

Abstract
Background
Despite the established association between obesity and cancer risk, it remains unclear whether visceral obesity is causally related to cancer risk and whether it is more pro-oncogenic than total body fat.
Methods
We conducted two-sample Mendelian randomization (MR) analysis to assess the causal effects of visceral adipose tissue (VAT) on six common cancers. For exposure data, 221 genetic variants associated with the predicted volume of VAT in 325 153 Europeans from UK Biobank were used as instrumental variables. Genetic association data of six common cancers (breast, lung, colorectal, ovarian, pancreatic and prostate cancers) were obtained from large-scale consortia with an average of 19 576 cases and 43 272 controls. We performed univariable MR with five MR methods [inverse-variance weighted (IVW), MR-Egger regression, weighted median, MR-Pleiotropy Residual Sum and Outlier (MR-PRESSO) and Radial MR] and multivariable MR to estimate the effect of VAT independent of body mass index (BMI). Finally, we performed a series of sensitivity analyses as validation of primary MR results.
Results
Two associations survived the false discovery rate correction for multiple testing (q-value < 0.05): in IVW, the odds ratios (95% CIs) per unit increase in genetically determined VAT were 1.65 (1.03 to 2.62) for pancreatic cancer and 1.47 (1.20 to 1.82) for lung squamous-cell carcinoma, respectively, which showed the same directions and overlapped confidence intervals with MR-Egger regression and weighted median results. There were no outlier variants identified by MR-PRESSO and no evidence supporting the presence of heterogeneity and pleiotropy in sensitivity analyses, although with wider confidence intervals that included the null, multivariable MR results for these two cancers showed the same directions and similar effect sizes as in IVW, which were independent of the effect from BMI. There was no evidence for a causal effect of VAT on the risk of other types of cancer.
Conclusion
Our findings suggest that lifelong exposure to elevated volumes of VAT might increase the risk of pancreatic cancer and lung squamous-cell carcinoma, highlighting the importance of revealing the underlying mechanisms for intervention targets.
Keywords: Mendelian randomization, visceral adipose tissue, cancers, causal inference
Key Messages.
We conducted a systematic two-sample Mendelian randomization (MR) analysis to estimate the causal effects of viscera adipose tissue (VAT) on six common cancers.
Univariable and multivariable MR results suggested that genetically determined VAT might increase the risk of pancreatic cancer and lung squamous-cell carcinoma, which were independent of the effect from body mass index.
Future studies are needed to clarify the non-linear relationships between VAT and cancer risks.
Introduction
The prevalence of excess body weight and the associated cancer burden have been rising worldwide. Epidemiologic studies have shown that obesity, measured by body mass index (BMI), is associated with 13 different types of cancers.1 However, BMI is an indirect indicator and does not reflect the difference between fat and lean body mass, nor does it reflect the location of adipose (i.e. central, peripheral or in the organ at risk). It is known that central adiposity, primarily referring to visceral adipose tissue (VAT), is more harmful than adipose from other locations,2 resulting in a metabolic, hormonal and inflammatory milieu that features tumour promotion.3 An increasing number of studies indicated that VAT represents a risk factor for metabolic disorders as well as some types of cancers.4–6 Accurate measurement of VAT depends on imaging methods such as magnetic resonance imaging (MRI) and computed tomography (CT), limiting its broad application to the general population. Therefore, previous studies were largely limited by small sample sizes. Moreover, due to their observational nature, these studies were likely subject to residual confounding and reverse causation, restricting their ability for causal inference.
In contrast to observational studies with the above limitations, Mendelian randomization (MR) offers an approach to efficiently and reliably investigate the potential causal relationships between increased VAT and cancer risks. MR is considered as ‘nature’s randomized control trial’,7 using genetic variants robustly associated with the exposure of interest to explore causal effects on the outcomes,8 which can therefore address the limitations above in observational studies. In this study, we performed two-sample MR analyses to evaluate the causal effects of VAT on the risk of different cancers and whether the estimates were independent of BMI.
Methods
Study design
The flow chart of our study design is shown in Figure 1. First, we identified genetic variants as instrumental variables (IVs) for VAT. Second, we collected the summary data containing all single-nucleotide polymorphisms (SNPs) from the large-scale genome-wide association studies (GWASs) for cancers. Third, we performed univariable two-sample MR with five MR methods, including inverse-variance weighted (IVW), MR-Egger regression, weighted median, MR-Pleiotropy Residual Sum and Outlier (MR-PRESSO) and Radial regression of MR (Radial MR). Fourth, we conducted a series of sensitivity analyses and multivariable MR (to adjust for BMI). Finally, we compared our MR results with observational studies by performing a systematic review.
Figure 1.
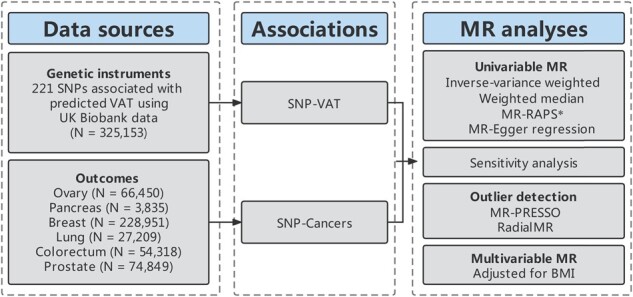
Study design *Only in the Supplementary material. MR, Mendelian randomization; SNP, single-nucleotide polymorphism; VAT, visceral adipose issue; BMI, body mass index; MR-RAPS, MR-Robust adjusted profile score; MR-PRESSO, MR-Pleiotropy Residual Sum and Outlier.
Selection of genetic predictors of VAT
UK Biobank recruited >500 000 individuals aged 37–73 years across the UK between 2006 and 2010. It aimed to identify the phenotypic and health-related information by following up participants over time. All participants gave written informed consent for data collection, analysis and record linkage. A recent study constructed two sub-cohorts to predict VAT in UK Biobank: one was called the VAT-training data set measured by dual energy x-ray absorptiometry (DXA, GE Healthcare Lunar iDXA scanner) and used to create prediction models; and the other was called the VAT-application data set, in which VAT was calculated according to the prediction models [coefficient of determination = 0.76 (0.74 to 0.78)]. After screening and quality control, a total of 4198 and 325 153 participants enrolled in the training data set for model construction and application data set for genome-wide association (GWA) analyses, respectively.9 In total, 11 predictors (age, menopause status in females, waist circumference, hip circumference, height, weight, and impedance of left arm and leg, right arm and leg, and whole body) distributed on 20 different linear and interaction terms (age × weight, waist circumference × weight, etc.) were included in the prediction models. Two reduced prediction models (menopause status, hip circumference and five bioelectrical impedance predictors were omitted in males, and age, menopause status, height, right arm and right leg impedance were omitted in females), which included only regression terms with P-values < 0.05, were developed for use in the clinic, whereas the two full models included all terms. Overall, the training and application data sets had similar characteristics, and the median depot of VAT was ∼2.5 times larger in males than females. GWA analyses for predicted VAT were performed using linear regression models in males (N = 164 004) and females (N = 161 149) separately, and the sex-combined associations were subsequently computed using a fixed-effect meta-analysis. GWAS summary data for predicted VAT are available at https://www.ebi.ac.uk/gwas/downloads/summary-statistics (Study Accession ID: GCST008744 for combined sexes, GCST008743 for males only and GCST008742 for females only).
Among the SNPs available in each GWAS summary data set, we selected SNPs robustly associated with VAT as IVs (P < 5 × 10−8, IV Assumption 1, Figure 2). To minimize the influence of linkage disequilibrium (LD), which may bias the results of randomized allele allocation, a stringent condition (LD threshold of r2 < 0.001 and distance located 10 000 kb apart from each other) was set to ensure that the genetic instruments selected for VAT were conditionally independent from each other. F-statistic represents the strength of the relationship between IVs and VAT. Generally, F > 10 may attenuate bias produced by weak IVs.10
Figure 2.

Core assumptions of Mendelian randomization. SNP, single-nucleotide polymorphism; VAT, visceral adipose tissue; IV, instrumental variable; BMI, body mass index.
Similarly, we extracted BMI GWAS summary data for combined sexes from a meta-analysis of GWASs including 681 275 participants11 and sex-specific data from another meta-analysis of GWASs including 152 893 males and 171 977 females,12 respectively. These data were from the Genetic Investigation of ANthropometric Traits (GIANT) consortium (https://portals.broadinstitute.org/collaboration/giant/index.php/GIANT_consortium_data_files).
Selection of cancer outcomes
We collected summary data of six common types of cancers from large-scale consortia: breast cancer from Breast Cancer Association Consortium (BCAC),13 lung cancer from International Lung Cancer Consortium (ILCCO),14 colorectal cancer from Genetic Epidemiology Research in Adult Health and Aging (GERA),15 ovarian cancer from Ovarian Cancer Association Consortium (OCAC),16 pancreatic cancer from Pancreatic Cancer Cohort Consortium (PANSCAN)17 and prostate cancer from Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome (PRACTICAL).15 Summary statistics of the largest available GWAS were extracted from the MR-Base database.18 The participants had an identical genetic background (European ancestry) and, to our knowledge, there was no sample overlap between the exposure and outcome GWASs.
Comparison with observational studies
To compare the MR results with observational results reported by previous epidemiological studies, we searched the electronic databases of PubMed, Medline and Embase from database inception to 15 October 2021, with no language restrictions, for studies in humans of the associations between VAT volume and cancer incidence for six cancer types: colorectal (colon and rectum), lung (adenocarcinoma and squamous-cell carcinoma) and pancreatic cancers for combined sexes; breast (pre-menopausal and post-menopausal) and ovarian cancers for females; and prostate cancer for males. Our core search consisted of terms related to VAT (visceral adipose tissue, VAT and visceral fat), combined with the terms for each cancer type (Supplementary Table S1, Supplementary Figure S1 and see Supplementary Methods, available as Supplementary data at IJE online, for the details of review protocol).
Statistical analysis
Two-sample MR
As shown in Figure 2, we estimated the causal effect of VAT on cancers using a classic MR model: βcausal effect = βZY/βZX (βZX and βZY represent the regression coefficient of SNPs on VAT and cancers, respectively).8,19 Ideally, a valid instrument should satisfy three assumptions (Figure 2): (i) must be truly associated with VAT (in this study, defined as the genetic association with P < 5 × 10−8); (ii) not associated with confounders of VAT and cancers; and (iii) should only be associated with the cancers through VAT.
To evaluate the causal effects of VAT on cancer risk by combining multiple SNPs, we conducted a two-sample Mendelian randomization20 analysis using four primary methods, including IVW,21 MR-Egger regression,22 weighted median23 and MR-PRESSO.24 The IVW is a conventional method to obtain an MR estimate performing a meta-analysis of each Wald ratio for multiple SNPs. The weighted median estimator makes the median effect of SNPs, allowing ≤50% of the invalid SNPs. The MR-Egger regression, with a relaxed criterion, allows the presence of horizontal pleiotropy across SNPs. It requires the InSIDE (Instrument Strength Independent of Direct Effect) assumption to be satisfied.22 However, it has less power and provides wider confidence intervals than the IVW. The MR-PRESSO regresses the SNP–outcome estimates against the SNP–exposure estimates to test for outlier SNPs and outputs a corrected MR estimate. In addition, we used Radial regression of MR (Radial MR) as an alternative method of MR-PRESSO to identify outlier SNPs.25
When examining the effects of VAT on sex-specific cancers such as ovarian cancer, breast cancer and prostate cancer, we used the VAT GWAS results from the same sex as the exposure GWAS data. For example, we used the VAT GWAS results from women in the analysis for breast cancer. For other cancers, sex-combined GWAS results for VAT were used. All results were corrected for multiple testing using the false discovery rate (FDR) method and FDR q-values were provided.
MR sensitivity analyses
We evaluated the heterogeneity of the results using the Cochran’s Q-test26 and detected the potential presence of horizontal pleiotropy using the MR-Egger intercept tests. We also performed the leave-one-out analysis by eliminating SNPs one by one and recomputing the effect. Once heterogeneity or horizontal pleiotropy was noted, we recomputed IVW and MR-Egger estimates after removing the outlier SNPs identified by MR-PRESSO or Radial MR.
Multivariable MR
MR analysis adjusted for potential confounders has a distinct advantage in favour of specifying the independent effect of VAT on the outcome. As BMI is highly correlated with VAT, and BMI has been reported to be related to several cancers,27–29 we additionally used multivariable MR (MVMR) analysis to estimate the direct causal effects of VAT on the risk of six cancers independently of the effect from BMI.
Based on the analyses above, we took the IVW results as the primary causal effect estimates and considered the consistency of the results across all MR methods. In this study, we defined the evidence for a potential causal effect when the following criteria were met: (i) one of the IVW and MVMR results had an FDR q-value < 0.05; (ii) IVW and MVMR showed the same effect direction and overlapped confidence intervals; (iii) other MR methods showed the same effect direction and similar effect sizes to IVW and MVMR; and (iv) there was no evidence of horizontal pleiotropy (i.e. P-value for Egger intercept > 0.05).
MR analyses were performed in R (version 4.0.4) with R packages ‘vroom’, ‘tidyr’, ‘tibble’, ‘dplyr’, ‘TwoSampleMR’,18 ‘MR-PRESSO’,24 ‘RadialMR’25 and ‘MVMR’.30 FDR q-values were estimated using the R package ‘fdrtool’.
Results
Participant characteristics and instruments
The characteristics of the participants from UK Biobank, GIANT and consortia of cancer outcomes are shown in Table 1. We selected 221, 96 and 70 SNPs as instruments for predicted VAT (Supplementary Tables S2–S4, available as Supplementary data at IJE online) in combined sexes, males and females, respectively. The F-statistic ranged from 901.13 to 1260.80, reflecting strong instrument strength. We also selected 490, 30 and 37 BMI-associated SNPs for combined sexes, males and females, respectively, to perform multivariable MR analysis.
Table 1.
Characteristics of cancer consortia and UK Biobank data sets
| Variables | Consortium | SNPs* | Cases/controls | Sample size | Population |
|---|---|---|---|---|---|
| Exposure | |||||
| VAT (sex-combined) | UK Biobank | 221 | Not relevant | 325 153 | European-ancestry |
| VAT (male) | UK Biobank | 96 | Not relevant | 164 004 | European-ancestry |
| VAT (female) | UK Biobank | 70 | Not relevant | 161 149 | European-ancestry |
| Outcomes | |||||
| Ovarian cancer | OCAC | 70 | 25 509/40 941 | 66 450 | European-ancestry |
| Low-grade mucinous | OCAC | 70 | 1149/40 941 | 42 090 | European-ancestry |
| Invasive mucinous | OCAC | 70 | 1417/40 941 | 42 358 | European-ancestry |
| Low-grade serous | OCAC | 70 | 1012/40 941 | 41 953 | European-ancestry |
| High-grade serous | OCAC | 70 | 13 037/40 941 | 53 978 | European-ancestry |
| Endometrioid | OCAC | 70 | 2810/40 941 | 43 751 | European-ancestry |
| Clear cell | OCAC | 70 | 1366/40 941 | 42 090 | European-ancestry |
| Pancreatic cancer | PANSCAN | 118 | 1896/1939 | 3835 | European-ancestry |
| Breast cancer | BCAC | 70 | 122 977/105 974 | 228 951 | European-ancestry |
| ER+ | BCAC | 70 | 69 501/105 974 | 175 475 | European-ancestry |
| ER– | BCAC | 70 | 21 468/105 974 | 127 442 | European-ancestry |
| Lung cancer | ILCCO | 206 | 11 348/15 861 | 27 209 | European-ancestry |
| Adenocarcinoma | ILCCO | 206 | 3442/14 894 | 18 336 | European-ancestry |
| Squamous-cell carcinoma | ILCCO | 206 | 3275/15 038 | 18 313 | European-ancestry |
| Colorectal cancer | GERA | 172 | 3793/50 525 | 54 318 | European-ancestry |
| Prostate cancer | PRACTICAL | 96 | 46 939/27 910 | 74 849 | European-ancestry |
Numbers for the exposure represent the total number of VAT instrumental SNPs; numbers for the outcomes represent the number of VAT instrumental SNPs (either sex-combined or sex-specific, whichever is the most appropriate) available in each outcome GWAS.
VAT, visceral adipose tissue; BMI, body mass index; SNP, single-nucleotide polymorphism; ER, oestrogen receptor; GIANT, the Genetic Investigation of ANthropometric Traits; BCAC, Breast Cancer Association Consortium; ILCCO, International Lung Cancer Consortium; GERA, Genetic Epidemiology Research in Adult Health and Aging; OCAC, Ovarian Cancer Association Consortium; PANSCAN, Pancreatic Cancer Cohort Consortium; PRACTICAL, Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome; GWAS, genome-wide association study.
Estimation of causal effects of VAT on cancers
Univariable two-sample MR results
Table 2 shows the results of univariable MR analysis for the effect of increased VAT on cancer risks. IVW results showed that genetically increased VAT was associated with a higher risk of pancreatic cancer [odds ratio (OR) = 1.65, 95% CI = 1.03 to 2.62], total lung cancer (OR = 1.24, 95% CI = 1.06 to 1.45) and its subtype lung squamous-cell carcinoma (OR = 1.47, 95% CI = 1.20 to 1.82). Of these, the results from other MR methods were largely consistent with the IVW results for pancreatic cancer (P < 0.05 in both MR-Egger regression and weighted median). The association between increased VAT and lung squamous-cell carcinoma showed similar effect sizes and overlapped confidence intervals across different univariable MR methods. Subsequently, the IVW results for pancreatic cancer, total lung cancer and lung squamous-cell carcinoma survived the multiple testing correction (FDR q-value < 0.05). There was little evidence to support an association between genetically increased VAT and other cancer types.
Table 2.
Two-sample Mendelian randomization results for the effect of visceral adipose tissue on the risk of different types of cancer
| Outcomes | Methods | Odds ratio (95% CI) | P-value | q-valuea | Q-statistics | P h | Egger intercept | Pintercept |
|---|---|---|---|---|---|---|---|---|
| Ovarian cancer | MR-Egger | 1.13 (0.67–1.89) | 6.62E-01 | 1.66E-01 | 75.56 | 8.49E-02 | –0.001 (–0.015–0.015) | 9.90E-01 |
| Inverse-variance weighted | 1.12 (0.96–1.31) | 1.44E-01 | 1.13E-01 | 75.56 | 9.94E-02 | |||
| Weighted median | 1.05 (0.85–1.30) | 6.32E-01 | 5.25E-01 | |||||
| MR-PRESSO | 1.12 (0.97–1.30) | 1.38E-01 | 1.49E-01 | |||||
| Ovarian cancer | MR-Egger | 0.74 (0.14–3.84) | 7.23E-01 | 1.74E-01 | 68.79 | 2.31E-01 | 0.023 (–0.031–0.061) | 5.14E-01 |
| (Low-mucinous) | Inverse-variance weighted | 1.26 (0.78–2.03) | 3.52E-01 | 1.64E-01 | 69.27 | 2.46E-01 | ||
| Weighted median | 0.91 (0.44–1.89) | 7.94E-01 | 5.81E-01 | |||||
| Ovarian cancer | MR-Egger | 2.85 (0.67–12.10) | 1.61E-01 | 9.69E-02 | 65.77 | 3.15E-01 | –0.027 (–0.067–0.014) | 1.97E-01 |
| (Invasive mucinous) | Inverse-variance weighted | 1.14 (0.74–1.74) | 5.54E-01 | 1.85E-01 | 67.60 | 2.92E-01 | ||
| Weighted median | 1.65 (0.89–3.05) | 1.11E-01 | 1.92E-01 | |||||
| Ovarian cancer | MR-Egger | 1.91 (0.31–11.78) | 4.88E-01 | 1.45E-01 | 72.57 | 1.47E-01 | –0.025 (–0.076–0.026) | 3.44E-01 |
| (Low-serous) | Inverse-variance weighted | 0.82 (0.48–1.40) | 4.69E-01 | 1.78E-01 | 73.65 | 1.48E-01 | ||
| Weighted median | 0.93 (0.42–2.03) | 8.49E-01 | 5.98E-01 | |||||
| Ovarian cancer | MR-Egger | 0.84 (0.44–1.59) | 5.97E-01 | 1.59E-01 | 79.80 | 4.46E-02 | 0.007 (–0.011–0.025) | 4.48E-01 |
| (High-serous) | Inverse-variance weighted | 1.07 (0.88–1.29) | 5.03E-01 | 1.81E-01 | 80.57 | 4.74E-02 | ||
| Weighted median | 1.20 (0.92–1.55) | 1.74E-01 | 2.35E-01 | |||||
| MR-PRESSO | 1.08 (0.90–1.30) | 4.08E-01 | 2.71E-01 | |||||
| Ovarian cancer | MR-Egger | 1.80 (0.57–5.66) | 3.20E-01 | 1.16E-01 | 79.17 | 4.93E-02 | –0.002 (–0.034–0.031) | 9.13E-01 |
| (Endometrioid) | Inverse-variance weighted | 1.39 (1.00–1.94) | 4.97E-02 | 5.62E-02 | 79.19 | 5.87E-02 | ||
| Weighted median | 1.38 (0.86–2.21) | 1.78E-01 | 2.37E-01 | |||||
| Ovarian cancer | MR-Egger | 4.28 (0.94–19.58) | 6.55E-02 | 8.01E-02 | 71.17 | 1.75E-01 | –0.035 (–0.077–0.008) | 1.14E-01 |
| (Clear cell) | Inverse-variance weighted | 1.30 (0.83–2.04) | 2.49E-01 | 1.45E-01 | 74.17 | 1.38E-01 | ||
| Weighted median | 1.73 (0.91–3.29) | 9.73E-02 | 1.92E-01 | |||||
| Pancreatic cancer | MR-Egger | 6.19 (1.57–24.45) | 1.05E-02 | 3.79E-02* | 115.39 | 4.46E-01 | –0.030 (–0.05–0.001) | 4.72E-02 |
| Inverse-variance weighted | 1.65 (1.03–2.62) | 3.53E-02 | 4.80E-02* | 119.47 | 3.69E-01 | |||
| Weighted median | 2.23 (1.10–4.51) | 2.63E-02 | 1.92E-01 | |||||
| Breast cancer | MR-Egger | 0.66 (0.42–1.02) | 6.69E-02 | 8.05E-02 | 111.33 | 4.98E-06 | 0.013 (0.001–0.024) | 3.62E-02 |
| Inverse-variance weighted | 1.05 (0.94–1.17) | 4.00E-01 | 1.70E-01 | 121.02 | 4.76E-07 | |||
| Weighted median | 1.11 (0.98–1.26) | 9.09E-02 | 1.16E-01 | |||||
| MR-PRESSO | 1.06 (0.95–1.17) | 4.49E-01 | 1.92E-01 | |||||
| Breast cancer | MR-Egger | 0.67 (0.42–1.09) | 1.11E-01 | 9.10E-02 | 93.65 | 4.86E-04 | 0.012 (–0.001–0.025) | 6.46E-02 |
| (ER+) | Inverse-variance weighted | 1.05 (0.93–1.19) | 4.09E-01 | 1.71E-01 | 99.94 | 1.45E-04 | ||
| Weighted median | 1.02 (0.88–1.17) | 8.19E-01 | 5.89E-01 | |||||
| MR-PRESSO | 0.98 (0.88–1.10) | 7.61E-01 | 3.98E-01 | |||||
| