

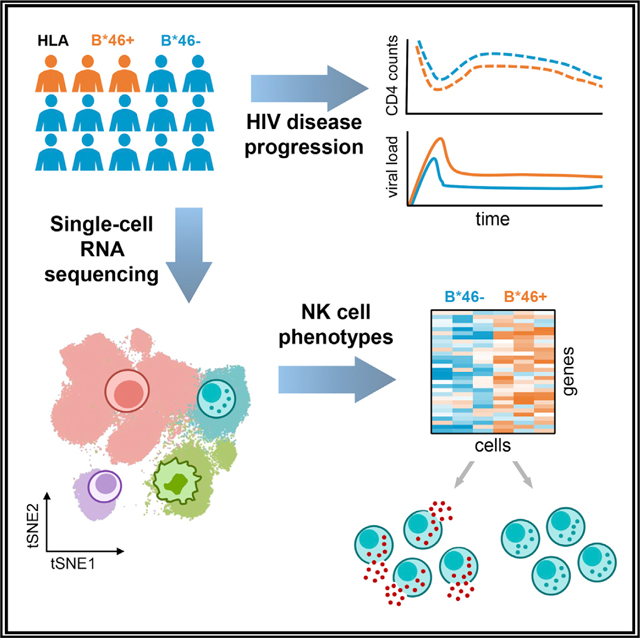
SUMMARY
Human leukocyte antigen (HLA) alleles have been linked to HIV disease progression and attributed to differences in cytotoxic T lymphocyte (CTL) epitope representation. These findings are largely based on treatment-naive individuals of European and African ancestry. We assessed HLA associations with HIV-1 outcomes in 1,318 individuals from Thailand and found HLA-B*46:01 (B*46) associated with accelerated disease in three independent cohorts. B*46 had no detectable effect on HIV-specific T cell responses, but this allele is unusual in containing an HLA-C epitope that binds inhibitory receptors on natural killer (NK) cells. Unbiased transcriptomic screens showed increased NK cell activation in people with HIV, without B*46, and simultaneous single-cell profiling of surface proteins and transcriptomes revealed a NK cell subset primed for increased responses in the absence of B*46. These findings support a role for NK cells in HIV pathogenesis, revealed by the unique properties of the B*46 allele common only in Asia.
Graphical Abstract
In brief
Li et al. show that B*46, the most frequent HLA-B allele in Thailand, is associated with HIV disease progression in three independent cohorts. This may result from differences in NK cell populations, specifically a subset with an activated phenotype that was found to be absent in people with HLA-B*46.
INTRODUCTION
Human leukocyte antigen (HLA) is a significant determinant in the host adaptive immune responses influencing human immunodeficiency virus (HIV) disease progression (Bashirova et al., 2011; Collins et al., 2020). Class I and class II genes comprising the HLA cluster encode the classical HLA class I (HLA-A, HLA-B, and HLA-C) and class II (HLA-DR, HLA-DQ, and HLA-DP) molecules, which are identified for their role in antigen presentation to CD8+ and CD4+ T cells, respectively. HLA class I molecules also function as ligands for activating and inhibitory killer immunoglobulin-like receptors (KIRs) on natural killer (NK) cells. HLA class I and class II genes are highly polymorphic, and HLA-B is recognized as the most diverse of all genes in the human genome. Variations in the HLA alleles have been shown to correlate with the differences in disease outcome for pathogens, including HIV. Initially, candidate gene studies demonstrated alleles such as B*57 and B*27 were protective against HIV disease progression (Gao et al., 2001; Kaslow et al., 1996; Migueles et al., 2000). Effects of HLA on HIV outcomes were further validated when significant associations were identified between HLA-B and HIV viral load (VL), as well as acquired immunodeficiency syndrome (AIDS) progression, by the first genome-wide association study (GWAS) in people living with HIV (PLWH) (Fellay et al., 2007). This was closely followed by validation from other large GWAS and candidate gene studies (Apps et al., 2013; McLaren et al., 2015; Pelak et al., 2010; International HIV Controllers Study et al., 2010). The mechanisms of HLA associations are likely to be, at least in part, through the effect on antigen presentation that triggers specific adaptive, cell-mediated, and humoral immune responses enabling effective viral control. This is supported by the in vivo observation of predictable patterns of viral escape mutation in defined cytotoxic T lymphocyte (CTL) epitopes and, for the most potent epitopes, reversion of these viral sequences upon transmission of the virus to a host lacking the restricting HLA allotype (Friedrich et al., 2004; Leslie et al., 2004).
The majority of the association studies described were identified in large multicentric longitudinal natural infection cohorts, conducted prior to the initiation of antiretroviral therapy (ART), among participants of primarily European and African American ancestry. There are very few cohorts with longitudinal measurements of HIV VL and CD4+ T cell counts (CD4 counts) among ART-naive individuals since the successful implementation of a universal test-and-treat (UTT) strategy, where ART is started following HIV diagnosis regardless of CD4 count and WHO clinical stage. As a consequence, there is a lack of consensus regarding HLA alleles that may influence HIV outcomes among individuals from other geographic regions, including Asia. Thailand accounts for 9% of the Asia-Pacific region’s total population of PLWH and has one of the highest HIV prevalence rates in the region (avert.org). Although the epidemic is in decline, HIV incidence remains high among key affected groups, with young people particularly at risk. Given the large burden of HIV in Thailand, there has been a concerted effort by the government to advance HIV prevention interventions and research, including vaccination. Incidentally, the only HIV vaccine trial to show a modest efficacy was conducted in Thailand (Rerks-Ngarm et al., 2009). Among those enrolled in the RV144 study, specific HLA class II alleles were observed to modulate the binding antibody responses that were identified as immune correlates of risk (Haynes et al., 2012; Prentice et al., 2015). In fact, differences in host HLA may have been one of the contributing factors of non-efficacy in the HVTN 702 HIV vaccine trial that employed a similar canarypox/protein regimen in South Africa (Gray et al., 2021). Moreover, in the context of natural susceptibility, there have been no consistent and reproducible findings of HLA with HIV acquisition in populations of European and African ancestry (McLaren and Carrington, 2015), and there still remains a knowledge gap in Asian populations.
Only very recently, with increasing use of next generation sequencing (NGS) technologies, has the diversity of HLA genes themselves been explored at high resolution in Thailand, which has been shown to be distinct when compared with European, African, and neighboring Asian populations (Geretz et al., 2018; Prentice et al., 2014). These prior population genetic studies facilitated this in-depth HLA disease association analysis, using contemporary cohorts with participants of Thai Asian ancestry. Given the potential impact of various HLA alleles on HIV acquisition and pathogenesis, these results will better inform susceptibility and disease outcomes, which are important for considering treatment and/or vaccine effectiveness, as well as contextualize human host determinants influencing the epidemiology of HIV. We present herein a comprehensive investigation of HLA associations and their function with HIV disease outcomes, including acquisition and disease progression in multiple cohorts from an Asian population.
RESULTS
HLA alleles do not associate with HIV acquisition
The association of HLA class I alleles on acquisition was examined among participants from the Bangkok men who have sex with men (MSM) Cohort Study (BMCS), a high HIV incidence cohort of MSM, where HIV outcomes were followed quarterly for up to 60 months (van Griensven et al., 2013). HLA genotypes were determined for a subset of enrolled participants (n = 678), and alleles with frequencies >5% were included in the analyses. Associations between acquisition and HLA alleles were investigated using a case-control study design, including seroincident cases (n = 274) and controls from the same cohort that remained negative even after repeated exposure to HIV (n = 404). None of the 31 tested HLA alleles associated significantly with HIV acquisition (Table S1).
HLA-B*46:01 associates with increased disease progression in treatment-naive cohorts
To investigate HLA class I associations with disease progression, we examined the association of HLA alleles with HIV progression outcomes in a discovery cohort and then validated findings in a second independent cohort (Rerks-Ngarm et al., 2013; van Griensven et al., 2013). We genotyped all 114 PLWH from the RV152 study that enrolled HIV seroincident participants from the RV144 HIV vaccine trial in Thailand (Rerks-Ngarm et al., 2009). We first examined the untreated participants from the placebo arm (n = 65) for HLA associations as part of the discovery cohort. A total of 31 HLA class I alleles with a frequency >5% were selected for further investigation. HIV disease progression outcomes examined included time to decline (in months) of CD4 counts (<350 cells/mm3) prior to ART initiation, time to ART initiation, and levels of VL (copies/mL) prior to ART initiation. Time to CD4 decline was significantly faster among individuals with specific HLA alleles A*02:07 (hazard ratio [HR] = 3.9, p = 0.003, q = 0.028); B*46:01 (HR = 5.01, p = 0.0001, q = 0.004); and C*01:02 (HR = 3.9, p = 0.0005, q = 0.007) (Tables 1 and S2A; Figures 1A and 1B). The HLA-B*46:01 allele was also significantly associated with decreased time to ART initiation (HR = 4.11, p = 0.001, q = 0.04) and higher VL in this pre-UTT cohort (geometric mean ratio [GMR] = 2.74, p = 0.007, q = 0.20) (Figure 1C; Tables 1, S2B, and S2C). We next examined the effect of these three alleles using univariate analyses with HIV disease outcomes in a second independent cohort from a subset of seroincident cases diagnosed during follow-up in the BMCS. Among the subset consisting of HIV incident cases with known last pre-ART visits (n = 207), the rate of CD4+ T cell decline to <350 cells/mm3 and VL burden were significantly greater in individuals with the B*46:01 and A*02:07 alleles but not in those with C*01:02 (Tables 1 and S2D). Using a multivariate model to simultaneously test independent effects of these two alleles in the discovery cohort, only the B*46:01 allele association with HIV progression remained statistically significant (Table S2E). Further analyses in the vaccine arm of the RV152 study (n = 49) showed the association of B*46:01 with disease progression in the same direction for all three outcomes, but it remained significant only for one (Table S2F).
Table 1.
Specific HLA alleles associated with increased HIV progression in two independent ART-naive cohorts
| Discovery cohort: RV152 (n = 65) | |||||
|---|---|---|---|---|---|
|
|
|||||
| HIV outcome | HLA allele | n (%) | HR | 95% CI | p value (q valuea) |
|
| |||||
| Time to CD4 <350 cells/mm3 | A*02:07 | 12 (18) | 3.9 | 1.6, 9.47 | 0.003 (0.028)c |
| B*46:01 | 20(31) | 5.01 | 2.19, 11.47 | 0.0001 (0.004)c | |
| C*01:02 | 24 (37) | 3.9 | 1.82, 8.34 | 0.0005 (0.007)c | |
|
| |||||
| GMR | 95% CI | p value (q valuea) | |||
|
| |||||
| VL copies/mL | A*02:07 | 12(18) | 1.59 | 0.59, 4.24 | 0.36 (0.98) |
| B*46:01 | 20(31) | 2.74 | 1.32, 5.66 | 0.007 (0.20)c | |
| C*01:02 | 24 (37) | 2.24 | 1.16, 4.34 | 0.02 (0.26) | |
|
| |||||
| Validation cohort: BMCS (n = 207) | |||||
|
|
|||||
| n (%) | HR | 95% CI | p value (q valueb) | ||
|
| |||||
| Time to CD4 < 350 cells/mm3 | A*02:07 | 35 (17) | 1.53 | 1.01, 2.33 | 0.047 (0.07)c |
| B*46:01 | 43(21) | 1.5 | 1.00, 2.25 | 0.048 (0.07)c | |
| C*01:02 | 51 (25) | 1.33 | 0.91, 1.95 | 0.14(0.14) | |
|
| |||||
| GMR | 95% CI | p value (q valueb) | |||
|
| |||||
| VL copies/mL | A*02:07 | 35 (17) | 2.92 | 1.36, 6.3 | 0.007 (0.02)c |
| B*46:01 | 43(21) | 2.09 | 1.02, 4.28 | 0.044 (0.07)c | |
| C*01:02 | 51 (25) | 1.68 | 0.86, 3.3 | 0.13(0.13) | |
See also Table S2.
HR, hazard ratio; GMR, geometric mean ratio; CI, confidence interval.
q value was adjusted for multiple comparisons (31 common HLA class I alleles) to control for false discovery rate (FDR). The covariates age, sex, baseline behavioral risk, and the calendar year of HIV-1 infection diagnosis (2003–2005, 2006, 2007, 2008, and 2009) were adjusted in all RV152 data analyses. CD4 counts were also adjusted in the RV152 VL analysis.
q value was adjusted for three HLA alleles to validate the RV152 findings. Age as a covariate was adjusted in the BMCS data analyses.
Significant HLA associations (p < 0.05, q < 0.2)
Figure 1. HLA-B*46:01 associates with increased HIV disease progression, but not acquisition, in three independent cohorts.

(A) HLA-B*46:01 had the strongest association with CD4+ T cell decline in the RV152 discovery cohort (n = 65) after adjusting for multiple tests. *p < 0.05, q < 0.2.
(B) CD4 decline was significantly faster in people with B*46:01 in the same cohort.
(C) VL was also significantly higher in the presence of B*46:01 in the same study (n = 65).
(D) Absolute CD4 counts (cells/mm3) were lower following HIV infection among people with the HLA-B*46:01 in the discovery, as well as two additional cohorts (n = 798). These significant differences were observed in three independent studies including RV152 (n = 65), BMCS (n = 207) and RV254 (n = 526) (***p < 0.001, *p < 0.05, Mann-Whitney U test). See also Tables S1 and S2.
