(D) Flow-cytometry analysis of BrdU+ SSPCs treated for 24 h or 48 h with veh or AMA with or without 10 nM FK866 (n = 8).
(E) Metabolite levels of pyruvate, lactate, alanine, and glycerol-3-phosphate (glycerol-3-P) after treatment for 3 days (n = 8).
(F) Glycolytic flux (n = 3).
(G) Fractional contribution of 13C6-glucose to lactate, alanine, and glycerol-3-P (n = 8).
(H) Metabolite levels of citrate, α-ketoglutarate (αKG), succinate (suc), fumarate (fum), malate, and 2-hydroxyglutarate (2HG) after treatment for 3 days (n = 11–15).
(I) Fractional contribution of 13C5-glutamine to citrate, αKG, suc, fum, malate, and 2HG (n = 8).
(J) Schematic representation of 13C5-glutamine labeling patterns. Empty circles indicate 12C, black circles denote 13C derived from oxidative metabolism, and gray circles denote 13C derived from reductive metabolism.
(K and L) Oxidative (K) and reductive (L) contribution of 13C5-glutamine to suc, fum, malate, and citrate (n = 8).
(M) Fractional contribution of 13C6-glucose to citrate, αKG, suc, fum, malate, and 2HG (n = 8).
(N) Schematic representation of 13C6-glucose labeling patterns. Empty circles indicate 12C, black circles denote 13C entering TCA cycle via pyruvate dehydrogenase (PDH), and gray circles denote 13C entering TCA cycle via pyruvate carboxylase (PC).
(O and P) M + 2 (O) and M + 3 (P) contribution of 13C6-glucose to citrate, αKG, suc, fum, and malate (n = 8).
(Q and R) Pcx mRNA levels (Q, n = 3) and immunoblot detection of PC and β-actin (R). Representative image of three experiments is shown.
(S) Schematic representation of [3,4]-13C2-glucose labeling of TCA cycle intermediates. Empty circles indicate 12C, black circles denote 13C entering TCA cycle via PC activity.
(T) M + 1 contribution of [3,4]-13C2-glucose to citrate, malate, fum, and suc.
Data are means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 versus veh-treated cells (paired two-tailed Student’s t test), $p < 0.05 (two-way ANOVA). See also Figure S3.
To unravel the mechanism behind the metabolic flexibility of SSPCs we first analyzed cellular energy levels, as the ETC is a major source of ATP production. AMA treatment resulted in an energy deficit as evidenced by reduced energy charge, increased AMP/ATP ratio, and higher levels of activated AMPK (p-AMPKT172) (Figures S3A–S3C). Despite this energy deficit, cell proliferation of SSPCs was normal, suggesting that the energy deficit did not restrict proliferation.
Research in tumor cells has revealed that ETC inhibition can reduce proliferation because of impaired conversion of NADH to NAD+ at complex I, resulting in decreased NAD+ levels (Diehl et al., 2019; Gui et al., 2016; Luengo et al., 2020). By blocking complex III of the ETC, AMA may indirectly inhibit the electron transport from complex I, thereby reducing NAD+ regeneration (Fendt et al., 2013) (Figures S3I and S3J). NAD+ levels are linked to proliferation, as this co-factor is critical for the synthesis of oxidized macromolecules such as nucleotides and amino acids (Bao et al., 2016; Birsoy et al., 2015; Diehl et al., 2019; Garcia-Bermudez et al., 2018; Gui et al., 2016; Sullivan et al., 2015), and NAD+ is also required for histone deacetylation by sirtuins and for poly(ADP-ribose) polymerase activity (Kennedy et al., 2016). We observed that NADH levels increased in AMA-treated mPDCs from 6 h on, resulting in a decreased NAD+/NADH ratio (Figures S3D and S3E). Consequently, NAD+ levels decreased briefly upon AMA treatment but, interestingly, were restored to levels similar to or even higher than the ones observed in vehicle-treated cells at 24 h (Figure 2B), resulting in an increased total NAD(H) pool (Figure S3F). In line with these results, NAD+ levels were also preserved after Myx treatment (Figures S3G and S3H). These temporary changes in NAD+ levels correspond to the proliferation dynamics, suggesting a link between maintenance of NAD+ levels and proliferation in AMA-treated SSPCs.
We next investigated how SSPCs preserved NAD+ levels upon ETC blockade. The increase in the total NAD(H) pool upon AMA treatment suggests enhanced NAD synthesis. NAD+ can be synthesized in the salvage pathway, where nicotinamide, released in NAD+-dependent signaling reactions or taken up from the environment, is converted first to nicotinamide mononucleotide by nicotinamide phosphoribosyltransferase (NAMPT) and then to NAD+ (Kennedy et al., 2016; Thapa and Dallmann, 2020). We found increased Nampt expression in AMA-treated cells (Figure 2C) and investigated its importance for proliferation by using the NAMPT inhibitor FK866. Adding FK866 for 24 h reduced bromodeoxyuridine (BrdU) incorporation in AMA-treated cells, whereas this decrease was only observed after 48 h in vehicle-treated cells (Figure 2D), indicating that AMA-treated SSPCs rely more on NAD+ synthesis for proliferation.
Together, these data indicate that SSPCs increase the NAD+ pool when mitochondrial respiration is blocked.
Glycerol-3-phosphate biosynthesis contributes to NAD+ regeneration in AMA-treated SSPCs
In addition to NAD+ synthesis, we hypothesized that AMA-treated SSPCs upregulated NAD+ regeneration pathways (Figures S3I and S3J) and focused first on glycolysis and its side pathways. AMA treatment increased glycolysis, as evidenced by increased glucose uptake, lactate levels, and lactate efflux, higher lactate/glucose ratio and glycolytic flux, and enhanced contribution of 13C6-glucose to lactate (Figures 2E–2G and S3K–S3M). This increase in glycolysis likely prevented an even larger energy deficit but also caused AMA-treated cells to become highly dependent on extracellular glucose as glucose deprivation for 24 h reduced their survival (Figure S3N).
Next to increased glycolysis, we observed adaptations in side pathways of glycolysis upon AMA treatment, as 13C6-glucose contributed more to glycerol-3-phosphate (glycerol-3-P) and alanine, which was associated with a manifest increase in glycerol-3-P levels (Figures 2E and 2G). The conversion of the glycolysis intermediate dihydroxyacetone phosphate to glycerol-3-P by glycerol-3-P dehydrogenase (GPDH) involves the oxidation of NADH to NAD+, and thus contributes to NAD+ regeneration, as recently reported (Figure S3J) (Liu et al., 2021). The synthesis of alanine from pyruvate via transamination converts simultaneously glutamate to α-ketoglutarate (αKG) and can thus bypass glutamate dehydrogenase, which needs NAD+ as co-factor. However, this transaminase reaction consumes pyruvate that could otherwise be converted to lactate and regenerate NAD+.
Thus, the increase in the glycerol-3-P pathway likely contributes to NADH/NAD+ redox homeostasis by increasing NAD+ regeneration.
Rerouting of the TCA cycle and reverse SDH activity in AMA-treated cells
As the TCA cycle is a major source of NADH production (Figure S3I) and the electron transfer from NADH to complex I of the ETC is indirectly inhibited by AMA, we expected a decrease in the levels of TCA cycle intermediates in AMA-treated SSPCs. We found reduced citrate levels (−80%), but an increase in αKG (2-fold), malate (2-fold), fumarate (3-fold), and especially succinate (11-fold), as well as in the αKG derivative 2HG (8-fold) (Figure 2H).
To understand these unexpected changes in TCA cycle metabolites, we analyzed glutamine and glucose metabolism, two important nutrients for TCA cycle activity in SSPCs (Figure S3O). Glutamine uptake was not changed (Figure S3P), but the contribution of 13C5-glutamine to αKG, and especially to succinate, was increased in AMA-treated cells (Figure 2I). More precisely, the oxidative contribution of 13C5-glutamine to 13C4-succinate was increased, but the contribution to 13C4-fumarate, 13C4-malate, and 13C4-citrate was decreased in AMA-treated cells (Figures 2J and 2K), indicating a block of succinate oxidation. It is likely that SDH is indirectly inhibited, as it is part of ETC complex II and cannot further transfer electrons to complex III upon AMA treatment (Vasan et al., 2020) (Figure S3J). However, reductive carboxylation of 13C5-glutamine was strongly increased in AMA-treated cells with higher contribution to 13C5-citrate, 13C3-malate, and 13C3-fumarate (Figure 2L). Surprisingly, we also detected 13C3-succinate in AMA-treated cells but none in controls, suggesting conversion of fumarate to succinate and thus reversal of SDH activity (Figure 2L). 13C5-glutamine also contributed more to 2HG upon AMA treatment (Figure 2I). These changes in glutamine metabolism in AMA-treated SSPCs can contribute to NADH/NAD+ balance as follows: first, less NADH is produced in the TCA cycle as the oxidative arm is partially blocked at SDH, leading to decreased conversion of malate to oxaloacetate (OAA); and second, NAD+ is regenerated by the increase in reductive carboxylation flux, with conversion of OAA to malate and by the enhanced conversion of αKG to 2HG (Figure S3J).
Oxidative glucose metabolism was also altered upon AMA treatment, as the contribution of 13C6-glucose to citrate and αKG was decreased, whereas its contribution to malate and fumarate was similar and to succinate even increased (Figure 2M), suggesting altered entry and/or metabolism of glucose in the TCA cycle. 13C6-glucose-derived pyruvate can enter the TCA cycle either via pyruvate dehydrogenase (PDH) that decarboxylates 13C3-pyruvate to 13C2-acetyl-coenzyme A (CoA) (M + 2 labeling), or via pyruvate carboxylase (PC) that adds an unlabeled CO2 to 13C3-pyruvate to form four-carbon 13C3-OAA (M + 3 labeling; Figure 2N). We found that M + 2 labeling of citrate, αKG, succinate, malate, and fumarate was manifestly decreased upon AMA treatment (Figure 2O), indicative of decreased PDH-mediated TCA cycle anaplerosis. On the other hand, M + 3 labeling of malate, fumarate, and succinate was increased whereas M + 3 labeling of citrate and αKG was decreased (Figure 2P), suggesting that glucose enters the TCA cycle through PC and is then converted not to citrate and αKG but to malate, fumarate, and succinate (Figure S3J).
The enhanced PC activity in AMA-treated cells was further confirmed by increased PC mRNA and protein levels (Figures 2Q and 2R), and by [3,4]-13C2-glucose tracing. This approach labels glucose-derived pyruvate M + 1 and can only label TCA cycle intermediates when pyruvate is metabolized via PC, as the 13C label is lost when pyruvate is converted via PDH prior to entering the TCA cycle (Figure 2S). This method also excludes the possibility that 13C6-glucose-derived M + 3 labeling is due to a second TCA turn. We found that M + 1 labeling of malate, fumarate, and succinate was increased in AMA-treated cells and that M + 1 citrate labeling was decreased (Figure 2T), proving that PC activity is increased.
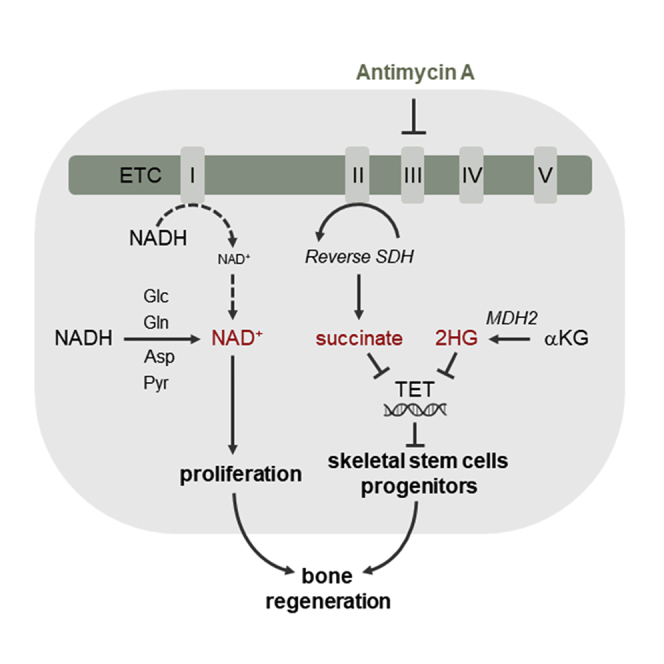
This rerouted glucose metabolism in the TCA cycle contributes to NAD+ levels in two ways (Figure S3J). First, the conversion of glucose-derived pyruvate to OAA via PC and not to acetyl-CoA through PDH allows glucose to enter the TCA cycle without consuming NAD+ in the PDH-mediated reaction. Second, OAA is not converted to αKG via citrate, thus reducing NAD+ consumption in the TCA cycle. Rather, OAA is metabolized to malate by malate dehydrogenase (MDH), which regenerates NAD+ instead of producing NADH. These data show that, next to glutamine, glucose is metabolized to succinate by reverse SDH activity in AMA-treated SSPCs, indicating that fumarate is acting as an electron acceptor receiving electrons from ubiquinol. The reverse SDH activity will thus allow some electron transfer from complex I and conversion of NADH to NAD+.
Extracellular pyruvate promotes glucose-metabolizing pathways that regenerate NAD+
In tumor cells, the decrease in proliferation by ETC inhibition can be rescued by increasing extracellular pyruvate that functions as an electron acceptor when converted to lactate by lactate dehydrogenase (LDH) (Gui et al., 2016; Sullivan et al., 2015). Similar to tumor cells, 13C3-pyruvate was mainly metabolized to lactate, thereby converting NADH to NAD+, and minimally to malate and citrate in AMA-treated SSPCs (Figure 3A). However, pyruvate deprivation further reduced the proliferation of AMA-treated tumor cells (Figures S4A–S4H), whereas it had no effect on proliferation, or on NAD+ levels, of AMA-treated SSPCs (Figures 3G and 3H).
Figure 3.

Extracellular pyruvate and MDH2 control NAD+ regeneration
(A) Fractional contribution of 13C3-pyruvate to lactate, malate, and citrate in cells treated with vehicle (veh) or 10 μM AMA for 3 days (n = 4).
(B–E) Fractional contribution of 13C6-glucose to lactate (B, n = 8), malate (C, n = 8), alanine (D, n = 8) and glycerol-3-phosphate (glycerol-3-P; E, n = 8) in cells cultured with or without 1 mM pyruvate.
(F) Glycerol-3-P levels in cells cultured with or without pyruvate (n = 8).
(G) NAD+ levels in cells cultured in either full medium (1 mM pyruvate, 0.22 mM aspartate) or without pyruvate, aspartate, or combination (n = 4).
(H) Flow-cytometry analysis of BrdU+ cells cultured in either full medium or without pyruvate, aspartate, or combination (n = 4).
(I) Fractional contribution of 13C4-aspartate to malate, fumarate (fum), succinate (suc), and α-ketoglutarate (αKG) after treatment for 3 days (n = 4).
(J) Schematic representation of 13C4-aspartate labeling patterns. Empty circles indicate 12C, black circles denote 13C labeling pattern with conversion of oxaloacetate to malate, and gray circles denote 13C labeling pattern with conversion of oxaloacetate to citrate.
(K) M + 2 and M + 4 contribution of 13C4-aspartate to suc (n = 4).
(L) NAD+ levels in cells cultured in full medium or without pyruvate and aspartate (Pyr/Asp), supplemented or not with 1 mM α-ketobutyrate (AKB) (n = 4).
(M) Flow-cytometry analysis of BrdU+ cells treated for 24 h with veh or AMA and cultured in full or no Pyr/Asp medium, supplemented or not with AKB (n = 4).
(N and O) Malate dehydrogenase2 (Mdh2) mRNA levels (N, n = 4) and immunoblot detection of MDH2 and β-actin (O) in cells transduced with control (Mdh2CO) or Mdh2-specific shRNA (Mdh2KD). Representative image of three experiments is shown.
(P) NAD+ levels in veh- and AMA-treated Mdh2CO and Mdh2KD cells cultured with or without pyruvate for 3 days (n = 4).
(Q) Flow-cytometry analysis of BrdU+Mdh2CO and Mdh2KD cells treated for 24 h with veh or AMA and cultured with or without pyruvate (n = 4).
Data are means ± SD. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 versus veh-treated cells (paired two-tailed Student’s t test). $p < 0.05 (two-way ANOVA), §p < 0.05 (three-way ANOVA). See also Figures S4–S6.
We hypothesized that AMA-treated SSPCs preserve NAD+/NADH balance upon pyruvate withdrawal by adapting glucose and/or glutamine metabolism. Pyruvate deprivation of AMA-treated cells caused 13C6-glucose to contribute more to lactate and less to malate and alanine (Figures 3B–3D). The decreased alanine transaminase activity reduced not only alanine secretion but also the contribution of 13C5-glutamine to αKG and 2HG, resulting in decreased αKG and 2HG levels (Figures S5A–S5D). Of note, malate levels were not significantly changed in pyruvate-deprived AMA-treated cells (Figure S5E), suggesting complementary action by another nutrient. Important with respect to NAD+/NADH balance, 13C6-glucose contributed more to glycerol-3-P, resulting in a further increase in glycerol-3-P levels in pyruvate-deprived AMA-treated cells (Figures 3E and 3F).
These data indicate that extracellular pyruvate is mainly converted to lactate, thereby contributing to NAD+ regeneration in AMA-treated cells, but it also allows glucose to be converted to alanine and to malate. The results also suggest that AMA-treated SSPCs, but not tumor cells, can metabolically adapt to pyruvate deprivation by increasing the conversion of glucose to glycerol-3-P to maintain their NAD+/NADH balance.
MDH2 controls NAD+ regeneration
In many tumor cells, NAD+ is required for de novo aspartate synthesis, as aspartate uptake is limited and aspartate is essential for nucleotide synthesis and, thus, proliferation (Birsoy et al., 2015; Garcia-Bermudez et al., 2018; Sullivan et al., 2015). In contrast, SSPCs consumed extracellular aspartate (Figure S5F) and 13C4-aspartate contributed to TCA cycle metabolites malate and fumarate (Figures 3I and S3J) at baseline. AMA treatment increased aspartate uptake and decreased the 13C4-aspartate contribution to αKG, but manifestly increased the 13C4-aspartate contribution to 13C4-succinate (22.6% ± 1.37% versus 1.88% ± 0.08%; AMA versus vehicle; Figures 3I–3K), suggesting that aspartate is not metabolized in the TCA cycle via OAA to citrate but rather to fumarate and finally to succinate by reverse SDH activity.
Given the high contribution of aspartate to TCA cycle metabolites via reverse SDH activity, we hypothesized that aspartate metabolism is important for the NADH/NAD+ balance and proliferation of AMA-treated SSPCs, especially during pyruvate deprivation, and we therefore performed combined pyruvate and aspartate (Pyr/Asp) deprivation. Of note, depletion of only aspartate did not affect NAD+ levels or proliferation of SSPCs cultured with or without AMA. In contrast, Pyr/Asp withdrawal decreased NAD+ levels and BrdU incorporation in AMA-treated SSPCs (Figures 3G and 3H). In comparison, aspartate deprivation of AMA-treated tumor cells did not further decrease the reduced proliferation observed in normal and especially in pyruvate-deprived conditions (Figures S4A–S4H). Thus, extracellular pyruvate is critical for AMA-treated tumor cells but less so for AMA-treated SSPCs, as SSPCs can regenerate electron acceptors by using other nutrients such as aspartate.
The combined Pyr/Asp deprivation resulted in decreased malate levels, suggesting decreased MDH2 activity and, thus, reduced NAD+ regeneration. This decrease in MDH2 activity is also supported by the reduced 2HG levels (Figure S5G), as MDH2 can convert αKG to 2HG, thereby regenerating NAD+ (Intlekofer et al., 2017; Ye et al., 2018) (Figure S3J). Pyr/Asp-deprived AMA-treated cells could not compensate for the reduced MDH2 activity by enhancing LDH activity, as lactate levels were not changed, nor by GPDH activity, as glycerol-3-P levels did not additionally increase compared with the pyruvate-deprived condition (Figures S5H and S5I).
To confirm the link between NAD+ levels and proliferation, we added α-ketobutyrate (AKB), which can be used by intracellular dehydrogenases to regenerate NAD+ from NADH (Sullivan et al., 2015). Adding AKB to AMA-treated cells, cultured in Pyr/Asp-deprived medium, prevented the decrease in NAD+ levels and BrdU incorporation (Figures 3L and 3M), indicating that maintaining NAD+ levels is important to ensure proliferation upon AMA treatment.
Thus, MDH2 activity is important for NAD+ regeneration and proliferation in AMA-treated cells, by promoting the conversion not only of glucose and aspartate to malate but also of glutamine-derived αKG to 2HG (Figure S3J). To prove this hypothesis, we inactivated Mdh2 using short hairpin RNA (shRNA) (Mdh2KD) (Figures 3N and 3O). However, MDH2 deletion did not affect NAD+ levels or BrdU incorporation in AMA-treated SSPCs (Figures 3P and 3Q), possibly because the enormous increase in 2HG levels (14-fold) was only reduced by 50% (Figure S5J), thereby allowing sufficient NAD+ regeneration. Production of 2HG from αKG is indirectly supported by pyruvate uptake in AMA-treated SSPCs (Figures S5B and S5C), as pyruvate promotes the conversion of glucose to alanine and concurrently of glutamate to αKG. We therefore cultured AMA-treated Mdh2KD cells without pyruvate and observed decreased NAD+ levels and proliferation (Figures 3P and 3Q), indicating that the combination of pyruvate uptake and MDH2 activity is critical to preserve proliferation in AMA-treated SSPCs. Similar to the Pyr/Asp-deprived condition, GPDH activity could not compensate for MDH2 knockdown, as glycerol-3-P levels did not additionally increase compared with the pyruvate-deprived condition, thus explaining the decrease in NAD+ levels (Figure S5K).
