

Abstract
While contemporary changes in feeding preferences have been documented in phytophagous insects, the mechanisms behind these processes remain to be fully clarified. In this context, the insect gut microbiome plays a central role in adaptation to novel host plants. The cucurbit frugivorous fruit fly Zeugodacus cucurbitae (Diptera, Tephritidae) has occasionally been reported on “unconventional” host plants from different families, including Solanaceae. In this study, we focus on wild parental (F0) adults and semiwild first filial (F1) larvae of Z. cucurbitae from multiple sites in La Réunion and explore how the gut microbiome composition changes when this fly is feeding on a noncucurbit host (Solanum melongena). Our analyses show nonobvious gut microbiome responses following the F0–F1 host shift and the importance of not just diet but also local effects, which heavily affected the diversity and composition of microbiomes. We identified the main bacterial genera responsible for differences between treatments. These data further stress the importance of a careful approach when drawing general conclusions based on laboratory populations or inadequately replicated field samples.
Keywords: gut microbiota, host shift, plant–insect interactions, Tephritidae, wild populations
We investigated how the gut microbiome composition changes in a cucurbit‐feeding fly Zeugodacus cucurbitae when it feeds on atypical solanaceous hosts instead of typical cucurbit hosts. Wild parental (F0) adults and semiwild first filial (F1) larvae of Z. cucurbitae collected from two locations in Réunion Island showed heterogeneous microbiome responses across host plants. This study shows how local processes and host plants can strongly affect the composition of the insect microbiome and the importance of using adequately sampled populations.
1. INTRODUCTION
Insects are the most diverse group of Eukaryotes (Forister et al., 2015) with a vast variety of species being phytophagous and functionally classified in polyphagous, oligophagous, and monophagous when feeding on plants from multiple families, a single family or a single species, respectively (A. R. Clarke, 2017). Contemporary host plant feeding preferences are generally well defined but shifts in host plant preferences have been reported in a variety of insects, such as lepidopterans, beetles, and grasshoppers (Adams et al., 2013; Brown et al., 2014; Rosenberger et al., 2018; Singer & Parmesan, 2021; Sword et al., 2005). The shift toward novel hosts results in ecological niche expansion and subsequent adaptation to the new host can promote genetic divergence between populations, possibly with the evolution of host races and eventually of new species (Feder et al., 1988; Tilmon, 2008). Host races have been reported in a variety of insects, such as beetles and grasshoppers (Lefort et al., 2014; Sword et al., 2005), and a classical textbook example of the evolution of host races is Rhagoletis pomonella (Diptera, Tephritidae), where host‐shifts from hawthorn to apples have led to the evolution of genetically divergent populations with different feeding preferences (Feder et al., 1988). Host plant shift or expansion may also favor geographic range expansion (and vice versa) due to the possibility of occupying novel ecological niches and a wider geographic distribution (Hood et al., 2020; Lefort et al., 2014; Rosenberger et al., 2018; Singer & Parmesan, 2021). These processes are of major significance in agronomy and conservation biology as they can promote the emergence of new invasive species and agricultural pests (Brown et al., 2014; Lefort et al., 2014; Lu et al., 2011). The host shift of R. pomonella is considered the main cause of the expansion of this species in the Northwest Pacific (Hood et al., 2020).
While changes in insect feeding preferences have been documented in phytophagous insects, the mechanisms behind these processes remain to be clarified. In this context, the insect gut microbiome plays a central role in adaptation to novel host plants as it is of crucial importance for the complex interactions with insect metabolic pathways, which ultimately affect insect fitness (Hammer & Bowers, 2015; Zilber‐Rosenberg & Rosenberg, 2008). The gut microbiome of phytophagous insects can help to (1) break down the complex polysaccharides of the host plant cell wall, and (2) supplement nitrogen, vitamins, and sterols to nutritionally poor diets (Ben‐Yosef et al., 2010, 2014; Douglas, 2009), and (3) detoxify host plant allelochemicals (Hammer & Bowers, 2015) and insecticides (Ishigami et al., 2021; Kikuchi et al., 2012). For example, the symbiotic bacterium “Candidatus Erwinia dacicola” is essential for the metabolism of larvae of the olive fly Bactrocera oleae (Rossi, 1790) (Diptera, Tephritidae) as it allows them to feed on unripe olives rich in allelochemicals (Ben‐Yosef et al., 2015; Pavlidi et al., 2017). The geographic range expansion of the kudzu bug (Megacopta cribaria, Hemiptera: Plataspidae) in the United States has been related to genomic mutations in its symbiont Ishikawaella, which allowed the insect to attack soybean as a novel host plant (Brown et al., 2014). Likewise, the invasive spread of some bark beetle species (Coleoptera: Scolytinae) has been associated with compositional changes in their microbial and fungal symbionts (Adams et al., 2013; Lu et al., 2011).
Tephritid fruit flies are a diverse group of flies with herbivorous larvae, with several species being notorious agricultural pests (Norrbom et al., 1999). Recent studies have started to investigate the importance of their gut microbiomes. However, as most of these studies target laboratory colonies with depauperate microbiomes, their results might not be fully representative of what happens in the field (Augustinos et al., 2019; Morrow et al., 2015). For example, laboratory lines of the olive fly, Bactrocera oleae often lack “Candidatus Erwinia dacicola” despite this symbiont is crucial for larval survival and development on the olive host (Augustinos et al., 2019; Ben‐Yosef et al., 2015; Kounatidis et al., 2009). Conversely, Acetobacter, Morganella, and Paenibacillus can be found in laboratory lines of B. oleae, while these bacteria are not a relevant component of the gut microbiome of wild populations (Kounatidis et al., 2009). In this respect, De Cock et al. (2020) reported that gut microbiomes of wild tephritid agricultural pests are highly heterogeneous and stressed the importance of local effects in shaping gut microbiome diversity.
In this study, we focus on wild parental (F0) adults and semiwild (F1) larvae of the frugivorous fruit fly Zeugodacus cucurbitae (Coquillett, 1899) (Diptera, Tephritidae). This widespread agricultural pest is commonly found in South and East Asia, Africa, and Hawaii (De Meyer et al., 2015; Virgilio et al., 2010). It recently expanded from East to West Africa and the islands in the Indian Ocean including La Réunion (De Meyer et al., 2015; Delatte et al., 2019). Larvae of this oligophagous species typically feed on Cucurbitaceae, but occasionally they also attack a variety of “unconventional” host plants from different families, including Solanaceae (De Meyer et al., 2015; Dhillon et al., 2005; Hafsi et al., 2016; Moquet et al., 2021). This, together with observed geographic range expansion in recent decades (De Meyer et al., 2015), raises concerns about the invasion risk of this species, which might be polyphagous rather than oligophagous.
The objective of this study is to explore how the gut microbiome composition of the cucurbit‐feeder Z. cucurbitae changes when it feeds on a noncucurbit host plant and how gut microbiome assemblages could facilitate the use of novel host plants in this phytophagous agricultural pest.
2. MATERIALS AND METHODS
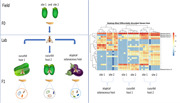
2.1. Experimental design, wet and dry laboratory procedures
In August 2019, infested, wild ivy gourd fruits (Coccinia grandis L. Voigt) were collected in La Réunion at two sites: Bassin Plat (−21.321457, 55.485044, “BP”) and Manapany (−21.374674, 55.598343, “M”). They were brought to the laboratories at CIRAD (Centre de coopération internationale en recherche agronomique pour le développement) in Saint‐Pierre. Collected fruits were placed in plastic boxes that were covered with fine‐mesh clothes and contained sand as a substrate for pupation. The fruits were kept in a climatic chamber (25 ± 1°C; 80 ± 10% HR; 12:12 light:dark photoperiod, with artificial light) until pupation. Boxes were regularly inspected for the presence of pupae. Pupae were transferred to 30 × 30 × 30 cm cages in the same climatic chamber. A few days later, adults emerged from the pupae. Adults were fed ad libitum with a diet of sugar and hydrolyzed yeast and had access to a wet sponge as a water source. We waited 3–4 weeks until females were mature and mated before the experiment was started. For each of the two sites, 12 adult gravid F0 females were randomly assigned to three experimental cages (four females per cage), each containing a single fruit of C. grandis L. (Cucurbitaceae, “Co”), or of Cucumis sativus L. (Cucurbitaceae host, “Cu”), or Solanum melongena L. (Solanaceae host, “So”). The females were let to oviposit and the resulting six groups of third instar F1 larvae (one group for each combination of site and host), were subsampled and subjected to gut microbiome profiling. For each fruit, we considered five replicated microbiome profiles. Each profile was represented by pooled DNA extracts from five individual larvae (following preliminary standardization of individual DNA concentrations). Due to the relatively large dimensions of the target fruits and the relatively limited proportion of plant tissue affected by larval activity, we assume that the extent of horizontal bacterial transmission across larvae within fruits was relatively limited and did not substantially bias the microbial patterns observed. To evaluate whether microbiome differences between larvae reflect differences between adults, we also profiled the microbiomes of four F0 adult females from each parental group. All specimens were preserved in 100% EtOH at −80°C before microbiome profiling. As De Cock et al. (2019) did not observe significant differences between whole body and gut microbiome profiles, we profiled whole body DNA extracts from larvae and adults. Laboratory procedures followed De Cock et al. (2020) unless indicated otherwise. Larval microbiome profiles were obtained by pooling DNA extracts from five individual larvae, while adult F0 females were individually profiled. Metagenomic library preparation and sequencing were outsourced to Macrogen (https://www.macrogen.com). For library preparation, the Nextera XT kit was used (target: V3–V4 regions of 16S rRNA, insert size: ca. 464 bp, primers: 341 F and 805 R (Takahashi et al., 2014). Libraries were sequenced on an Illumina MiSeq. 150 PE platform (300 bp paired‐end sequencing). A negative and positive control (ZymoBIOMICS Microbial Community Standard D6300) were included to check for artifacts in library preparation and sequencing. Results for the mock community can be found in Table A1.
