

Abstract
Background
Carcass backfat thickness (BFT), carcass lean percentage (CLP) and carcass fat percentage (CFP) are important to the commercial pig industry. Nevertheless, the genetic architecture of BFT, CLP and CFP is still elusive. Here, we performed a genome-wide association study (GWAS) based on specific-locus amplified fragment sequencing (SLAF-seq) to analyze seven fatness-related traits, including five BFTs, CLP, and CFP on 223 four-way crossbred pigs.
Results
A total of 227, 921 highly consistent single nucleotide polymorphisms (SNPs) evenly distributed throughout the genome were used to perform GWAS. Using the mixed linear model (MLM), a total of 20 SNP loci significantly related to these traits were identified on ten Sus scrofa chromosomes (SSC), of which 10 SNPs were located in previously reported quantitative trait loci (QTL) regions. On SSC7, two SNPs (SSC7:29,503,670 and rs1112937671) for average backfat thickness (ABFT) exceeded 1% and 10% Bonferroni genome-wide significance levels, respectively. These two SNP loci were located within an intron region of the COL21A1 gene, which was a protein-coding gene that played an important role in the porcine backfat deposition by affecting extracellular matrix (ECM) remodeling. In addition, based on the other three significant SNPs on SSC7, five candidate genes, ZNF184, ZNF391, HMGA1, GRM4 and NUDT3 were proposed to influence BFT. On SSC9, two SNPs for backfat thickness at 6–7 ribs (67RBFT) and one SNP for CLP were in the same locus region (19 kb interval). These three SNPs were located in the PGM2L1 gene, which encoded a protein that played an indispensable role in glycogen metabolism, glycolysis and gluconeogenesis as a key enzyme. Finally, one significant SNP on SSC14 for CLP was located within the PLBD2 gene, which participated in the lipid catabolic process.
Conclusions
A total of two regions on SSC7 and SSC9 and eight potential candidate genes were found for fatness-related traits in pigs. The results of this GWAS based on SLAF-seq will greatly advance our understanding of the genetic architecture of BFT, CLP, and CFP traits. These identified SNP loci and candidate genes might serve as a biological basis for improving the important fatness-related traits of pigs.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12864-022-08827-8.
Keywords: Pigs, SLAF-seq, GWAS, Carcass backfat thickness, Carcass lean percentage, Carcass fat percentage
Background
Carcass backfat thickness (BFT), carcass lean percentage (CLP), and carcass fat percentage (CFP) are complex quantitative traits and the most important economic factors in the pig industry. The excessive deposition of fat in pigs leads to low feed efficiency and is not favored by consumers. It is well known that CLP is strongly negatively correlated with BFT [1, 2]. In the past few years, researchers have worked on increasing carcass lean percentage and decreasing backfat thickness by advanced molecular breeding methods to improve production efficiency. To date, 3,402 QTLs and 265 QTLs associated with fatness traits and carcass lean percentage have been accumulated in the pig QTL database (http://www.animalgenome.org/cgi-bin/QTLdb/index, Dec 27, 2021).
Domestic pigs show large phenotypic variation, which is ascribed to approximately 10,000 years of natural and artificial selection [3]. At present, Chinese native pigs differ from Western commercial pigs in terms of their fatness phenotypes. Western commercial breeds, such as Large White, Landrace, and Duroc with fast growth and a high lean percentage are widely distributed all over the world. Conversely, Chinese indigenous breeds, such as the Saba pig, are fat-type pig breeds without intensive artificial selection. The native Saba pig is well known for its high prolificacy, superior meat quality, and strong resistance to harsh environments, which is widely distributed in Yunnan Province, China. However, the shared disadvantage of the indigenous breeds containing Saba pig is the excessive deposition of fat resulting in a slow growth rate and low feed conversion rate. In general, Chinese native pigs have a lean percentage of less than 45%, which is extremely different from Western commercial pigs, which typically have a lean percentage of more than 60% [4]. Using Chinese and Western pig breeds as parents, the hybrid offspring show different extreme phenotypes in fatness and more genetic variation.
Currently, remarkable advances in fatness-related traits have been made, and many related QTLs and genes have been reported [5–12]. However, identifying exact quantitative trait loci locations and new candidate genes is still a challenge. For complex traits, such as BFT, CLP and CFP, large-scale analysis is necessary to detect trait-associated SNPs. Genome-wide association study (GWAS) [13] represents a powerful approach to correlate SNPs and functional genes with quantitative traits, and has been widely applied in important economic traits of pigs, including carcass [14–19], meat quality [14, 15, 19, 20], growth [15, 21], immunity [22], and reproductive traits [23]. For species with larger genomes, such as pigs, the cost of GWAS using whole-genome sequencing (WGS) is still high at present. However, GWAS based on SNP array technology can only detect known SNP loci but not new loci. In view of these limitations, specific-locus amplified fragment sequencing (SLAF-seq) was developed, which is based on a reduced representation library and high-throughput sequencing. This technique has several distinguishing characteristics: deep sequencing, reduced sequencing costs, optimized marker efficiency, and applicability to large populations [24]. Based on the reference genome, a pre-experiment of SLAF-seq is carried out to select the appropriate combination of enzyme digestion, so as to produce a sufficient number of tags covering the whole genome and effectively avoid repeated sequences. The number of chosen fragments can be used for individualized research purposes, so as to maintain the balance between tag density and population size [24]. GWAS based on SLAF-seq was successfully used to identify SNPs associated with important economic traits in chickens, ducks, geese, and rabbits [25–30]. Additionally, genotyping and genetic structure analysis were also successfully applied in pigs using the SLAF-seq genotyping method [31, 32].
Here, we examined 223 four-way crossbreds with Landrace, Yorkshire, Duroc and Saba pigs as the hybrid parents (Saba pigs as the hybrid females) raised under the same environmental conditions for BFT, CLP and CFP traits. Subsequently, SLAF-seq was employed to perform GWAS and recover potential alleles controlling these traits. To our knowledge, this was the first SLAF-seq-based GWAS to identify SNP loci and candidate genes linked to porcine economic traits. The results provided a basis for the molecular marker-assisted breeding and improvement of the fatness-related traits in pigs.
Results
Phenotype description and correlation among traits
Table 1 summarized the statistical information on the seven fatness-related traits. The mean values for backfat thickness at the shoulder (SBFT), backfat thickness at the last rib (LRBFT), backfat thickness at the last lumbar (LBFT), average backfat thickness (ABFT), backfat thickness at 6–7 ribs (67RBFT), CLP, and CFP were 4.4609 cm, 2.8903 cm, 2.8371 cm, 3.399 cm, 3.6014 cm, 54.4451% and 27.1509%, respectively. All trait distributions basically conformed to the normal distribution (Fig. S1). The phenotypic correlation coefficients for the seven traits were shown in Fig. S2. Significantly positive correlations were found among five BFT traits (r > 0, p < 0.001), between five BFT traits and CFP (r > 0, p < 0.001). Five BFT traits and CFP were significantly negatively correlated with CLP (r < 0, p < 0.001). Furthermore, CLP showed the strongest negatively correlated with CFR (r = -0.86, p < 0.001), while ABFT showed the strongest positive correlations with SBFT, LRBFT and LBFT (r = 0.85, p < 0.001).
Table 1.
Descriptive statistics of seven fatness-related traits
| Traits | Na | Minb | Maxc | Mean | SDd | CVe |
|---|---|---|---|---|---|---|
| Backfat thickness at the shoulder, SBFT (cm) | 223 | 2.000 | 7.334 | 4.4609 | 0.9780 | 21.9442 |
| Backfat thickness at last rib, LRBFT (cm) | 223 | 1.188 | 5.540 | 2.8903 | 0.7842 | 27.1320 |
| Backfat thickness at last lumbar, LBFT (cm) | 223 | 0.974 | 6.232 | 2.8371 | 0.8176 | 28.8182 |
| Average backfat thickness, ABFT (cm) | 223 | 1.827 | 6.248 | 3.3990 | 0.7334 | 21.5764 |
| Backfat thickness at 6–7 ribs, 67RBFT (cm) | 223 | 0.986 | 5.652 | 3.6014 | 0.8214 | 22.8071 |
| Carcass lean percentage, CLP (%) | 223 | 40.14 | 70.55 | 54.4451 | 4.4748 | 8.2189 |
| Carcass fat percentage, CFP (%) | 223 | 10.97 | 39.37 | 27.1509 | 5.1876 | 19.1065 |
a Number of samples
b Minimum
c Maximum
d Standard deviation
e Coefficient of variation
Identification of SLAFs and SNPs
According to the selection principle of the enzyme digestion scheme, two restriction enzymes, RsaI and HaeIII, were selected as enzyme combinations for developing SLAF tags, and the sequence with the length of 314–344 bp was defined as SLAF tags. A total of 1,109.92 million reads were obtained from all individuals. Average Q30 and GC contents were 90.74% and 44.83%, respectively. Similar to the number of expected SLAFs, a total of 1,552,377 SLAF tags (an average of 331,608 SLAFs for each individual) were identified from all individuals with sequencing to an 11.94 average depth. Furthermore, 245,734 SLAFs were identified across the whole genome through genomic mapping, of which 230,239 polymorphic SLAF tags (Table S1). In addition, Oryza sativa indica was used as a control during sequencing. The results showed that the percentage of digestion normally and paired-end mapped reads of control were 90.77% and 95.4%, respectively, indicating that the SLAF-seq process was normal. The density distribution of SLAFs was calculated throughout the pig genome and was shown in Fig. 1A.
Fig. 1.

SLAF and SNP density distribution on chromosomes of the pig genome. A The number of SLAFs within 1 Mb window size. B The number of SNPs within 1 Mb window size. The horizontal axis (X-axis) shows the chromosome length (Mb). Color index indicates the number of labels
After genomic mapping and SNP calling, a total of 10,784,484 SNPs were discovered using all individuals. A series of quality control filtering of SNPs was performed to identify 227,921 highly consistent SNPs used in the further analysis based on the selection criteria (integrity > 0.8; MAF > 0.05). The density distribution of SNPs was calculated throughout the pig genome and was shown in Fig. 1B. Almost all of the genome’s non-overlapped 1 Mb regions contained SNPs, which indicated that the data was reliable.
Genome-wide association study and identification of candidate genes
As population stratification might affect GWAS, quantile–quantile (Q-Q) plots of all traits were drawn. The observed P-value calculated by the association study fit the expected ones, which suggested that the population stratification was well-corrected and the association analysis using MLM was reliable. The Q-Q plot of each trait was shown following the Manhattan plot of the corresponding trait (Fig. 2). A total of 20 SNPs were identified as significant (P ≤ 1.0 × 10–5) for the traits investigated (Table 2). The phenotypic variation explained (PVE) by the significant SNPs was from 4.588 to 16.855. Among the significant SNPs, only one SNP on SSC7 (SSC7:29,503,670 for ABFT) exceeded the 1% genome-wide significance level (P = 3.26 × 10–8). Another SNP in proximity to this SNP (rs320451735 for ABFT) exceeded the 10% genome-wide significance level (P = 2.21 × 10–7). Among the detected SNPs, four, three, two, seven, five, six and four SNPs, were significantly associated with SBFT, LRBFT, LBFT, ABFT, 67RBFT, CLP and CFP traits, respectively. These SNPs detected were distributed in ten Sus scrofa chromosomes (SSC), including SSC3, SSC7, SSC8, SSC9, SSC10, SSC11, SSC12, SSC13, SSC14 and SSC15. In this study, 33 genes located within 100 kb upstream and downstream of these significant SNPs were considered potential candidate genes (Table 2).
Fig. 2.
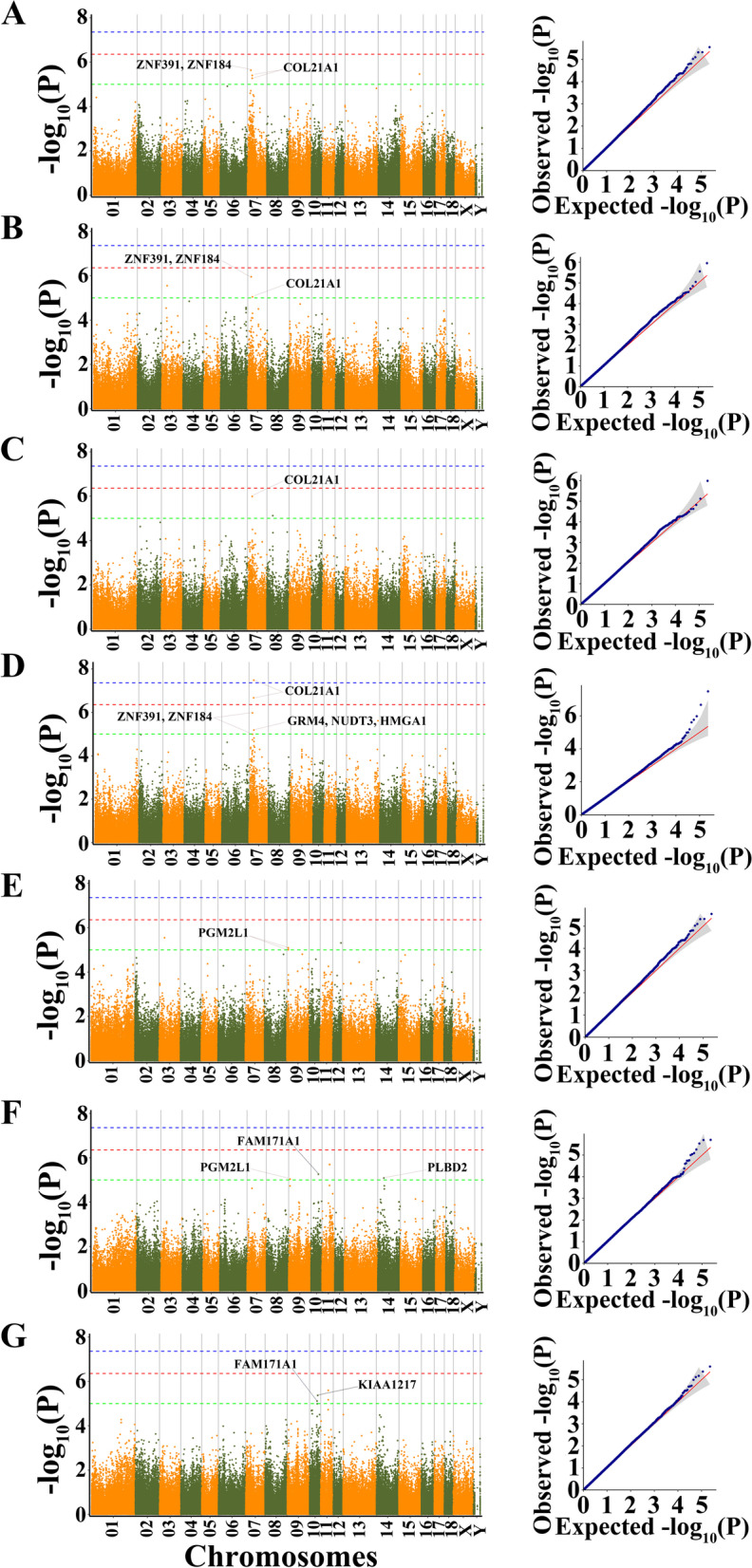
Manhattan plots and QQ plots for seven fatness-related traits using MLM model of GEMMA software. A SBFT B LRBFT C LBFT D ABFT E 67RBFT F CLP G CFP. Negative log10 (P) values of the filtered high-quality SNPs were plotted against their genomic positions. The dashed lines of green, orange and blue correspond to the Bonferroni-corrected thresholds of P = 1.00 × 10–5 (-log10(P) = 5), P = 4.39 × 10–7(-log10(P) = 6.36) and P = 4.39 × 10–8 (-log10(P) = 7.36), respectively
Table 2.
The significant SNPs and candidate genes for seven fatness-related traits
| Traita | SNPb | Pos (bp)c | MAFd | P-valuee | -log10Pf | Allele | PEV (%)g | Genesh | Distancei |
|---|---|---|---|---|---|---|---|---|---|
| SBFT | SSC7:21,392,136 | 0.11 | 2.31 × 10–6 | 5.64 | T/C | 11.182 | POM121L2 | U:50,008 | |
| ZNF391 | Intron | ||||||||
| ZNF184 | D:21,214 | ||||||||
| rs1112937671 | SSC7:29,486,003 | 0.12 | 5.29 × 10–6 | 5.28 | T/C | 10.845 | COL21A1 | Intron | |
| SSC7:29,503,670 | 0.14 | 3.94 × 10–6 | 5.4 | T/C | 10.549 | COL21A1 | Intron | ||
| rs319334375 | SSC15:117,022,248 | 0.2 | 3.42 × 10–6 | 5.47 | T/C | 8.982 | BARD1 | D:25,591 | |
| ENSSSCG00000048197 | U:30,544 | ||||||||
| LRBFT | rs320451735 | SSC7:21,466,553 | 0.12 | 1.11 × 10–6 | 5.96 | C/A | 15.435 | ZNF391 | U:66,090 |
| ZNF184 | U:31,328 | ||||||||
| SSC7:29,503,670 | 0.14 | 8.98 × 10–6 | 5.05 | T/C | 12.749 | COL21A1 | Intron | ||
| rs332294996 | SSC3:34,308,396 | 0.06 | 2.80 × 10–6 | 5.55 | G/A | 8.402 | NA | NA | |
| LBFT | SSC7:29,503,670 | 0.14 | 1.02 × 10–6 | 5.99 | T/C | 14.006 | COL21A1 | Intron | |
| rs322063364 | SSC8:36,710,706 | 0.08 | 7.50 × 10–6 | 5.12 | G/T | 11.22 | NA | NA | |
| ABFT | AEMK02000449.1:179,574 | 0.12 | 1.73 × 10–6 | 5.76 | C/G | 6.138 | OR4P4L | U:56,372 | |
| OR4C13L | U:69,382 | ||||||||
| OR5D14 | D:96,469 | ||||||||
| OR5D13L | D:83,741 | ||||||||
| SSC7:21,392,136 | 0.11 | 1.01 × 10–5 | 5.00 | T/C | 12.82 | POM121L2 | U:50,008 | ||
| ZNF391 | Intron | ||||||||
| ZNF184 | D:21,214 | ||||||||
| rs320451735 | SSC7:21,466,553 | 0.12 | 1.05 × 10–6 | 5.98 | C/A | 14.59 | ZNF391 | U:66,090 | |
| ZNF184 | U:31,328 | ||||||||
| rs1112937671 | SSC7:29,486,003 | 0.12 | 2.12 × 10–7* | 6.67* | T/C | 15.416 | COL21A1 | Intron | |
| SSC7:29,503,670 | 0.14 | 3.26 × 10–8 ** | 7.49** | T/C | 16.855 | COL21A1 | Intron | ||
| rs341689410 | SSC7:30,292,654 | 0.14 | 6.58 × 10–6 | 5.18 | G/A | 12.635 | GRM4 | U:13,533 | |
| SMIM29 | D:36,896 | ||||||||
| NUDT3 | D:27,653 | ||||||||
| HMGA1 | D:27,800 | ||||||||
| rs706606912 | SSC13:197,751,407 | 0.08 | 2.46 × 10–6 | 5.61 | G/A | 8.815 | MRPS6 | Intron | |
| SLC5A3 | U:32,711 | ||||||||
| 67RBFT | AEMK02000449.1:179,574 | 0.12 | 4.84 × 10–6 | 5.32 | C/G | 7.11 | OR4P4L | U:56,372 | |
| OR4C13L | U:69,382 | ||||||||
| OR5D14 | D:96,469 | ||||||||
| OR5D13L | D:83,741 | ||||||||
| rs341161412 | SSC9:8,763,434 | 0.38 | 8.00 × 10–6 | 5.1 | C/G | 10.151 | LIPT2 | D:74,668 | |
| KCNE3 | D:47,288 | ||||||||
| PGM2L1 | UPS | ||||||||
| P4HA3 | U:95,660 | ||||||||
| ENSSSCG00000042110 | U:60,242 | ||||||||
| rs343149423 | SSC9:8,763,503 | 0.38 | 9.62 × 10–6 | 5.02 | A/G | 10.01 | LIPT2 | D:74,599 | |
| KCNE3 | D:47,219 | ||||||||
| PGM2L1 | UPS | ||||||||
| P4HA3 | U:95,729 | ||||||||
| ENSSSCG00000042110 | U:60,311 | ||||||||
| rs332294996 | SSC3:34,308,396 | 0.06 | 2.81 × 10–6 | 5.55 | G/A | 5.416 | NA | NA | |
| rs344553616 | SSC12:50,896,936 | 0.11 | 4.80 × 10–6 | 5.32 | G/A | 9.537 | PITPNM3 | 3’UTR | |
| PIMREG | DOWNS | ||||||||
| AIPL1 | D:6,502 | ||||||||
| CLP | AEMK02000598.1:813,208 | 0.38 | 2.95 × 10–6 | 5.53 | T/C | 14.179 | OR1J4L | DOWNS | |
| OR1J4L | D:88,150 | ||||||||
| SSC9:8,744,721 | 0.08 | 9.18 × 10–6 | 5.04 | C/T | 13.264 | LIPT2 | D:93,381 | ||
| KCNE3 | D:66,001 | ||||||||
| PGM2L1 | Intron | ||||||||
| P4HA3 | U:76,947 | ||||||||
| ENSSSCG00000042110 | U:41,529 | ||||||||
| SSC14:38,533,013 | 0.25 | 8.13 × 10–6 | 5.09 | G/A | 4.588 | SDS | U:19,133 | ||
| LHX5 | U:75,836 | ||||||||
| SDSL | U:48,858 | ||||||||
| PLBD2 | Intron | ||||||||
| DTX1 | D:27,847 | ||||||||
| RASAL1 | D:65,256 | ||||||||
| rs320036825 | SSC10:46,524,739 | 0.45 | 5.52 × 10–6 | 5.26 | G/A | 8.98 | FAM171A1 | Intron | |
| rs329489266 | SSC11:47,266,057 | 0.5 | 2.01 × 10–6 | 5.7 | C/G | 11.239 | NA | NA | |
| rs81261044 | SSC11:47,266,119 | 0.42 | 1.98 × 10–6 | 5.7 | G/A | 10.719 | NA | NA | |
| CFP | rs320036825 | SSC10:46,524,739 | 0.45 | 7.61 × 10–6 | 5.12 | G/A | 10.873 | FAM171A1 | Intron |
| rs329489266 | SSC11:47,266,057 | 0.5 | 6.72 × 10–6 | 5.17 | C/G | 9.643 | NA | NA | |
| rs81261044 | SSC11:47,266,119 | 0.42 | 2.51 × 10–6 | 5.6 | G/A | 10.736 | NA | NA | |
| SSC10:50,888,840 | 0.4 | 4.26 × 10–6 | 5.37 | T/G | 9.072 | KIAA1217 | Intron |
a Description of the traits is in Table 1SBFT Backfat thickness at the shoulder LRBFT Backfat thickness at the last rib LBFT Backfat thickness at the last lumbar ABFT Average backfat thickness 67RBFT Backfat thickness at 6–7 ribs CLP Carcass lean percentage CFP Carcass fat percentage
b SNP rs ID from Ensembl
c Positions of the significant SNP according to the Sus Scrofa Build 11.1 assembly SSC Sus Scrofa chromosome
d Minor Allele Frequency
e Genome-wide significant associations are underlined e,f*and** represented the 10% and 1% genome-wide significance, respectively
g Phenotypic Variation Explain
h The gene located with 100 kb upstream and downstream of the significant SNP
i UPS Upstream (5’ of the gene) DOWNS Downstream (3’ of the gene) U/D represented the gene located upstream or downstream of the SNP (Intergenic region)
Interestingly, nine significant SNPs were identified to be associated with at least two fatness-related traits (Table S2). Among them, four SNPs on SSC7 were identified to be associated with at least two BFTs (Table S2). Two SNPs (SSC7:21,392,136 and rs320451735) on SSC7 have located apart 74.417 kb each other, while another two (rs1112937671 and SSC7:29,503,670) on SSC7 have located apart 17.667 kb each other, which meant that the responsible gene may locate near here. The results showed that the nearest genes of the SSC7:21,392,136 and rs320451735 were ZNF184 and ZNF391, while rs1112937671 and SSC7:29,503,670 were located within an intron region of the COL21A1 gene (Table S2, Table 2). Furthermore, a significant SNP (rs341689410) on SSC7 had located downstream 13.533 kb, upstream 27.653 kb and 27.8 kb of GRM4, NUDT3 and HMGA1, respectively. On SSC9, two adjacent SNPs (rs341161412 and rs343149423) associated with 67RBFT and another adjacent SNP (SSC9:8,744,721) related to CLP were located in a region between 8.744 Mb and 8.763 Mb (19 kb interval). These three SNPs were located in the PGM2L1 gene. (Table 2). In addition, it was interesting that two significant SNPs on SSC11 and one significant SNP on SSC10 were found to have pleiotropic effects on CLP and CFP. The locus on SSC10 was located in the intron region of the FAM171A1 gene. Nevertheless, there were no genes found nearby the two loci on SSC11 (Table S2, Table 2). Finally, two significant SNPs (SSC14:38,533,013 for CLP and SSC10:50,888,840 for CFP) were located in the intron region of PLBD2 and KIAA1217, respectively (Table 2).
Comparison with previously mapped QTL in pigs
To evaluate whether QTLs associated with seven fatness-related traits in this study replicate any previously known QTLs, the Pig Quantitative Trait Locus (QTL) Database (Pig QTLdb, https://www.animalgenome.org/cgi-bin/QTLdb/SS/index) was searched based on SNP and QTL locations. A total of 20 SNPs were identified in the study, of which 10 SNPs were located in previously reported QTL regions in pigs. The remaining 10 SNPs had not been included in any previously reported QTLs that were associated with BFT, CLP and CFP of pigs. On SSC7, a total of five SNPs significantly associated with BFT were found, which were located in a region from 21.39 to 30.29 Mb (8.9 Mb interval). This region was located within 20 previously reported QTLs associated with BFT. Among five significant SNPs, three adjacent SNPs (from 29.49 to 30.29 Mb, 0.8 Mb interval) were located within the 26 BFT-related QTLs that had been previously reported. On SSC9, a total of three adjacent SNPs, including two SNPs associated with 67RBFT and one SNP associated with CLP were located in a region between 8,745 and 8,764 kb (19 kb interval), which was located in one formerly acknowledged QTL (from 0.1 to 11.1 Mb) associated with LRBFT and CLP. Besides, one SNP locus (rs344553616) on SSC12 associated with 67RBFT was located within a known QTL region (47.9–59.4 Mb) related to backfat thickness at 10 ribs (10RBFT), another locus (rs319334375) on SSC15 associated with SBFT was located within a formerly reported QTL (57.5–120.1 Mb) related to SBFT. These results were shown in Table S3.
GO annotation of candidate genes
The result of GO annotation showed that ZNF184 and ZNF391 were mainly involved in regulation of transcription, DNA-template and nucleic acid binding. The cellular components of COL21A1 were collagen trimer and extracellular matrix. PGM2L1 mainly participated in carbohydrate metabolic process, phosphorylation and glucose-1,6-bisphosphate synthase activity. The PLBD2 gene was involved in lipid catabolic process and hydrolase activity. GO annotation results of other genes were shown in Table S4.
Discussion
Comparison of SLAF-seq with other genotyping methods
In the present study, we performed a GWAS based on SLAF-seq to screen and select candidate SNPs for fatness-related traits on 223 four-way crossbreds with Landrace, Yorkshire, Duroc and Saba pigs as the hybrid parents. A total of 1,552,377 SLAFs were predicted, and 10,784,484 SNPs were obtained (11.94-fold sequencing depth), and 227, 921 highly consistent SNPs were evenly distributed over the entire genome (As shown in Fig. 1B, almost all of the genome’s non-overlapped 1 Mb regions contained SNPs) were identified to perform GWAS, which were nearly three times the number of SNPs on Illumina PorcineSNP80 Genotyping BeadChip. SLAF-seq has a more significant advantage for genome-wide association studies compared with SNP arrays and can produce more information on genomic variation and allow the detection of novel SNPs on the genome. Currently, SLAF-seq was successfully applied to pig genotyping and identified a large number of new mutation sites [31, 32]. Besides, SLAF-seq can be applied not only to species with a reference genome but also to those without a reference genome. So far, SLAF-seq was successfully used to create a genetic map for common carp (Cyprinus carpio L.), soybean and orchardgrass [24, 33, 34]. However, compared with WGS, which is another major genotyping method that has been used over the last several years, SLAF-seq as a reduced representation sequencing method hasn’t covered the whole genome SNPs. For large populations, WGS is prohibitively expensive, while SLAF-seq can reduce sequencing costs. In the study, the pre-experiment of SLAF-seq was based on the pig reference genome to screen the appropriate enzyme digestion combination (RsaI and HaeIII), which ensured the number and depth of SLAF tags covering the whole genome (Table S1, Fig. 1A), and effectively avoided repeated sequences. Considering the huge differences in genomic variation between Chinese and Western pig breeds, SLAF-seq used in the GWAS study for large populations was a better choice in the current higher sequencing cost and had great potential for further study in more pig breeds. Therefore, the SLAF-seq method could be considered a more competitive choice in pig genome research at present.
Comparison of QTLs identified in this study with findings of previous studies
In the study, a genome-wide association study was performed for seven fatness-related traits to identify significant SNPs in pigs. Of the 20 significant SNPs associated with BFT, CLP and CFP traits found in four-way crossbred pigs in our study, except for ten significant SNPs that had not been reported in previous studies, other SNPs were located in previously reported QTLs. On SSC7, a total of five SNPs significantly associated with BFTs were located in a region (from 21.39 to 30.29 Mb, 8.9 Mb interval), which was located within 20 previously reported QTLs associated with BFT (Table S3), which span more than 16.4 Mb. We further narrowed the interval of QTLs for BFT in the study. Among five significant SNPs, three adjacent SNPs (from 29.49 to 30.29 Mb, 0.8 Mb interval) were located in the 26 BFT-related QTLs that had been previously reported. Qiao et al. found that the strongest association was between a 750 kb region (between 34. 67 and 35. 42 Mb for Sscrofa10.2) on SSC7 and backfat thickness at the first rib [7]. Gozalo-Marcilla et al. found significant genome-wide associations with backfat thickness for 13 SNPs in genomic regions on SSC7 at 30 Mb (30.10–30.89 Mb) in three lines [35]. Thus, the 8.9 Mb region (between 21.39 and 30.29 Mb) on SSC7, especially the 0.8 Mb region (between 29.49 and 30.29 Mb) might be important QTLs related to BFT. On SSC9, there were three adjacent SNPs, including two SNPs associated with 67RBFT and one SNP associated with CLP, located in the region of 8,745 to 8,764 kb (19 kb interval), which was located in one previously reported QTL (from 0.1 to 11.1 Mb) associated with LRBFT and CLP. Gozalo-Marcilla et al. also found significant genome-wide associations with backfat thickness for 51 SNPs in genomic regions on SSC11 at 8 Mb (7.03–9.57 Mb) in four lines [35].
Candidate genes for fatness-related traits
The GWAS result showed that four significant SNPs on SSC7 were associated with at least two BFTs. The nearest genes of two SNPs (SSC7:21,392,136 and rs320451735) were zinc finger protein 184 (ZNF184) and zinc finger protein 391 (ZNF391). The result of GO annotation showed that ZNF184 and ZNF391 participated in regulation of transcription, DNA-template and nucleic acid binding. As is known, zinc-finger proteins (ZFPs) represent the largest transcription factor family in mammals [36]. These two genes are members of the ZFP transcription factor family, which are essential for controlling a variety of growth and development processes through nucleic acid binding and transcription activation [37]. An increasing number of ZFPs involved in adipogenesis had been discovered, such as zinc finger protein 423 (Zfp423) [38, 39], Zfp467 [40, 41], Zfp521 [42], ZNF395 [43]. Besides, a large number of ZFPs played roles in preadipocyte differentiation, such as ZNF638 [44], GATA2 [45], GATA3 [45] and SLUG [46], which belong to the subfamily of C2C2-type zinc finger proteins, which are characterized by a highly conserved zinc finger DNA binding domain. It is known that ZNF184 and ZNF391 also are the C2H2-type zinc finger transcriptional factors. According to their structure and function, it was inferred that ZNF184 and ZNF391 might be involved in regulating adipogenesis as C2H2-type transcriptional factors. In addition, another two SNPs (rs1112937671 and SSC7:29,503,670) on SSC7 were located within an intron region of collagen type XXI alpha 1 chain (COL21A1). The result of GO annotation showed that the cellular component of COL21A1 was collagen trimer and extracellular matrix (ECM). It has been reported that COL21A1 is VAdomain-containing collagen with a domain structure and which is a part and the smallest of the FACIT family of collagen expressed in various tissues [47, 48]. By connecting them to other matrix components or cells, the co-expression of collagen XXI and collagen I in tissues and muscles plays a significant role in the organization of interstitial collagen fibrils [47, 49]. The collagen protein has an important role in ECM remodeling [50], which is associated with the modulation of adipogenesis during adipose tissue expansion [51]. Several research showed that collagen I genes (COL1A1 and COL1A2) influenced porcine fat deposition by affecting ECM remodeling [52, 53]. Thus, it was inferred that COL21A1 might play an important role in porcine backfat deposition by affecting ECM remodeling and should be considered a strong candidate gene for porcine BFT traits.
Furthermore, a significant SNP (rs341689410) on SSC7 had located upstream 13.5 kb, downstream 27.6 kb and 27.8 kb of glutamate metabotropic receptor 4 (GRM4), nudix hydrolase 3 (NUDT3) and high mobility group protein HMG-I (HMGA1), respectively. GO annotation result showed that GRM4 was involved in activation of MAPK activity (Table S4). The MAPK pathway was demonstrated in numerous studies to be crucial for adipogenesis [54, 55]. So GRM4 might play an important role in adipogenesis by activation of MAPK activity. A study found that variants of NUDT3 have been linked to alterations in human body mass index values [56]. As above, the two genes might be potential candidate genes for the SSC7 locus. Besides, given that HMGA1 is functionally related to fat metabolism and that several of its variations have been linked to the backfat thickness [7], it may be a leading candidate gene for the locus. Some research suggested that HMGA1 could act as an IGF1 activity regulator to control the uptake of glucose [57] and could bind to PPARG, a crucial regulator of adipocyte differentiation and glucose homeostasis [58]. On SSC9, two adjacent SNPs (rs341161412 and rs343149423) associated with 67RBFT and another adjacent SNP (SSC9:8,744,721) related to CLP were located in a region between 8.744 and 8.763 Mb (19 kb). These three SNPs were located in the phosphoglucomutase 2 like 1 (PGM2L1) gene. GO annotation result showed that PGM2L1 was mainly involved in carbohydrate metabolic process, phosphorylation and glucose-1,6-bisphosphate synthase activity. It is known that PGM2L1, a member of a distinct family of α-phosphohexomutases widely distributed in prokaryotes, is able to produce glucose-1,6-bisphosphate, a crucial metabolic regulator. According to a study, PGM2L1 encodes a crucial enzyme that is essential for glycogen metabolism, glycolysis, and gluconeogenesis [59]. Therefore, PGM2L1 should be considered a potential candidate gene for BFT and CLP traits.
For the traits of CLP and CFP, one significant SNP locus (rs320036825) onSSC10 was located in an intron region of family with sequence similarity 171 member A1 (FAM171A1). The result of GO annotation showed that the FAM171A1 gene participated in regulation of cell shape (Table S4). The FAM171A1 gene is a member of the family of sequence similarities, which is responsible for encoding the APCN/FAM171A1 protein. APCN/FAM171A1 is an evolutionarily conserved 98 kDa transmembrane type I glycoprotein, which is expressed in various cells and participates in the regulation of cytoskeleton dynamics, thereby regulating cell shape [60]. A study found that the related gene FAM110B was associated with the lean meat percentage of pigs [12]. According to its function and extensive expression in various cells, perhaps FAM171A1 could be considered a potential candidate gene for the porcine CLP and CFP traits. Additionally, one significant SNP on SSC14 associated with CLP was located within the intron region of phospholipase B-like 2 (PLBD2). GO annotation result indicated that the PLBD2 gene participated in lipid catabolic process. Wang et al. found that PLBD2 was predominantly involved in the lipid catabolic process, which was consistent with the favoured selection for fatness in Chinese pigs [61]. Thus, the PLBD2 gene should be considered a strong candidate gene for porcine CLP trait. Finally, one SNP (SSC10:50,888,840) related to CFP trait was located in an intron region of KIAA1217. A study found that the KIAA1217 gene showed differences in promoter methylation and mRNA expression in the omental visceral adipose tissue between non-obese and obese individuals [62]. Perhaps, the KIAA1217 gene might also be used as a potential candidate gene for the porcine CFP trait.
However, these identified loci and genes need to be further verified in more pig populations, and their functions also need to be validated by more biological experiments in pigs.
Conclusions
We performed GWAS based on SLAF-seq for seven fatness-related traits in 223 four-way crossbred pigs. The sequencing results showed that the SLAF-seq method could be considered a more competitive choice in pig genome research at present. Two regions on SSC7 (from 21.39 to 30.29 Mb, 8.9 Mb) and SSC9 (from 8,745 to 8,764 kb, 19 kb) were found to be associated with fatness-related traits. Furthermore, a total of eight candidate genes, including COL21A1, ZNF184, ZNF391, HMGA1, GRM4, NUDT3, PGM2L1, and PLBD2 were proposed for fatness-related traits. The GWAS using SLAF-seq data provided new insight into the genetic characteristics of fatness-related traits in pigs. Moreover, these identified SNP loci and candidate genes might serve as a biological basis for improving these important fatness-related traits of pigs.
Materials and methods
Experimental animals and phenotypes
A total of 223 four-way crossbred pigs (108 males and 115 females, DSYLS) used in this study were produced with 7 hybrid boars (Duroc × Saba, DS) and 37 hybrid sows (Yorkshire × (Landrace × Saba), YLS) as the parents. All the animals were reared under the same nutritional and environmental conditions until slaughtered (105.25 ± 15.75 kg) at the pigs and broilers breeding farm in Chuxiong City, Yunnan Province, China. Ear tissue samples were collected from 223 crossbreds.
In the study, the studied phenotypes were BFT, CLP and CFP traits. BFT traits included backfat thickness at the shoulder (SBFT), backfat thickness at the last rib (LRBFT), backfat thickness at the last lumbar (LBFT), average backfat thickness (ABFT), and backfat thickness at 6–7 ribs (67RBFT). The SBFT was the backfat thickness at the thickest point over the shoulder. The LRBFT was the backfat thickness at the last rib. The LBFT was the backfat thickness at the last lumbar. The 67RBFT was the backfat thickness between the 6th and 7th ribs. SBFT, LRBFT, LBFT and 67RBFT were measured using the vernier caliper. The ABFT was calculated as the average of three measurements: SBFT, LRBFT, and LBFT. The left side of each carcass was dissected by separating bone, muscle, fat, and skin. Each component was individually weighed and recorded as bone weight (BW), muscle weight (MW), fat weight (FW) and skin weight (SW). CLP and CFP were calculated as follows:
The MEANS procedure of SAS (SAS Institute, Inc., Cary, NC) was used to generate descriptive statistics for studied traits. The sample distribution was visualized as a frequency distribution histogram using the R package “ggpubr”. The phenotypic correlation was visualized as a correlation heatmap by the R function “PerformanceAnalytics”.
SLAF library construction and sequencing
Genomic DNA was extracted from ear tissue samples. Total DNA was extracted by the phenol–chloroform extraction method, with concentration and purity measured using the NanodropTM 2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA) and electrophoresis. A simulated restriction enzyme digestion was carried out on the current pig genome (Sscrofa 11.1, ftp://ftp.ensembl.org/pub/release-102/) to identify expected SLAF yield, avoid repetitive SLAFs, and obtain the relatively uniform distribution of restriction fragments in the genome. As a result, genomic DNA was digested with RsaI and HaeIII restriction enzyme combinations. Meanwhile, to assess the experimental procedure, Oryza sativa indica (http://rapdb.dna.affrc.go.jp/) was used as a control for evaluating the effectiveness of enzyme digestion and paired-end mapped reads. In brief, SLAF library construction and sequencing for each individual was conducted as described previously [24] with slight modifications: DNA fragments of 314–344 base pair (bp) were selected as SLAFs and used for paired-end sequencing by Illumina HiSeq 2500 system (Illumina, Inc., San Diego, CA, USA) at Beijing Biomarker Technologies Corporation. Raw sequencing reads were identified by dual-indexing [63] and classified to each sample.
Genome mapping, SNP calling and filtering
Raw paired-end reads were mapped to the pig reference genome (Sscrofa 11.1_102) using BWA software [64]. Local realignments were conducted, and SNPs were detected using GATK software [65]. To ensure the accuracy of the SNPs identified using GATK, SAMtools software also was used to detect SNPs [66]. The intersection of SNPs detected using the two methods was designated as the final SNPs. Ultimately, highly consistent SNPs were obtained for GWAS analysis by filtering according to minor allele frequency (MAF: 0.05) and integrity (int: 0.8) using PLINK 2 [67].
Genome-wide association study (GWAS)
Total filtered SNPs (integrity > 0.8, MAF > 0.05) detected from 223 accessions were used for GWAS. GEMMA software [68] was used for association analysis between traits and SNPs. The mixed linear model (MLM) formula of GEMMA software was as follows:
where y was the phenotype, X was the genotype, W (fixed effect) was the matrix of population structure calculated by the ADMIXTURE software [69], and Z was the matrix of kinship relationship calculated using GCTA software [70], Wα and Xβ were fixed effects, Zμ and ε were random effects. Finally, an association result could be obtained for each variation site. Since the Bonferroni correction (BC) method [68] for multiple testing was too conservative and only a few significant SNPs were detected, markers with adjusted − log10 (P) ≥ 5 (control threshold) were regarded to be significant SNPs for fatness-related traits. Based on the number of SNPs analyzed (n = 227,921), the threshold p-value for genome-wide 1% and 10% significance were 4.39 × 10–8 (0.01/227,921) and 4.39 × 10–7 (0.1/227,921), respectively. Considering the complexity of the traits, markers that passed the threshold score or above the threshold − log10(P) were held to be significantly associated with the target trait.
Identification and functional enrichment analysis of candidate genes
Based on the reference [71–73], the genes within 100 kb up- or down-stream of significant associated SNPs were considered trait-associated potential candidate genes. The relevant information of genes in 100 kb windows surrounding each significant SNP was downloaded from the Ensembl Sscrofa11.1 database (www.ensembl.org). GO annotation of candidate genes was then performed using Gene Ontology Consortium (http://geneontology.org).
Supplementary Information
Additional file 1: Figure S1. Frequency distribution histogram for seven fatness-related traits, including five BFTs, CLP and CFP. A SBFT. B LRBFT. C LBFT. D ABFT. E 67RBFT. F CLP. G CFP.
Additional file 2: Figure S2. The phenotypic correlation for seven fatness-related traits, including five BFTs, CLP and CFP. The values in the box represented the phenotypic correlation of the traits. Negative values represented negative correlation, and positive values represented positive correlation. *significant at P<0.05, **significant at P<0.01, ***significant at P<0.001. All of the phenotypic correlation coefficients were significant with P < 0.05.
Additional file 3: Table S1. Distribution of SLAF tags and polymorphism SLAF tags on Sus Scrofa chromosomes.
Additional file 4: Table S2. The information of shared SNPs for fatness-related traits.
Additional file 5: Table S3. Comparison of significant SNPs with previously reported QTLs from the pig QTL database and newly significant SNPs for fatness-related traits.
Additional file 6: Table S4. The description and GO annotation of the gene with 100 kb upstream and downstream of the significant SNP.
Acknowledgements
Not applicable.
Abbreviations
- GWAS
Genome-wide association study
- SLAF-seq
Specific-locus amplified fragment sequencing
- SNP
Single nucleotide polymorphism
- QTL
Quantitative trait loci;
- BFT
Carcass backfat thickness
- SBFT
Backfat thickness at the shoulder
- LRBFT
Backfat thickness at the last rib
- LBFT
Backfat thickness at the last lumbar
- ABFT
Average backfat thickness
- 67RBFT
Backfat thickness at 6–7 ribs
- CLP
Carcass lean percentage
- CFP
Carcass fat percentage
- SSC
Sus scrofa Chromosome
- MAF
Minor Allele Frequency
- Q-Q plot
Quantile–quantile plot
- MLM
Mixed linear model
- PVE
Phenotypic variation explain
- GO
Gene ontology
- WGS
Whole-genome sequencing
- ZFP
Zinc-finger protein
- ECM
Extracellular matrix
Authors’ contributions
SXL and YCP conceived and designed the experiments. DWY, XYW, XXD and MLL determined the phenotypic data and collected the sample. HYW performed the experiment and processed and analyzed the data. HS and QC assisted with the processing of data. HYW and XYW wrote the manuscript that was subsequently revised by YCP and SXL. All authors have read and approved the final manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (U1402266), Yunnan Swine Industry Technology System Program (2020KJTX0016) and Yunnan Province Important National Science & Technology Specific Projects (202102AE090039). These funding agencies played no role in the design of the study, data collection, analysis and interpretation, or in writing the manuscript.
Availability of data and materials
The Genome sequencing raw data was deposited in NCBI’s SRA database (https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi? view = studies&f = study&term = &go = Go; Accession: SRP376933).
Declarations
Ethics approval and consent to participate
All animals used in this study were used according to the guidelines for the care and use of experimental animals established by the Ministry of Agriculture and Rural Affairs of China. The ethics committee of Yunnan Agricultural University (YNAU, Kunming, China) approved the entire study. We confirmed that the study was conducted in accordance with ARRIVE guidelines 2.0 [74].
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Huiyu Wang and Xiaoyi Wang contributed equally to this work.
Contributor Information
Yuchun Pan, Email: panyuchun1963@aliyun.com.
Shaoxiong Lu, Email: shxlu_ynau@163.com.
References
- 1.Hoa VB, Seo HW, Seong PN, Cho SH, Kang SM, Kim YS, Moon SS, Choi YM, Kim JH, Seol KH. Back-fat thickness as a primary index reflecting the yield and overall acceptance of pork meat. Anim Sci J. 2021;92(1):e13515. doi: 10.1111/asj.13515. [DOI] [PubMed] [Google Scholar]
- 2.Knecht D, Duziński K. The effect of sex, carcass mass, back fat thickness and lean meat content on pork ham and loin characteristics. Arch Anim Breed. 2016;59(1):51–57. doi: 10.5194/aab-59-51-2016. [DOI] [Google Scholar]
- 3.Ramos-Onsins SE, Burgos-Paz W, Manunza A, Amills M. Mining the pig genome to investigate the domestication process. Heredity (Edinb) 2014;113(6):471–484. doi: 10.1038/hdy.2014.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.China national commission of animal genetic resources. Animal genetic resources in China pigs. Beijing: Chinese Agriculture Press; 2011.
- 5.Ai H, Ren J, Zhang Z, Ma J, Guo Y, Yang B, Huang L. Detection of quantitative trait loci for growth- and fatness-related traits in a large-scale White Duroc × Erhualian intercross pig population. Anim Genet. 2012;43(4):383–391. doi: 10.1111/j.1365-2052.2011.02282.x. [DOI] [PubMed] [Google Scholar]
- 6.Jiang Y, Tang S, Wang C, Wang Y, Qin Y, Wang Y, Zhang J, Song H, Mi S, Yu F, et al. A genome-wide association study of growth and fatness traits in two pig populations with different genetic backgrounds. J Anim Sci. 2018;96(3):806–816. doi: 10.1093/jas/skx038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qiao R, Gao J, Zhang Z, Li L, Xie X, Fan Y, Cui L, Ma J, Ai H, Ren J, et al. Genome-wide association analyses reveal significant loci and strong candidate genes for growth and fatness traits in two pig populations. Genet Sel Evol. 2015;47:17. doi: 10.1186/s12711-015-0089-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo Y, Huang Y, Hou L, Ma J, Chen C, Ai H, Huang L, Ren J. Genome-wide detection of genetic markers associated with growth and fatness in four pig populations using four approaches. Genet Sel Evol. 2017;49(1):21. doi: 10.1186/s12711-017-0295-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Z, Chen Z, Diao S, Ye S, Wang J, Ning G, Yuan X, Chen Z, Zhang H, Li J. Identifying the complex genetic architecture of growth and fatness traits in a Duroc pig population. J Integr Agr. 2021;20(6):1607–1614. doi: 10.1016/S2095-3119(20)63264-6. [DOI] [Google Scholar]
- 10.Guo Y, Qiu H, Xiao S, Wu Z, Yang M, Yang J, Ren J, Huang L. A genome-wide association study identifies genomic loci associated with backfat thickness, carcass weight, and body weight in two commercial pig populations. J Appl Genet. 2017;58(4):499–508. doi: 10.1007/s13353-017-0405-6. [DOI] [PubMed] [Google Scholar]
- 11.Yang Q, Wu P, Wang K, Chen D, Zhou J, Ma J, Li M, Xiao W, Jiang A, Jiang Y. SNPs associated with body weight and backfat thickness in two pig breeds identified by a genome-wide association study. Genomics. 2019;111(6):1583–1589. doi: 10.1016/j.ygeno.2018.11.002. [DOI] [PubMed] [Google Scholar]
- 12.Zhou S, Ding R, Meng F, Wang X, Zhuang Z, Quan J, Geng Q, Wu J, Zheng E, Wu Z, et al. A meta-analysis of genome-wide association studies for average daily gain and lean meat percentage in two Duroc pig populations. BMC Genomics. 2021;22(1):12. doi: 10.1186/s12864-020-07288-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JP, Hirschhorn JN. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9(5):356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 14.Herault F, Damon M, Cherel P, Le Roy P. Combined GWAS and LDLA approaches to improve genome-wide quantitative trait loci detection affecting carcass and meat quality traits in pig. Meat Sci. 2018;135:148–158. doi: 10.1016/j.meatsci.2017.09.015. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y, Zhang J, Gong H, Cui L, Zhang W, Ma J, Chen C, Ai H, Xiao S, Huang L, et al. Genetic correlation of fatty acid composition with growth, carcass, fat deposition and meat quality traits based on GWAS data in six pig populations. Meat Sci. 2019;150:47–55. doi: 10.1016/j.meatsci.2018.12.008. [DOI] [PubMed] [Google Scholar]
- 16.Eusebi PG, Gonzalez-Prendes R, Quintanilla R, Tibau J, Cardoso TF, Clop A, Amills M. A genome-wide association analysis for carcass traits in a commercial Duroc pig population. Anim Genet. 2017;48(4):466–469. doi: 10.1111/age.12545. [DOI] [PubMed] [Google Scholar]
- 17.Chen D, Wu P, Yang Q, Wang K, Zhou J, Yang X, Jiang A, Shen L, Xiao W, Jiang Y. Genome-wide association study for backfat thickness at 100 kg and loin muscle thickness in domestic pigs based on genotyping by sequencing. Physiol Genomics. 2019;51(7):261–266. doi: 10.1152/physiolgenomics.00008.2019. [DOI] [PubMed] [Google Scholar]
- 18.Li LY, Xiao SJ, Tu JM, Zhang ZK, Zheng H, Huang LB, Huang ZY, Yan M, Liu XD, Guo YM. A further survey of the quantitative trait loci affecting swine body size and carcass traits in five related pig populations. Anim Genet. 2021;52(5):621–632. doi: 10.1111/age.13112. [DOI] [PubMed] [Google Scholar]
- 19.Sato S, Uemoto Y, Kikuchi T, Egawa S, Kohira K, Saito T, Sakuma H, Miyashita S, Arata S, Kojima T, et al. SNP- and haplotype-based genome-wide association studies for growth, carcass, and meat quality traits in a Duroc multigenerational population. BMC Genet. 2016;17:60. doi: 10.1186/s12863-016-0368-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X, Xiong X, Yang J, Zhou L, Yang B, Ai H, Ma H, Xie X, Huang Y, Fang S, et al. Genome-wide association analyses for meat quality traits in Chinese Erhualian pigs and a Western Duroc × (Landrace × Yorkshire) commercial population. Genet Sel Evol. 2015;47:44. doi: 10.1186/s12711-015-0120-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruan D, Zhuang Z, Ding R, Qiu Y, Zhou S, Wu J, Xu C, Hong L, Huang S, Zheng E, et al. Weighted single-step GWAS identified candidate genes associated with growth traits in a Duroc pig population. Genes (Basel). 2021;12(1):117. doi: 10.3390/genes12010117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dauben CM, Pröll-Cornelissen MJ, Heuß EM, Appel AK, Henne H, Roth K, Schellander K, Tholen E, Große-Brinkhaus C. Genome-wide associations for immune traits in two maternal pig lines. BMC Genomics. 2021;22(1):1–15. doi: 10.1186/s12864-021-07997-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang Y, Tang S, Xiao W, Yun P, Ding X. A genome-wide association study of reproduction traits in four pig populations with different genetic backgrounds. Asian-Australas J Anim sci. 2020;33(9):1400. doi: 10.5713/ajas.19.0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun X, Liu D, Zhang X, Li W, Liu H, Hong W, Jiang C, Guan N, Ma C, Zeng H, et al. SLAF-seq: an efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE. 2013;8(3):e58700. doi: 10.1371/journal.pone.0058700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li F, Liu J, Liu W, Gao J, Lei Q, Han H, Yang J, Li H, Cao D, Zhou Y. Genome-wide association study of body size traits in Wenshang Barred chickens based on the specific-locus amplified fragment sequencing technology. Anim Sci J. 2021;92(1):e13506. doi: 10.1111/asj.13506. [DOI] [PubMed] [Google Scholar]
- 26.Melak S, Wang Q, Tian Y, Wei W, Zhang L, Elbeltagy A, Chen J. Identification and validation of marketing weight-related SNP markers using SLAF sequencing in male Yangzhou Geese. Genes (Basel). 2021;12(8):1203. doi: 10.3390/genes12081203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang W, Zhang T, Zhang G, Wang J, Han K, Wang Y, Zhang Y. Genome-wide association study of antibody level response to NDV and IBV in Jinghai yellow chicken based on SLAF-seq technology. J Appl Genet. 2015;56(3):365–373. doi: 10.1007/s13353-014-0269-y. [DOI] [PubMed] [Google Scholar]
- 28.Wang WH, Wang JY, Zhang T, Wang Y, Zhang Y, Han K. Genome-wide association study of growth traits in Jinghai Yellow chicken hens using SLAF-seq technology. Anim Genet. 2019;50(2):175–176. doi: 10.1111/age.12346. [DOI] [PubMed] [Google Scholar]
- 29.Xi Y, Xu Q, Huang Q, Ma S, Wang Y, Han C, Zhang R, Wang J, Liu H, Li L. Genome-wide association analysis reveals that EDNRB2 causes a dose-dependent loss of pigmentation in ducks. BMC Genomics. 2021;22(1):381. doi: 10.1186/s12864-021-07719-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang X, Deng F, Wu Z, Chen SY, Shi Y, Jia X, Hu S, Wang J, Cao W, Lai SJ. A genome-wide association study identifying genetic variants associated with growth, carcass and meat quality traits in rabbits. Animals (Basel). 2020;10(6):1068. doi: 10.3390/ani10061068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Z, Wei S, Li H, Wu K, Cai Z, Li D, Wei W, Li Q, Chen J, Liu H, et al. Genome-wide genetic structure and differentially selected regions among Landrace, Erhualian, and Meishan pigs using specific-locus amplified fragment sequencing. Sci Rep. 2017;7(1):10063. doi: 10.1038/s41598-017-09969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qin M, Li C, Li Z, Chen W, Zeng Y. Genetic diversities and differentially selected regions between Shandong indigenous pig breeds and western pig breeds. Front Genet. 2020;10:1351. doi: 10.3389/fgene.2019.01351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qi Z, Huang L, Zhu R, Xin D, Liu C, Han X, Jiang H, Hong W, Hu G, Zheng H, et al. A high-density genetic map for soybean based on specific length amplified fragment sequencing. PLoS ONE. 2014;9(11):e114349. doi: 10.1371/journal.pone.0114349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao X, Huang L, Zhang X, Wang J, Yan D, Li J, Tang L, Li X, Shi T. Construction of high-density genetic linkage map and identification of flowering-time QTLs in orchardgrass using SSRs and SLAF-seq. Sci Rep. 2016;6(1):29345. doi: 10.1038/srep29345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gozalo-Marcilla M, Buntjer J, Johnsson M, Batista L, Diez F, Werner CR, Chen CY, Gorjanc G, Mellanby RJ, Hickey JM, et al. Genetic architecture and major genes for backfat thickness in pig lines of diverse genetic backgrounds. Genet Sel Evol. 2021;53(1):76. doi: 10.1186/s12711-021-00671-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ganss B, Jheon A. Zinc finger transcription factors in skeletal development. Crit Rev Oral Biol Med. 2004;15(5):282–297. doi: 10.1177/154411130401500504. [DOI] [PubMed] [Google Scholar]
- 37.Leon O, Roth M. Zinc fingers: DNA binding and protein-protein interactions. Biol Res. 2000;33(1):21–30. doi: 10.4067/S0716-97602000000100009. [DOI] [PubMed] [Google Scholar]
- 38.Gupta RK, Arany Z, Seale P, Mepani RJ, Ye L, Conroe HM, Roby YA, Kulaga H, Reed RR, Spiegelman BM. Transcriptional control of preadipocyte determination by Zfp423. Nature. 2010;464(7288):619–623. doi: 10.1038/nature08816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang Y, Das AK, Yang QY, Zhu MJ, Du M. Zfp423 promotes adipogenic differentiation of bovine stromal vascular cells. PLoS ONE. 2012;7(10):e47496. doi: 10.1371/journal.pone.0047496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quach JM, Walker EC, Allan E, Solano M, Yokoyama A, Kato S, Sims NA, Gillespie MT, Martin TJ. Zinc finger protein 467 is a novel regulator of osteoblast and adipocyte commitment. J Biol Chem. 2011;286(6):4186–4198. doi: 10.1074/jbc.M110.178251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.You L, Pan L, Chen L, Chen J-Y, Zhang X, Lv Z, Fu D. Suppression of zinc finger protein 467 alleviates osteoporosis through promoting differentiation of adipose derived stem cells to osteoblasts. J Transl Med. 2012;10(1):1–11. doi: 10.1186/1479-5876-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kang S, Akerblad P, Kiviranta R, Gupta RK, Kajimura S, Griffin MJ, Min J, Baron R, Rosen ED. Regulation of early adipose commitment by Zfp521. PLoS Biol. 2012;10(11):e1001433. doi: 10.1371/journal.pbio.1001433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hasegawa R, Tomaru Y, de Hoon M, Suzuki H, Hayashizaki Y, Shin JW. Identification of ZNF395 as a novel modulator of adipogenesis. Exp Cell Res. 2013;319(3):68–76. doi: 10.1016/j.yexcr.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 44.Meruvu S, Hugendubler L, Mueller E. Regulation of adipocyte differentiation by the zinc finger protein ZNF638. J Biol Chem. 2011;286(30):26516–26523. doi: 10.1074/jbc.M110.212506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tong Q, Dalgin G, Xu H, Ting CN, Leiden JM, Hotamisligil GS. Function of GATA transcription factors in preadipocyte-adipocyte transition. Science. 2000;290(5489):134–138. doi: 10.1126/science.290.5489.134. [DOI] [PubMed] [Google Scholar]
- 46.Perez-Mancera PA, Bermejo-Rodríguez C, González-Herrero I, Herranz M, Flores T, Jiménez R, Sánchez-García I. Adipose tissue mass is modulated by SLUG (SNAI2) Hum Mol Genet. 2007;16(23):2972–2986. doi: 10.1093/hmg/ddm278. [DOI] [PubMed] [Google Scholar]
- 47.Fitzgerald J, Bateman JF. A new FACIT of the collagen family: COL21A1. FEBS Lett. 2001;505(2):275–280. doi: 10.1016/S0014-5793(01)02754-5. [DOI] [PubMed] [Google Scholar]
- 48.Mohamad Shah NS, Sulong S, Wan Sulaiman WA, Halim AS. Two novel genes TOX3 and COL21A1 in large extended Malay families with nonsyndromic cleft lip and/or palate. Mol Genet Genom Med. 2019;7(5):e635. doi: 10.1002/mgg3.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chou MY, Li HC. Genomic organization and characterization of the human type XXI collagen (COL21A1) gene. Genomics. 2002;79(3):395–401. doi: 10.1006/geno.2002.6712. [DOI] [PubMed] [Google Scholar]
- 50.Chun TH, Hotary KB, Sabeh F, Saltiel AR, Allen ED, Weiss SJ. A pericellular collagenase directs the 3-dimensional development of white adipose tissue. Cell. 2006;125(3):577–591. doi: 10.1016/j.cell.2006.02.050. [DOI] [PubMed] [Google Scholar]
- 51.Choe SS, Huh JY, Hwang IJ, Kim JI, Kim JB. Adipose tissue remodeling: its role in energy metabolism and metabolic disorders. Front Endocrinol (Lausanne) 2016;7:30. doi: 10.3389/fendo.2016.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Poklukar K, Candek-Potokar M, Vrecl M, Batorek-Lukac N, Fazarinc G, Kress K, Stefanski V, Skrlep M. Adipose tissue gene expression of entire male, immunocastrated and surgically castrated pigs. Int J Mol Sci. 2021;22(4):1768. doi: 10.3390/ijms22041768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang H, Wang X, Li M, Wang S, Chen Q, Lu S. Identification of key sex-specific pathways and genes in the subcutaneous adipose tissue from pigs using WGCNA method. BMC Genomic Data. 2022;23:35. doi: 10.1186/s12863-022-01054-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aouadi M, Laurent K, Prot M, Le Marchand-Brustel Y, Binetruy B, Bost F. Inhibition of p38MAPK increases adipogenesis from embryonic to adult stages. Diabetes. 2006;55(2):281–289. doi: 10.2337/diabetes.55.02.06.db05-0963. [DOI] [PubMed] [Google Scholar]
- 55.Zhang D, Wu W, Huang X, Xu K, Zheng C, Zhang J. Comparative analysis of gene expression profiles in differentiated subcutaneous adipocytes between Jiaxing Black and Large White pigs. BMC Genomics. 2021;22:61. doi: 10.1186/s12864-020-07361-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, Lango Allen H, Lindgren CM, Luan J, Magi R, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42(11):937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Iiritano S, Chiefari E, Ventura V, Arcidiacono B, Possidente K, Nocera A, Nevolo MT, Fedele M, Greco A, Greco M, et al. The HMGA1-IGF-I/IGFBP system: a novel pathway for modulating glucose uptake. Mol Endocrinol. 2012;26(9):1578–1589. doi: 10.1210/me.2011-1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kubota N, Terauchi Y, Miki H, Tamemoto H, Yamauchi T, Komeda K, Satoh S, Nakano R, Ishii C, Sugiyama T, et al. PPAR gamma mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol Cell. 1999;4(4):597–609. doi: 10.1016/S1097-2765(00)80210-5. [DOI] [PubMed] [Google Scholar]
- 59.Neumann N, Friz S, Forchhammer K. Glucose-1, 6-bisphosphate, a key metabolic regulator, is synthesized by a distinct family of α-phosphohexomutases widely distributed in prokaryotes. mBio. 2022:e01469–22. [DOI] [PMC free article] [PubMed]
- 60.Rasila T, Saavalainen O, Attalla H, Lankila P, Haglund C, Hölttä E, Andersson LC. Astroprincin (FAM171A1, C10orf38): a regulator of human cell shape and invasive growth. Am J Pathol. 2019;189(1):177–189. doi: 10.1016/j.ajpath.2018.09.006. [DOI] [PubMed] [Google Scholar]
- 61.Wang C, Wang H, Zhang Y, Tang Z, Li K, Liu B. Genome-wide analysis reveals artificial selection on coat colour and reproductive traits in Chinese domestic pigs. Mol Ecol Resour. 2015;15(2):414–424. doi: 10.1111/1755-0998.12311. [DOI] [PubMed] [Google Scholar]
- 62.Keller M, Hopp L, Liu X, Wohland T, Rohde K, Cancello R, Klos M, Bacos K, Kern M, Eichelmann F, et al. Genome-wide DNA promoter methylation and transcriptome analysis in human adipose tissue unravels novel candidate genes for obesity. Mol Metab. 2017;6(1):86–100. doi: 10.1016/j.molmet.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol. 2013;79(17):5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44(7):821–824. doi: 10.1038/ng.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88(1):76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yin T, Halli K, König S. Direct genetic effects, maternal genetic effects, and maternal genetic sensitivity on prenatal heat stress for calf diseases and corresponding genomic loci in German Holsteins. J Dairy Sci. 2022;105(8):6795–6808. doi: 10.3168/jds.2022-21804. [DOI] [PubMed] [Google Scholar]
- 72.Jinfeng G, Wenwu X, Yan Z, Shuhe Z, Xinqin L, Hongguo C, Tao Z, Yong T, Chunqin W, Lizhi L, et al. Genome-wide association study of egg-laying traits and egg quality in LingKun chickens. Front Vet Sci. 2022;9:877739. doi: 10.3389/fvets.2022.877739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang R, Gao X, Yang J, Kong X. Genome-wide association study to identify favorable SNP allelic variations and candidate genes that control the timing of spring bud flush of tea (Camellia sinensis) using SLAF-seq. J Agric Food Chem. 2019;67(37):10380–10391. doi: 10.1021/acs.jafc.9b03330. [DOI] [PubMed] [Google Scholar]
- 74.Percie du Sert N, Ahluwalia A, Alam S, Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl U, Emerson M, et al. Reporting animal research: Explanation and elaboration for the ARRIVE guidelines 20. PLoS Biol. 2020;18(7):e3000411. doi: 10.1371/journal.pbio.3000411. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Frequency distribution histogram for seven fatness-related traits, including five BFTs, CLP and CFP. A SBFT. B LRBFT. C LBFT. D ABFT. E 67RBFT. F CLP. G CFP.
Additional file 2: Figure S2. The phenotypic correlation for seven fatness-related traits, including five BFTs, CLP and CFP. The values in the box represented the phenotypic correlation of the traits. Negative values represented negative correlation, and positive values represented positive correlation. *significant at P<0.05, **significant at P<0.01, ***significant at P<0.001. All of the phenotypic correlation coefficients were significant with P < 0.05.
Additional file 3: Table S1. Distribution of SLAF tags and polymorphism SLAF tags on Sus Scrofa chromosomes.
Additional file 4: Table S2. The information of shared SNPs for fatness-related traits.
Additional file 5: Table S3. Comparison of significant SNPs with previously reported QTLs from the pig QTL database and newly significant SNPs for fatness-related traits.
Additional file 6: Table S4. The description and GO annotation of the gene with 100 kb upstream and downstream of the significant SNP.
Data Availability Statement
The Genome sequencing raw data was deposited in NCBI’s SRA database (https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi? view = studies&f = study&term = &go = Go; Accession: SRP376933).