Overall, metabolism of extracellular pyruvate and MDH2 activity are closely interconnected to enable NAD+ regeneration and proliferation in SSPCs with ETC dysfunction.
DHODH activity is preserved, allowing pyrimidine synthesis
We next investigated nucleotide synthesis in more detail, given the importance of both NAD+ and aspartate for this metabolic process. In contrast to several observations in tumor cells (Birsoy et al., 2015; Diehl et al., 2019; Sullivan et al., 2015), the decrease in NAD+ and proliferation in Pyr/Asp-deprived or Pyr-deprived Mdh2KD AMA-treated SSPCs was not linked to a defect in nucleotide synthesis. Indeed, nucleotide levels were not changed and supplementing nucleotides (Table S1) did not rescue the proliferation defect (Figures S6A–S6C), suggesting that NAD+ levels regulate proliferation in SSPCs by mechanisms other than nucleotide synthesis. These data indicate that nucleotides can still be synthesized in AMA-treated cells and that DHODH, an enzyme involved in pyrimidine synthesis and indirectly linked to the ETC, is not fully inhibited. DHODH generates orotate from dihydroorotate by transferring electrons to ubiquinone, resulting in the generation of ubiquinol, which is oxidized back to ubiquinone by complex III (Figure S3I). Inhibiting complex III by AMA treatment is thus expected to indirectly block DHODH activity. We observed a decreased orotate/dihydroorotate ratio in AMA-treated cells, indicating that the reaction catalyzed by DHODH was impaired (Figure S6D), but we also noted increased levels of dihydroorotate and orotate, suggesting that DHODH can still produce orotate from dihydroorotate (Figures S6E and S6F). A likely explanation for the preserved, although reduced, DHODH activity is that the reverse SDH activity favors electron transfer from ubiquinol to complex II, allowing ubiquinone to accept electrons from DHODH and pyrimidine synthesis to continue. Indeed, adding atpenin A5 (Atp), an inhibitor of SDH, together with AMA, decreased the contribution of 13C4-aspartate to orotate and reduced orotate levels, indicating that DHODH activity depends on SDH activity in AMA-treated cells (Figures S6G and S6H).
Together, these data indicate that skeletal progenitors can maintain their proliferation when complex III is blocked, in part by preserving DHODH activity, likely because of reverse SDH activity.
Metabolic changes increase TET activity and preserve SSPCs
Since AMA-treated SSPCs not only preserved proliferation but also showed improved stemness (Figures 1 and S2), we questioned whether the AMA-induced metabolic changes also supported stemness properties. The metabolic rerouting of the TCA cycle resulted in a significant decrease in the αKG/succinate and αKG/2HG ratios, after both short- and long-term AMA treatment (Figures 4A and 4B), as well as after adding Myx (Figures S7A and S7B).
Figure 4.

Metabolic changes increase TET activity and preserve SSPCs
(A and B) Ratio of α-ketoglutarate (αKG) to succinate (suc) and of αKG to 2-hydroxyglutarate (2HG) in cells treated with vehicle (veh) or 10 μM AMA for 3 days (A, n = 15) or 3 weeks (B, n = 7).
(C) Immunoblot of HIF-1α with Lamin A/C as loading control in cells treated with veh or AMA for 3 days (n = 3). IOX2, an inhibitor of HIF prolyl hydroxylase 2, was used as positive control. Representative image is shown.
(D) Immunoblot of dimethylated lysine 9 of histone H3 (H3K9me2), trimethylated lysine 27 of histone H3 (H3K27me3), and dimethylated lysine 79 of histone H3 (H3K79me2), with total histone 3 (H3) and Ponceau red as loading control (n = 3). Representative image is shown.
(E) Dot blot assay of 5-hydroxymethylcytosine (5hmC) in cells treated with veh or AMA for 3 days, with methylene blue staining as loading control. Representative images of four experiments are shown.
(F) Percentage of 5hmC of total cytosine (n = 4).
(G) Dot blot assay of 5hmC in cells treated with veh or AMA in the presence or absence of 0.5 mM ascorbic acid (Asc A) with methylene blue staining as loading control. Representative images of three experiments are shown.
(H–J) Number of colonies formed (H, n = 8), and percentage of SSCs (I, n = 5) and pre-BCSPs (J, n = 5) in cells treated with veh or AMA in the presence or absence of Asc A.
(K and L) αKG, succinate, and 2HG levels (K, n = 4) and ratio of αKG to succinate and of αKG to 2HG (L, n = 4) in cells treated with veh or 500 nM atpenin A5 (Atp).
(M) Dot blot assay of 5hmC in veh- or Atp-treated cells with methylene blue staining as loading control. Representative images of four experiments are shown.
(N–P) Number of colonies formed (N, n = 6), and percentage of SSCs (O, n = 5) and pre-BCSPs (P, n = 5) in cells treated with veh (control) or Atp.
(Q) Tet1, Tet2, and Tet3 mRNA levels in periosteal cells (n = 9).
(R) Tet2 mRNA levels in control (Tet2CO) or Tet2 knockdown (Tet2KD) periosteal cells (n = 3).
(S) Dot blot assay of 5hmC in Tet2CO and Tet2KD cells with methylene blue staining as loading control. Representative images of three experiments are shown.
(T–V) Number of colonies formed (T, n = 6–7), and percentage of SSCs (U, n = 5–6) and pre-BCSPs (V, n = 5–6) in Tet2CO and Tet2KD cells.
Data are means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 versus veh (two-tailed Student’s t test). ##p < 0.01 (one-way ANOVA), $p < 0.05 (two-way ANOVA).
The accumulation of succinate and 2HG relative to αKG might affect the activity of αKG-dependent dioxygenases, including prolyl hydroxylases that regulate hypoxia-inducible factor α (HIF-α), JmjC domain-containing histone demethylases, and Ten-eleven translocation (TET) DNA demethylases (Intlekofer and Finley, 2019). These enzymes use αKG as co-factor but are inhibited by fumarate, succinate, and 2HG, which all increased in AMA-treated cells. We did not observe changes in HIF-1α levels (Figure 4C) or global histone methylation (Figure 4D). On the other hand, 5-hydroxymethylcytosine (5hmC) levels were reduced in AMA-treated cells (Figures 4E and 4F), suggesting inhibition of TET activity and decreased DNA demethylation. To investigate whether inhibition of TET enzymes was important for SSPC properties, we used three different approaches. First, we treated SSPCs with ascorbic acid, which is known to activate TET enzymes (Blaschke et al., 2013). The addition of ascorbic acid increased 5hmC levels in AMA- and vehicle-treated cells (Figure 4G) and blocked the AMA-induced increase in SSPCs, as evidenced by the reduced number of colonies formed (Figure 4H) and decreased percentage of SSCs and pre-BCSPs (Figures 4I and 4J). Second, we investigated the effect of the SDH inhibitor Atp. Atp treatment increased succinate levels but had no effect on 2HG, and resulted in a decreased αKG/succinate ratio (Figures 4K and 4L), which correlated with a drop in 5hmC levels (Figure 4M). Accordingly, cells expanded with Atp formed more colonies and contained a higher percentage of SSCs and pre-BCSPs (Figures 4N–4P), supporting the link between αKG/succinate ratio, 5hmC levels, and maintenance of SSPCs. Third, to confirm the importance of TET inhibition for SSPC properties, we genetically inactivated Tet2, the most expressed DNA demethylase (Figure 4Q). The decreased Tet2 expression resulted in reduced 5hmC levels (Figures 4R and 4S) and was associated with an increased capacity to form colonies and a higher percentage of SSCs and pre-BCSPs (Figures 4T–4V).
We next investigated the changes in DNA methylation and demethylation induced by AMA treatment. To this end, DNA was isolated and hybridized to Beadchip microarrays after either bisulfite (BS) conversion to uncover the aggregate levels of 5mC and 5hmC, or after oxidative bisulfite (OxBS) conversion to uncover 5mC levels. Comparison of the OxBS and BS signals revealed that, out of the 282,160 CpGs investigated, 70,325 showed significant levels of 5hmC. The mean estimated 5hmC level at these sites was 2.18% (Figures S7C and S7D), and, as expected, 5hmC was detected mostly at regions with intermediate levels of DNA methylation (Booth et al., 2012). Among these 70,325 CpGs, 8,473 showed DNA hypermethylation and 4,919 showed DNA hypomethylation (1% false discovery rate), indicating that AMA treatment predominantly induces DNA hypermethylation (Figure 5A). Importantly, 5hmC levels were significantly reduced at CpGs showing hypermethylation, suggesting that the observed AMA-induced DNA hypermethylation is due to reduced DNA demethylation activity (Figure 5B). Notably, among the 300 most significantly hypermethylated CpGs, 36 were located in protein-coding gene promoters, including several genes involved in differentiation (Cxcr6, Runx1, Runx2, Runx3, and Wnt10b; Figures 5C–5H).
Figure 5.

AMA treatment induces DNA hypermethylation
(A) Volcano plot showing differential methylation at CpGs in cells treated with vehicle (veh) or 10 μM AMA for 10 days (n = 3). Probes showing a significant false discovery rate of <1%. DNA hypomethylation and DNA hypermethylation are highlighted in blue and red, respectively. AMA treatment preferentially induced DNA hypermethylation (Fisher’s exact test, p < 10−16).
(B) 5hmC estimate at CpGs of veh- or AMA-treated cells (n = 3).
(C–H) Examples of AMA-induced DNA hypermethylation at CpGs located in the protein-coding gene promoters of Cxcr6 (C), Runx1 (D), Runx2 (E and F), Runx3 (G), and Wnt10b (H) (n = 3).
Data are means ± SD. @@@p < 0.001 (Wilcoxon signed rank test with continuity correction). See also Figure S7.
AMA-induced metabolic changes thus preserve SSPCs during expansion by inhibiting TET DNA demethylase activity.
MDH2 activity is critical for preserving SSPCs
We hypothesized that the decreased αKG/succinate and αKG/2HG ratios upon AMA treatment were mediated by the observed adaptations in TCA cycle-related glucose, glutamine, and aspartate metabolism (Figures 2I, 2M, and 3I). We focused on MDH2, as this enzyme both converts glucose- and aspartate-derived OAA to malate, which is then metabolized to succinate by reverse SDH activity, and also converts glutamine-derived αKG to 2HG, and hence contributes to the increase in succinate and 2HG (Figure S3J). Accordingly, MDH2 inactivation suppressed the AMA-induced increase in fumarate, succinate, and 2HG levels, while it further enhanced αKG levels (Figure 6A), thereby normalizing αKG/succinate and αKG/2HG ratios (Figure 6B). In addition, MDH2 inactivation prevented the AMA-induced decrease in 5hmC levels and inhibited the increase in colony formation and percentage of SSCs and pre-BCSPs (Figures 6C–6F).