B*46:01 also associates with decreased CD4 counts during acute HIV infection
We explored the HLA-B*46:01 association with disease progression in a cohort of people newly diagnosed with acute HIV infection (AHI) in Thailand (RV254) (Ananworanich et al., 2013; De Souza et al., 2015). Following AHI diagnosis, analyzing a single baseline VL and CD4 count available for each participant prior to ART initiation (n = 526) found no significant association between B*46:01 and the baseline VL measurement; however, B*46:01 was significantly associated with lower baseline CD4 counts (beta = −0.03, p = 0.046) (Figure 1D; Table S2G). Similar associations were also observed in the previous natural history cohorts (RV152: beta = −0.19, p = 0.0006; BMCS: beta = −0.06, p = 0.055) (Figure 1D; Table S2G). These findings point to a strong association of B*46:01 with loss of CD4 counts in multiple independent cohorts. NGS high-resolution analyses in all three independent cohorts from Thailand revealed the presence of only the B*46:01 subtype, and we will refer to this allele as B*46 hereafter.
HIV-specific T cell responses detected do not differ in the presence of HLA-B*46
The strongest evidence for the effects of HLA on HIV pathogenesis observed in European and African cohorts have been via the recognition of presented viral epitopes by CD8+ T cells. Therefore, we assessed interferon-gamma (IFN-γ) responses in 99 HIV-infected individuals from RV152 by assessing ELIspot responses to overlapping peptides spanning HIV envelope (Env) and Gag. Since there were no significant differences in HLA-A and HLA-B frequencies between the participants in the placebo or vaccine arm, we combined the individuals from the two groups for subsequent T cell analyses (Table S3A). Positive responses to at least one peptide were observed in over 50% of the study group and were predominantly to Gag. For either HIV protein, comparing the frequency of responses between participants with or without the B*46 allele showed no significant differences, suggesting that functional T cell responses targeting these HIV proteins may not account for the worse outcomes of infection observed in B*46+ individuals (Figure 2A). We also looked for peptides for which CTL responses associated with common class I alleles (frequency >5% in this cohort) in the same dataset. We did not find any B*46-associated epitopes that were significant, but this was also the case for multiple other alleles, although we did observe a very high frequency of significant HLA-B*15:02-associated viral epitopes targeting Gag and Env (Figure 2B) that were not significantly more frequent in individuals with or without B*46 (Table S3B).
Figure 2. HIV Env or Gag proteins were not differentially detected by T cell responses in HLA-B*46+ versus HLA-B*46– individuals.

(A) There was no significant difference in the frequency of functional T cell responses to HIV Env or Gag peptides detected by ELIspot when compared between participants that were B*46+ or B*46− in RV152 (n = 99).
(B) For all alleles with frequencies greater than 5% in the Thai population, the number of Gag and Env peptides for which a response was significantly associated with each allele using Fisher’s exact test is shown. See also Table S3.
Altered NK cell phenotype in B*46+ individuals with HIV
Although the strongest evidence for the effects of other HLA on HIV pathogenesis have been via CD8+ T cells, the B*46 allele is notable for displaying unique characteristics in terms of binding to NK cell receptors. The B*46 allele is a genetic recombinant of the B*15:01 and C*01:02 alleles, where alignment shows the B*46 is largely homologous to B*15:01 but with an 11-aminoacid region in exon 2, which is homologous to HLA-C*01:02 (Figure 3A; Abi-Rached et al., 2010). Importantly, the B*46 recombinant pattern observed includes HLA-C epitopes that mediate binding to KIR expressed on NK cells. The crystal structure of B*46, with mapped contact sites that were identified to confer binding of HLA-C to KIR2DL3 (Moradi et al., 2021), emphasizes that the HLA residues corresponding to HLA-C in HLA-B*46 are those which contribute substantially to KIR binding (Figure 3B). HLA-B*46 binds to the variable KIR2DL2/2DL3 (Moesta et al., 2008). KIR2DL2 was present in 42% of the samples in the RV152 discovery cohort but did not associate with an effect on time to CD4 <350 or VL independently or in the presence of B*46 (Figures S1A and S1B). Therefore KIR2DL3, which was present in almost all individuals (95%), may modulate NK cell function via recognition of B*46.
Figure 3. The HLA-B*46:01 allele influences NK cell activity during HIV disease pathogenesis.

(A) Sequence alignment of B*46:01 with the parental B*15:01 and C*01:02 alleles. The sequences matching HLA-C are shown in red font. Asterisks below the sequences represent identical residues, spaces indicate mismatches with C*01:02, while periods are mismatches with B*15:01.
(B) The crystal structure of HLA-B*46:01 is shown with heavy-chain sequence corresponding to B*15 (teal) and to C*01 (blue), as well as b2m (gray) and the presented peptide (red). The molecular backbone is shown for most residues, with all atoms depicted for HLA residues that contact KIR2DL3 in its solved cocrystal structure with HLA-C*07 (Moradi et al., 2021). Of these contact residues, three within heavy-chain positions 66–76 that correspond to HLA-C residues in HLA-B*46:01 are annotated in yellow.
(C) Pathway enrichment analyses of longitudinal RNA-seq samples were performed among people with AHI in the RV217 cohort (n = 7). Genes expressed in sorted NK, CD8+ T, CD4+ T, and B cells were ranked based on differential expression between B*46+ and B*46− individuals, with the number of enriched pathways reported for each genotype at four time points. Statistical significance was determined by chi-square test.
(D) Relative gene expression of all enriched genes in the significant pathways in NK cells when comparing B*46+ versus B*46 people at VL setpoint during AHI. The log2-fold change in expression relative to the mean gene expression in B*46+ donors is shown.
See also Table S4 and Figures S2–S4.
We next investigated which cell types showed differences in gene expression among HIV-infected individuals that carried or lacked HLA-B*46. Whole transcriptome RNA-seq profiling was performed on major PBMC populations sorted from longitudinal sampling of 7 viremic subjects of the RV217 prospective acute infection cohort at four different time points including preinfection, pre-peak, peak, and set point (Robb et al., 2016; Figure S2). We identified a higher distribution of significantly enriched gene signatures (p < 9.3e13) in the absence of the B*46 allele for NK cells at the VL set point, which was not seen for the other cell subsets including CD4+ T cells, CD8+ T cells, and B cells (Figure 3C). Permutation analysis indicated that 22 or more pathways enriched in B*46− versus B*46+ people occurred in fewer than 0.7% of 1,000 permutations and is unlikely to occur by chance (Figure S3). Genes from 24 of 26 pathways enriched in NK cells showed higher relative expression in the B*46− group compared with the B*46+ group (Figure 3D, Table S4). The top significant enriched module (M11.0) was present at the set-point time point among sorted NK cells (NES = 2.8, p < 0.001, q < 0.001) and contained genes related to degranulation, cytokine signaling, and inflammatory responses (Figure S4A). The second significant pathway enriched at set point was the M37.0 module (NES = 2.2, p < 0.001, q < 0.001) with genes related to infection and immune activation (Figure S4B). Overall, the observed expression of these pathways suggests a more activated NK cell phenotype in the absence of B*46 (Figure S4). That this unbiased screen detected the presence of B*46 to have the greatest effect on gene expression in NK cells among infected individuals, compared with other major cell types, supports the interpretation that the epidemiological association we observed between B*46 and loss of HIV control may be mediated by an effect of NK cells.
Single-cell transcriptomics uncovers NK cell subsets that demonstrate expression profiles which associate with HLA-B*46
To investigate with increased granularity the differences in NK cells that might shed light on how B*46 influences HIV disease outcomes, we used cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) to simultaneously profile both transcriptomes and 63 cell surface markers from the same single cells. Since greater differences in enriched pathways between the B*46+ versus B*46– groups had been identified at VL set point, compared with other time points of HIV infection, we hypothesized that the B*46-associated phenotype would be most apparent when viremia was suppressed and not during acute infection. We therefore generated CITE-seq data from peripheral blood for 18 treated PLWH. Cells were first clustered using the protein markers, and for each of these major populations, differentially expressed genes (DEGs) were identified between people with and without B*46, of which the population with the greatest frequency of DEGs was NK cells (Figure 4A). The results remained the same when NK cells were identified based on the CD56 marker alone (Figure S5A). The two major known NK cell populations, CD56dim and CD56hi subsets, were corroborated by inspection of CD16 (FCGR3A), CD56 (NCAM1), and CD57 (B3GAT1) protein and gene expression (Figure S5B). There were no significant differences in frequencies of NK cell clusters between the B*46+ and B*46− groups (Figures S5C and S5D). We next subclustered the NK cell compartment based on all proteins measured. With further clustering CD56hi cells remained distinct (cluster 3 with top expressing markers GZMK, XCL1, SELL, CD56, CD62L, CD27), but the larger group of CD56dim cells divided into three clusters (0–2) (Figures 4B and 4C). Cluster 0 was differentiated most strongly by markers GZMH, CCL5, and CD95; cluster 1 by GZMH, KLRC2, CD45RA, and CD16; and cluster 2 by FCER1G, KLRB1, CD38, and CD161. There were again no significant differences in frequencies of NK cell clusters 0–3 between the B*46+ and B*46− groups (Figures S5E and S5F). We performed differential expression analyses within the four clusters to identify significant genes and proteins that were either up- or downregulated in the presence or absence of B*46. The top DEG in all clusters associating with lower expression in the B*46+ group was HLA-B (Figure 4D). This was supported by surface antibody markers detecting lower expression of HLA class I as one of the top hits in the B*46+ group for clusters 0–2 (Figure 4E). To identify the biological nature of the perturbations associating with B*46 in each of the four clusters, we performed gene set enrichment analyses (GSEAs) on predefined collections of genes and found four significantly enriched pathways in the B*46 group in cluster 2, which were all related to immune activation (q < 0.01) (Figure 4F). Leading edge analyses of overlapping enriched genes in the pathways from cluster 2 identified CD3E, LCK, and ZAP70 in the B*46− group (Figure 4G). The pathways identified in cluster 2 were not significantly enriched in the B*46− group in the combined CD56dim NK cell population (NES = −1.24–1.43, FDR > 0.01). Although we confirmed the presence of the B*46-associated cluster using scRNA-seq during acute infection, we did not identify HLA-B as the top DEG or observe significant enrichment of pathways in the cluster when grouped by B*46 (Figures S6A and S6B). Thus, in PLWH we detected a subset of CD56dim NK cells, differentiated by both gene and protein markers, that demonstrates a more activated phenotype in B*46-treated individuals.
Figure 4. Single-cell CITE-seq data show significant differences in NK cell clusters associating with the presence of HLA-B*46.

(A) CITE-seq data from treated PLWH (n = 18) identified NK cells as the population with the most DEG, comparing B*46+ versus B*46− groups.
(B) Unsupervised clustering identified four NK cell clusters based on protein markers.
(C) Top gene or protein markers differentiating the four NK clusters. Statistical significance is denoted for comparisons between one cluster with the combined three other clusters. Gene and protein expression of top differential markers comparing B*46+ versus B*46− groups. ****p < 0.0001.
(D and E) Most differentially expressed (D) gene (HLA-B) and (E) protein (HLA class I expression using W6/32 antibody) associating with the presence of B*46 in all clusters. Statistical significance is denoted for comparisons between the groups within a specific cluster. Statistical significance for gene and protein clusters was determined by the Wilcoxon rank-sum test and likelihood ratio test implemented in the R Seurat package, respectively. ****p < 0.0001.
(F) Significant core enriched gene expression pathways associating with differences between B*46+ and B*46− PLWH in cluster 2. NES indicates the absolute GSEA normalized enrichment score.
(G) Enrichment scores for the core genes in signatures enriched in B*46− NK cells.See also Figures S5 and S6.
A specific NK cell population, but not the phenotype associating with B*46, is present prior to HIV infection
To investigate if differences in the NK cell population are only observed in the context of HIV infection, we next performed CITE-seq in people without HIV (PWOH) (n = 9). Unsupervised clustering revealed three subpopulations (0–2 clusters) of NK cells (Figure 5A). These included the CD56hi and two CD56dim NK cells populations of which cluster 1 expressed the same features which identified it as the cluster that displayed an activated phenotype associating with absence of B*46 in treated PLWH. These cluster markers include FCER1G, KLRB1, CD38, and CD161 (Figure 5B). The top DEG between B*46+ and B*46− individuals was HLA-B, which showed decreased expression in all NK clusters for people with B*46, as was seen for the treated PLWH (Figures 5C and 5D). Based on surface antibody staining HLA class I was only differentially expressed in cluster 0 (Figure 5E). However when GSEA was performed, we did not observe enrichment of pathways in the B*46− group that were enriched in this cluster at the ART time point. Thus, we identified a subset of CD56dim NK cells that is present prior to infection, which develops a phenotype of increased activation after infection in the absence of B*46, that may underlie the decreased acute CD4 counts and accelerated disease progression observed in B*46+ individuals.
Figure 5. Single-cell CITE-seq data reveal that the same NK population that exhibited a distinct transcriptional phenotype associated with B*46 after infection is also present in the absence of HIV infection.

(A) CITE-seq data from PWOH (n = 9) identified three NK cell clusters.
(B) Gene and protein markers of the specific NK cell cluster differentiated by B*46 during HIV infection in the current three NK cell populations. Statistical significance is denoted for comparisons between one cluster with the combined two other clusters. ****p < 0.0001.
(C) HLA-B is the most differentially expressed gene comparing B*46+ versus B*46− groups.
(D and E) (D) Gene (HLA-B) and (E) protein (HLA class I expression using W6/32 antibody) expression in all clusters were higher in the B*46− group. Statistical significance is denoted for comparisons between the groups within a specific cluster. Statistical significance for gene and protein clusters were determined by the Wilcoxon rank-sum and likelihood ratio tests, respectively. ****p < 0.0001.
DISCUSSION
HIV infection is typically associated with an acute viral syndrome, followed by a prolonged generally asymptomatic period eventually leading to clinical AIDS. The kinetics of disease progression from AHI to AIDS, in the absence of treatment, can vary between individuals from 1 year to more than 35 years for some rare individuals referred to as elite controllers (Goulder and Walker, 2012). Host genetic variations, specifically HLA alleles, have been shown to influence HIV disease progression and outcomes, but these associations were often identified from analyses of large natural history cohorts comprising participants of European or African ancestry. In Asian populations, reproducible HLA associations with HIV outcomes have not previously been observed, potentially due to a lack of cohort collections of sufficient sample size, differences in ART initiation criteria, and understudied genetic diversity (Gandhi et al., 2016; Wei et al., 2015). To address this, we systematically genotyped HLA class I genes at high resolution from independent Thai cohorts and evaluated associations with HIV acquisition and disease progression in ART-naive PLWH. Significant associations were validated separately in independent cohorts to demonstrate reproducibility of observed associations.
We were unable to identify significant associations of common HLA variants among Thai participants with HIV acquisition, consistent with prior investigations in non-Asian populations. We did identify significant associations of HLA-A*02:07, B*46:01, and C*01:02 with indicators of HIV disease progression outcomes prior to ART initiation, including time to decline of CD4 count to <350 cells/mm3, time until meeting criteria for ART initiation, and VL in the primary discovery cohort. While B*46 and C*01:02 are in strong positive linkage disequilibrium (D′ = 1.0, p < 0.001), the association of the HLA-C allele on disease progression did not validate in a replication cohort. Only alleles A*02:07 and B*46 were associated with indicators of HIV disease progression in a second independent Thai cohort of treatment-naive people with HIV. When testing for an independent effect, only the B*46 association remained significant in a multivariate analysis. The B*46 allele is unique in several regards. The B*46 serotype does not trace from an African lineage, distinguishing it from all other known B serotypes. Instead, the B*46 haplotype resulted from a recombination event between B*15:01 and C*01:02 alleles and likely occurred more recently in Southeast Asian populations (Abi-Rached et al., 2010; Hilton et al., 2017). B*46:01 is not only the most common HLA-B allele but also the only B*46 subtype present in Thailand, and it is found wherever Asian wet-rice farming peoples have traveled (Geretz et al., 2018; Prentice et al., 2014). It has been reported to associate with protection from leprosy and observed to show a much larger peptidome and to bind with significantly higher affinity to Mycobacterium leprae-derived peptides, compared with the B*46 parent alleles (Abi-Rached et al., 2010; Hilton et al., 2017; Wang et al., 1999). Most HLA class I A and B allotypes carry a unique public epitope (Bw4 or Bw6), of which the Bw4 epitope binds to three domain KIR receptors on NK cells. HLA-C, however, possesses either a C1 or C2 epitope that binds to two domain KIR. HLA-B*46 is one of the few B alleles that does not carry the Bw epitope, but it carries the C1 epitope from the parental allele C*01:02 (Moesta et al., 2008). As a result, HLA-B*46 is a ligand for KIR2DL2/L3 and considered a strong educator of 2DL3 expressing NK cells (Barber et al., 1996; Yawata et al., 2008; Zemmour and Parham, 1992).
Our analyses demonstrated an association of B*46 with HIV disease progression outcomes in two independent cohorts, and we also observed a significant association of B*46 with decreased absolute CD4 counts in an AHI cohort having one measurement prior to ART. The effect of B*46 on CD4 counts was also significant in our initial discovery and validation cohorts examining longitudinal specimens, and clinical data prior to ART initiation and lower CD4 counts are established to associate with increased disease progression and mortality (Hogg et al., 2001). Although previous guidance for ART initiation was linked to CD4 counts of less than 200 or 350 cells/mm3, current test-and-treat guidelines indicate ART should be initiated soon after diagnosis, regardless of CD4 count (WHO, 2015). Consequently, early diagnosis, treatment initiation, and sustained ART adherence may be particularly important for improving long-term HIV disease outcomes among persons carrying the B*46 allele (associated with lower CD4 count in ART-naive individuals) and may be a consideration for the UTT program in regions where this allele is prevalent.
Decreased CD4 counts during HIV infection result from a failure of virologic control (Ho et al., 1995), and the underlying mechanism for this in B*46+ people warrants further study. We did not observe any effect of B*46 on HIV-specific T cell responses detected by screening of overlapping peptides spanning Env or Gag by IFN-γ ELIspot assay. This assay used peptides of 15 residues and can detect both CD4 and CD8 T cell responses. Frequencies of T cell responses were no different between donors with or without B*46. Further, although no individual peptides showed an association with a CTL response when people carrying versus lacking B*46 were compared, this was not significantly different from other alleles of similar frequency. Interestingly, the highest number of peptides for which a response associated with any allele was B*15:02, which differs by only 5-amino-acid residues from B*15:01, which is the major parental allele of B*46:01. However, none of the 10 peptides that demonstrated a response associated with the presence of B*15:02 showed significant association for a response with the presence of B*46. This suggests the allele differences are sufficient to be functionally significant, and indeed specifically B*15:02, but not B*15:01 or B*46:01, has been shown to associate with hypersensitivity to carbamazepine in people of Asian ancestry (Chung et al., 2004). Together, these observations argue that rather than influencing T cells, the KIR epitopes of B*46 and their effects on NK cells may contribute to the larger effect of this allele on disease progression. Due to strong LD, all B*46 carriers also carry the C*01 type, resulting in the expression of at least two C1 ligands recognized by KIR2DL2/3 receptors. HLA-B molecules are expressed at levels around 10–20 times higher than HLA-C molecules, suggesting that B*46 would predominate when compared with C*01 in providing C1 epitopes for KIR2DL2/3, and the functional consequences of the higher expression level of this KIR ligand (B*46) on NK cells are not well established (Apps et al., 2015). A study of HLA-KIR pairs on HIV outcomes in a Thai cohort showed increased mortality and VL in people with B*46 and KIR2DL2 (Mori et al., 2018). Although this study reported a combined HLA and KIR association, they did not show a direct effect of B*46 on HIV outcomes. By contrast, we observed a strong association of B*46 with VL and other HIV disease outcomes, irrespective of KIR2DL2. Our interpretation is that any effect of B*46 binding KIR in individual cells likely occurs far upstream of the phenotypes we are now observing, where subsequent influence on development of the NK cell compartment as a whole and then response to viral infection in the context of broad innate and adaptive interactions have likely disconnected KIR expression from the NK phenotypes that we are documenting in B*46+ individuals. Since almost all participants from these Thai cohorts possessed KIR2DL3, we surmise this may mediate effects of B*46 on the NK compartment to negatively impact viral control in the host.
We found evidence for NK cell differences in individuals with B*46, consistent with alternative mechanisms for more rapid disease progression. In the context of infection, HIV-1 encoded Nef and Vpu proteins downregulate different HLA class I molecules, to subvert CTL responses which are primarily restricted by HLA-A/B, while preserving inhibition of NK cells for which C1 can act as a ligand for inhibitory receptors KIR2DL2/3 (Apps et al., 2016; Cohen et al., 1999; Collins et al., 1998). Although the Nef protein downregulates all HLA-B allotypes, including B*46, on CD4+ T cells infected in vitro, protein expression of HLA-B remains 2–5 times higher than HLA-C (Apps et al., 2015). Among individuals carrying the B*46 allele, this residual expression of a C1 epitope could inhibit NK cell killing of infected CD4+ T cells, leading to increased viral replication and CD4+ T cell decline. In support of this hypothesis, we observed an effect on NK function in treatment-naive AHI individuals carrying B*46. We found that there were more significant enriched pathways in sorted NK cells from B*46− compared with B*46+ individuals at the HIV VL set point, indicating that NK cells are differentially responsive in individuals carrying the B*46 allele. In comparison, there were no differences in other cell subsets, including CD4+ T cells, CD8+ T cells, and B cells. We hypothesized that the changes in NK cells are more dramatic when viremia is at a steady state and confirmed these findings using scRNA-seq.
Single-cell analyses in PLWH supported the hypothesis of NK inhibition in B*46+ individuals but revealed a possibility of differences in the NK compartment in the absence of active viral replication, which could potentially impact disease progression when virus is encountered. Single-cell RNA-seq in participants during AHI (peak viremia) revealed a very high expression of interferon-stimulated genes due to increased VL and did not reveal any differences in the presence of B*46. Given that transcriptomic differences in bulk-sorted NK cells during HIV infection were mainly observed at the VL set point, we hypothesized that differences in the B*46 phenotype would be apparent when viremia was suppressed. In an unbiased approach comparing effects of B*46 in individuals with effective treatment, NK cells showed the highest number of DEG. These differences were driven due to DEGs in the CD56dim NK cell population. To delineate biological perturbations at the granular level, further NK cell subcluster analyses identified variation in the expression of HLA-B/class I as the strongest host factor that differentiated B*46+ versus B*46− people. Variations in HLA expression, based on alleles and their impact on HIV disease progression, have been reported for HLA-A and HLA-C but not for HLA-B (Apps et al., 2013; Ramsuran et al., 2018; Thomas et al., 2009). Although the difference in HLA-B*46 expression may be a contributing factor to the genetic findings, we performed gene set pathway analyses to avoid confounding effects linked to a single gene. GSEA defined a subset of CD56dim NK cells with an increased activation phenotype in people without B*46. The enriched subset was marked by increased expression of HLA-B, HLA-DRB1, GZMH, and class I associated with lymphocyte activation and cytotoxicity. The activation phenotype was driven by leading edge genes LCK (lck) and zeta-chain-associated protein kinase-70 (ZAP-70) that encode intracellular kinases involved in NK activation signaling downstream of canonical immunoreceptor tyrosine-based activation motifs (ITAMs) receptors (Lanier, 2008; Ting et al., 1995). Modulation of this cluster demonstrates parallels to contrasting markers of NK cell activation (CD38) and inhibition (CD161 [KLRB1]) previously reported for CD56dim NK cell populations (Lanier et al., 1994; Sconocchia et al., 1999), but the phenotype is only observed in a subset of this larger NK cell population. This NK population was also observed in people with no HIV infection, although the activated phenotype was not present. Overall, our single-cell in vivo protein and mRNA data showed a phenotypic difference in NK cell profiles between people with and without HLA-B*46, revealed after HIV infection in the absence of viremia, where a lack of the activated NK cell population identified in people with B*46 may underlie their worse outcomes in HIV infection.
Remarkably, more than 25 years after the first description of specific HLA allele associations with HIV outcomes in multiple cohorts, we report that the presence of the HLA-B*46 allele likely impacts HIV outcomes among Asian populations, suggesting a role for NK cell inhibition in loss of HIV control (Kaslow et al., 1996). More broadly, understanding what contributed to B*46 expansion in Asia and its effects in other diseases may be helpful in uncovering the role of NK cell functions in vivo. Given that HLA associations also differ based on HIV subtypes, investigations of HLA associations with virus escape and infection outcomes with the major subtype that is spreading rapidly though Asia, CRF01_AE, are also warranted.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Rasmi Thomas (rthomas@hivresearch.org).
Materials availability
The study did not generate unique reagents.
Data and code availability
RNA-seq data has been deposited at GEO and are publicly available as of the date of publication. Accession number is listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| anti-Human CD1c (Mouse monoclonal) | 3ioLegend | Cat# 331547; Clone L161; RRID: AB_2800871 |
| anti-Human CD163 (Mouse monoclonal) | 3ioLegend | Cat# 333637; Clone GHI/61; RRID: AB_2810510 |
| anti-Human CD141 (Mouse monoclonal) | 3ioLegend | Cat# 344125; Clone M80; RRID: AB_2810541 |
| anti-Human CD11a (Mouse monoclonal) | 3ioLegend | Cat# 350617; Clone TS2/4; RRID: AB_2800935 |
| anti-Human CD197 (Mouse monoclonal) | 3ioLegend | Cat# 353251;Clone G043H7; RRID: AB_2800943 |
| anti-Human CD14 (Mouse monoclonal) | 3ioLegend | Cat# 301859; Clone M5E2; RRID:AB_2800736 |
| anti-Human CD16 (Mouse monoclonal) | BioLegend | Cat# 302065; Clone 3G8; RRID: AB_2800738 |
| anti-Human CD19 (Mouse monoclonal) | BioLegend | Cat# 302265; Clone HIB19; RRID: AB_2800741 |
| anti-Human CD45RO (Mouse monoclonal) | BioLegend | Cat# 304259; Clone UCHL1; RRID: AB_2800766 |
| anti-Human CD2 (Mouse monoclonal) | BioLegend | Cat# 309231; Clone TS1/8; RRID: AB_2810464 |
| anti-Human CD138 (Mouse monoclonal) | BioLegend | Cat# 356539; Clone MI15; RRID: AB_2810567 |
| anti-Human CD303 (Mouse monoclonal) | BioLegend | Cat# 354241; Clone 201A; RRID: AB_2814295 |
| anti-Human CD56 (Mouse monoclonal) | BioLegend | Cat# 362559; Clone5.1h11; RRID: AB_2801002 |
| anti-Human CD4 (Mouse monoclonal) | BioLegend | Cat# 300567; Clone RPA-T4; RRID: AB_2800725 |
| anti-Human CD3 (Mouse monoclonal) | BioLegend | Cat# 300479; Clone UCHT1; RRID: AB_2800723 |
| anti-Human CD45RA (Mouse monoclonal) | BioLegend | Cat# 304163; Clone HI100; RRID: AB_2800764 |
| anti-Human CD39 (Mouse monoclonal) | BioLegend | Cat# 328237;Clone A1; RRID: AB_2800853 |
| anti-Human CD279 (Mouse monoclonal) | BioLegend | Cat# 329963;Clone EH12.2H7; RRID: AB_2800862 |
| anti-Human CD8 (Mouse monoclonal) | BioLegend | Cat# 344753; Clone SK1; RRID: AB_2800922 |
| anti-Human CD27 (Mouse monoclonal) | BioLegend | Cat# 302853; Clone O323; RRID: AB_2800747 |
| anti-Human CD20 (Mouse monoclonal) | BioLegend | Cat# 302363; Clone 2H7; RRID: AB_2800743 |
| anti-Human HLA-A/B/C (Mouse monoclonal) | BioLegend | Cat# 311449; Clone W6/32; RRID: AB_2800816 |
| anti-Human IgM (Mouse monoclonal) | BioLegend | Cat# 314547; Clone MHM-88; RRID: AB_2800835 |
| anti-Human CD127 (Mouse monoclonal) | BioLegend | Cat# 351356; Clone A019D5; RRID: AB_2800937 |
| anti-Human CD195 (Rat monoclonal) | BioLegend | Cat# 359137; Clone J418F1; RRID: AB_2810570 |
| anti-Human HLA-DR (Mouse monoclonal) | BioLegend | Cat# 307663; Clone L243; RRID: AB_2800795 |
| anti-Human IgG (Fc) (Rat monoclonal) | BioLegend | Cat# 410727; Clone M1310G05; RRID: AB_2801087 |
| anti-Human TCR Vd2 (Mouse monoclonal) | BioLegend | Cat# 331435; Clone B6; RRID: AB_2800864 |
| anti-Human TCR Va7.2 (Mouse monoclonal) | BioLegend | Cat# 351735;Clone 3C10; RRID: AB_2810556 |
| anti-Human TCR Va24-Ja18 (Mouse monoclonal) | BioLegend | Cat# 342925; Clone 6B11; RRID: AB_2810539 |
| anti-Human TCR g/d (Mouse monoclonal) | BioLegend | Cat# 331231; Clone B1; RRID AB_2814199 |
| anti-Human TCR Vg9 (Mouse monoclonal) | BioLegend | Cat# 331313; Clone B3; RRID: AB_2814203 |
| anti-Human CD7 (Mouse monoclonal) | BioLegend | Cat# 343127; Clone CD7–6B7; RRID: AB_2800914 |
| anti-Human CD11c (Mouse monoclonal) | BioLegend | Cat# 371521; Clone S-HCL-3; RRID: AB_2801018 |
| anti-Human CD185 (Mouse monoclonal) | BioLegend | Cat# 356939,;Clone J252D4; RRID: AB_2800968 |
| anti-Human CD1d (Mouse monoclonal) | BioLegend | Cat# 350319; Clone 51.1; RRID: AB_2800934 |
| anti-Human IgD (Mouse monoclonal) | BioLegend | Cat# 348245; Clone IA6–2; RRID: AB_2810553 |
| anti-Human CD11b (Mouse monoclonal) | BioLegend | Cat# 301359; Clone ICRF44; RRID: AB_2800732 |
| anti-Human CD62L (Mouse monoclonal) | BioLegend | Cat# 304851; Clone DREG-56; RRID: AB_2800770 |
| anti-Human CD66a/c/e (Mouse monoclonal) | BioLegend | Cat# 342325; Clone ASL-32; RRID: AB_2810538 |
| anti-Human CD15 (Mouse monoclonal) | BioLegend | Cat# 323053; Clone W6D3; RRID: AB_2800847 |
| anti-Human CD32 (Mouse monoclonal) | BioLegend | Cat# 303225; Clone FUN-2; RRID: AB_2814129 |
| anti-Human CD57 (Mouse monoclonal) | BioLegend | Cat# 393321; Clone QA17A04; RRID: AB_2801030 |
| anti-Human CD73 (Mouse monoclonal) | BioLegend | Cat# 344031; Clone AD2; RRID: AB_2800916 |
| anti-Human CD123 (Mouse monoclonal) | BioLegend | Cat# 306045; Clone 6H6; RRID: AB_2800789 |
| anti-Human Mouse IgG1, k Isotype Ctrl (Mouse monoclonal) | BioLegend | Cat# 400187; Clone MOPC-21; RRID: AB_2888921 |
| anti-Human Mouse IgG2a, k Isotype Ctrl (Mouse monoclonal) | BioLegend | Cat# 400293; Clone MOPC-173; RRID: AB_2888922 |
| anti-Human Mouse IgG2b, k Isotype Ctrl (Mouse monoclonal) | BioLegend | Cat# 400381; Clone MPC-11; RRID: AB_2888923 |
| anti-Human Rat IgG2b, k Isotype Ctrl (Rat monoclonal) | BioLegend | Cat# 400677; Clone RTK4530: RRID: AB_2894967 |
| anti-Human CD28 (Mouse monoclonal) | BioLegend | Cat# 302963; Clone CD28.2; RRID:AB_2800751 |
| anti-Human CD161 (Mouse monoclonal) | BioLegend | Cat# 339947; Clone HP-3G10; RRID: AB_2810532 |
| anti-Human CD95 (Mouse monoclonal) | BioLegend | Cat# 305651; Clone DX2; RRID: AB_2800787 |
| anti-Human CD38 (Mouse monoclonal) | BioLegend | Cat# 303543; Clone HIT2; RRID: AB_2800758 |
| anti-Human KLRG1 (Syrian hamster monoclonal) | BioLegend | Cat# 138433; Clone 2F1/KLRG1; RRID: AB_2800649 |
| anti-Human CD25 (Mouse monoclonal) | BioLegend | Cat# 302649; Clone BC96; RRID: AB_2800745 |
| anti-Human CD41 (Mouse monoclonal) | BioLegend | Cat# 303739; Clone HIP8; RRID: AB_2814134 |
| anti-Human CD184 (Mouse monoclonal) | BioLegend | Cat# 306533; Clone 12G5; RRID: AB_2800791 |
| anti-Human CD137 (Mouse monoclonal) | BioLegend | Cat# 309839; Clone 4B4–1; RRID: AB_2800807 |
| anti-Human CD24 (Mouse monoclonal) | BioLegend | Cat# 311143; Clone ML5; RRID: AB_2800813 |
| anti-Human CD274 (Mouse monoclonal) | BioLegend | Cat# 329751; Clone 29E.2A3; RRID: AB_2800860 |
| anti-Human CD96 (Mouse monoclonal) | BioLegend | Cat# 338423; Clone NK92.39; RRID: AB_2810531 |
| anti-Human CD194 (Mouse monoclonal) | BioLegend | Cat# 359425; Clone L291H4; RRID: AB_2800988 |
| anti-Human CD152 (Mouse monoclonal) | BioLegend | Cat# 369621; Clone BNI3; RRID: AB_2801015 |
| Hashl: anti-Human CD298 S b2-microglobulin (Mouse monoclonals) | BioLegend | Cat# 394661; Clone LNH-94; 2M2; RRID: AB_2801031 |
| Hash2: anti-Human CD298 S b2-microglobulin (Mouse monoclonals) | BioLegend | Cat# 394663; Clone LNH-94; 2M2; RRID: AB_2801032 |
| Hash3: anti-Human CD298 S b2-microglobulin (Mouse monoclonals) | BioLegend | Cat# 394665; Clone LNH-94; 2M2; RRID: AB_2801033 |
| Hash4: anti-Human CD298 S b2-microglobulin (Mouse monoclonals) | BioLegend | Cat# 394667; Clone LNH-94; 2M2; RRID: AB_2801034 |
| Hash5: anti-Human CD298 S b2-microglobulin (Mouse monoclonals) | BioLegend | Cat# 394669; Clone LNH-94; 2M2; RRID: AB_2801035 |
| Hash6: anti-Human CD298 S b2-microglobulin (Mouse monoclonals) | BioLegend | Cat# 394671; Clone LNH-94; 2M2; RRID: AB_2820042 |
| Hash7: anti-Human CD298 S b2-microglobulin (Mouse monoclonals) | BioLegend | Cat# 394673; Clone LNH-94; 2M2; RRID: AB_2820043 |
| Hash8: anti-Human CD298 S b2-microglobulin (Mouse monoclonals) | BioLegend | Cat# 394675; Clone LNH-94; 2M2, RRID: AB_2820044 |
| Hash9: anti-Human CD298 S b2-microglobulin (Mouse monoclonals) | BioLegend | Cat# 394677; Clone LNH-94; 2M2; RRID: AB_2820045 |
| Hash10: anti-Human CD298 S b2-microglobulin (Mouse monoclonals) | BioLegend | Cat# 394679; Clone LNH-94; 2M2; RRID: AB_2820046 |
| Mouse anti-human CD3 FITC | Becton Dickinson | Cat# 555332; Clone UCHT1; RRID: AB_395739 |
| Mouse anti-human CD8 PerCP-eF710 | eBiosciences | Cat# 46–0087-42; Clone SK1; RRID: AB_1834411 |
| Mouse anti-human CD14 V500 | Becton Dickinson | Cat# 562693; Clone M5E2; RRID: AB_2737727 |
| Mouse anti-human CD45RO eF650NC | eBiosciences | Cat# 95–0457-42; Clone UCHL1; RRID: AB_1603247 |
| Mouse anti-human CD4 APC | Becton Dickinson | Cat# 561841; Clone RPAT4; RRID: AB_395739 |
| Mouse anti-human CD45RA APC-H7 | Becton Dickinson | Cat# 560674; Clone HI100; RRID: AB_1727497 |
| Mouse anti-human CD19 PE-Cy5 | Invitrogen | Cat# MHCD1918; Clone SJ25-C1; RRID: AB_10373840 |
| Mouse anti-human CD56 PE-Cy7 | Becton Dickinson | Cat# 335809; Clone NCAM16.2; RRID: AB_399984 |
| Mouse IgG isotype control | Caltag | Cat# 10400C; RRID: AB_2532980 |
|
| ||
| Biological samples | ||
|
| ||
| PBMC from MHRP RV217 donors | M. Robb | Robb et al., 2016 |
| PBMC from MHRP RV254 donors | D. Hsu | Ananworanich et al., 2013; De Souza et al., 2015 |
| PBMC from MHRP RV152 donors | A. Schuetz | Rerks-Ngarm et al., 2013 |
| PBMC from CDC BMCS donors | A. Hickey | van Griensven et al., 2013 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Cell Staining Buffer | BioLegend | Cat# 420201 |
| TruStain FcX Blocking Reagent, human | BioLegend | Cat# 422301 |
| Acridine Orange | ThermoFisher Scientific | Cat# A3568 |
| Ethidium Homodimer | ThermoFisher Scientific | Cat# E1169 |
| Glycerin (Glycerol), 50% (v/v) | Ricca Chemical Co | Cat# 3290–16 |
| Buffer EB | Qiagen | Cat# 19086 |
| Tween 20, 10% (v/v) | Bio-Rad | Cat# 1610781 |
| SPRIselect beads | Beckman Coulter | Cat# 823318 |
| Guava ViaCount Reagent | Millipore Corp | Cat# 4000–0041 |
| Live/Dead Fixable Aqua | ThermoFisher | Cat# L34957 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Chromium Single Cell 5’ Library & Gel Bead kit, v2 | 10x Genomics | Cat# 1000006 |
| Chromium Single Cell 5’ Feature Barcode Library kit | 10x Genomics | Cat# 1000080 |
| Single Index kit N, Set A | 10x Genomics | Cat# 1000212 |
| Single Index kit T, Set A | 10x Genomics | Cat# 1000213 |
| PhiX Control kit, v3 | Illumina | Cat# FC-110–3001 |
| High Sensitivity DNA kit | Agilent | Cat# 5067–4626 |
| Qubit dsDNA HS Assay kit | ThermoFisher Scientific | Cat# Q32854 |
| SMART-Seq v4 UltraLow Input RNA kit | Takara | Cat# 634891 |
| Single Cell RNA Purification kit | Norgen Biotek | Cat# 51800 |
| Nextera XT DNA Sample Preparation kit | Illumina | Cat# FC-131–1096 |
| ERCC RNA Spike-in Mix 1 | Life Technologies | Cat# 4456740 |
| NovaSeq 6000 S4 Reagent kit, 300 cycles, v1.5 | Illumina | Cat# 20028312 |
| MiSeq Reagent Nano kit, 500 cycles, v2 | Illumina | Cat# MS-103–1003 |
|
| ||
| Deposited data | ||
|
| ||
| RNA-seq analysis of human peripheral blood cells collected at multiple timepoints during the course of acute HIV-1 infection (RV217) | This paper | GEO: GSE192384; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE192384 |
| R code and data files for figures | This paper | https://doi.org/10.6084/m9.figshare.c.5728400 |
|
| ||
| Software and algorithms | ||
|
| ||
| FlowJo | FlowJo, LLC | https://www.flowjo.com/ |
| bcl2fastq | Illumina | https://support.illumina.com/downloads/bcl2fastq-conversion-software-v2–20.html |
| featureCounts | Liao et al., 2014 | http://subread.sourceforge.net/ |
| Cell Ranger | Zheng et al., 2017 | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads |
| R | https://www.r-project.org/ | |
| DSB | Mulè et al., 2022 | https://github.com/niaid/dsb |
| edgeR | Robinson et al., 2010 | https://bioconductor.riken.jp/packages/devel/bioc/html/edgeR.html |
| geepack | H0jsgaard et al., 2006 | https://cran.r-project.org/web/packages/geepack/index.html |
| Seurat | Hao et al., 2021 | https://cran.r-project.org/web/packages/Seurat/index.html |
| GSEA | Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/index.jsp |
| Metascape | Zhou et al., 2019 | https://metascape.org |
| Arlequin | Excoffier and Lischer, 2010 | http://cmpg.unibe.ch/software/arlequin35/Arl35Downloads.html |
All original R code are available in the figshare repository and is publicly available as of the date of publication. DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subjects
HLA genetic associations with HIV outcomes were assessed independently in three cohort studies from Thailand, including RV152 (Rerks-Ngarm et al., 2013), BMCS (van Griensven et al., 2013), and RV254 (Ananworanich et al., 2013; De Souza et al., 2015). RV152: The discovery cohort examined included men and women heterosexual participants 18–30 years of age from the RV144 HIV vaccine trial conducted in Thailand who were HIV-1 negative at the first placebo or candidate vaccine visit and acquired HIV-1 infection during the 42 months of follow-up (Rerks-Ngarm et al., 2009). The time of HIV acquisition was defined as the mid-point between the last HIV-negative and first HIV-positive result. All participants from the RV144 vaccine trial that seroconverted during follow up were enrolled in the RV152 long-term post-infection follow-up study to evaluate post-infection endpoints with an assessment every six months. A total of 114 HIV-positive participants (65 placebos and 49 vaccine recipients) were enrolled in the RV152 long-term post-infection study and had clinical data, including time to decline of CD4 count (<350 cells/mm3), time until meeting criteria for ART initiation, and longitudinal VL measurements. Study follow-up visits were scheduled at 0, 1, 3, and 6 months and every 3 months thereafter. After month 12, CD4 counts and VL were obtained at 6-month intervals until reaching a CD4 count <350 cells/mm3 or ART was initiated, at which time CD4 counts and VL were obtained every 3 months. BMCS: The second independent validation cohort, the Bangkok men who have sex with men cohort study (BMCS) enrolled participants ages 18–65 at the Silom Community Clinic Bangkok, Thailand between 2006–2010. HIV/STI testing, clinical assessments, and survey of risk information for participants were conducted every 4 months for up to 60 months of follow up (van Griensven et al., 2013). Incident HIV cases diagnosed during the BMCS were followed once per month for the first 8 months, month 12, and month 16 following diagnosis of HIV. PBMCs from participants diagnosed with HIV during follow up (n = 274) and seronegative participants (at study exit) that could be re-consented for the collection of PBMCs for genetic testing (n = 404) were included in these analyses. Clinical measurements available included seroconversion date, CD4 counts, HIV VL, and date of ART initiation. Individuals who initiated ART were censored at their last visit pre-ART. The estimated date of infection (EDI) was defined as the median between the date of last negative and first positive HIV test. RV254: The RV254 cohort (SEARCH 010) enrolled predominantly male Thai participants ages 18–70 with AHI (Fiebig stages 1–5), and explored the impact of early ART on immune responses and HIV disease progression (Ananworanich et al., 2013; De Souza et al., 2015). Participants had one baseline measurement of HIV VL and CD4 counts (2–3 days after diagnosis) prior to ART initiation. We selected 526 participants from this study for HLA analyses. For scRNA-seq analyses PBMCs from 18 matched participants (1:2) treated during AHI and 6 matched participants (1:2) during AHI (Fiebig stage 3) were selected from people with and without HLA-B*46. RV217: This study enrolled HIV-negative men and women participants 18–50 years of age to follow progression of the early stages of acute HIV infection (Robb et al., 2016). Blood from participants was collected twice weekly for HIV nucleic acid testing. Incident HIV cases were followed twice weekly for the first 4 weeks after detection and enrolled into a second phase that included intensive longitudinal study of AHI, including viremia and host responses. ART was initiated promptly as indicated by the clinical team as per the Thai national guidelines. Longitudinal samples from 7 male participants from 4 different time points (HIV negative, pre-peak, peak and setpoint) were selected for this study.
All participants from the aforementioned human studies provided informed consent and use of samples for research was approved by ethical review boards at the Walter Reed Army Institute of Research, the Thai Ministry of Public Health, the Royal Thai Army Medical Department, Mahidol University Faculty of Tropical Medicine, Chulalongkorn University Faculty of Medicine.
METHOD DETAILS
HLA and KIR genotyping
High-resolution typing of class I HLA-A, -B, and -C loci was performed on all selected participants from the different cohorts using either DNA sequence-based typing (SBT) or NGS HLA genotyping methods. SBT was carried out by PCR amplification and sequencing of exons 2 and 3 as described previously (Lazaro et al., 2013; Prentice et al., 2014). NGS HLA genotyping was performed by sequencing full-length HLA genes as described previously (Ehrenberg et al., 2014; Ehrenberg et al., 2018). HLA alleles with frequencies > 5% are denoted at the 1–2 field level for each cohort. Linkage disequilibrium between specific HLA alleles was computed using the Arlequin software v3.5 (Excoffier and Lischer, 2010). KIR genotyping was performed as described previously (Prentice et al., 2014).
HIV-specific peptide screening by IFN-γ ELIspot Matrix assay
HIV peptides were mapped using an IFN-γ ELIspot matrix format assay as described previously (Rerks-Ngarm et al., 2009). In brief, 96-well hydrophobic membrane-bottom plates (Millipore) were coated overnight at 4°C with anti-human IFN-γ mAb (final concentration of 5 μg/ml (Mabtech). PBMC were resuspended in complete medium and plated at a concentration of 1 × 105 cells per well. A panel of 26 pools of 9–13 (total of 165) peptides spanning the 92TH023 gp160 and 22 pools of 10–11 (total 120) peptides encompassing the LAI Gag protein was added to single wells of cells in a matrix format, and the plates were incubated overnight at 37°C in 5% CO2. Wells containing cells and media only were supplemented with the equivalent concentration of DMSO as the peptide pools and served as negative controls. PHA was used as a positive control, and if there were sufficient cells, CMV pp65 and the CMV, EBV, and influenza peptide pool also were included in the assay as additional positive controls. Cells plus PHA were tested in triplicate wells. Negative controls were performed in quadruplicate. After incubation at 37°C in 5% CO2 for 20–24 h, cells were removed by washing with PBS/0.05% Tween 20 (Sigma-Aldrich). Captured IFN-γ was detected by incubation for 2 h at 37°C with biotinylated anti-human IFN-γ mAb (Mabtech) at a concentration of 2 μg/ml in PBS/0.5% BSA. After incubation, plates were washed with PBS/0.05% Tween 20, and avidin-peroxidase complex (Vectastain Elite Kit) was added for 1 h at room temperature. Unbound complex was removed by washing, and peroxidase staining was performed using 3-amino-9-ethylcarbazole substrate (Vectastain AEC Kit) according to the manufacturer’s instructions. Spots were counted with a CTL analyzer and software (version 4.0.19; CTL Analyzers). Results are expressed as spot-forming cells (SFCs) /106 PBMCs. A positive IFN-γ response was defined as ≥ 55 SFCs /106 PBMCs (uncorrected) and ≥ 4 times the average of the DMSO-treated wells. The PHA response had to be positive for an assay to be valid. The median number (range) of SFCs /106 for PBMCs treated with media plus DMSO only for the 99 participants’ PBMCs was 0 (0–15) and the mean (standard deviation) was 1. Data are shown as corrected (media only SFC subtracted) SFC/106 PBMC. Each peptide was represented twice within the Gag and Env ELIspot matrices, therefore, to reduce additive effects from other members of the pool the minimum SFC/106 PBMC for the two wells was used as the SFC/106 PBMC for that peptide. This method may inflate the breadth of the peptide response peptides as T cells that respond to 11-mer overlap regions occurring between consecutive peptides are counted as responding to two peptides. We restricted our analyses to HLA class I associations that favor detection of CD8 T cell responses, using 15-mer peptides overlapping by 11 amino acids. Comparisons between HLA B*46 presence and absence were made using Fisher’s exact test for categorical peptide response analyses. For all alleles with frequencies greater than 5% in the Thai population, HLA specific CTL responses were analyzed with each HLA allele using Fisher’s exact test.
RNA sequencing
Cryopreserved PBMC were thawed in media containing 20% fetal bovine serum. Cell counts and viabilities were assessed using Guava ViaCount reagent and a Guava PCA (Millipore Sigma). Cells were washed and stained with Aqua Live/Dead stain (Molecular Probes), washed, and blocked using normal mouse IgG (Caltag). The cells were surface stained for CD3 FITC (clone UCHT1, Becton Dickinson [BD]), CD8 PerCP-eF710 (SK1, eBiosciences), CD14 V500 (M5E2, BD), CD45RO eF650NC (UCHL1, eBiosciences), CD4 APC (RPAT4, BD), CD45RA APC-H7 (HI100, BD), CD19 PE-Cy5 (SJ25-C1, Invitrogen), and CD56 PE-Cy7 (NCAM16.2, BD) (Figure S2). The cells were then washed and sorted/analyzed on the BD FACSAria II SORP Cell Sorter. Total RNA from sorted cell subsets was extracted using the Single Cell RNA Purification kit (Norgen Biotek Corp). RNA was prepared for NGS RNA sequencing (RNAseq) using the SMART-Seq technology as described previously (Ehrenberg et al., 2019; Picelli et al., 2014; Ramsköld et al., 2012; Shangguan et al., 2021). Briefly, cDNA was generated per manufacturer’s instructions from 2.5 ng of RNA, final exogenous ERCC RNA spike-in dilutions of 1:190,000, and 11 cycles of library PCR amplification. A total of 300 ng of cDNA was input template for final library processing using the Nextera XT kit (Illumina) per manufacturer’s instructions.
CITE-seq single cell RNA sequencing
Total PBMC from whole blood obtained from 18 RV254 participants 48 weeks after ART initiation were processed for CITE-seq on the 10x Genomics platform as described previously (Shangguan et al., 2021). Briefly, cell counts and viability were assessed by both the Countess II FL (Thermo Fisher) and trypan blue staining. Donor cells were hashed with TotalSeq-C anti-human Hashtag antibodies (BioLegend) and stained with a cocktail comprised of 63 TotalSeq-C antibodies targeting surface-expressed proteins. Cells from batches consisting of 9 or 10 samples were loaded into each of four Chromium chip wells before loading into the Chromium instrument. Cell mRNA gene expression (GEX) and antibody-derived tag (ADT) libraries were subsequently generated using the Chromium Next GEM Single Cell 5’ Library and Gel Bead Kit v1.1 (10x Genomics) according to the manufacturer’s instructions. Pooled libraries were quantitated with the MiSeq Nano v2 reagent cartridge (Illumina) and sequenced on an Illumina NovaSeq 6000 using an S4 reagent cartridge (2×100 bp)(Illumina). A modified version of the scRNA-seq assay was performed in RV254 participants at the acute HIV time point (n = 6) without feature barcodes.
QUANTIFICATION AND STATISTICAL ANALYSIS
HLA associations with HIV acquisition
Logistic regression was used to evaluate the association between HLA alleles and HIV acquisition in the BMCS natural history cohort, using a case-control study design, including 274 seroincident cases (with EDI defined as the median between the date of last negative and first positive HIV test) and 404 seronegative controls (participants who were at high risk of HIV incidence, but remained HIV negative). Age was adjusted in each regression model.
HLA association with HIV disease progression outcomes
In the discovery RV152 cohort, 31 HLA alleles that passed the allele frequency threshold were tested for associations with HIV disease progression. Cox regression models were used to assess whether HLA alleles were associated with time to CD4 count <350 cells/mm3 and time to meeting criteria for ART initiation. Time to CD4 <350 cells/mm3 was defined as the number of months from the date of seroconversion, to when CD4 counts dropped below 350 cells/mm3, or were censored to the last date of follow up in the study for those whose CD4 counts remained above 350 cells/mm3 during the study follow up period. Similarly, time to ART initiation was also defined as time measured in months from the date of seroconversion to the date of ART initiation for those who started ART, or were censored to the last date of follow up for those who did not start treatment during the study follow up period. The median survival time was estimated using the Kaplan-Meier method. Generalized estimating equations with multiple imputation (MIGEEs) to handle missingness due to either missing visits or ART initiation were employed using the geepack R package to assess the association of HLA alleles with longitudinal pre-ART plasma HIV VL over the planned post-infection diagnosis visit time points (<1, 1, 3, 6, 9, 12, 18, 24, 30, 36, 42, 48, and 54 months) (Højsgaard et al., 2006). Post-infection diagnosis visit time points at 60 months and after, which were included in the primary post-infection analyses (Rerks-Ngarm et al., 2013), were excluded because insufficient observations were made at those time points to enable subgroup analyses. The centered analysis times in polynomial terms up to the sixth order were also included in the models, since time from HIV diagnosis was a significant predictor of VL. A first-order autoregressive covariance matrix was used to account for the correlation of VL over time within individuals. Geometric mean ratio (GMR), mean, standard deviation (SD) and 95% confidence interval (CI) were presented for each HLA allele. Age, sex, baseline behavioral risk, and the calendar year of HIV-1 infection diagnosis (2003–2005, 2006, 2007, 2008–2009) were adjusted in the Cox analyses and each regression model as described previously (Rerks-Ngarm et al., 2013). CD4 counts were also adjusted in the RV152 VL analysis. Similarly, KIR and HLA & KIR combinations were analyzed with the above disease progression outcomes in the RV152 discovery cohort for the KIR2DL2 and combined HLA-B*46–KIR2DL2 genotypes.
Survival analysis was performed using Cox proportional hazard model to validate whether the previously significant HLA alleles modified disease progression with time to CD4 count <350 cells/mm3 or initiation of ART in the BMCS cohort. Individuals who initiated ART were censored at their last visit prior to treatment. The estimated date of infection (EDI) was defined as the median between the date of last negative (LN) and first positive (FP) HIV test. Time from EDI to the first instance of either outcome was calculated in months. The proportionality assumption for the Cox model was acceptable on the basis of the test of independence between the scaled Schoenfeld residuals and time. Linear regression was employed to assess the association of significant HLA alleles with the average VL over all available time points before ART initiation. All plasma VL measurements were log10-transformed to normalize their distribution. GMR, mean, SD and 95% CI were presented for each HLA allele. Both the Cox and linear regression models were adjusted for age.
HLA associations with CD4 counts
The association of specific HLA alleles with baseline CD4 counts prior to ART initiation was assessed using linear regression in the RV254 cohort. The model was adjusted with covariate age, sex, Fiebig stage and baseline viral load. All baseline measurements were log10-transformed to normalize their distribution. Assessment of model diagnostics (Q-Q plot for normality, residuals vs. fitted values for homoscedasticity, leverage plots for influential observations, variance inflation factors for multicollinearity) showed that the assumptions of the linear models were reasonable after removing four outliers identified by Cook’s Distance in RV254. Similar linear regression analysis was performed in the BMCS and RV152 cohorts. The first CD4 count in RV152 and BMCS were used as described previously (Leelawiwat et al., 2018; Rerks-Ngarm et al., 2013). This included the first CD4 count after HIV diagnosis in RV152, or within 120 days from EDI in BMCS. For the BMCS study, the model included adjustment only for age (Leelawiwat et al., 2018), while RV152 models included age, sex, baseline behavioral risk, and the calendar year of HIV-1 infection diagnosis (2003–2005, 2006, 2007, 2008–2009) as described previously (Rerks-Ngarm et al., 2013).
HLA alleles at 1 or 2 field resolution with an observed frequency of >5% were tested for all associations with HIV acquisition and disease progression. A two-sided p < 0.05 and q < 0.2 were considered to be significant for all HLA genetics analyses described above (Benjamini and Hochberg, 1995).
Bulk RNA-seq analyses
Sequence alignments were performed as previously described (Ehrenberg et al., 2019; Shangguan et al., 2021), but modified to use Subread featureCounts v1.6.4 for counting (Liao et al., 2014). The Trimmed Mean of M-values method (TMM), as implemented in the R package edgeR v3.34.0, was used for normalization with the limma voom model (Robinson et al., 2010). The general linear model (GLM) pipeline in edgeR was used to identify differentially expressed genes between the B*46 + and - groups within each time point. Enriched gene signatures were identified by the Gene Set Enrichment Analysis (GSEA) method using the blood transcription modules (BTM) and the GSEA software (version 4.1.0) (Li et al., 2014; Subramanian et al., 2005). For each GSEA comparison, we tested whether gene sets were significantly enriched in the presence or absence of HLA-B*46 in all four sorted PBMC populations. Transcriptome-wide signatures were considered significantly enriched using a threshold of false discovery rate (FDR) <0.01. Permutation analyses was performed to assess the robustness of the GSEA results in NK cells. GSEA was performed on samples from the 7 participants at the setpoint time point after randomly assigning them into two groups of 3 and 4 samples (representing B*46+ and samples, respectively) 1000 times. For each of the 1000 iterations the difference in the number of significantly enriched pathways between the two assigned groups were computed. The distribution of these differences were used to determine the probability of identifying a difference greater than or equal to the observed difference. Functional annotation of genes from the M11.0 signature at the pre-peak and setpoint time points was performed using Metascape with default parameters (database v20220101) (Zhou et al., 2019). The differential expression of core enriched genes in all signatures significantly enriched in NK cells at setpoint was visualized in a heatmap showing the log2 fold-change of gene expression for each sample relative to the mean expression of the gene in B*46+ donors.
Single cell data processing
FASTQ generation, read alignment, gene counting, and cell calling was performed with CellRanger 3.1.0 (10x Genomics), using human genome reference (GRCh38 Ensembl annotation v98). Analysis of gene and ADT data was performed in Seurat v4.0.5 (Hao et al., 2021; Zheng et al., 2017). An average of approximately 17,600 estimated cells were recovered per lane. The average number of genes per cell was 1331 and the average number of unique molecular identifiers was 3623. The mean read depth per cell was approximately 37,500–72,500. The minimum fraction of reads mapped to the genome was 82%. Average sequencing saturation for RNA and protein (including hashing antibodies) was 78% (ranging between 76–87%) and 55% (54–58%), respectively. The cell hashing ADT counts were centered log ratio (CLR) normalized. Hash counts were also CLR normalized and demultiplexed using HTODemux in Seurat in order to assign cell barcodes to specific donors. The non-hash feature barcode ADT data were normalized using the Denoised and Scaled by Background (DSB) method (Kotliarov et al., 2020; Mulè et al., 2022). After the cells were filtered to remove hash multiplets and cells with mitochondrial gene expression percentages > 10%, cells belonging to the same donors were merged. For the new donor-specific Seurat objects, gene expression was normalized with NormalizeData, and prepared for integration using FindVariableFeatures, ScaleData, and RunPCA. Integration features were selected and the gene expression data was integrated across donors using rpca and reference-based integration (one donor per ADT batch was selected as a ‘‘reference’’). The ADT data were scaled with ADT batch regressed and used for PCA. Genes and proteins upregulated and belonging to specific clusters in 64,020 cells were identified by FindAllMarkers in Seurat. Similarly, FindMarkers was also used to identify genes or proteins that were up- or down-regulated between the B*46+ and - cells within each cluster. Significant results from the FindMarkers analyses were defined as features that had a log2 fold change > 0.25, adjusted p < 0.05, and expression in at least 10% of cells in either the B*46+ or - group. Significance of differential expression was tested by Wilcoxon Rank Sum test for RNA features, and Logistic Regression for regressing the ADT batch for the protein features. The cells comprising the NK CD56dim and NK CD56hi clusters were extracted as a subset for additional QC and reclustering: ADT and RNA features and clusters were used to identify and remove nonNK cells from the NK subset. Four clusters generated from ADT data at a resolution of 0.5 were used as the final clusters for DEG analysis. In each of the four clusters identified in Seurat, genes expressed in at least 10% of cells in the B*46+ and - groups were ranked using their fold change between the two groups, and enrichment of gene signatures from BTM pathways in NK cells was examined using the pre-ranked GSEA algorithm. Signatures that were significantly enriched (q < 0.01) were subjected to a Leading Edge GSEA analysis to determine genes contributing most towards their enrichment scores. CITE-seq data generated previously in healthy RV306 participants without HIV infection (n = 9) was analyzed as described above and stratified further by the HLA-B*46 genotype (Shangguan et al., 2021). Similar analyses were performed for RV254 scRNA-seq data (n = 6) but without the ADT data analyses; the RV254 ART dataset was used as a reference to project cluster labels in Seurat.
All descriptive and inferential statistical analyses were performed using R 3.5.1 (or later) software packages. Custom R scripts were used for visualization.
Supplementary Material
Highlights.
HLA-B*46 associates with accelerated HIV disease
progression and reduced CD4 counts
HLA-B*46 is unique in conferring unusual stimulation of NK cell receptors
In people without B*46, CITE-seq reveals subsets of NK cells with distinct phenotypes
These differences in innate immunity may confer protection upon HIV infection
ACKNOWLEDGMENTS
We would like to thank the volunteers and staff of the RV152, RV254/SEARCH 010, BMCS, and RV217 cohorts. We thank Dr. Denise Hsu, MHRP for helpful comments. The views expressed are those of the authors and should not be construed to represent the positions or views of the US Army or the US Department of Defense (DOD), the US Centers for Disease Control and Prevention, and/or the US Public Health Service and/or the US Government. This work was supported by a cooperative agreement (W81XWH-07-2-0067) between the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc., the DOD, and CDC/DHAP. This research was funded in part by the US National Institute of Allergy and Infectious Disease.
The RV254/SEARCH 010 is supported by cooperative agreements (WW81XWH-18-2-0040) between the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc. and the US Department of Defense (DOD); by an intramural grant from the Thai Red Cross AIDS Research Centre; and, in part, by the Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institute of Health (DAIDS, NIAID, NIH) (grant AAI20052001). We are grateful to the Thai Government Pharmaceutical Organization (GPO), ViiV Healthcare, Gilead Sciences, and Merck for providing the antiretroviral medications for this study. Material has been reviewed by the Walter Reed Army Institute of Research, and there is no objection to its presentation and/or publication. The investigators have adhered to the policies for protection of human subjects as prescribed in AR 70-25.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.chom.2022.06.005.
REFERENCES
- Abi-Rached L, Moesta AK, Rajalingam R, Guethlein LA, and Parham P. (2010). Human-specific evolution and adaptation led to major qualitative differences in the variable receptors of human and chimpanzee natural killer cells. PLoS Genet. 6, e1001192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananworanich J, Fletcher JL, Pinyakorn S, van Griensven F, Vandergeeten C, Schuetz A, Pankam T, Trichavaroj R, Akapirat S, Chomchey N, et al. (2013). A novel acute HIV infection staging system based on 4th generation immunoassay. Retrovirology 10, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apps R, Del Prete GQ, Chatterjee P, Lara A, Brumme ZL, Brockman MA, Neil S, Pickering S, Schneider DK, Piechocka-Trocha A, et al. (2016). HIV-1 Vpu mediates HLA-C downregulation. Cell Host Microbe 19, 686–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apps R, Meng Z, Del Prete GQ, Lifson JD, Zhou M, and Carrington M. (2015). Relative expression levels of the HLA class-I proteins in normal and HIV-infected cells. J. Immunol 194, 3594–3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apps R, Qi Y, Carlson JM, Chen H, Gao X, Thomas R, Yuki Y, Del Prete GQ, Goulder P, Brumme ZL, et al. (2013). Influence of HLA-C expression level on HIV control. Science 340, 87–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber LD, Percival L, Valiante NM, Chen L, Lee C, Gumperz JE, Phillips JH, Lanier LL, Bigge JC, Parekh RB, et al. (1996). The inter-locus recombinant HLA-b*4601 has high selectivity in peptide binding and functions characteristic of HLA-C. J. Exp. Med 184, 735–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashirova AA, Thomas R, and Carrington M. (2011). HLA/KIR restraint of HIV: surviving the fittest. Annu. Rev. Immunol 29, 295–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc 57, 289–300. [Google Scholar]
- Chung WH, Hung SI, Hong HS, Hsih MS, Yang LC, Ho HC, Wu JY, and Chen YT (2004). Medical genetics: a marker for Stevens-Johnson syndrome. Nature 428, 486. [DOI] [PubMed] [Google Scholar]
- Cohen GB, Gandhi RT, Davis DM, Mandelboim O, Chen BK, Strominger JL, and Baltimore D. (1999). The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 10, 661–671. [DOI] [PubMed] [Google Scholar]
- Collins DR, Gaiha GD, and Walker BD (2020). CD8(+) T cells in HIV control, cure and prevention. Nat. Rev. Immunol 20, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins KL, Chen BK, Kalams SA, Walker BD, and Baltimore D. (1998). HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 391, 397–401. [DOI] [PubMed] [Google Scholar]
- De Souza MS, Phanuphak N, Pinyakorn S, Trichavaroj R, Pattanachaiwit S, Chomchey N, Fletcher JL, Kroon ED, Michael NL, Phanuphak P, et al. (2015). Impact of nucleic acid testing relative to antigen/antibody combination immunoassay on the detection of acute HIV infection. AIDS 29, 793–800. [DOI] [PubMed] [Google Scholar]
- Ehrenberg PK, Geretz A, Baldwin KM, Apps R, Polonis VR, Robb ML, Kim JH, Michael NL, and Thomas R. (2014). High-throughput multiplex HLA genotyping by next-generation sequencing using multi-locus individual tagging. BMC Genomics 15, 864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenberg PK, Geretz A, and Thomas R. (2018). High-throughput contiguous full-length next-generation sequencing of HLA class I and II genes from 96 donors in a single MiSeq run. Methods Mol. Biol 1802, 89–100. [DOI] [PubMed] [Google Scholar]
- Ehrenberg PK, Shangguan S, Issac B, Alter G, Geretz A, Izumi T, Bryant C, Eller MA, Wegmann F, Apps R, et al. (2019). A vaccine-induced gene expression signature correlates with protection against SIV and HIV in multiple trials. Sci. Transl. Med 11, eaaw4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, and Lischer HE (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour 10, 564–567. [DOI] [PubMed] [Google Scholar]
- Fellay J, Shianna KV, Ge D, Colombo S, Ledergerber B, Weale M, Zhang K, Gumbs C, Castagna A, Cossarizza A, et al. (2007). A whole-genome association study of major determinants for host control of HIV-1. Science 317, 944–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich TC, Dodds EJ, Yant LJ, Vojnov L, Rudersdorf R, Cullen C, Evans DT, Desrosiers RC, Mothé BR, Sidney J, et al. (2004). Reversion of CTL escape-variant immunodeficiency viruses in vivo. Nat. Med 10, 275–281. [DOI] [PubMed] [Google Scholar]
- Gandhi RT, Bosch RJ, Rangsin R, Chuenchitra T, Sirisopana N, Kim JH, Robb ML, Vejbaesya S, Paris RM, and Nelson KE (2016). HLA Class I alleles associated with mortality in Thai military recruits with HIV-1 CRF01_AE infection. AIDS Res. Hum. Retrovir 32, 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Nelson GW, Karacki P, Martin MP, Phair J, Kaslow R, Goedert JJ, Buchbinder S, Hoots K, Vlahov D, et al. (2001). Effect of a single amino acid change in MHC class I molecules on the rate of progression to AIDS. N. Engl. J. Med 344, 1668–1675. [DOI] [PubMed] [Google Scholar]
- Geretz A, Ehrenberg PK, Bouckenooghe A, Fernández Viña MA, Michael NL, Chansinghakule D, Limkittikul K, and Thomas R. (2018). Full-length next-generation sequencing of HLA class I and II genes in a cohort from Thailand. Hum. Immunol 79, 773–780. [DOI] [PubMed] [Google Scholar]
- Goulder PJ, and Walker BD (2012). HIV and HLA class I: an evolving relationship. Immunity 37, 426–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray GE, Bekker LG, Laher F, Malahleha M, Allen M, Moodie Z, Grunenberg N, Huang Y, Grove D, Prigmore B, et al. (2021). Vaccine efficacy of ALVAC-HIV and bivalent Subtype C gp120-MF59 in adults. N. Engl. J. Med 384, 1089–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes BF, Gilbert PB, McElrath MJ, Zolla-Pazner S, Tomaras GD, Alam SM, Evans DT, Montefiori DC, Karnasuta C, Sutthent R, et al. (2012). Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N. Engl. J. Med 366, 1275–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton HG, McMurtrey CP, Han AS, Djaoud Z, Guethlein LA, Blokhuis JH, Pugh JL, Goyos A, Horowitz A, Buchli R, et al. (2017). The intergenic recombinant HLA-B *46:01 Has a Distinctive Peptidome that Includes KIR2DL3 Ligands. Cell Rep. 19, 1394–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, and Markowitz M. (1995). Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373, 123–126. [DOI] [PubMed] [Google Scholar]
- Hogg RS, Yip B, Chan KJ, Wood E, Craib KJ, O’Shaughnessy MV, and Montaner JS (2001). Rates of disease progression by baseline CD4 cell count and viral load after initiating triple-drug therapy. JAMA 286, 2568–2577. [DOI] [PubMed] [Google Scholar]
- Højsgaard S, Halekoh U, and Yan J. (2006). The R package geepack for generalized estimating equations. Journal of Statistical Software 34 (1). [Google Scholar]
- International HIV Controllers Study, Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PI, Walker BD, Ripke S, Brumme CJ, Pulit SL, et al. (2010). The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science 330, 1551–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaslow RA, Carrington M, Apple R, Park L, Muñoz A, Saah AJ, Goedert JJ, Winkler C, O’Brien SJ, Rinaldo C, et al. (1996). Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nat. Med 2, 405–411. [DOI] [PubMed] [Google Scholar]
- Kotliarov Y, Sparks R, Martins AJ, Mule MP, Lu Y, Goswami M, Kardava L, Banchereau R, Pascual V, Biancotto A, et al. (2020). Broad immune activation underlies shared set point signatures for vaccine responsiveness in healthy individuals and disease activity in patients with lupus. Nat. Med 26, 618–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier LL (2008). Up on the tightrope: natural killer cell activation and inhibition. Nat. Immunol 9, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier LL, Chang C, and Phillips JH (1994). Human NKR-P1A. A disulphide-linked homodimer of the C-type lectin superfamily expressed by a subset of NK and T lymphocytes. J. Immunol 153, 2417–2428. [PubMed] [Google Scholar]
- Lazaro A, Tu B, Yang R, Xiao Y, Kariyawasam K, Ng J, and Hurley CK (2013). Human leukocyte antigen (HLA) typing by DNA sequencing. Methods Mol. Biol 1034, 161–195. [DOI] [PubMed] [Google Scholar]
- Leelawiwat W, Pattanasin S, Sriporn A, Wasinrapee P, Kongpechsatit O, Mueanpai F, Tongtoyai J, Holtz TH, and Curlin ME (2018). Association between HIV genotype, viral load and disease progression in a cohort of Thai men who have sex with men with estimated dates of HIV infection. PLoS One 13, e0201386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, Feeney M, Tang Y, Holmes EC, Allen T, Prado JG, et al. (2004). HIV evolution: CTL escape mutation and reversion after transmission. Nat. Med 10, 282–289. [DOI] [PubMed] [Google Scholar]
- Li S, Rouphael N, Duraisingham S, Romero-Steiner S, Presnell S, Davis C, Schmidt DS, Johnson SE, Milton A, Rajam G, et al. (2014). Molecular signatures of antibody responses derived from a systems biology study of five human vaccines. Nat. Immunol 15, 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- McLaren PJ, and Carrington M. (2015). The impact of host genetic variation on infection with HIV-1. Nat. Immunol 16, 577–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren PJ, Coulonges C, Bartha I, Lenz TL, Deutsch AJ, Bashirova A, Buchbinder S, Carrington MN, Cossarizza A, Dalmau J, et al. (2015). Polymorphisms of large effect explain the majority of the host genetic contribution to variation of HIV-1 virus load. Proc. Natl. Acad. Sci. USA 112, 14658–14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migueles SA, Sabbaghian MS, Shupert WL, Bettinotti MP, Marincola FM, Martino L, Hallahan CW, Selig SM, Schwartz D, Sullivan J, et al. (2000). HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc. Natl. Acad. Sci. USA 97, 2709–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moesta AK, Norman PJ, Yawata M, Yawata N, Gleimer M, and Parham P. (2008). Synergistic polymorphism at two positions distal to the ligand-binding site makes KIR2DL2 a stronger receptor for HLA-C than KIR2DL3. J. Immunol 180, 3969–3979. [DOI] [PubMed] [Google Scholar]
- Moradi S, Stankovic S, O’Connor GM, Pymm P, MacLachlan BJ, Faoro C, Retiè re C, Sullivan LC, Saunders PM, Widjaja J, et al. (2021). Structural plasticity of KIR2DL2 and KIR2DL3 enables altered docking geometries atop HLA-C. Nat. Commun 12, 2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, Wichukchinda N, Miyahara R, Rojanawiwat A, Pathipvanich P, Miura T, Yasunami M, Ariyoshi K, and Sawanpanyalert P. (2018). Impact of HLA allele-KIR pairs on disease outcome in HIV-infected Thai population. J. Acquir. Immune Defic. Syndr 78, 356–361. [DOI] [PubMed] [Google Scholar]
- Mulè MP, Martins AJ, and Tsang JS (2022). Normalizing and denoising protein expression data from droplet-based single cell profiling. Nat. Commun 13, 2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelak K, Goldstein DB, Walley NM, Fellay J, Ge D, Shianna KV, Gumbs C, Gao X, Maia JM, Cronin KD, et al. (2010). Host determinants of HIV-1 control in African Americans. J. Infect. Dis 201, 1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Bjö rklund AK, Winberg G, Sagasser S, and Sandberg R. (2014). Full-length RNA-seq from single cells using Smartseq2. Nat. Protoc 9, 171–181. [DOI] [PubMed] [Google Scholar]
- Prentice HA, Ehrenberg PK, Baldwin KM, Geretz A, Andrews C, Nitayaphan S, Rerks-Ngarm S, Kaewkungwal J, Pitisuttithum P, O’Connell RJ, et al. (2014). HLA class I, KIR, and genome-wide SNP diversity in the RV144 Thai phase 3 HIV vaccine clinical trial. Immunogenetics 66, 299–310. [DOI] [PubMed] [Google Scholar]
- Prentice HA, Tomaras GD, Geraghty DE, Apps R, Fong Y, Ehrenberg PK, Rolland M, Kijak GH, Krebs SJ, Nelson W, et al. (2015). HLA class II genes modulate vaccine-induced antibody responses to affect HIV-1 acquisition. Sci. Transl. Med 7, 296ra112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsköld D, Luo S, Wang YC, Li R, Deng Q, Faridani OR, Daniels GA, Khrebtukova I, Loring JF, Laurent LC, et al. (2012). Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol 30, 777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsuran V, Naranbhai V, Horowitz A, Qi Y, Martin MP, Yuki Y, Gao X, Walker-Sperling V, Del Prete GQ, Schneider DK, et al. (2018). Elevated HLA-A expression impairs HIV control through inhibition of NKG2A-expressing cells. Science 359, 86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rerks-Ngarm S, Paris RM, Chunsutthiwat S, Premsri N, Namwat C, Bowonwatanuwong C, Li SS, Kaewkungkal J, Trichavaroj R, Churikanont N, et al. (2013). Extended evaluation of the virologic, immunologic, and clinical course of volunteers who acquired HIV-1 infection in a phase III vaccine trial of ALVAC-HIV and AIDSVAX B/E. J. Infect. Dis 207, 1195–1205. [DOI] [PubMed] [Google Scholar]
- Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, Premsri N, Namwat C, de Souza M, Adams E, et al. (2009). Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med 361, 2209–2220. [DOI] [PubMed] [Google Scholar]
- Robb ML, Eller LA, Kibuuka H, Rono K, Maganga L, Nitayaphan S, Kroon E, Sawe FK, Sinei S, Sriplienchan S, et al. (2016). Prospective study of acute HIV-1 infection in adults in East Africa and Thailand. N. Engl. J. Med 374, 2120–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sconocchia G, Titus JA, Mazzoni A, Visintin A, Pericle F, Hicks SW, Malavasi F, and Segal DM (1999). CD38 triggers cytotoxic responses in activated human natural killer cells. Blood 94, 3864–3871. [PubMed] [Google Scholar]
- Shangguan S, Ehrenberg PK, Geretz A, Yum L, Kundu G, May K, Fourati S, Nganou-Makamdop K, Williams LD, Sawant S, et al. (2021). Monocyte-derived transcriptome signature indicates antibody-dependent cellular phagocytosis as a potential mechanism of vaccine-induced protection against HIV-1. eLife 10, e69577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas R, Apps R, Qi Y, Gao X, Male V, O’HUigin C, O’Connor G, Ge D, Fellay J, Martin JN, et al. (2009). HLA-C cell surface expression and control of HIV/AIDS correlate with a variant upstream of HLA-C. Nat. Genet 41, 1290–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting AT, Dick CJ, Schoon RA, Karnitz LM, Abraham RT, and Leibson PJ (1995). Interaction between lck and syk family tyrosine kinases in Fc gamma receptor-initiated activation of natural killer cells. J. Biol. Chem 270, 16415–16421. [DOI] [PubMed] [Google Scholar]
- van Griensven F, Thienkrua W, McNicholl J, Wimonsate W, Chaikummao S, Chonwattana W, Varangrat A, Sirivongrangson P, Mock PA, Akarasewi P, et al. (2013). Evidence of an explosive epidemic of HIV infection in a cohort of men who have sex with men in Thailand. AIDS 27, 825–832. [DOI] [PubMed] [Google Scholar]
- Wang LM, Kimura A, Satoh M, and Mineshita S. (1999). HLA linked with leprosy in southern China: HLA-linked resistance alleles to leprosy. Int. J. Lepr. Other Mycobact. Dis 67, 403–408. [PubMed] [Google Scholar]
- Wei Z, Liu Y, Xu H, Tang K, Wu H, Lu L, Wang Z, Chen Z, Xu J, Zhu Y, et al. (2015). Genome-wide association studies of HIV-1 host control in ethnically diverse Chinese populations. Sci. Rep 5, 10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO; World Health Organization (WHO). (2015). Guideline on When to Start Antiretroviral Therapy and on Pre-exposure Prophylaxis for HIV (Geneva: World Health Organization; ). https://www.ncbi.nlm.nih.gov/books/NBK327115/. [PubMed] [Google Scholar]
- Yawata M, Yawata N, Draghi M, Partheniou F, Little AM, and Parham P. (2008). MHC class I-specific inhibitory receptors and their ligands structure diverse human NK-cell repertoires toward a balance of missing self-response. Blood 112, 2369–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemmour J, and Parham P. (1992). Distinctive polymorphism at the HLA-C locus: implications for the expression of HLA-C. J. Exp. Med 176, 937–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat. Commun 8, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data has been deposited at GEO and are publicly available as of the date of publication. Accession number is listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| anti-Human CD1c (Mouse monoclonal) | 3ioLegend | Cat# 331547; Clone L161; RRID: AB_2800871 |
| anti-Human CD163 (Mouse monoclonal) | 3ioLegend | Cat# 333637; Clone GHI/61; RRID: AB_2810510 |
| anti-Human CD141 (Mouse monoclonal) | 3ioLegend | Cat# 344125; Clone M80; RRID: AB_2810541 |
| anti-Human CD11a (Mouse monoclonal) | 3ioLegend | Cat# 350617; Clone TS2/4; RRID: AB_2800935 |
| anti-Human CD197 (Mouse monoclonal) | 3ioLegend | Cat# 353251;Clone G043H7; RRID: AB_2800943 |
| anti-Human CD14 (Mouse monoclonal) | 3ioLegend | Cat# 301859; Clone M5E2; RRID:AB_2800736 |
| anti-Human CD16 (Mouse monoclonal) | BioLegend | Cat# 302065; Clone 3G8; RRID: AB_2800738 |
| anti-Human CD19 (Mouse monoclonal) | BioLegend | Cat# 302265; Clone HIB19; RRID: AB_2800741 |
| anti-Human CD45RO (Mouse monoclonal) | BioLegend | Cat# 304259; Clone UCHL1; RRID: AB_2800766 |
| anti-Human CD2 (Mouse monoclonal) | BioLegend | Cat# 309231; Clone TS1/8; RRID: AB_2810464 |
| anti-Human CD138 (Mouse monoclonal) | BioLegend | Cat# 356539; Clone MI15; RRID: AB_2810567 |
| anti-Human CD303 (Mouse monoclonal) | BioLegend | Cat# 354241; Clone 201A; RRID: AB_2814295 |
| anti-Human CD56 (Mouse monoclonal) | BioLegend | Cat# 362559; Clone5.1h11; RRID: AB_2801002 |
| anti-Human CD4 (Mouse monoclonal) | BioLegend | Cat# 300567; Clone RPA-T4; RRID: AB_2800725 |
| anti-Human CD3 (Mouse monoclonal) | BioLegend | Cat# 300479; Clone UCHT1; RRID: AB_2800723 |
| anti-Human CD45RA (Mouse monoclonal) | BioLegend | Cat# 304163; Clone HI100; RRID: AB_2800764 |
| anti-Human CD39 (Mouse monoclonal) | BioLegend | Cat# 328237;Clone A1; RRID: AB_2800853 |
| anti-Human CD279 (Mouse monoclonal) | BioLegend | Cat# 329963;Clone EH12.2H7; RRID: AB_2800862 |
| anti-Human CD8 (Mouse monoclonal) | BioLegend | Cat# 344753; Clone SK1; RRID: AB_2800922 |
| anti-Human CD27 (Mouse monoclonal) | BioLegend | Cat# 302853; Clone O323; RRID: AB_2800747 |
| anti-Human CD20 (Mouse monoclonal) | BioLegend | Cat# 302363; Clone 2H7; RRID: AB_2800743 |
| anti-Human HLA-A/B/C (Mouse monoclonal) | BioLegend | Cat# 311449; Clone W6/32; RRID: AB_2800816 |
| anti-Human IgM (Mouse monoclonal) | BioLegend | Cat# 314547; Clone MHM-88; RRID: AB_2800835 |
| anti-Human CD127 (Mouse monoclonal) | BioLegend | Cat# 351356; Clone A019D5; RRID: AB_2800937 |
| anti-Human CD195 (Rat monoclonal) | BioLegend | Cat# 359137; Clone J418F1; RRID: AB_2810570 |
| anti-Human HLA-DR (Mouse monoclonal) | BioLegend | Cat# 307663; Clone L243; RRID: AB_2800795 |
| anti-Human IgG (Fc) (Rat monoclonal) | BioLegend | Cat# 410727; Clone M1310G05; RRID: AB_2801087 |
| anti-Human TCR Vd2 (Mouse monoclonal) | BioLegend | Cat# 331435; Clone B6; RRID: AB_2800864 |
| anti-Human TCR Va7.2 (Mouse monoclonal) | BioLegend | Cat# 351735;Clone 3C10; RRID: AB_2810556 |
| anti-Human TCR Va24-Ja18 (Mouse monoclonal) | BioLegend | Cat# 342925; Clone 6B11; RRID: AB_2810539 |
| anti-Human TCR g/d (Mouse monoclonal) | BioLegend | Cat# 331231; Clone B1; RRID AB_2814199 |
| anti-Human TCR Vg9 (Mouse monoclonal) | BioLegend | Cat# 331313; Clone B3; RRID: AB_2814203 |
| anti-Human CD7 (Mouse monoclonal) | BioLegend | Cat# 343127; Clone CD7–6B7; RRID: AB_2800914 |
| anti-Human CD11c (Mouse monoclonal) | BioLegend | Cat# 371521; Clone S-HCL-3; RRID: AB_2801018 |
| anti-Human CD185 (Mouse monoclonal) | BioLegend | Cat# 356939,;Clone J252D4; RRID: AB_2800968 |
| anti-Human CD1d (Mouse monoclonal) | BioLegend | Cat# 350319; Clone 51.1; RRID: AB_2800934 |
| anti-Human IgD (Mouse monoclonal) | BioLegend | Cat# 348245; Clone IA6–2; RRID: AB_2810553 |
| anti-Human CD11b (Mouse monoclonal) | BioLegend | Cat# 301359; Clone ICRF44; RRID: AB_2800732 |
| anti-Human CD62L (Mouse monoclonal) | BioLegend | Cat# 304851; Clone DREG-56; RRID: AB_2800770 |
| anti-Human CD66a/c/e (Mouse monoclonal) | BioLegend | Cat# 342325; Clone ASL-32; RRID: AB_2810538 |
| anti-Human CD15 (Mouse monoclonal) | BioLegend | Cat# 323053; Clone W6D3; RRID: AB_2800847 |
| anti-Human CD32 (Mouse monoclonal) | BioLegend | Cat# 303225; Clone FUN-2; RRID: AB_2814129 |
| anti-Human CD57 (Mouse monoclonal) | BioLegend | Cat# 393321; Clone QA17A04; RRID: AB_2801030 |
| anti-Human CD73 (Mouse monoclonal) | BioLegend | Cat# 344031; Clone AD2; RRID: AB_2800916 |
| anti-Human CD123 (Mouse monoclonal) | BioLegend | Cat# 306045; Clone 6H6; RRID: AB_2800789 |
| anti-Human Mouse IgG1, k Isotype Ctrl (Mouse monoclonal) | BioLegend | Cat# 400187; Clone MOPC-21; RRID: AB_2888921 |
| anti-Human Mouse IgG2a, k Isotype Ctrl (Mouse monoclonal) | BioLegend | Cat# 400293; Clone MOPC-173; RRID: AB_2888922 |
| anti-Human Mouse IgG2b, k Isotype Ctrl (Mouse monoclonal) | BioLegend | Cat# 400381; Clone MPC-11; RRID: AB_2888923 |
| anti-Human Rat IgG2b, k Isotype Ctrl (Rat monoclonal) | BioLegend | Cat# 400677; Clone RTK4530: RRID: AB_2894967 |
| anti-Human CD28 (Mouse monoclonal) | BioLegend | Cat# 302963; Clone CD28.2; RRID:AB_2800751 |
| anti-Human CD161 (Mouse monoclonal) | BioLegend | Cat# 339947; Clone HP-3G10; RRID: AB_2810532 |
| anti-Human CD95 (Mouse monoclonal) | BioLegend | Cat# 305651; Clone DX2; RRID: AB_2800787 |
| anti-Human CD38 (Mouse monoclonal) | BioLegend | Cat# 303543; Clone HIT2; RRID: AB_2800758 |
| anti-Human KLRG1 (Syrian hamster monoclonal) | BioLegend | Cat# 138433; Clone 2F1/KLRG1; RRID: AB_2800649 |
| anti-Human CD25 (Mouse monoclonal) | BioLegend | Cat# 302649; Clone BC96; RRID: AB_2800745 |
| anti-Human CD41 (Mouse monoclonal) | BioLegend | Cat# 303739; Clone HIP8; RRID: AB_2814134 |
| anti-Human CD184 (Mouse monoclonal) | BioLegend | Cat# 306533; Clone 12G5; RRID: AB_2800791 |
| anti-Human CD137 (Mouse monoclonal) | BioLegend | Cat# 309839; Clone 4B4–1; RRID: AB_2800807 |
| anti-Human CD24 (Mouse monoclonal) | BioLegend | Cat# 311143; Clone ML5; RRID: AB_2800813 |
| anti-Human CD274 (Mouse monoclonal) | BioLegend | Cat# 329751; Clone 29E.2A3; RRID: AB_2800860 |
| anti-Human CD96 (Mouse monoclonal) | BioLegend | Cat# 338423; Clone NK92.39; RRID: AB_2810531 |
| anti-Human CD194 (Mouse monoclonal) | BioLegend | Cat# 359425; Clone L291H4; RRID: AB_2800988 |
| anti-Human CD152 (Mouse monoclonal) | BioLegend | Cat# 369621; Clone BNI3; RRID: AB_2801015 |
| Hashl: anti-Human CD298 S b2-microglobulin (Mouse monoclonals) | BioLegend | Cat# 394661; Clone LNH-94; 2M2; RRID: AB_2801031 |
| Hash2: anti-Human CD298 S b2-microglobulin (Mouse monoclonals) | BioLegend | Cat# 394663; Clone LNH-94; 2M2; RRID: AB_2801032 |
| Hash3: anti-Human CD298 S b2-microglobulin (Mouse monoclonals) | BioLegend | Cat# 394665; Clone LNH-94; 2M2; RRID: AB_2801033 |
| Hash4: anti-Human CD298 S b2-microglobulin (Mouse monoclonals) | BioLegend | Cat# 394667; Clone LNH-94; 2M2; RRID: AB_2801034 |
| Hash5: anti-Human CD298 S b2-microglobulin (Mouse monoclonals) | BioLegend | Cat# 394669; Clone LNH-94; 2M2; RRID: AB_2801035 |
| Hash6: anti-Human CD298 S b2-microglobulin (Mouse monoclonals) | BioLegend | Cat# 394671; Clone LNH-94; 2M2; RRID: AB_2820042 |
| Hash7: anti-Human CD298 S b2-microglobulin (Mouse monoclonals) | BioLegend | Cat# 394673; Clone LNH-94; 2M2; RRID: AB_2820043 |
| Hash8: anti-Human CD298 S b2-microglobulin (Mouse monoclonals) | BioLegend | Cat# 394675; Clone LNH-94; 2M2, RRID: AB_2820044 |
| Hash9: anti-Human CD298 S b2-microglobulin (Mouse monoclonals) | BioLegend | Cat# 394677; Clone LNH-94; 2M2; RRID: AB_2820045 |
| Hash10: anti-Human CD298 S b2-microglobulin (Mouse monoclonals) | BioLegend | Cat# 394679; Clone LNH-94; 2M2; RRID: AB_2820046 |
| Mouse anti-human CD3 FITC | Becton Dickinson | Cat# 555332; Clone UCHT1; RRID: AB_395739 |
| Mouse anti-human CD8 PerCP-eF710 | eBiosciences | Cat# 46–0087-42; Clone SK1; RRID: AB_1834411 |
| Mouse anti-human CD14 V500 | Becton Dickinson | Cat# 562693; Clone M5E2; RRID: AB_2737727 |
| Mouse anti-human CD45RO eF650NC | eBiosciences | Cat# 95–0457-42; Clone UCHL1; RRID: AB_1603247 |
| Mouse anti-human CD4 APC | Becton Dickinson | Cat# 561841; Clone RPAT4; RRID: AB_395739 |
| Mouse anti-human CD45RA APC-H7 | Becton Dickinson | Cat# 560674; Clone HI100; RRID: AB_1727497 |
| Mouse anti-human CD19 PE-Cy5 | Invitrogen | Cat# MHCD1918; Clone SJ25-C1; RRID: AB_10373840 |
| Mouse anti-human CD56 PE-Cy7 | Becton Dickinson | Cat# 335809; Clone NCAM16.2; RRID: AB_399984 |
| Mouse IgG isotype control | Caltag | Cat# 10400C; RRID: AB_2532980 |
|
| ||
| Biological samples | ||
|
| ||
| PBMC from MHRP RV217 donors | M. Robb | Robb et al., 2016 |
| PBMC from MHRP RV254 donors | D. Hsu | Ananworanich et al., 2013; De Souza et al., 2015 |
| PBMC from MHRP RV152 donors | A. Schuetz | Rerks-Ngarm et al., 2013 |
| PBMC from CDC BMCS donors | A. Hickey | van Griensven et al., 2013 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Cell Staining Buffer | BioLegend | Cat# 420201 |
| TruStain FcX Blocking Reagent, human | BioLegend | Cat# 422301 |
| Acridine Orange | ThermoFisher Scientific | Cat# A3568 |
| Ethidium Homodimer | ThermoFisher Scientific | Cat# E1169 |
| Glycerin (Glycerol), 50% (v/v) | Ricca Chemical Co | Cat# 3290–16 |
| Buffer EB | Qiagen | Cat# 19086 |
| Tween 20, 10% (v/v) | Bio-Rad | Cat# 1610781 |
| SPRIselect beads | Beckman Coulter | Cat# 823318 |
| Guava ViaCount Reagent | Millipore Corp | Cat# 4000–0041 |
| Live/Dead Fixable Aqua | ThermoFisher | Cat# L34957 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Chromium Single Cell 5’ Library & Gel Bead kit, v2 | 10x Genomics | Cat# 1000006 |
| Chromium Single Cell 5’ Feature Barcode Library kit | 10x Genomics | Cat# 1000080 |
| Single Index kit N, Set A | 10x Genomics | Cat# 1000212 |
| Single Index kit T, Set A | 10x Genomics | Cat# 1000213 |
| PhiX Control kit, v3 | Illumina | Cat# FC-110–3001 |
| High Sensitivity DNA kit | Agilent | Cat# 5067–4626 |
| Qubit dsDNA HS Assay kit | ThermoFisher Scientific | Cat# Q32854 |
| SMART-Seq v4 UltraLow Input RNA kit | Takara | Cat# 634891 |
| Single Cell RNA Purification kit | Norgen Biotek | Cat# 51800 |
| Nextera XT DNA Sample Preparation kit | Illumina | Cat# FC-131–1096 |
| ERCC RNA Spike-in Mix 1 | Life Technologies | Cat# 4456740 |
| NovaSeq 6000 S4 Reagent kit, 300 cycles, v1.5 | Illumina | Cat# 20028312 |
| MiSeq Reagent Nano kit, 500 cycles, v2 | Illumina | Cat# MS-103–1003 |
|
| ||
| Deposited data | ||
|
| ||
| RNA-seq analysis of human peripheral blood cells collected at multiple timepoints during the course of acute HIV-1 infection (RV217) | This paper | GEO: GSE192384; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE192384 |
| R code and data files for figures | This paper | https://doi.org/10.6084/m9.figshare.c.5728400 |
|
| ||
| Software and algorithms | ||
|
| ||
| FlowJo | FlowJo, LLC | https://www.flowjo.com/ |
| bcl2fastq | Illumina | https://support.illumina.com/downloads/bcl2fastq-conversion-software-v2–20.html |
| featureCounts | Liao et al., 2014 | http://subread.sourceforge.net/ |
| Cell Ranger | Zheng et al., 2017 | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads |
| R | https://www.r-project.org/ | |
| DSB | Mulè et al., 2022 | https://github.com/niaid/dsb |
| edgeR | Robinson et al., 2010 | https://bioconductor.riken.jp/packages/devel/bioc/html/edgeR.html |
| geepack | H0jsgaard et al., 2006 | https://cran.r-project.org/web/packages/geepack/index.html |
| Seurat | Hao et al., 2021 | https://cran.r-project.org/web/packages/Seurat/index.html |
| GSEA | Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/index.jsp |
| Metascape | Zhou et al., 2019 | https://metascape.org |
| Arlequin | Excoffier and Lischer, 2010 | http://cmpg.unibe.ch/software/arlequin35/Arl35Downloads.html |
All original R code are available in the figshare repository and is publicly available as of the date of publication. DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.