After verifying the quality of reads with FastQC (Andrews, 2014), we used the DADA2 pipeline (Callahan et al., 2016) to remove primers and truncate reads (to a final length of 260 bp for forward reads and 240 bp for reverse reads resulting in ca. 36 bp overlap) and to fit the parametric error model for the identification and filtering of sequencing errors (2 × 106 reads used for model fitting). Filtering was based on maxEE = 1 in DADA2 (the complete DADA2 pipeline is available on GitHub and in Zenodo at https://doi.org/10.5281/zenodo.6810766). For error estimation, the first two million sequences were used in the error model construction. Before pairing forward and reverse reads and filtering out chimeras, unique amplicon sequence variants (ASVs; 100% unique sequence identity) were extracted using the Bayesian classifier method of DADA2. The Silva v.132 database was used for the taxonomic classification of ASVs (percentage of identity = 97% similarity, p‐min‐consensus = 0.51). Chloroplast and mitochondrial sequences were removed using the R package decontam (Davis et al., 2018).
Core (stable associates) bacterial genera and ASVs were identified using the abundance–ubiquity method with a 50% minimal ubiquity threshold. This statistic evaluates whether a bacterial taxon is not more abundant than expected for its ubiquity. Significant deviations from expectation indicate that a bacterial taxon is not a stable core member (Hester et al., 2016). Statistical analyses were performed in R unless stated otherwise. Permutational analysis of variance (PERMANOVA) was conducted with PRIMER v7 (K. R. Clarke & Gorley, 2015) using 9999 unrestricted permutations of raw data).
2.2. Microbiome diversity and predictive functional profiling
Three α diversity metrics were calculated: the Abundance coverage estimator (ACE) to assess ASV richness, the Inverse Simpson index (ISI) to assess ASV evenness, and Faith's phylogenetic diversity (FPD) to investigate phylogenetic richness. To evaluate differences in microbial α diversity between larvae raised on different host plants and/or between sites, we used two‐way ANOVAs (Underwood, 1997) which for F1 larvae included Site (BP and M) as a random factor and Host Plant (C. grandis, C. sativus, and S. melongena) as a fixed orthogonal factor and for F0 adults included Site (BP and M) as a random factor and Parental Group (groups 1, 2, and 3) as a random factor nested in Site. ANOVA was implemented using the GAD package (Sandrini‐Neto & Camargo, 2015). Count data from which diversity metrics were calculated were not normalized as all rarefaction plots reached a plateau (Figure A1). To ensure homoscedasticity, a log transformation was applied to the ISI and a fourth root transformation was applied to the ACE and FPD. Cochran's C tests were used to test for homogeneity of variances with the GAD package (Fox, 2006). Pairwise comparisons were done by using an F test with Holm correction for multiple comparisons implemented in the phia package (De Rosario‐Martinez, 2013).
Before calculating β diversities, we first removed all ASVs that occurred in only one sample (Chakrabarti et al., 2016; L. J. Clarke et al., 2019) and we normalized counts by transforming them into proportions to represent community structure (McKnight et al., 2019; McMurdie & Holmes, 2014).
Generalized UniFrac distances using the d5 matrix and unweighted UniFrac distances were calculated as β diversity metrics (Chen et al., 2012). As UniFrac distances take into account the phylogenetic relationships, we constructed a midpoint‐rooted maximum likelihood tree of the bacterial relationships using a general time‐reversible substitution model in the program Fasttree (Price et al., 2009). Bacterial 16S sequences were aligned with the DECIPHER algorithm (Wright, 2015).
Differences in microbiome β diversity between larvae raised on different host plants and/or between sites were tested using a two‐way PERMANOVA (Anderson, 2017) with Site (BP and M) as a random factor and host plant (C. grandis, C. sativus, and S. melongena) as a fixed orthogonal factor. The false discovery rate (FDR) correction (Benjamini & Hochberg, 1995) with experiment‐wise p < 0.05 was used to correct for multiple testing. PERMANOVA was also used to estimate the components of variance explained by each factor. Differences between larvae raised on different host plants were visualized with a principal coordinate analysis (PCoA) and 95% confidence ellipses were drawn using the ggplot2 package (Wickham & Chang, 2016).
To test for a differential abundance of microbial genera (i.e., genera with relatively more sequences assigned to them) among larvae raised on different host plants and from different sites, we used ALDEx2 (Fernandes et al., 2013). ASVs that could not be classified were assigned to distinct, unidentified genera. Genera that showed differential abundance between two treatments with an effect size difference between 1 and −1 were filtered out to reduce the false positive rate (Gloor, 2015). Significance was assessed by both the Welch t test and the Wilcoxon rank‐sum test followed by FDR correction with experiment‐wise p < 0.05 as FDR is better suited for exploratory analyses (Lee & Lee, 2018). We selected 23 genera that showed the greatest differences in ALDEx2 to visualize patterns in heatmaps using the pheatmap package (Kolde, 2015). Clustering of sites and diet sources was done using Euclidean distances. For eight of these genera, we constructed boxplots with ggplot and arranged them in a single figure using the ggarrange function in the ggpubr package (Kassambra & Kassambra, 2020).
3. RESULTS
The MiSeq Illumina run yielded more than 9.1 × 106 reads (mean per sample = 95,128.91; SD = 15,800.33). After filtering, demultiplexing, merging, and chimera removal, about 3.6 × 106 reads and 2030 unique ASVs were identified. The latter was then assigned to 404 identified genera.
3.1. Microbiome composition of adult Z. cucurbitae
Enterobacter, Klebsiella, and Citrobacter were identified as core genera sensu Hester et al. (2016). In general, microbiomes of adult flies had a mean ASV richness of 26.12 (SD = 8.12) and a mean FPD of 5.97 (SD = 1.01) for adult microbiomes. They were also more uneven (more dominated by few abundant taxa) with an average ISI of 4.95 (SD = 2.25). No differences in any diversity metric were found between F0 adults from different sites or different parental groups (Table A2). PERMANOVA of parental F0 adult fly microbiomes (Tables A3 and A4) revealed significant differences between parental groups when species presence/absence (unweighted UniFrac distances) was considered (with one out of six significant post hoc comparisons). However, when relative abundances were taken into account and more weight was given to highly abundant taxa (generalized UniFrac distances), the F0 parental groups were not significantly different from each other. No significant differences between sites were found for both distances (Table A3). The PCoA plots (Figure A2), could not resolve distinct parental clusters for both unweighted and generalized UniFrac distances.
3.2. Microbiome composition of larval Z. cucurbitae
Across all host plants, we identified seven core genera (Hester et al., 2016): Acinetobacter, Enterobacter, Klebsiella, Paenibacillus, Pseudomonas, Stenotrophomonas, and Sphingobacterium. Larval microbiomes showed a high ASV richness (mean = 145.36, SD = 75.56) but low phylogenetic diversity (mean = 13.51, SD = 4.58). Despite their high ASV richness, larval microbiomes were dominated by very few ASV as the ISI had a mean of 7.97 (SD = 6.21). The log‐transformed ISI did not show consistent patterns across sites or host plants (Figure A3 and Table A5). There was a significant interaction between the host plant and site in the fourth root transformed ACE, the log‐transformed ISI, and the fourth root transformed FPD (Table 1). The α diversity indices did not show consistent patterns across sites and host plants. ASV richness as estimated by ACE and FPD showed significantly higher values for Co in M, but not in BP. Conversely, evenness showed significantly higher values for So in BP, but not in M (Figure A3 and Table A5).
Table 1.
Analysis of variance on α diversity metrics (abundance coverage estimator, inverse Simpson index, and Faith's phylogenetic diversity) calculated from semiwild F1 larvae from different host plants (Coccinia grandis, Cucumis sativus, Solanum melongena) and sites (Basin Plat and Manapany).
| Abundance coverage estimator | df | Mean squares | Pseudo F | p Value | Effect |
|---|---|---|---|---|---|
| Site (Si) | 1 | 0.010 | 0.132 | 0.718 | Random |
| Host plant (Ho) | 2 | 0.255 | 0.219 | 0.820 | Fixed |
| Si × Ho | 2 | 1.167 | 14.494 | 0.000*** | |
| Residual | 24 | 0.080 | |||
| Transformation = Fourth root | |||||
| C = 0.365 n.s. | |||||
| Inverse Simpson index | df | Mean squares | Pseudo F | p Value | Effect |
|---|---|---|---|---|---|
| Site (Si) | 1 | 1.408 | 9.484 | 0.005** | Random |
| Host plant (Ho) | 2 | 2.433 | 3.896 | 0.204 | Fixed |
| Si × Ho | 2 | 0.624 | 4.206 | 0.027* | |
| Residual | 24 | 0.148 | |||
| Transformation = Log | |||||
| C = 0.385 n.s. | |||||
| Faith's phylogenetic diversity | df | Mean squares | Pseudo F | p Value | Factor |
|---|---|---|---|---|---|
| Site (Si) | 1 | 0.002 | 0.216 | 0.645 | Random |
| Host plant (Ho) | 2 | 0.054 | 0.444 | 0.692 | Fixed |
| Si × Ho | 2 | 0.122 | 9.254 | 0.001** | |
| Residual | 24 | 0.013 | |||
| Transformation = Fourth root | |||||
| C = 0.602** | |||||
Abbreviations: C, Cochran's C; n.s., not significant.
p < 0.05;
p < 0.01;
p < 0.001.
PERMANOVA of the generalized and unweighted UniFrac distances revealed a significant interaction for β diversity between host plant and site (Table 2). However, pairwise comparisons did not provide additional information as all were significant (Table A6). PERMANOVA of the unweighted UniFrac distances showed that the host diet explained 21% of variation, the site explained 18.8% of variation, and 33% of variation was explained by their interaction. For the generalized UniFrac distances, 18% of variation was explained by site, 13.6% of variation by host diet, and 27% by their interaction.
Table 2.
Permutational analysis of variance on (A) unweighted UniFrac and (B) generalized UniFrac distances calculated from semiwild F1larvae from different host plants (Coccinia grandis, Cucumis sativus, and Solanum melongena) and sites (Basin Plat and Manapany).
| df | Mean squares | Pseudo F | p Value | Effect | |
|---|---|---|---|---|---|
| (A) Unweighted UniFrac distances | |||||
| Site (Si) | 1 | 0.573 | 13.086 | 0.000*** | Random |
| Host plant (Ho) | 2 | 1.026 | 1.750 | 0.031* | Fixed |
| Si × Ho | 2 | 0.586 | 13.378 | 0.000*** | |
| Residual | 24 | 0.043 | |||
| Total | 29 | ||||
| (B) Generalized UniFrac distances | |||||
| Site (Si) | 1 | 0.5129 | 23.6740 | 0.0001*** | Random |
| Host plant (Ho) | 2 | 0.5764 | 1.4745 | 0.1157 n.s. | Fixed |
| Si × Ho | 2 | 0.3909 | 18.0409 | 0.0001*** | |
| Residual | 24 | 0.0217 | |||
| Total | 29 | ||||
Abbreviation: n.s., not significant.
* p < 0.05;
p < 0.01;
p < 0.001.
The PCoA on presence/absence data (unweighted UniFrac distances) explains 46.15% of variation and suggests a similarity between larvae feeding on different host plants (Figure 1); however, this pattern is not recovered in the PCoA on weighted abundancies (generalized UniFrac distances, 59.03% of variation).
Figure 1.
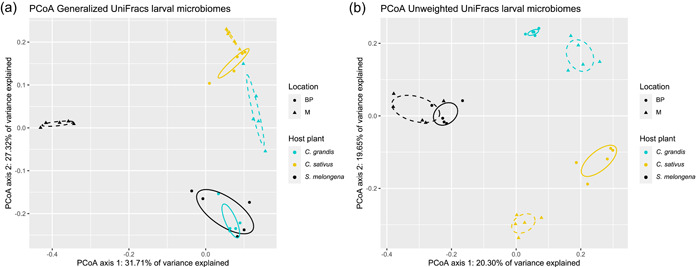
Principal coordinates analysis (PCoA) of microbial communities of semiwild F1 larvae of Zeugodacus cucurbitae from different host plants (Coccinia grandis [C. grandis], Cucumis sativus [C. sativus], Solanum melongena [S. melongena]) and sites (BP: Basin Plat and M: Manapany) as calculated from (a) generalized UniFrac and (b) unweighted UniFrac distances.
Several microbial genera showed a significant differential abundance between larvae raised on different host plants (Figures 2 and 3 and Supporting Information table at https://doi.org/10.5281/zenodo.6811204). Ketogulonicigenium, Devosia, Salmonella, Allorhizobium, and three putative unidentified genera from the family Enterobacteriaceae genera were more abundant on Co than on Cu and So. Most of the genera with higher abundance in So larvae belonged to the Order Enterobacterales, with Rahnella being detected in So larvae only. Compared to Cu larvae, larvae on So also showed enrichment in a few genera belonging to the Acetobacteriaceae, the class Bacilli, and the genus Pseudomonas. Larvae raised on Cu showed a higher abundance of genera belonging to the bacterial families Clostridia and Negativicutes from the phylum Firmicutes and of the genus Leucobacter. An additional three genera of the Clostridia were more abundant in Cu than in Co larvae. In contrast, taxa belonging to Acetobacteraceae and the genus Pseudomonas were less abundant in larvae raised on Cu compared to larvae from other host plants.
Figure 2.
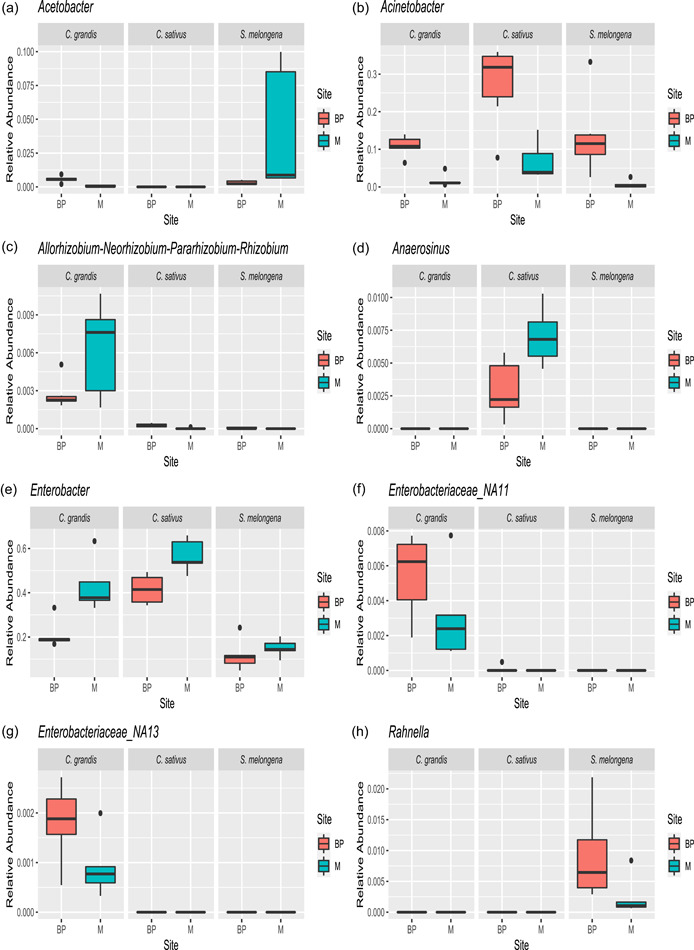
ALDEx2. Relative abundances (%) of bacterial genera in semiwild F1 larvae. Results are shown for eight genera (including two unidentified genera of Enterobacteriaceae) with the highest contribution to differences across host plants. BP, Basin Plat; M, Manapany. From left to right and top to bottom: (a) Acetobacter, (b) Acinetobacter, (c) Allorhizobium‐Neorhizobium‐Pararhizobium‐Rhizobium, (d) Anaerosinus, (e) Enterobacter, (f) Enterobacteriaceae_NA11, (g) Enterobacteriaceae_NA13, (h) Rahnella.
Figure 3.
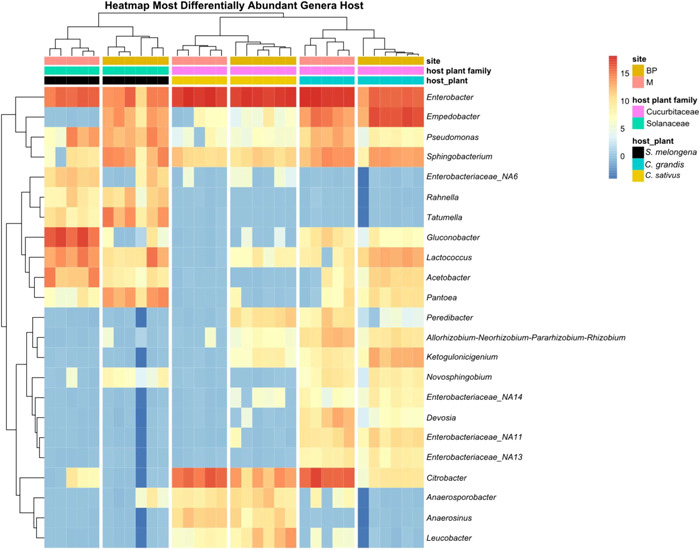
Differential abundances of bacterial genera in semiwild F1 larvae across sites (BP: Basin Plat and M: Manapany) and host plants (Coccinia grandis [C. grandis], Cucumis sativus [C. sativus], and Solanum melongena [S. melongena]). Host plant families are indicated in different colors). The red, yellow, and blue colors show the relative abundance of a bacterial genus from high to low. Relative abundances are expressed as centered log‐ratio transformations of the count data.
Between sites, only the genus Comamonas showed a pattern of differentiation, as its relative abundance was 8.5× lower in larvae from M than in larvae from BP. Within host plant treatments, more bacterial genera showed differential abundance between M and BP (Supporting Information table at https://doi.org/10.5281/zenodo.6811204).
4. DISCUSSION
Although our observations are largely consistent with other tephritid studies, some of our results are not in line with patterns reported in earlier studies (Andongma et al., 2015; Augustinos et al., 2019; Choudhary et al., 2021; De Cock et al., 2020; Morrow et al., 2015), but this is not surprising considering the high variability already described in the microbiome diversity of the closely related genera Bactrocera and Zeugodacus (De Cock et al., 2020). Part of the variability reported for the microbiome patterns of Bactrocera/Zeugodacus and more in general for frugivorous tephritids can certainly be related to the different lab rearing and experimental conditions reported in the literature (Asimakis et al., 2019; Augustinos et al., 2019; Ras et al., 2017; Sacchetti et al., 2019). However, discrepancies can also be observed between studies targeting wild or semiwild populations (e.g., Choudhary et al., 2021; De Cock et al., 2020). For example, previous research on wild populations of Z. cucurbitae suggested a close association with Ochrobactrum (Choudhary et al., 2021; De Cock et al., 2020), while this bacterial genus has an inconsistent presence across our samples.
One of the objectives of this study was to explore changes in the microbiome composition of a cucurbit‐feeder fly feeding on a noncucurbit host (S. melongena, Solanaceae) with the expectation that the shift to a noncucurbit diet would produce major and consistent changes in its microbiome. This only partially happened. First, changes in the microbiome composition related to host plant diet mostly concerned less abundant taxa as differences were observed mainly from the analysis of presence/absence data (unweighted UniFrac distances) rather than from the weighted abundances (generalized UniFrac distances). This is consistent with recent studies on termites and wood‐eating cockroaches that also found that dietary shifts mainly resulted in changes in rare microbes while abundant microbial taxa remained generally stable (Benjamino et al., 2018; Pérez‐Cobas et al., 2015). It suggests that low‐abundant species might have a key role in host plant adaptation. Second, our results show that local factors other than diet, have a deep impact on the microbiome diversity of Z. cucurbitae and more strongly contribute to the variability of patterns observed. Regardless of the complex interactions between diet and local‐geographical factors, we need to consider that this study mainly focused on F1 larvae. So, one explanation for these results might be that several generations are required before reaching stable and consistent microbiome assemblages in flies shifting to a novel “atypical” diet. A recent study in the whitefly Bemisia tabaci (Hemiptera, Aleyrodidae), for example, found that an initial host switch from watermelon to the less suitable host pepper did not result in major changes in microbiome composition and structure (Santos‐Garcia et al., 2020). Yet, major microbiome changes did occur in subsequent generations. Moreover, the same study also showed that the first generation following the host shift had a lower survival rate, which increased again in subsequent generations, suggesting that the microbiota was involved in longer‐term adaptation to the new host plant. Similarly, in the diamondback moth (Plutella xylostella, Lepidoptera, Plutellidae) a host shift to novel pea hosts resulted in major microbiome changes only in later generations, not in the first generation (Yang et al., 2020).
Another hypothesis for the lack of straightforward relationships between diet and microbiome composition is that microbiome changes mainly involve bacterial taxa that serve important metabolic functions but which are so rare that they remain undetected. Indeed, rare members of microbial communities sometimes perform key functions in these communities (Jousset et al., 2017). Desulfosporinus, for example, represents only 0.006% of reads detected in microbial peatland communities and yet it contributes the most to sulfate reduction (Pester et al., 2010). Likewise, the capacity of freshwater microbial communities to degrade pollutants is severely reduced when rare taxa disappear (Delgado‐Baquerizo et al., 2016). So, rare taxa may support a community with a wide range of metabolic functions that might only be important under specific circumstances (Jousset et al., 2017) such as the use of an unconventional host plant species.
We also need to consider that changes in host plant use might not necessarily translate into major compositional changes in microbiome assemblages but rather result in changes in the gene expression patterns of the “holobiont” sensu Margulis and Fester (1991) (i.e., the insect and its microbiota living on the host plant), which is the central unit of symbiogenesis and evolution (Guerrero et al., 2013). Symbiont microbial pectinases complement the insect endogenous cellulases and xylanases in herbivorous beetles (Cassidinae) and the pectinolytic range of symbiotic bacteria of the genus Stammera has been associated with the diversity of host plants that can be attacked by these beetles (Salem et al., 2020). Accordingly, the flexibility of insect and microbiome gene expression patterns could provide a complementary/alternative explanation to the complex relationships observed between microbiome assemblages, feeding preferences, and range expansion of Z. cucurbitae.
Additionally, differences in microbiome composition and structure between larvae feeding on the noncucurbit host could have also been affected by differences in their parental microbiomes, as significant heterogeneity was detected between the microbiomes of the parental lines and since in tephritids, at least part of the microbiome is vertically transmitted (Behar et al., 2008).
One potential drawback of our study is that we did not investigate the effect of captivity on the fly microbiome, especially with regard to vertical transmission from adults to larvae. Although we used wild populations for our experiment, our setup required the breeding of one generation in captivity. Previous studies have already shown that captive conditions can affect the microbiomes of tephritids (Asimakis et al., 2019; Augustinos et al., 2019; Morrow et al., 2015; Ras et al., 2017; Sacchetti et al., 2019). Moreover, changes in the larval microbiome due to captivity can occur in even relatively short periods. Majumder et al. (2022), for example, already detected changes in larval microbiomes after a single generation in captivity. Interestingly, changes in the adult microbiome took more generations before they started to occur. This suggests that rapid changes in the larval microbiome due to captivity are more due to environmental differences between the captive and natural environment experienced by the larvae (not the adult), such as diet, rather than due to a loss of vertically transmitted symbionts or genetic changes (Majumder et al., 2020). Indeed, prior studies have shown that the larval microbiome undergoes significant changes when larvae are reared on an artificial diet rather than a more natural diet. Likewise, studies on zoo animals have shown that exposing animals to more natural conditions and bacterial sources keeps their microbiome more similar to the microbiomes of their wild relatives (Loudon et al., 2014). Because of this, we do not consider this a major drawback of our study as larvae in our experiment were reared on a natural diet and exposed to a more natural environment, reducing, therefore, the impact of captivity on larval microbiomes and keeping them more representative of the natural conditions.
5. CONCLUSIONS
This study describes the effects of parent–offspring host switches in a cucurbit feeder fly and reveals complex microbiome responses in wild populations. As in De Cock et al. (2020), our results stress the importance of local effects on microbiome diversity and composition and the impact that factors such as diet can have on the microbiome. We identified the main bacterial genera responsible for the patterns observed. The high local‐scale variability and its interaction with diet shifts revealed the importance of proper spatial replication in microbiome research targeting wild/semiwild tephritid flies and provide a cautionary tale on general inferences drawn from laboratory populations.
AUTHOR CONTRIBUTIONS
Wouter Hendrycks: formal analysis (lead); funding acquisition (supporting); investigation (supporting); visualization (lead); writing – original draft (lead); writing – review & editing (equal). Hélène Delatte: conceptualization (equal); methodology (equal); project administration (equal); resources (equal); supervision (equal); writing – original draft (supporting); writing – review & editing (equal). Laura Moquet: investigation (supporting); project administration (supporting); supervision (equal); writing – original draft (supporting); writing – review & editing (equal). Kostas Bourtzis: conceptualization (supporting); funding acquisition (equal); writing – original draft (supporting); writing – review & editing (equal). Nele Mullens: formal analysis (supporting); investigation (lead); visualization (supporting); writing – original draft (supporting); writing – review & editing (equal). Marc De Meyer: conceptualization (equal); funding acquisition (supporting); methodology (equal); resources (equal); supervision (supporting); writing – original draft (supporting); writing – review & editing (equal). Thierry Backeljau: conceptualization (supporting); funding acquisition (equal); writing – original draft (supporting); writing – review & editing (equal). Massimiliano Virgilio: conceptualization (equal); formal analysis (supporting); funding acquisition (supporting); methodology (equal); resources (equal); supervision (equal); writing – original draft (supporting); writing – review & editing (equal).
CONFLICT OF INTEREST
None declared.
ETHICS STATEMENT
None required.
ACKNOWLEDGMENTS
We thank the Joint Experimental Molecular Unit of RMCA and RBINS for technical and conceptual feedback and stimulating discussions. We would also like to thank Kwinten Hendrycks for helping with the graphical design of the figures. This study was supported by funds from a “Bijzonder Onderzoeksfonds” grant from the University of Antwerp, a “Fonds voor wetenschappelijk onderzoek” Ph.D. fellowship of the FWO (11G9221N), and was supported by the International Atomic Energy Agency (IAEA, Vienna) through the technical contract n. 20876 “Comparative Microbiomics of African Fruit Flies” (CMAFF).
APPENDIX A.
See Figures A1, A2, A3 and Tables A1, A2, A3, A4, A5, A6
Figure A1.
Rarefaction plot showing relationships between sampling depth and the number of bacterial amplicon sequence variants detected.
Figure A2.
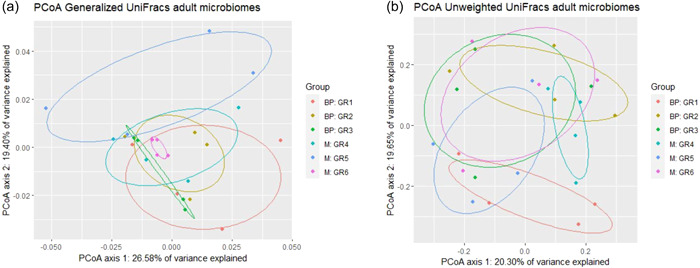
Principal coordinates analysis (PCoA) of microbial communities of wild parental (F0) adults of Zeugodacus cucurbitae using either (a) Generalized UniFrac distances or (b) Unweighted UniFrac distances. BP_Gr1 = F0 adults emerging from Coccinia grandis [C. grandis] and ovipositing on Solanum melongena from Bassin Plat (BP), BP_Gr2 = on C. grandis from BP, BP_Gr3 = on Cucumis sativus from BP, M_Gr1 = on S. melongena from Manapany (M), NDM_Gr2 = on C. grandis from M, M_Gr3 = on C. sativus from M. The 95% confidence ellipses are shown for each group.
Figure A3.
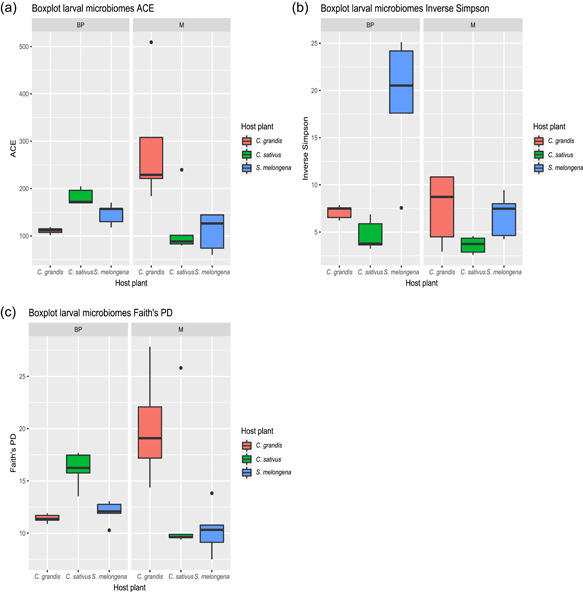
α Diversity metrics: (a) abundance coverage estimator (ACE), (b) inverse Simpson index, and (c) Faith's phylogenetic diversity (PD), calculated from semiwild F1 larvae from different host plants (Coccinia grandis [C. grandis], Cucumis sativus [C. sativus], and Solanum melongena [S. melongena]) and sites (BP: Basin Plat and M: Manapany).
Table A1.
Mock community standard results showing the taxa detected in the mock community and their relative abundance compared to their expected relative abundance.
| Species | Relative abundance | Zymo expected 16S rRNA copy (theoretical) |
|---|---|---|
| Lactobacillus fermentum | 14.1 | 18.4 |
| Salmonella entericaor unclassified | 2.8 | 10.4 |
| Escherichia colior unclassified | 30 | 10.1 |
| Pseudomonas aeruginosa | 17.5 | 4.2 |
| Enterococcus faecalis | 13.4 | 9.9 |
| Bacillus subtilis | 9.6 | 17.4 |
| Listeria monocytogenesor unclassified | 2.7 | 14.1 |
| Staphylococcus aureus | 10 | 15.5 |
| Lactobacillales NA | 0.01 | |
| Enterobacteriaceae NA | 0.005 |
Table A2.
Analysis of variance on α diversity metrics (abundance coverage estimator, inverse Simpson index, and Faith's phylogenetic diversity) calculated from F0 adult microbiomes from different host plants (Coccinia grandis, Cucumis sativus, and Solanum melongena) and sites (BP: Basin Plat and M: Manapany).
| df | Mean squares | Pseudo F | p Value | Effect | |
|---|---|---|---|---|---|
| Abundance coverage estimator | |||||
| Site: Si | 1 | 0.671 | 0.012 | 0.917 n.s. | Random |
| F0 parental group: Gr(Si) | 4 | 55.126 | 0.651 | 0.633 n.s. | Random |
| Residual | 18 | 84.582 | |||
| Transformation = none | |||||
| C = 0.4241 n.s. | |||||
| Inverse Simpson index | |||||
| Site: Si | 1 | 0.001 | 0.000 | 0.993 n.s. | Random |
| F0 parental group: Gr(Si) | 2 | 12.790 | 2.759 | 0.059 n.s. | Random |
| Residual | 18 | 0.028 | ‐ | ||
| Transformation = none | |||||
| C = 0.475 n.s. | |||||
| Faith's phylogenetic diversity | |||||
| Site: Si | 1 | 1.069 | 0.631 | 0.471 n.s. | Random |
| F0 parental group: Gr(Si) | 2 | 1.693 | 1.687 | 0.197 n.s. | Random |
| Residual | 18 | 1.003 | ‐ | ||
| Transformation = none | |||||
| C = 0.172 n.s. | |||||
Abbreviations: C, Cochran's C; df, degree of freedom; n.s., not significant.
Table A3.
Permutational analysis of variance on (A) unweighted UniFrac and (B) generalized UniFrac distances calculated for microbiomes from parental F0 adults from two sites (BP: Basin Plat and M: Manapany).
| (A) Unweighted UniFrac distances | df | Mean squares | Pseudo F | p Value | Effect |
|---|---|---|---|---|---|
| Site: Si | 1 | 0.353 | 1.351 | 0.223 n.s. | Random |
| F0 parental group: Gr(Si) | 4 | 0.262 | 1.819 | 0.005** | Random |
| Residual | 18 | 0.144 | |||
| Total | 23 |
| (B) Generalized UniFrac distances | df | Mean squares | Pseudo F | p Value | Factor |
|---|---|---|---|---|---|
| Site: Si | 1 | 0.003 | 1.752 | 0.098 n.s. | Random |
| F0 parental group: Gr(Si) | 4 | 0.001 | 1.154 | 0.259 n.s. | Random |
| Residual | 18 | 0.002 | |||
| Total | 23 |
Abbreviations: df, degree of freedom; F0, wild parental; n.s., not significant.
p < 0.01.
Table A4.
A posteriori comparisons between (unweighted UniFrac distances, see Table A3 (A)) between microbiomes of parental F0 adults from two sites (BP: Basin Plat and M: Manapany). Comparisons are shown between groups of adults used to infest three different fruits (Co: Coccinia grandis, Cu: Cucumis sativus, So: Solanum melongena)
| Unweighted UniFrac distances post hoc comparison | ||||
|---|---|---|---|---|
| Site | Comparison | Average dissimilarity | t Value | p Value |
| BP | So–Co | 0.632 | 1.865 | 0.030* |
| BP | So–Cu | 0.602 | 1.546 | 0.056 n.s. |
| BP | Co–Cu | 0.523 | 1.161 | 0.206 n.s. |
| M | So–Co | 0.495 | 0.841 | 0.743 n.s. |
| M | So–Cu | 0.572 | 1.193 | 0.174 n.s. |
| M | Co–Cu | 0.596 | 1.246 | 0.111 n.s. |
p < 0.05.
Table A5.
A posteriori comparisons between α diversity metrics (abundance coverage estimator, inverse Simpson index, and Faith's phylogenetic diversity) calculated from semiwild; F1 larvae feeding on different host plants (Co: Coccinia grandis, Cu: Cucumis sativus, and So: Solanum melongena) and sites (BP: Basin Plat and M: Manapany).
| Site | Host plant | Estimate | df | Sum of squares | F value | p Value |
|---|---|---|---|---|---|---|
| Abundance coverage estimator | ||||||
| BP | So–Co | 0.232 | 1 | 0.134 | 1.669 | 0.625 |
| BP | So–Cu | −0.205 | 1 | 0.105 | 1.303 | 0.625 |
| BP | Co–Cu | −0.436 | 1 | 0.477 | 5.923 | 0.091 |
| M | So–Co | −0.867 | 1 | 1.882 | 23.361 | 0.000*** |
| M | So–Cu | −0.056 | 1 | 0.006 | 0.082 | 0.776 |
| M | Co–Cu | 0.816 | 1 | 1.665 | 20.664 | 0.000*** |
| Inverse Simpson index | ||||||
| BP | So–Co | 0.903 | 1 | 2.040 | 13.739 | 0.005** |
| BP | So–Cu | 1.367 | 1 | 4.676 | 31.491 | 0.000*** |
| BP | Co–Cu | 0.464 | 1 | 0.538 | 3.629 | 0.137 |
| M | So–Co | −0.035 | 1 | 0.003 | 0.021 | 0.885 |
| M | So–Cu | 0.601 | 1 | 0.903 | 6.083 | 0.063 |
| M | Co–Cu | 0.636 | 1 | 1.013 | 6.826 | 0.061 |
| Faith's phylogenetic diversity | ||||||
| BP | So–Co | 0.036 | 1 | 0.003 | 0.243 | 0.625 |
| BP | So–Cu | −0.127 | 1 | 0.040 | 3.069 | 0.277 |
| BP | Co–Cu | −0.163 | 1 | 0.066 | 5.044 | 0.136 |
| M | So–Co | −0.321 | 1 | 0.258 | 19.522 | 0.001** |
| M | So–Cu | −0.080 | 1 | 0.016 | 1.207 | 0.565 |
| M | Co–Cu | 0.241 | 1 | 0.146 | 11.018 | 0.014* |
Abbreviations: df, degree of freedom; n.s., not significant.
p < 0.05;
p < 0.01;
p < 0.001.
Table A6.
A posteriori comparisons between (A) unweighted UniFrac and (B) generalized UniFrac distances (see Table 2) calculated from semiwild F1 larvae feeding on different host plants (Co: Coccinia grandis, Cu: Cucumis sativus, and So: Solanum melongena) and sites (BP: Basin Plat and M: Manapany).
| Population | Host plant | Average dissimilarity | t Value | p Value |
|---|---|---|---|---|
| (A) Unweighted UniFrac distances post hoc comparison | ||||
| BP | So–Co | 0.547 | 5.733 | 0.008** |
| BP | So–Cu | 0.636 | 4.678 | 0.008** |
| BP | Co–Cu | 0.614 | 4.494 | 0.008** |
| M | So–Co | 0.685 | 3.644 | 0.007** |
| M | So–Cu | 0.618 | 4.260 | 0.008** |
| M | Co–Cu | 0.637 | 3.961 | 0.006** |
| (B) Generalized UniFrac distances post hoc comparison | ||||
| BP | So–Co | 0.373 | 3.677 | 0.008** |
| BP | So–Cu | 0.493 | 4.005 | 0.008** |
| BP | Co–Cu | 0.467 | 4.868 | 0.008** |
| M | So–Co | 0.582 | 5.206 | 0.007** |
| M | So–Cu | 0.532 | 8.334 | 0.008** |
| M | Co–Cu | 0.369 | 3.394 | 0.006** |
Abbreviation: n.s., not significant.
p < 0.01;
Hendrycks, W. , Delatte, H. , Moquet, L. , Bourtzis, K. , Mullens, N. , De Meyer, M. , Backeljau, T. , & Virgilio, M. (2022). Eating eggplants as a cucurbit feeder: Dietary shifts affect the gut microbiome of the melon fly Zeugodacus cucurbitae (Diptera, Tephritidae). MicrobiologyOpen, 11, e1307. 10.1002/mbo3.1307
DATA AVAILABILITY STATEMENT
All raw sequence read data are available from the European Nucleotide Archive under the accession number PRJEB49793: https://www.ebi.ac.uk/ena/browser/view/PRJEB49793. Sample data set, sample metadata, and codes for analysis are available on GitHub: https://github.com/wouterhendrycks/tephritid_microbiome_host_switch_project and in Zenodo: https://doi.org/10.5281/zenodo.6810766. Supporting Information table (Differential abundance analysis of bacterial genera between larval microbiomes from larvae between different host plants and between different sites using ALDEx2) is available in the Zenodo repository at https://doi.org/10.5281/zenodo.6811204).
REFERENCES
- Adams, A. S. , Aylward, F. O. , Adams, S. M. , Erbilgin, N. , Aukema, B. H. , Currie, C. R. , Suen, G. , & Raffa, K. F. (2013). Mountain pine beetles colonizing historical and naïve host trees are associated with a bacterial community highly enriched in genes contributing to terpene metabolism. Applied and Environmental Microbiology, 79(11), 3468–3475. 10.1128/AEM.00068-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, M. J. (2017). Permutational multivariate analysis of variance (PERMANOVA), Wiley Statsref: Statistics Reference Online (pp. 1–15). John Wiley & Sons. 10.1002/9781118445112.stat07841 [DOI] [Google Scholar]
- Andongma, A. A. , Wan, L. , Dong, Y. C. , Li, P. , Desneux, N. , White, J. A. , & Niu, C. Y. (2015). Pyrosequencing reveals a shift in symbiotic bacteria populations across life stages of Bactrocera dorsalis . Scientific Reports, 5, 9470. 10.1038/srep09470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews, S. (2014). FastQC: A quality control tool for high‐throughput sequence data. Babraham Institute. Retrieved March 19, 2020, from http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- Asimakis, E. D. , Khan, M. , Stathopoulou, P. , Caceres, C. , Bourtzis, K. , & Tsiamis, G. (2019). The effect of diet and radiation on the bacterial symbiome of the melon fly, Zeugodacus cucurbitae (Coquillett). BMC Biotechnology, 19(Suppl 2), 88. 10.1186/s12896-019-0578-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustinos, A. A. , Tsiamis, G. , Cáceres, C. , Abd‐Alla, A. M. M. , & Bourtzis, K. (2019). Taxonomy, diet, and developmental stage contribute to the structuring of gut‐associated bacterial communities in tephritid pest species. Frontiers in Microbiology, 10, 2004. 10.3389/fmicb.2019.02004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar, A. , Jurkevitch, E. , & Yuval, B. (2008). Bringing back the fruit into fruit fly‐bacteria interactions. Molecular Ecology, 17(5), 1375–1386. 10.1111/j.1365-294X.2008.03674.x [DOI] [PubMed] [Google Scholar]
- Benjamini, Y. , & Hochberg, Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B (Methodological), 57(1), 289–300. [Google Scholar]
- Benjamino, J. , Lincoln, S. , Srivastava, R. , & Graf, J. (2018). Low‐abundant bacteria drive compositional changes in the gut microbiota after dietary alteration. Microbiome, 6(1), 86. 10.1186/s40168-018-0469-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐Yosef, M. , Aharon, Y. , Jurkevitch, E. , & Yuval, B. (2010). Give us the tools and we will do the job: Symbiotic bacteria affect olive fly fitness in a diet‐dependent fashion. Proceedings of the Royal Society B: Biological Sciences, 277(1687), 1545–1552. 10.1098/rspb.2009.2102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐Yosef, M. , Pasternak, Z. , Jurkevitch, E. , & Yuval, B. (2014). Symbiotic bacteria enable olive flies (Bactrocera oleae) to exploit intractable sources of nitrogen. Journal of Evolutionary Biology, 27(12), 2695–2705. 10.1111/jeb.12527 [DOI] [PubMed] [Google Scholar]
- Ben‐Yosef, M. , Pasternak, Z. , Jurkevitch, E. , & Yuval, B. (2015). Symbiotic bacteria enable olive fly larvae to overcome host defences. Royal Society Open Science, 2, 150170. 10.1098/rsos.150170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, A. M. V. , Huynh, L. Y. , Bolender, C. M. , Nelson, K. G. , & McCutcheon, J. P. (2014). Population genomics of a symbiont in the early stages of a pest invasion. Molecular Ecology, 23(6), 1516–1530. 10.1111/mec.12366 [DOI] [PubMed] [Google Scholar]
- Callahan, B. J. , McMurdie, P. J. , Rosen, M. J. , Han, A. W. , Johnson, A. J. A. , & Holmes, S. P. (2016). DADA2: High‐resolution sample inference from Illumina amplicon data. Nature Methods, 13(7), 581–583. 10.1038/nmeth.3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti, A. , Siddharth, J. , Lauber, C. L. , Membrez, M. , Betrisey, B. , Loyer, C. , Chou, C. J. , Pataky, Z. , Golay, A. , & Parkinson, S. J. (2016). Resolving microbial membership using Abundance and Variability in Taxonomy (AVIT). Scientific Reports, 6, 31655. 10.1038/srep31655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Bittinger, K. , Charlson, E. S. , Hoffmann, C. , Lewis, J. , Wu, G. D. , Collman, R. G. , Bushman, F. D. , & Li, H. (2012). Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics, 28(16), 2106–2113. 10.1093/bioinformatics/bts342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary, J. S. , Naaz, N. , Prabhakar, C. S. , Das, B. , Singh, A. K. , & Bhatt, B. P. (2021). High taxonomic and functional diversity of bacterial communities associated with melon fly, Zeugodacus cucurbitae (Diptera: Tephritidae). Current Microbiology, 78(2), 611–623. 10.1007/s00284-020-02327-2 [DOI] [PubMed] [Google Scholar]
- Clarke, A. R. (2017). Why so many polyphagous fruit flies (Diptera: Tephritidae)? A further contribution to the “generalism” debate. Biological Journal of the Linnean Society, 120, 245–257. [Google Scholar]
- Clarke, K. R. , & Gorley, R. N. (2015). PRIMER v7: User manual/tutorial. PRIMER‐E Ltd. [Google Scholar]
- Clarke, L. J. , Suter, L. , King, R. , Bissett, A. , & Deagle, B. E. (2019). Antarctic krill are reservoirs for distinct southern ocean microbial communities. Frontiers in Microbiology, 10, 3226. 10.3389/fmicb.2018.03226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, N. M. , Proctor, D. M. , Holmes, S. P. , Relman, D. A. , & Callahan, B. J. (2018). Simple statistical identification and removal of contaminant sequences in marker‐gene and metagenomics data. Microbiome, 6(1), 226. 10.1186/s40168-018-0605-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cock, M. , Virgilio, M. , Vandamme, P. , Augustinos, A. , Bourtzis, K. , Willems, A. , & De Meyer, M. (2019). Impact of sample preservation and manipulation on insect gut microbiome profiling. A test case with fruit flies (Diptera, Tephritidae). Frontiers in Microbiology, 10, 2833. 10.3389/fmicb.2019.02833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cock, M. , Virgilio, M. , Vandamme, P. , Bourtzis, K. , De Meyer, M. , & Willems, A. (2020). Comparative microbiomics of tephritid frugivorous pests (Diptera: Tephritidae) from the field: A tale of high variability across and within species. Frontiers in Microbiology, 11, 1890. 10.3389/fmicb.2020.01890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Meyer, M. , Delatte, H. , Mwatawala, M. , Quilici, S. , Vayssières, J. F. , & Virgilio, M. (2015). A review of the current knowledge on Zeugodacus cucurbitae (Coquillett) (Diptera, Tephritidae) in Africa, with a list of species included in Zeugodacus . ZooKeys, 540, 539–557. 10.3897/zookeys.540.9672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rosario‐Martinez, H. (2013). R package “Phia.” Retrieved October 13, 2020, from https://cran.r-project.org/package=phia
- Delatte, H. , De Meyer, M. , & Virgilio, M. (2019). Genetic structure and range expansion of Zeugodacus Cucurbitae (Diptera: Tephritidae) in Africa. Bulletin of Entomological Research, 109(6), 713–722. 10.1017/S0007485319000026 [DOI] [PubMed] [Google Scholar]
- Delgado‐Baquerizo, M. , Giaramida, L. , Reich, P. B. , Khachane, A. N. , Hamonts, K. , Edwards, C. , Lawton, L. A. , & Singh, B. K. (2016). Lack of functional redundancy in the relationship between microbial diversity and ecosystem functioning. Journal of Ecology, 104(4), 936–946. 10.1111/1365-2745.12585 [DOI] [Google Scholar]
- Dhillon, M. K. , Singh, R. , Naresh, J. S. , & Sharma, H. C. (2005). The melon fruit fly, Bactrocera cucurbitae: A review of its biology and management. Journal of Insect Science, 5(1), 1–16. 10.1093/jis/5.1.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas, A. E. (2009). The microbial dimension in insect nutritional ecology. Functional Ecology, 23(1), 38–47. 10.1111/j.1365-2435.2008.01442.x [DOI] [Google Scholar]
- Feder, J. L. , Chilcote, C. A. & Bush, G. L. (1988). Genetic differentiation between sympatric host races of the apple magot fly Rhagoletis pomonella . Nature, 366, 61–64. [Google Scholar]
- Fernandes, A. D. , Macklaim, J. M. , Linn, T. G. , Reid, G. , & Gloor, G. B. (2013). ANOVA‐Like Differential Expression (ALDEx) analysis for mixed population RNA‐Seq. PLOS One, 8(7), e67019. 10.1371/journal.pone.0067019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forister, M. L. , Novotny, V. , Panorska, A. K. , Baje, L. , Basset, Y. , Butterill, P. T. , Cizek, L. , Coley, P. D. , Dem, F. , Diniz, I. R. , Drodz, P. , Fox, M. , Glassmire, A. E. , Hazen, R. , Hrcek, J. , Jahner, J. P. , Kaman, O. , Kozubowski, T. J. , Kursar, T. A. , … Dyer, L. A. (2015). The global distribution of diet breadth in insect herbivores. Proceedings of the National Academy of Sciences of the United States of America, 112(2), 442–447. 10.1073/pnas.1423042112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox, J. (2006). The car package. Retrieved October 13, 2020, from http://cran.r-project.org/web/packages/car
- Gloor, G. (2015). Aldex2: Anova‐Like Differential Expression tool for compositional data. ALDEX Manual Modular, 20, 1–11. bioconductor.org/packages/devel/bio/vignettes/ALDEx2/inst/doc/ALDEx2_vignette.html [Google Scholar]
- Guerrero, R. , Margulis, L. , & Berlanga, M. (2013). Symbiogenesis: The holobiont as a unit of evolution. International Microbiology, 16(3), 133–143. 10.2436/20.1501.01.188 [DOI] [PubMed] [Google Scholar]
- Hafsi, A. , Facon, B. , Ravigné, V. , Chiroleu, F. , Quilici, S. , Chermiti, B. , & Duyck, P. F. (2016). Host plant range of a fruit fly community (Diptera: Tephritidae): Does fruit composition influence larval performance? BMC Ecology, 16(1), 40. 10.1186/s12898-016-0094-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer, T. J. , & Bowers, M. D. (2015). Gut microbes may facilitate insect herbivory of chemically defended plants. Oecologia, 179(1), 1–14. 10.1007/s00442-015-3327-1 [DOI] [PubMed] [Google Scholar]
- Hester, E. R. , Barott, K. L. , Nulton, J. , Vermeij, M. J. A. , & Rohwer, F. L. (2016). Stable and sporadic symbiotic communities of coral and algal holobionts. ISME Journal, 10(5), 1157–1169. 10.1038/ismej.2015.190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood, G. R. , Powell, T. H. Q. , Doellman, M. M. , Sim, S. B. , Glover, M. , Yee, W. L. , Goughnour, R. B. , Mattsson, M. , Schwarz, D. , & Feder, J. L. (2020). Rapid and repeatable host plant shifts drive reproductive isolation following a recent human‐mediated introduction of the apple maggot fly, Rhagoletis pomonella . Evolution, 74(1), 156–168. 10.1111/evo.13882 [DOI] [PubMed] [Google Scholar]
- Ishigami, K. , Jang, S. , Itoh, H. & Kikuchi, Y. (2021). Insecticide resistance governed by gut symbiosis in a rice pest, Cletus punctiger, under laboratory conditions. Biology Letters, 17, 20200780. 10.1098/rsbl.2020.0780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jousset, A. , Bienhold, C. , Chatzinotas, A. , Gallien, L. , Gobet, A. , Kurm, V. , Küsel, K. , Rillig, M. C. , Rivett, D. W. , Salles, J. F. , Van Der Heijden, M. G. A. , Youssef, N. H. , Zhang, X. , Wei, Z. , & Hol, G. W. H. (2017). Where less may be more: How the rare biosphere pulls ecosystems strings. ISME Journal, 11(4), 853–862. 10.1038/ismej.2016.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassambara, A. & Kassambara, M. A. (2020). Package ‘ggpubr’. Retrieved April 8, 2022, from https://cran.r-project.org/web/packages/ggpubr/index.html
- Kikuchi, Y. , Hayatsu, M. , Hosokawa, T. , Nagayama, A. , Tago, K. , & Fukatsu, T. (2012). Symbiont‐mediated insecticide resistance. Proceedings of the National Academy of Sciences of the United States of America, 109(22), 8618–8622. 10.1073/pnas.1200231109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolde, R. (2015). R Pheatmap‐package “Pretty Heatmaps.” Retrieved June 16, 2020, from https://cran.r-project.org/package=pheatmap
- Kounatidis, I. , Crotti, E. , Sapountzis, P. , Sacchi, L. , Rizzi, A. , Chouaia, B. , Bandi, C. , Alma, A. , Daffonchio, D. , Mavragani‐Tsipidou, P. , & Bourtzis, K. (2009). Acetobacter tropicalis is a major symbiont of the olive fruit fly (Bactrocera oleae). Applied and Environmental Microbiology, 75(10), 3281–3288. 10.1128/AEM.02933-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. , & Lee, D. K. (2018). What is the proper way to apply the multiple comparison test? Korean Journal of Anesthesiology, 71(5), 353–360. 10.4097/kja.d.18.00242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefort, M. C. , Boyer, S. , De Romans, S. , Glare, T. , Armstrong, K. , & Worner, S. (2014). Invasion success of a scarab beetle within its native range: Host range expansion versus host‐shift. PeerJ, 2, 262. 10.7717/peerj.262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loudon, A. H. , Woodhams, D. C. , Parfrey, L. W. , Archer, H. , Knight, R. , McKenzie, V. , & Harris, R. N. (2014). Microbial community dynamics and effect of environmental microbial reservoirs on red‐backed salamanders (Plethodon cinereus). ISME Journal, 8, 830‐840. 10.1038/ismej.2013.200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, M. , Wingfield, M. J. , Gillette, N. , & Jiang‐Hua Sun, A. (2011). Do novel genotypes drive the success of an invasive bark beetle‐fungus complex? Implications for potential reinvasion. Ecology, 92(11), 2013–2019. [DOI] [PubMed] [Google Scholar]
- Majumder, R. , Sutcliffe, B. , Adnan, S. M. , Mainali, B. , Dominiak, B. C. , Taylor, P. W. , & Chapman, T. A. (2020). Artificial larval diet mediates the microbiome of Queensland fruit fly. Frontiers in Microbiology, 11, 576156. 10.3389/fmicb.2020.576156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder, R. , Taylor, P. W. , & Chapman, T. A. (2022). Dynamics of the Queensland fruit fly microbiome through the transition from nature to an established laboratory colony. Microbiome, 10, 291. 10.3390/microorganisms10020291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulis, L. & Fester, R. (1991). Symbiosis as a source of evolutionary innovation: Speciation and morphogenesis. MIT Press. [PubMed] [Google Scholar]
- McKnight, D. T. , Huerlimann, R. , Bower, D. S. , Schwarzkopf, L. , Alford, R. A. , & Zenger, K. R. (2019). Methods for normalizing microbiome data: An ecological perspective. Methods in Ecology and Evolution, 10(3), 389–400. 10.1111/2041-210X.13115 [DOI] [Google Scholar]
- McMurdie, P. J. , & Holmes, S. (2014). Waste not, want not: Why rarefying microbiome data is inadmissible. PLoS Computational Biology, 10(4), e1003531. 10.1371/journal.pcbi.1003531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moquet, L. , Payet, J. , Glenac, S. , & Delatte, H. (2021). Niche shift of tephritid species after the Oriental fruit fly (Bactrocera dorsalis) invasion in La Réunion. Diversity and Distributions, 27(1), 109–129. 10.1111/ddi.13172 [DOI] [Google Scholar]
- Morrow, J. L. , Frommer, M. , Shearman, D. C. A. , & Riegler, M. (2015). The microbiome of field‐caught and laboratory‐adapted Australian tephritid fruit fly species with different host plant use and specialisation. Microbial Ecology, 70(2), 498–508. 10.1007/s00248-015-0571-1 [DOI] [PubMed] [Google Scholar]
- Norrbom, A. L. , Carroll, L.E. , Thompson, F. C. , White, I. M. , & Freidberg, A. (1999). Systematic database of names. In: F. C. Thompson (Ed.) Fruit fly expert identification system and systematic information database (Vol. 9, pp. 65–251). Myia. [Google Scholar]
- Pavlidi, N. , Gioti, A. , Wybouw, N. , Dermauw, W. , Ben‐Yosef, M. , Yuval, B. , Jurkevich, E. , Kampouraki, A. , Van Leeuwen, T. , & Vontas, J. (2017). Transcriptomic responses of the olive fruit fly Bactrocera oleae and its symbiont Candidatus Erwinia dacicola to olive feeding. Scientific Reports, 7, 42633. 10.1038/srep42633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez‐Cobas, A. E. , Maiques, E. , Angelova, A. , Carrasco, P. , Moya, A. , & Latorre, A. (2015). Diet shapes the gut microbiota of the omnivorous cockroach Blattella germanica . FEMS Microbiology Ecology, 91(4), fiv022. 10.1093/femsec/fiv022 [DOI] [PubMed] [Google Scholar]
- Pester, M. , Bittner, N. , Deevong, P. , Wagner, M. , & Loy, A. (2010). A “rare biosphere” microorganism contributes to sulfate reduction in a peatland. ISME Journal, 4(12), 1591–1602. 10.1038/ismej.2010.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, M. N. , Dehal, P. S. , & Arkin, A. P. (2009). Fasttree: Computing large minimum evolution trees with profiles instead of a distance matrix. Molecular Biology and Evolution, 26(7), 1641–1650. 10.1093/molbev/msp077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ras, E. , Beukeboom, L. W. , Cáceres, C. , & Bourtzis, K. (2017). Review of the role of gut microbiota in mass rearing of the olive fruit fly, Bactrocera oleae, and its parasitoids. Entomologia Experimentalis et Applicata, 164(3), 237–256. 10.1111/eea.12609 [DOI] [Google Scholar]
- Rosenberger, D. W. , Venette, R. C. , & Aukema, B. H. (2018). Development of an aggressive bark beetle on novel hosts: Implications for outbreaks in an invaded range. Journal of Applied Ecology, 55(3), 1526–1537. 10.1111/1365-2664.13064 [DOI] [Google Scholar]
- Sacchetti, P. , Pastorelli, R. , Bigiotti, G. , Guidi, R. , Ruschioni, S. , Viti, C. , & Belcari, A. (2019). Olive fruit fly rearing procedures affect the vertical transmission of the bacterial symbiont Candidatus Erwinia dacicola. BMC Biotechnology, 19, 91. 10.1186/s12896-019-0582-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salem, H. , Kirsch, R. , Pauchet, Y. , Berasategui, A. , Fukumori, K. , Moriyama, M. , Cripps, M. , Windsor, D. , Fukatsu, T. , & Gerardo, N. M. (2020). Symbiont digestive range reflects host plant breadth in herbivorous beetles. Current Biology, 30(15), 2875–2886. 10.1016/j.cub.2020.05.043 [DOI] [PubMed] [Google Scholar]
- Sandrini‐Neto, L. & Camargo, M. G. (2015). R package: “GAD.” Retrieved October 13, 2020, from https://cran.r-project.org/web/packages/GAD/GAD.pdf
- Santos‐Garcia, D. , Mestre‐Rincon, N. , Zchori‐Fein, E. , & Morin, S. (2020). Inside out: Microbiota dynamics during host‐plant adaptation of whiteflies. ISME Journal, 14(3), 847–856. 10.1038/s41396-019-0576-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer, M. C. , & Parmesan, C. (2021). Colonizations cause diversification of host preferences: A mechanism explaining increased generalization at range boundaries expanding under climate change. Global Change Biology, 27(15), 3505–3518. 10.1111/gcb.15656 [DOI] [PubMed] [Google Scholar]
- Sword, G. A. , Joern, A. , & Senior, L. B. (2005). Host plant‐associated genetic differentiation in the snakeweed grasshopper, Hesperotettix viridis (Orthoptera: Acrididae). Molecular Ecology, 14(7), 2197–2205. 10.1111/j.1365-294X.2005.02546.x [DOI] [PubMed] [Google Scholar]
- Takahashi, S. , Tomita, J. , Nishioka, K. , Hisada, T. , & Nishijima, M. (2014). Development of a prokaryotic universal primer for simultaneous analysis of bacteria and Archaea using next‐generation sequencing. PLoS One, 9(8):e0105592. 10.1371/journal.pone.0105592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilmon, K. (2008). Specialization, speciation and radiation: The evolutionary biology of herbivorous insects. University of California Press. 10.1525/california/9780520251328.001.0001 [DOI] [Google Scholar]
- Underwood, A. J. (1997). Experiments in ecology: Their logical design and interpretation using analysis of variance. Cambridge University Press. [Google Scholar]
- Virgilio, M. , Delatte, H. , Backeljau, T. , & De Meyer, M. (2010). Macrogeographic population structuring in the cosmopolitan agricultural pest Bactrocera cucurbitae (Diptera: Tephritidae). Molecular Ecology, 19(13), 2713–2724. 10.1111/j.1365-294X.2010.04662.x [DOI] [PubMed] [Google Scholar]
- Wickham, H. & Chang, W. (2016). R package “ggplot2.” Retrieved May 10, 2020, from https://cran.r-project.org/web/packages/ggplot2
- Wright, E. S. (2015). DECIPHER: Harnessing local sequence context to improve protein multiple sequence alignment. BMC Bioinformatics, 16(1), 322. 10.1186/s12859-015-0749-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, F. Y. , Saqib, H. S. A. , Chen, J. H. , Ruan, Q. Q. , Vasseur, L. , He, W. Y. , & You, M. S. (2020). Differential profiles of gut microbiota and metabolites associated with host shift of Plutella xylostella . International Journal of Molecular Sciences, 21(17), 6283. 10.3390/ijms21176283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilber‐Rosenberg, I. , & Rosenberg, E. (2008). Role of microorganisms in the evolution of animals and plants: The hologenome theory of evolution. FEMS Microbiology Reviews, 32(5), 723–735. 10.1111/j.1574-6976.2008.00123.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All raw sequence read data are available from the European Nucleotide Archive under the accession number PRJEB49793: https://www.ebi.ac.uk/ena/browser/view/PRJEB49793. Sample data set, sample metadata, and codes for analysis are available on GitHub: https://github.com/wouterhendrycks/tephritid_microbiome_host_switch_project and in Zenodo: https://doi.org/10.5281/zenodo.6810766. Supporting Information table (Differential abundance analysis of bacterial genera between larval microbiomes from larvae between different host plants and between different sites using ALDEx2) is available in the Zenodo repository at https://doi.org/10.5281/zenodo.6811204).