Figure 6.

MDH2 activity is critical for preserving SSPCs
(A and B) Fumarate, succinate (suc), α-ketoglutarate (αKG), and 2-hydroxyglutarate (2HG) levels (A, n = 8) and ratios of αKG to suc and of αKG to 2HG (B, n = 8) in cells treated with vehicle (veh) or 10 μM AMA for 3 days and transduced with control (Mdh2CO) or Mdh2-specific shRNA (Mdh2KD).
(C) Dot blot assay of 5hmC in veh- and AMA-treated Mdh2CO and Mdh2KD cells with methylene blue staining as loading control. Representative images of four experiments are shown.
(D–F) Number of colonies formed (D, n = 4), and percentage of SSCs (E, n = 4) and pre-BCSPs (F, n = 4) in veh- and AMA-treated Mdh2CO and Mdh2KD cells.
Data are means ± SD. $p < 0.05 (two-way ANOVA).
Thus, AMA treatment of SSPCs decreases αKG/2HG and αKG/succinate ratios by a dual mechanism: first, by enhancing the conversion of glutamine to 2HG via MDH2 and second, by increasing the conversion of glucose and especially aspartate to malate via MDH2 and further to succinate via reverse SDH activity (Figure S7E). These metabolic changes inhibit TET activity, resulting in maintenance of SSPC properties.
Periosteal cells expanded with AMA form more bone in vivo
To validate that the preserved self-renewal and SSPC properties are physiologically relevant, we investigated whether AMA-treated cells showed increased bone formation after in vivo implantation. To this end, we used an ectopic model of direct bone formation in mice (Stegen et al., 2016). Histomorphometry showed that, 8 weeks after implantation, scaffolds seeded with AMA-treated mPDCs contained significantly more bone compared with the vehicle-treated condition (Figures 7A and 7B). To distinguish the implanted from the host-derived cells, we used mPDCs isolated from C57BL/6-ACTb-eGFP mice and found that in both AMA-treated and control conditions the new bone was formed by the implanted cells, as cells positive for the osteogenic marker Osterix co-expressed GFP (Figures 7C and 7D).
Figure 7.

Periosteal cells expanded with AMA form more bone in vivo
(A) H&E staining of ectopic implants, seeded with mPDCs treated with vehicle (veh) or 10 μM AMA for 3 weeks and implanted for 8 weeks (right panel is magnification of boxed area). b, bone; f, fibrous tissue; g, scaffold granule (n = 8).
(B) Quantification of the bone volume formed (n = 8).
(C and D) Osterix and Hoechst (DNA) staining of ectopic implants seeded with β-actin-GFP+ periosteal cells and implanted for 8 weeks (C, n = 4) with quantification of the percentage of β-actin-GFP+ cells within the Osterix+ population (D, n = 4). Magnifications of the boxed area show Osterix staining or merged pictures; white arrows point at β-actin-GFP+/Osterix+ cells.
(E) Viability of mPDCs, labeled prior to implantation with CellTracker Green CMFDA. Viability of CMFDA+ cells was determined after implantation for 3 days by annexin V-propidium iodide flow cytometry (n = 4).
(F–H) CD31 staining of ectopic implants, seeded with mPDCs and implanted for 8 weeks (F; right panel is magnification of boxed area; scale bars, 200 μm; n = 8), with quantification of the number of blood vessels per mm2 (G) and blood vessel surface area (H) (n = 8).
(I and J) Visualization (I, methylene blue staining) and quantification of colonies formed by CMFDA+/PI− cells isolated 3 days after implantation and cultured for 1 week (J, n = 6).
Data are means ± SD. ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 versus veh (unpaired two-tailed Student’s t test). Scale bars, 200 μm (A, C) and 1 cm (I).
The increased bone mass in implants containing AMA-treated mPDCs was not caused by increased survival of the implanted cells (Figure 7E) or enhanced vascularization (Figures 7F–7H). Instead, we observed a higher number of SSPCs in the AMA-treated condition, based on the following observation. Prior to implantation, donor cells were labeled with CellTracker CMFDA, and the surviving implanted cells (CMFDA+/PI−) were retrieved from the scaffold 3 days after implantation and seeded for colony formation. Again, the AMA-treated cells formed more colonies compared with controls (Figures 7I and 7J), indicating that this cell population continues to contain more SSPCs after in vivo implantation.
In conclusion, AMA treatment during expansion of SSPCs preserves their self-renewal and bone-forming capacity, resulting in better in vivo bone formation.
Discussion
In this study, we show that the metabolic plasticity of mouse SSPCs promotes their proliferation and self-renewal in response to ETC complex III inhibition. Mechanistically, several NAD+ regenerating pathways were induced to preserve proliferation, comprising changes in glycolysis side pathways that were highly interconnected with rerouting of the TCA cycle. MDH2 and reverse SDH activity proved essential not only for NAD+ regeneration that supported proliferation but also for the increased 2HG and succinate levels causing epigenetic changes by inhibiting TET DNA demethylase activity resulting in SSPC maintenance. These findings highlight the metabolic flexibility of stem cells and may aid in the development of metabolic strategies that prevent exhaustion of stem cells during in vitro expansion and improve cell-based regeneration.
Blocking ETC did not decrease the proliferation of SSPCs, whereas it reduced proliferation in the tested tumor cells, when cultured with identical levels of pyruvate and aspartate. SSPCs were able to preserve their NADH/NAD+ balance through combined adaptations in several metabolic pathways: NAD+ was regenerated by increased conversion of glucose to glycerol-3-P, less NADH was produced in the rerouted TCA cycle and, importantly, SDH activity was reversed, which enabled the transfer of electrons from complex I to ubiquinone and regeneration of NAD+. By reversing SDH activity, aspartate- or glucose-derived fumarate functions as a final electron acceptor in the ETC. MDH2 activity was important to maintain proliferation and regenerate NAD+ by converting aspartate- and glucose-derived OAA to malate and glutamine-derived αKG to 2HG, reactions that were favored by, but not dependent on, pyruvate uptake. Tumor cells, on the other hand, rely more on the uptake of pyruvate, which contributes to the synthesis of aspartate that is required for nucleotide synthesis, as aspartate uptake is often very limited in tumor cells (Birsoy et al., 2015; Diehl et al., 2019; Sullivan et al., 2015). The highly intertwined and rearranged metabolism of four major nutrients—glucose, glutamine, aspartate, and pyruvate—likely explains the ability of SSPCs to preserve proliferation when encountering ETC shutdown.
The response of SSPCs to ETC inhibition differs from tumor cells in a second aspect, as the correlation between NAD+ levels and proliferation cannot be explained by nucleotide levels, an observation that was also reported in hematopoietic stem cells (Anso et al., 2017). Further investigation is needed to understand how NAD+ levels ensure proliferation of AMA-treated SSPCs, but a possible mechanism is that NAD+ exerts its proliferation-supporting effect via sirtuin-dependent pathways, which modulate gene transcription and energy metabolism (Anderson et al., 2017).
The normal nucleotide levels in AMA-treated SSPCs can be explained by the reverse SDH activity, which converts glucose- and aspartate-derived fumarate to succinate and leads to a rearranged electron transfer. This reverse SDH activity might favor electron transfer from ubiquinol to complex II thereby regenerating ubiquinone, which then can accept electrons from DHODH and thus allows pyrimidine synthesis (Figure S3J). Until recently, reverse SDH has been poorly described and reported to be active during heart ischemia and in the hypoxic environment of the retina (Bisbach et al., 2020; Chouchani et al., 2014). A recent study, published during the review process of our manuscript, revealed that reverse SDH activity may be more common than expected, with tissues such as liver and kidney using fumarate as terminal electron acceptor independent of oxygen levels (Spinelli et al., 2021). The exact cell types, besides SSPCs, that are capable of activating this mechanism remain to be determined, as well as the conditions under which reverse SDH activity can occur.
ETC inhibition in SSPCs prevented their exhaustion, and we discovered that the combination of MDH2 and reverse SDH activity is the major driver of the metabolic adaptations. The increased 2HG and succinate levels, relative to αKG, induce the epigenetic changes that maintain stem cell properties. Whether a similar metabolic rewiring explains the connection between complex III and stemness observed in hematopoietic stem cells (Anso et al., 2017) needs additional exploration. Since preserved proliferation and stem cell properties in AMA-treated SSPCs are explained by the same metabolic adaptations, an appealing hypothesis is that the increased NADH levels are causing the metabolic reprogramming necessary not only to preserve proliferation but also to maintain stem cell properties. This concept on the driver function of NADH further expands on recent findings showing that it is the requirement for NAD+/NADH homeostasis rather than the demand for ATP that drives cells to switch to glycolysis (Luengo et al., 2020).
Altogether, our study shows that targeting skeletal progenitor metabolism is beneficial to control self-renewal and cell fate during expansion. Maintaining these in vitro properties is of interest to improve cell-based regenerative strategies, as evidenced by the increased bone formation observed when implanting AMA-treated cells in vivo. Preserving stem and progenitor properties by interfering with cellular metabolism is thus a promising approach, not only for bone regeneration but likely also for regenerative medicine in general. In addition, the described metabolic plasticity of SSPCs might reflect their capability to cope with changing conditions in the environment. Indeed, following fracture, skeletal progenitors in the periosteum start to proliferate despite the emerging local hypoxia. In normoxia, oxygen is considered a terminal electron acceptor, allowing electron flow through the ETC to persist, which is critical not only for energetics but also for biosynthetic processes. Based on our data, we speculate that hypoxic SSPCs might use fumarate as electron acceptor instead of oxygen by reversing SDH, thereby not only preventing reductive stress but also permitting nucleotide synthesis. In addition, hypoxia has been reported to increase 2HG levels and to inhibit TET activity (Intlekofer et al., 2015; Oldham et al., 2015; Thienpont et al., 2016). Taking our findings together, we propose that SSPCs have the metabolic flexibility to adapt to a hypoxic environment without loss of proliferation and SSPC properties and thereby support efficient fracture repair, although further research is needed.
Limitations of the study
This study has some limitations, as the metabolic plasticity of SSPC needs to be confirmed in human skeletal progenitors. In addition, it would be interesting to investigate which microenvironments in the skeleton favor these metabolic adaptations and thereby allow proliferation and self-renewal of SSPCs. Further mechanistic studies are required to understand how NAD+ levels affect SSPC proliferation, if not by changing nucleotide synthesis, and whether all of the identified NAD+ regenerating pathways are equally important for the phenotype.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-AMPKα | Cell Signaling Technology | Cat# 2532, RRID:AB_330331 |
| Rabbit monoclonal anti-phospho-AMPKα (Thr172) | Cell Signaling Technology | Cat# 2535 (40H9), RRID:AB_331250 |
| Mouse monoclonal anti-β-actin | Sigma-Aldrich | Cat# A5441 (AC-15), RRID:AB_476744 |
| Rabbit polyclonal anti-HIF1α | Novus Biologicals | Cat# NB100-449, RRID:AB_10001045 |
| Rabbit monoclonal anti-histone H3 | Cell Signaling Technology | Cat# 4499 (D1H2), RRID:AB_10544537 |
| Rabbit monoclonal anti-dimethyl-histone H3 (Lys9) | Abcam | Cat# ab176882, RRID:AB_2895140 |
| Rabbit monoclonal anti-trimethyl H3 (Lys27) | Abcam | Cat# ab192985, RRID:AB_2650559 |
| Rabbit polyclonal anti-dimethyl histone H3 (K79) | Abcam | Cat# ab3594, RRID:AB_91530 |
| Rabbit polyclonal anti-5-hydroxymethylcytosine | Active Motif | Cat# 39769, RRID:AB_10013602 |
| Goat polyclonal anti-Lamin A/C | Santa Cruz | Cat# sc-6215, RRID:AB_648152 |
| Rabbit polyclonal anti-MDH2 | Abcam | Cat# ab96193, RRID:AB_10679348 |
| Rabbit polyclonal anti-PC | Invitrogen | Cat# PA5-50101, RRID:AB_2635554 |
| Rat monoclonal anti-CD31 (PECAM-1), APC | eBioscience | Cat# 17-0311-80, RRID:AB_657736 |
| Rat anti-mouse CD31 | BD Pharmingen | Cat# 550274, RRID:AB_393571 |
| Rat monoclonal anti-CD45, APC | eBioscience | Cat# 17-0451-83, RRID:AB_469393 |
| Rat monoclonal anti-CD51 (Integrin alpha V), Biotin | eBioscience | Cat# 13-0512-82, RRID:AB_466477 |
| Rat monoclonal anti-CD90.2 (Thy-1.2), PE | eBioscience | Cat# 12-0903-81, RRID:AB_465778 |
| Rat monoclonal anti-CD105 (Endoglin), PE | eBioscience | Cat# 12-1051-81, RRID:AB_657526 |
| Rat monoclonal anti-CD200, PerCP-eFluor 710 | eBioscience | Cat# 46-5200-82, RRID:AB_10598213 |
| Rat monoclonal anti-CD249 (BP-1), PE | eBioscience | Cat# 12-5891-81, RRID:AB_466014 |
| Rabbit polyclonal anti-GFP | Invitrogen | Cat# A11122, RRID:AB_2307355 |
| Rabbit polyclonal anti-OSX | Santa Cruz | Cat# SC-22536-R, RRID:AB_831618 |
| Rabbit polyclonal anti-mouse-HRP | Dako | Cat# P0161, RRID:AB_2687969 |
| Rabbit polyclonal anti-goat-HRP | Dako | Cat# P0160, RRID:AB_2617143 |
| Goat polyclonal anti-rabbit-Alexa 546 | Invitrogen | Cat# A11010, RRID:AB_2534077 |
| Goat polyclonal anti-rabbit-alexa 488 | Invitrogen | Cat# A11008, RRID:AB_143165 |
| Goat polyclonal anti-rabbit-HRP | Dako | Cat# P0448, RRID:AB_2617138 |
| Goat polyclonal anti-rat-Biot | BD Pharmingen | Cat# 559286, RRID:AB_397214 |
| Streptavidin PE-Cyanine7 Conjugate | eBioscience | Cat# 25-4317-82, RRID:AB_10116480 |
| Rat monoclonal anti-TER-119, APC | eBioscience | Cat# 17-5921-83, RRID:AB_469474 |
| Chemicals, peptides, and recombinant proteins | ||
| 13C6-glucose | Cambridge Isotope Laboratories | Cat# CLM-1396 |
| 13C5-glutamine | Cambridge Isotope Laboratories | Cat# CLM-1822-H |
| 13C415N-aspartate | Cambridge Isotope Laboratories | Cat# CNLM-544-H |
| 3-isobutyl-1-methylxanthine (IBMX) | Sigma-Aldrich | Cat# CNLM-544-H |
| AKB | Sigma-Aldrich | Cat# K401 |
| Antimycin A | Sigma-Aldrich | Cat# A8674 |
| Aspartate | Sigma-Aldrich | Cat# A9256 |
| Atpenin A5 | Enzo | Cat# ALX-380-313 |
| cOmplete protease inhibitor cocktail | Roche | Cat# 11873580001 |
| Dexamethasone | Sigma-Aldrich | Cat# D4902 |
| D-(+)-Glucose | Sigma-Aldrich | Cat# G7528 |
| EmbryoMax Nucleoside mixture | Merck | Cat# ES-008-D |
| FK866 | Sigma-Aldrich | Cat# F8557 |
| β-Glycerophosphate disodium salt hydrate | Sigma-Aldrich | Cat# G9422 |
| Indomethacin | Sigma-Aldrich | Cat# I7378 |
| Insulin solution from bovine pancreas | Sigma-Aldrich | Cat# I0516 |
| L-Ascorbic Acid | Sigma-Aldrich | Cat# A4544 |
| L-aspartic acid | Sigma-Aldrich | Cat# A9256 |
| L-Glutamine | Gibco | Cat# 25030-024 |
| PhenylmethanesulfonylFluoride (PMSF) | Sigma-Aldrich | Cat# P7626 |
| Polybrene | Sigma-Aldrich | Cat# TR-1003 |
| PhosSTOP phosphatase inhibitor cocktail | Roche | Cat# 04906837001 |
| Puromycin | InvivoGen | Cat# ant-pr |
| Pyruvate | Gibco | Cat# 11360-039 |
| Recombinant human transforming growth factor-β1 | Peprotech | Cat# 100-21 |
| Superscript II Reverse Transcriptase | Thermo Fisher Scientific | Cat# 18064022 |
| SYBR GreenER qPCR SuperMix Universal | Thermo Fisher Scientific | Cat# 1178401K |
| Y-27632 (ROCK inhibitor) | Axon Medchem | Cat# 1683 |
| Critical commercial assays | ||
| Annexin V, APC conjugate (APC Annexin V) | Thermo Fisher Scientific | Cat# A35110 |
| BCA Protein Assay Reagent | Thermo Fisher Scientific | Cat# 23225 |
| CellTracker™ Green CMFDA Dye | Thermo Fisher Scientific | Cat# C7025 |
| CM-H2DCFDA | Thermo Fisher Scientific | Cat# C6827 |
| DNeasy Blood & Tissue Kit | QIAGEN | Cat# 69504 |
| FITC BrdU Flow Kit | BD Pharmingen | Cat# 559619 |
| Global 5-hmC Quantification Kit | Active Motif | Cat# 55018 |
| NucleoSpin RNA isolation kit | Machery Nagel | Cat# 740955 |
| Deposited data | ||
| Raw data | This manuscript; Mendeley | https://doi.org/10.17632/zc8f6f5s7n.1 |
| DNA methylation/demethylation | This manuscript; GEO | GSE198222 |
| Experimental models: Cell lines | ||
| 4T1 | ATCC | CRL-2539 RRID:CVCL_0125 |
| A549 | ATCC | CRL-185 RRID:CVCL_0023 |
| ROS 17/2.8 | Dr. G. Rodan University of Connecticut US |
RRID:CVCL_0508 |
| NOMO-1 | DSMZ | RRID:CVCL_1609 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6-ACTb-eGFP | Stiers et al., 2019 | N/A |
| Mouse: Tet2fl/fl | Moran-Crusio et al., 2011 | N/A |
| Oligonucleotides | ||
| Primer sequences for qRT-PCR - see Table S2 | N/A | N/A |
| Puro cre empty vector | Addgene | Cat#17408, RRID:Addgene_17408 |
| pLKO.1-puro Empty Vector Control Plasmid DNA | Sigma-Aldrich | Cat# SHC001 |
| Mdh2 shRNA: CCGGCGGAATGCAC TTACTTCTCTACTCGAGTAGAGAAGT AAGTGCATTCCGTTTTTG |
Sigma-Aldrich | Cat# SHCLNG-NM_008617 |
| Software and algorithms | ||
| Flowing Software | http://flowingsoftware.btk.fi/ | RRID:SCR_015781 |
| GraphPad Prism (version 8.3.0) | http://www.graphpad.com/ | RRID:SCR_002798 |
| ImageJ software | https://imagej.nih.gov/ij/ | RRID:SCR_003070 |
| Xcalibur Software | Thermo Fisher Scientific | RRID:SCR_014593 |
| Other | ||
| Alcian Blue 8GX | Sigma-Aldrich | Cat# A5268 |
| Alizarin Red S | Sigma-Aldrich | Cat# A5533 |
| BIOTRANS(+) nylon membrane | MP Biomedicals | Cat# ICN810205 |
| Collagenase type II | Gibco | Cat# 17101015 |
| Dispase | Gibco | Cat# 17105041 |
| Fetal bovine serum | Gibco | Cat# 10270106 |
| Low melting point agarose | Lonza | Cat# 50115 |
| New Methylene Blue N zinc chloride double salt | Sigma-Aldrich | Cat# 202096 |
| Oil red O | Sigma-Aldrich | Cat# O0625 |
| Penicillin/streptomycin | Gibco | Cat# 11548876 |
| Trypsin-EDTA (0.05%) | Gibco | Cat# 25300054 |
| Western Lightning Plus-ECL, Enhanced Chemiluminescence Substrate | PerkinElmer | Cat# NEL103E001EA |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Geert Carmeliet (geert.carmeliet@kuleuven.be).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Animals
C57BL/6, C57BL/6-ACTb-eGFP and Tet2fl/fl mice (Moran-Crusio et al., 2011) were bred in conventional conditions in our animal housing facility (Proefdierencentrum Leuven, Belgium) at 22°C under a 14-h light/10-h dark cycle, fed a normal chow diet (V1535; Ssniff GmbH, Soest, Germany). Rj:NMRI-Foxn1nu/nu mice were ordered from Janvier labs (Le Genest-Saint-Isle, France) and housed in the same conditions. Neonatal (5-day-old) C57BL/6 pups were used for the isolation of calvarial osteo-progenitors and growth plate chondrocytes. 8-week-old male and female C57BL/6, C57BL/6-ACTb-eGFP and Tet2fl/fl mice were used for the isolation of mPDCs and female Rj:NMRI-Foxn1nu/nu mice as host for the in vivo implantations. All animal experiments were conducted according to the regulations and with approval of the Animal Ethics Committee of the KU Leuven.
Cell lines
The four tumor cell lines (mouse female breast tumor cell line 4T1, human male lung carcinoma cell line A549, human female acute myeloid leukemia cell line NOMO-1 and rat osteosarcoma cell line ROS 17/2.8) were expanded in alpha-Minimum Essential Medium (αMEM) containing 5.5mM glucose, 1mM pyruvate and 0.22mM aspartate (Gibco, cat#32561) and supplemented with 2mM GlutaMAX, 10% FBS, 100U/ml penicillin and 100μg/ml streptomycin.
Primary cell culture
Cell isolation
To isolate murine periosteum-derived cells (mPDCs), tibiae and femora were dissected, surrounding muscles were removed and the epiphyses were embedded in 5% low melting point agarose (SeaPlaque; Lonza, Verviers, Belgium) (van Gastel et al., 2012). mPDCs were isolated from these bones, using a sequential digestion with a mixture of collagenase (3mg/ml) and dispase (4mg/ml; all from Gibco; Life Technologies, Ghent, Belgium) at 37°C in αMEM, containing 5.5mM glucose, 1mM pyruvate and 0.22 mM aspartate; supplemented with 2mM GlutaMAX, 100U/ml penicillin and 100μg/ml streptomycin (Gibco). Cells obtained after the first 10 min digest were discarded, whereas cells obtained after the second digest of 50 min were passed through a 70μm nylon mesh and plated in αMEM medium, supplemented with 2mM GlutaMAX, 10% FBS, 100U/ml penicillin and 100μg/ml streptomycin, referred as growth medium.
Primary growth plate chondrocytes were isolated as previously reported (Stegen et al., 2019). In brief, chondrocytes were isolated from growth plates of the proximal tibia and distal femur of 5-day-old mice. Isolated growth plates were pre-digested by constant agitation for 30 min at room temperature with collagenase type II (1mg/ml; Gibco) dissolved in growth medium. The remaining growth plate fragments were digested in collagenase type II (2mg/ml) for 3 h at 37°C. The primary chondrocytes obtained following the second digest were filtered through a 40μm nylon mesh and seeded at a density of 3 × 104 cells/cm2.
Calvarial osteo-progenitors were isolated from 5-day-old mice as described previously (van Gastel et al., 2020). The cells were obtained by 6 sequential 15 min digestions of calvariae in PBS containing collagenase type II (1mg/ml) and dispase (2mg/ml). Cells isolated in fractions 2–6 were pooled and cultured in growth medium. To differentiate the obtained osteoprogenitors into more mature osteoblasts, cells were seeded at 3.5 × 10³ cells/cm2 and cultured for 3 days in osteogenic differentiation medium consisting of growth medium supplemented with 10mM β-glycerophosphate and 50μg/ml L-ascorbic acid (both from Sigma-Aldrich), whereafter they were trypsinized and seeded for experiment.
Method details
Cell culture
Treatment conditions
Primary skeletal cells or tumor cell lines were cultured in the presence of antimycin A (AMA) or vehicle as control. After one week, the cells were seeded at a density of 5.5 × 10³ cells/cm2 to investigate proliferation and survival in the presence or absence of AMA.
Concerning other mPDC treatments, pharmacological inhibitors were added either the day after mPDC isolation or the day after seeding mPDCs at 5.5 × 10³ cells/cm2. When performing nutrient deprivation, αMEM deprived of glucose, pyruvate or aspartate (custom-made media, Gibco) was added the day after seeding mPDCs at 5.5 × 10³ cells/cm2. The list and concentrations of chemicals used in this study are shown in Table S1.
Colony formation
For the in vitro fibroblastic colony-forming unit (CFU-F) assay, mPDCs were seeded at very low density (10 cells/cm2) in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, cat#21969) containing high glucose (25mM) and supplemented with 15% FBS, 2mM glutamine, 100U/ml penicillin and 100μg/ml streptomycin (all from Gibco). After 10 days, the colonies were fixed with a solution of periodate-lysine-paraformaldehyde, stained with methylene blue (Sigma-Aldrich) and the number of colonies containing at least 20 cells was quantified (van Gastel et al., 2012).
Differentiation assays
For in vitro osteogenic differentiation, mPDCs were seeded at 25 × 10³ cells/cm2 in growth medium and cultured for 3 days to reach full confluence. Cells were then switched to osteogenic differentiation medium consisting of growth medium supplemented with 10mM β-glycerophosphate and 50μg/ml L-ascorbic acid (both from Sigma-Aldrich). After 10 days, cultures were stained for alkaline phosphatase (ALP) activity or mineral deposition (Alizarin Red) (Tournaire et al., 2020; van Gastel et al., 2012) or used for RNA isolation.
For chondrogenic differentiation, mPDCs were seeded at 25 × 10³ cells/cm2 in growth medium and switched after 3 days to chondrogenic differentiation medium consisting of growth medium supplemented with 10ng/ml recombinant human transforming growth factor-β 1 (Peprotech, Rocky Hill, CT, USA), 50μg/ml L-ascorbic acid and 20μM Y-27632 (ROCK inhibitor, Axon Medchem, Groningen, The Netherlands). After 10 days, cultures were stained for glycosaminoglycans (Alcian Blue 8GX; Sigma-Aldrich) (Tournaire et al., 2020; van Gastel et al., 2012) or used for RNA isolation.
For adipogenic differentiation, mPDCs were seeded 10 × 10³ cells/cm2 in growth medium for 3 days and then switched to adipogenic differentiation medium consisting of growth medium supplemented with 10nM dexamethasone, 50μM indomethacin, 10μg/ml insulin and 500nM IBMX (all from Sigma-Aldrich). After 10 days cells were stained for presence of lipid droplets (Oil Red O; Sigma-Aldrich) (Tournaire et al., 2020; van Gastel et al., 2012) or used for RNA isolation.
Genetic targeting
To silence Mdh2, mPDCs were transduced, in the presence of 8μg/ml polybrene (Sigma-Aldrich, Diegem, Belgium) with a lentivirus carrying the gene-targeting shRNA (Sigma-Aldrich) the day after isolation. A lentivirus containing an empty vector (Sigma-Aldrich) was used as negative control. To silence Tet2, cells isolated from Tet2fl/fl mice and wild-type (Tet2wt/wt) littermates were transduced with a lentivirus carrying a Puro.Cre empty vector (Addgene, Watertown, MA, USA). After 72h, puromycin (2μg/ml; InvivoGen, San Diego, CA, USA) was added to the culture medium for 48h. Sequences of the shRNAs are listed in Table S2.
Flow cytometry analysis and cell sorting
Stem cell and progenitor markers
Freshly isolated or expanded mPDCs were suspended in Hank’s Balanced Salt Solution (HBSS from Gibco) supplemented with 2% FBS and 2mM EDTA. The cells were incubated for 15 min at 4°C with specific antibodies at the indicated concentration (Table S3). The stromal fraction of freshly isolated mPDCs was defined as CD31−/CD45−/Ter119−/CD51+, the skeletal stem cells (SSCs) as CD31−/CD45−/Ter119−/CD51+/CD90−/CD105−/BP1−/CD200+ and the pre-bone/cartilage/stromal progenitors (pre-BCSPs) as CD31−/CD45−/Ter119−/CD51+/CD90−/CD105−/BP1−/CD200− (Chan et al., 2015).
Cell viability
In vitro and ex vivo cell viability was evaluated by Annexin V allophycocyanin (1/20; APC) conjugate and 1μg/ml Propidium Iodide (AnxV-PI, both from Invitrogen, Thermo Fisher Scientific). The AnxV−PI− cell population was considered as viable.
Cell proliferation
To investigate proliferation, 10μM BrdU was added to the mPDCs for 12h and to the tumor cell lines for 4h and its incorporation was measured by using the FITC BrdU Flow Kit (BD Pharmingen, Erembodegem, Belgium) according to the manufacturer’s instructions.
All flow cytometry analyses were performed on a Gallios Flow Cytometer (Beckman Coulter, Brea, USA) and analyzed by Flowing Software (University of Turku, Turku, Finland).
RNA extraction and quantitative real-time (qRT)-PCR
Total RNA was extracted using the Nucleospin RNA isolation Kit (Macherey-Nagel GmbH, Duren, Germany) according to the manufacturer’s instructions. Reverse transcription was performed using Superscript II Reverse Transcriptase (Invitrogen, Thermo Fisher Scientific) and the analysis of quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) was performed on the StepOnePlus Real-Time PCR System (Applied Biosystems, Thermo Fisher Scientific) using specific forward and reverse oligonucleotide primers with SYBR Green or with fluorescent dye (FAM) and quencher (TAMRA) when a probe was used (Table S2). Expression levels were normalized for the expression of the murine housekeeping gene hypoxanthine-guanine phosphoribosyl transferase (mHprt) and analyzed using the 2−ΔΔCT method.
Western blot
Total cell lysates were obtained by lysing the cells in 62.5mM Tris buffer (pH 6.8) containing 10% glycerol, 2% SDS, 1x cOmplete proteinase inhibitor cocktail (Roche, Basel, Switzerland) and 1x PhosSTOP phosphatase inhibitor cocktail (Roche). To obtain the nuclear protein fraction, cells were lysed in a hypotonic buffer (20mM Hepes pH 7.9, 10mM KCl, 1.5mM MgCl2, 1mM EDTA, 0.5% NP40, 1mM DTT, supplemented with 1mM Na3VO4, 20mM NaF, 1mM PMSF, 5μg/ml aprotinin, 5μg/ml leupeptin and 0.33μg/ml antipain) for 15 min at 4°C, followed by mechanical disruption of the cell membranes. Nuclei were pelleted from the lysates by centrifugation and the pellet was resuspended in a nuclear extraction buffer (50mM Hepes pH 7.9, 500mM NaCl, 1% NP40, supplemented with 1mM PMSF, 5μg/ml aprotinin, 5μg/ml leupeptin and 0.33μg/ml antipain), sonicated and incubated for 15 min at 4°C. Protein concentrations were determined with the PierceTM BCA Protein Assay Kit (Thermo Fisher Scientific).
Proteins were separated by SDS–PAGE under reducing conditions and transferred to a nitrocellulose membrane (GE Healthcare). Membranes were blocked with 5% dry milk or bovine serum albumin (Sigma-Aldrich) in Tris-buffered saline with 0.1% Tween-20 for 60 min at room temperature and incubated overnight at 4°C with primary antibodies (Table S3). Signals were detected by enhanced chemiluminescence (Western Lightning Plus ECL; PerkinElmer, Waltham, MA, USA) after incubation with appropriate HRP-conjugated secondary antibodies (Table S3). Protein levels were quantified relative to loading control.
DNA methylation/hydroxymethylation analysis
5-hydroxymethylcytosine (5hmC) dot blot
Genomic DNA was isolated using DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany). Isolated DNA (2μg) was denatured in 0.4M NaOH and 10mM EDTA for 10 min at 100°C. The samples were loaded on Biotrans(+) Transfer Membrane (MP Biomedical, Irvine, CA, USA) by using a Minifold vacuum-assisted dot blot apparatus (Whatman, GE Healthcare, Chicago, IL, USA). The membrane was rinsed with 2x saline sodium citrate buffer and DNA was immobilized by UV crosslinking using the Stratalinker 1800 (Stratagene, San Diego, CA, USA). The membrane was then blocked with 5% dry milk in Tris-buffered saline with 0.1% Tween 20 for 60 min at room temperature and incubated overnight at 4°C with primary anti-5hmC antibody (Table S3). Signals were detected by enhanced chemiluminescence (Western Lightning Plus ECL; PerkinElmer) after incubation with HRP-conjugated Goat Anti-Rabbit antibodies (Dako, Agilent Technologies, Santa Clara, CA, USA). Loading of spotted DNA was analyzed by staining the membrane with 0.02% methylene blue.
5hmC ELISA
To quantify 5hmC levels in genomic DNA, a Global 5hmC quantification kit (Active Motif, La Hulpe, Belgium) was used according to manufacturer’s instructions.
DNA methylation/demethylation analysis
To analyze DNA methylation/demethylation in 3 pools of mPDCs treated with vehicle or AMA for 10 days, DNA was extracted from cell lysates using silica columns following manufacturer’s instructions (DNeasy Blood and Tissue kit, Qiagen), followed by RNASE H and Proteinase K treatment and an additional purification using solid phase reversible immobilisation (AMPure XP, Beckman Coulter). DNA was next converted using the oxidative bisulfite or normal bisulfite method as described previously (De Borre and Branco, 2021), and hybridized to Illumina mouse EPIC Beadchip arrays (Life & Brain, Bonn, Germany). The resultant.idat files were analyzed using the minfi package. Methylation state distributions were analyzed as an initial quality control. Next, low-quality CpGs were excluded, before background correction. Raw data from bisulfite-chip and oxidative bisulfite-chip were each batch-normalized using inter-array quantile normalization, and converted to beta values using the ENmix R package. The Infinium Mouse Methylation v1.0 A1 manifest file was used for CpG annotation. CpGs showing significant levels of cytosine hydroxymethylation were identified using limma as those containing significantly more (1% false-discovery rate) signal following bisulfite-chip versus oxidative bisulfite-chip across all 6 samples. Differential DNA methylation between AMA- and veh-treated cells at probes showing significant 5hmC levels was determined by fitting multiple linear models by weighted or generalized least squares, using limma.
Metabolic analyses
Oxygen consumption rate (OCR)
OCR was quantified by an XF24 analyzer (Seahorse Bioscience, North Billerica, MA, USA) on cells seeded on Seahorse XF24 tissue culture plates (50 × 10³ cells per well) in unbuffered DMEM medium (D5030) supplemented with 5mM glucose and 2mM glutamine, pH7.4 (all from Gibco). OCR was measured every 6 min during 96 min. At 24 min Oligomycin A (1.24μM) was added to inhibit ATP synthase reducing OCR linked to cellular ATP production. The uncoupler 2,4-dinitrophenol (DNP) (100μM) was added at 48 min to evaluate the maximum respiration of the cells. 1μM AMA was finally added to the cells at 72 min (all from Sigma-Aldrich). Values were normalized to DNA content.
Lactate and glucose
Lactate and glucose concentrations in culture medium were measured using a Beckman Coulter analyzer AU640 (Beckman Coulter) and normalized to DNA content.
Glycolytic flux
To measure glycolytic flux, cells were incubated in growth medium containing 0.4μCi/ml [5-3H]-D-glucose (PerkinElmer). After 2h, the culture medium was transferred into glass vials sealed with rubber caps and 3H2O was captured in hanging wells containing a Whatman paper soaked with H2O over a period of 48h at 37°C. Radioactivity was determined in the paper by liquid scintillation counting and normalized to protein content.
Mass spectrometry
To measure metabolites and coenzymes, cells were rinsed with ice-cold 0.9% NaCl solution and scraped in ice-cold extraction buffer (80% methanol in AD, supplemented with 2μM d27 myristic acid). The metabolites were collected in the supernatant fraction and measured using liquid chromatography-mass spectrometry (LC-MS). For medium analysis, 10μL medium was added to 990μL extraction buffer. After an overnight storage at −80°C, the solution was centrifuged and the supernatant was analyzed by LC-MS. To trace the contribution of glucose, glutamine, pyruvate or aspartate to other metabolites, cells were cultured for 72h in glucose-free, glutamine-free, pyruvate-free or aspartate-free αMEM (custom media, Gibco) supplemented with dialyzed FBS and 13C6-glucose, [3,4]-13C2-glucose, 13C5-glutamine, 13C3-pyruvate or 13C4-15N-aspartate (all from Cambridge Isotope Laboratories, Tewksbury, MA, USA). The incorporation of carbons derived from glucose, glutamine, pyruvate or aspartate was analyzed using LC-MS. LC-MS runs were performed by the VIB Metabolomics Core (Leuven, Belgium), analyzed using Xcalibur Software (ThermoFisher) and normalized to protein content. When referring to metabolite contribution in figures, we show either (i) the fractional contribution, calculated based on the following equation (Buescher et al., 2015):
where n is the number of C atoms in the metabolite, i represents the different mass isotopomers and m refers to the abundance of a certain mass or (ii) relevant mass distribution vectors, specified in the figure legends.
Mouse model of bone regeneration
In order to investigate the in vivo osteogenic potential of mPDCs, an ectopic bone formation model was used (Stegen et al., 2016). Briefly, 1 × 106 mPDCs were seeded on a 3 × 3 × 3 mm3 calcium phosphate-collagen scaffold (NuOssTM collagen, ACE Surgical Supply, Brockton, MA, USA). After overnight incubation, scaffolds were implanted subcutaneously on the back of 8-week old Rj:NMRI-Foxn1nu/nu mice, near blood vessels. Mice were anesthetized by peritoneal injection of a mixture of 100mg/kg ketamine and 15mg/kg xylazine in saline. After 8 weeks, scaffolds were isolated and bone formation was assessed by (immuno)histochemical stainings on paraffin- or cryo-sections.
To study cell survival and colony formation of the donor cells after in vivo implantation, the cells were labeled with CellTrackerTM Green CMFDA (Invitrogen, Thermo Fisher Scientific) prior to seeding them on the scaffold. Three days after implantation, the scaffolds were isolated and the cells released using a mixture of 3mg/ml collagenase type II and 4mg/ml dispase in αMEM for 20 min. CD31−/CD45−/Ter119−/CMFDA+ cells were sorted by flow cytometry (BD FACSAria III, BD Biosciences, Erembodegem Belgium). Cell survival was investigated on sorted cells using AnxV-PI flow-cytometry analysis. Colony formation was analyzed by seeding isolated donor cells at 500 cells/cm2 in growth medium without addition of AMA and culturing them for one week.
(Immuno)histochemistry
Isolated scaffolds were fixed overnight in 2% paraformaldehyde at 4°C and decalcified in EDTA (0.5M; pH 7,4) for 14 days at 4°C. Scaffolds implanted with mPDCs derived from C57BL/6 mice were dehydrated, embedded in paraffin and sectioned at 4 μm. Scaffolds implanted with mPDCs derived from C57BL/6-ACTb-eGFP mice were embedded in NEG-50 frozen section medium (Richard-Allen Scientific, Thermo Fisher Scientific), and sectioned with a cryostat at 7μm. Hematoxylin and eosin (H&E) staining as well as immunohistochemical stainings were performed as described previously (Stegen et al., 2016; Stiers et al., 2019; Tournaire et al., 2020). Antibodies used are listed in Table S3. Images were taken on a Zeiss Axioplan 2 light microscope (Carl Zeiss Microscopy GmbH, Jena, Germany). Quantification of the newly formed bone in the implanted scaffolds was performed by manual outlining areas of bone and total tissue. The quantification of the area and number of blood vessels relative to the scaffold area was performed by counting or outlining CD31-positive vessels with a clear lumen. These histomorphometric analyses were performed using Axiovision software (v6.1.0). The quantification of cells that were double-positive for β-actin-GFP and Osterix was performed manually by using the ImageJ software (NIH, Bethesda, MD, USA; https://imagej.nih.gov/ij/).
Quantification and statistical analysis
Data are presented as mean ± SD and analyzed by two-tailed Student’s t-test or one-way, two-way, three-way ANOVA with Tukey-Kramer post-hoc test using the GraphPad Prism software (GraphPad Software, San Diego, CA, USA). The results of two-way and three-way ANOVA are shown in Table S4. The analysis of DNA methylation/demethylation was performed using Fisher’s exact test and Wilcoxon signed rank test with continuity correction. The number of biological replicates are represented by n values. For immunoblots, representative images are shown of at least three independent experiments. Differences were considered statistically significant at p < 0.05.
Acknowledgments
We wish to thank Ingrid Stockmans for technical assistance. G.C. acknowledges funding from Fund for Scientific Research – Flanders (FWO: G.0A42.16 and G.0B3418) and from KU Leuven (C24/17/077). S.L. is a doctoral fellow from the FWO (1S46318N). S.S. is a postdoctoral fellow from the FWO (12H5917N). G.R. has received consecutive PhD fellowships from Kom op tegen Kanker and FWO (1137117N and 1137119N). The Hercules Fund of the FWO is acknowledged for their financial support of the project I013518N. S.-M.F. acknowledges funding from the European Research Council under the ERC Consolidator Grant Agreement no. 771486 – MetaRegulation, FWO – Research Projects (G098120N, G088318N), KU Leuven – FTBO, and Fonds Baillet Latour. Schematics included in the figures were created using BioRender.com and Servier Medical Art templates.
Author contributions
G.T., N.v.G., and G.C. conceptualized and designed the study. G.T., S.L., and S.T. performed the in vitro experiments. G.T. and S.S. performed the in vivo experiments. K.M. performed histology. G.T., G.R., G.E., P.C., S.-M.F., B.T., and B.G. contributed to the design, execution, and interpretation of the metabolic or epigenetic experiments. G.T. and G.C. interpreted the data and wrote the manuscript. G.C. supervised the study.
Declaration of interests
The authors declare no competing interests.
Published: July 26, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111105.
Supplemental information
Data and code availability
-
•
The raw data of DNA methylation/demethylation have been deposited on GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Raw data of other experiments have been deposited on Mendeley and are publicly available as of the date of publication. DOI is listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
References
- Anderson K.A., Madsen A.S., Olsen C.A., Hirschey M.D. Metabolic control by sirtuins and other enzymes that sense NAD(+), NADH, or their ratio. Biochim. Biophys. Acta Bioenerg. 2017;1858:991–998. doi: 10.1016/j.bbabio.2017.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansó E., Weinberg S.E., Diebold L.P., Thompson B.J., Malinge S., Schumacker P., Liu X., Zhang Y., Shao Z., Steadman M., et al. The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat. Cell Biol. 2017;19:614–625. doi: 10.1038/ncb3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X.R., Ong S.E., Goldberger O., Peng J., Sharma R., Thompson D.A., Vafai S.B., Cox A.G., Marutani E., Ichinose F., et al. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. Elife. 2016;5:e10575. doi: 10.7554/eLife.10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco P., Cao X., Frenette P.S., Mao J.J., Robey P.G., Simmons P.J., Wang C.Y. The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat. Med. 2013;19:35–42. doi: 10.1038/nm.3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K., Wang T., Chen W., Freinkman E., Abu-Remaileh M., Sabatini D. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell. 2015;162:540–551. doi: 10.1016/j.cell.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisbach C.M., Hass D.T., Robbings B.M., Rountree A.M., Sadilek M., Sweet I.R., Hurley J.B. Succinate can shuttle reducing power from the hypoxic retina to the O2-rich pigment epithelium. Cell Rep. 2020;31:107606. doi: 10.1016/j.celrep.2020.107606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaschke K., Ebata K.T., Karimi M.M., Zepeda-Martínez J.A., Goyal P., Mahapatra S., Tam A., Laird D.J., Hirst M., Rao A., et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature. 2013;500:222–226. doi: 10.1038/nature12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth M.J., Branco M.R., Ficz G., Oxley D., Krueger F., Reik W., Balasubramanian S. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science. 2012;336:934–937. doi: 10.1126/science.1220671. [DOI] [PubMed] [Google Scholar]
- Buescher J.M., Antoniewicz M.R., Boros L.G., Burgess S.C., Brunengraber H., Clish C.B., DeBerardinis R.J., Feron O., Frezza C., Ghesquiere B., et al. A roadmap for interpreting (13)C metabolite labeling patterns from cells. Curr. Opin. Biotechnol. 2015;34:189–201. doi: 10.1016/j.copbio.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C., Seo E., Chen J., Lo D., McArdle A., Sinha R., Tevlin R., Seita J., Vincent-Tompkins J., Wearda T., et al. Identification and specification of the mouse skeletal stem cell. Cell. 2015;160:285–298. doi: 10.1016/j.cell.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani E.T., Pell V.R., Gaude E., Aksentijević D., Sundier S.Y., Robb E.L., Logan A., Nadtochiy S.M., Ord E.N.J., Smith A.C., et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colnot C. Skeletal cell fate decisions within periosteum and bone marrow during bone regeneration. J. Bone Miner. Res. 2009;24:274–282. doi: 10.1359/jbmr.081003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Borre M., Branco M.R. Oxidative bisulfite sequencing: an experimental and computational protocol. Methods Mol. Biol. 2021;2198:333–348. doi: 10.1007/978-1-0716-0876-0_26. [DOI] [PubMed] [Google Scholar]
- Diebold L.P., Gil H.J., Gao P., Martinez C.A., Weinberg S.E., Chandel N.S. Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nat. Metab. 2019;1:158–171. doi: 10.1038/s42255-018-0011-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl F.F., Lewis C.A., Fiske B.P., Vander Heiden M.G. Cellular redox state constrains serine synthesis and nucleotide production to impact cell proliferation. Nat. Metab. 2019;1:861–867. doi: 10.1038/s42255-019-0108-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendt S.M., Bell E.L., Keibler M.A., Olenchock B.A., Mayers J.R., Wasylenko T.M., Vokes N.I., Guarente L., Heiden M.G.V., Stephanopoulos G. Reductive glutamine metabolism is a function of the alpha-ketoglutarate to citrate ratio in cells. Nat. Commun. 2013;4:2236. doi: 10.1038/ncomms3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bermudez J., Baudrier L., La K., Zhu X.G., Fidelin J., Sviderskiy V.O., Papagiannakopoulos T., Molina H., Snuderl M., Lewis C.A., et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat. Cell Biol. 2018;20:775–781. doi: 10.1038/s41556-018-0118-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui D., Sullivan L., Luengo A., Hosios A., Bush L., Gitego N., Davidson S., Freinkman E., Thomas C., Vander Heiden M. Environment dictates dependence on mitochondrial complex I for NAD+ and aspartate production and determines cancer cell sensitivity to metformin. Cell Metab. 2016;24:716–727. doi: 10.1016/j.cmet.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer A., Dematteo R., Venneti S., Finley L., Lu C., Judkins A., Rustenburg A., Grinaway P., Chodera J., Cross J., Thompson C. Hypoxia induces production of L-2-hydroxyglutarate. Cell Metab. 2015;22:304–311. doi: 10.1016/j.cmet.2015.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer A.M., Finley L.W.S. Metabolic signatures of cancer cells and stem cells. Nat. Metab. 2019;1:177–188. doi: 10.1038/s42255-019-0032-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer A.M., Wang B., Liu H., Shah H., Carmona-Fontaine C., Rustenburg A.S., Salah S., Gunner M.R., Chodera J.D., Cross J.R., Thompson C.B. L-2-Hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nat. Chem. Biol. 2017;13:494–500. doi: 10.1038/nchembio.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy B.E., Sharif T., Martell E., Dai C., Kim Y., Lee P.W., Gujar S.A. NAD(+) salvage pathway in cancer metabolism and therapy. Pharmacol. Res. 2016;114:274–283. doi: 10.1016/j.phrs.2016.10.027. [DOI] [PubMed] [Google Scholar]
- Liu S., Fu S., Wang G., Cao Y., Li L., Li X., Yang J., Li N., Shan Y., Cao Y., et al. Glycerol-3-phosphate biosynthesis regenerates cytosolic NAD(+) to alleviate mitochondrial disease. Cell Metab. 2021;33:1974–1987.e9. doi: 10.1016/j.cmet.2021.06.013. [DOI] [PubMed] [Google Scholar]
- Luengo A., Li Z., Gui D.Y., Sullivan L.B., Zagorulya M., Do B.T., Ferreira R., Naamati A., Ali A., Lewis C.A., et al. Increased demand for NAD(+) relative to ATP drives aerobic glycolysis. Mol. Cell. 2020;81:691–707.e6. doi: 10.1016/j.molcel.2020.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt S.Y., Vander Heiden M.G. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- Moran-Crusio K., Reavie L., Shih A., Abdel-Wahab O., Ndiaye-Lobry D., Lobry C., Figueroa M., Vasanthakumar A., Patel J., Zhao X., et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham W., Clish C., Yang Y., Loscalzo J. Hypoxia-mediated increases in L-2-hydroxyglutarate coordinate the metabolic response to reductive stress. Cell Metab. 2015;22:291–303. doi: 10.1016/j.cmet.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira S.L., Grãos M., Rodrigues A.S., Anjo S.I., Carvalho R.A., Oliveira P.J., Arenas E., Ramalho-Santos J. Inhibition of mitochondrial complex III blocks neuronal differentiation and maintains embryonic stem cell pluripotency. PLoS One. 2013;8:e82095. doi: 10.1371/journal.pone.0082095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacchetti B., Funari A., Michienzi S., Di Cesare S., Piersanti S., Saggio I., Tagliafico E., Ferrari S., Robey P.G., Riminucci M., Bianco P. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell. 2007;131:324–336. doi: 10.1016/j.cell.2007.08.025. [DOI] [PubMed] [Google Scholar]
- Spinelli J.B., Rosen P.C., Sprenger H.G., Puszynska A.M., Mann J.L., Roessler J.M., Cangelosi A.L., Henne A., Condon K.J., Zhang T., et al. Fumarate is a terminal electron acceptor in the mammalian electron transport chain. Science. 2021;374:1227–1237. doi: 10.1126/science.abi7495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegen S., Laperre K., Eelen G., Rinaldi G., Fraisl P., Torrekens S., Van Looveren R., Loopmans S., Bultynck G., Vinckier S., et al. HIF-1α metabolically controls collagen synthesis and modification in chondrocytes. Nature. 2019;565:511–515. doi: 10.1038/s41586-019-0874-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegen S., van Gastel N., Eelen G., Ghesquière B., D'Anna F., Thienpont B., Goveia J., Torrekens S., Van Looveren R., Luyten F., et al. HIF-1α promotes glutamine-mediated redox homeostasis and glycogen-dependent bioenergetics to support postimplantation bone cell survival. Cell Metab. 2016;23:265–279. doi: 10.1016/j.cmet.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiers P., Stegen S., van Gastel N., Van Looveren R., Torrekens S., Carmeliet G. Inhibition of the oxygen sensor PHD2 enhances tissue-engineered endochondral bone formation. J. Bone Miner. Res. 2019;34:333–348. doi: 10.1002/jbmr.3599. [DOI] [PubMed] [Google Scholar]
- Sullivan L., Gui D., Hosios A., Bush L., Freinkman E., Vander Heiden M. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell. 2015;162:552–563. doi: 10.1016/j.cell.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapa M., Dallmann G. Role of coenzymes in cancer metabolism. Semin. Cell Dev. Biol. 2020;98:44–53. doi: 10.1016/j.semcdb.2019.05.027. [DOI] [PubMed] [Google Scholar]
- Thienpont B., Steinbacher J., Zhao H., D'Anna F., Kuchnio A., Ploumakis A., Ghesquière B., Van Dyck L., Boeckx B., Schoonjans L., et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature. 2016;537:63–68. doi: 10.1038/nature19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournaire G., Stegen S., Giacomini G., Stockmans I., Moermans K., Carmeliet G., van Gastel N. Nestin-GFP transgene labels skeletal progenitors in the periosteum. Bone. 2020;133:115259. doi: 10.1016/j.bone.2020.115259. [DOI] [PubMed] [Google Scholar]
- van Gastel N., Stegen S., Eelen G., Schoors S., Carlier A., Daniëls V.W., Baryawno N., Przybylski D., Depypere M., Stiers P.J., et al. Lipid availability determines fate of skeletal progenitor cells via SOX9. Nature. 2020;579:111–117. doi: 10.1038/s41586-020-2050-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gastel N., Torrekens S., Roberts S.J., Moermans K., Schrooten J., Carmeliet P., Luttun A., Luyten F.P., Carmeliet G. Engineering vascularized bone: osteogenic and proangiogenic potential of murine periosteal cells. Stem Cell. 2012;30:2460–2471. doi: 10.1002/stem.1210. [DOI] [PubMed] [Google Scholar]
- Varum S., Momčilović O., Castro C., Ben-Yehudah A., Ramalho-Santos J., Navara C.S. Enhancement of human embryonic stem cell pluripotency through inhibition of the mitochondrial respiratory chain. Stem Cell Res. 2009;3:142–156. doi: 10.1016/j.scr.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasan K., Werner M., Chandel N.S. Mitochondrial metabolism as a target for cancer therapy. Cell Metab. 2020;32:341–352. doi: 10.1016/j.cmet.2020.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye D., Guan K.L., Xiong Y. Metabolism, activity, and targeting of D- and L-2-hydroxyglutarates. Trends Cancer. 2018;4:151–165. doi: 10.1016/j.trecan.2017.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Xie C., Lin A.S., Ito H., Awad H., Lieberman J.R., Rubery P.T., Schwarz E.M., O'Keefe R.J., Guldberg R.E. Periosteal progenitor cell fate in segmental cortical bone graft transplantations: implications for functional tissue engineering. J. Bone Miner. Res. 2005;20:2124–2137. doi: 10.1359/JBMR.050806. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The raw data of DNA methylation/demethylation have been deposited on GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Raw data of other experiments have been deposited on Mendeley and are publicly available as of the date of publication. DOI is listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.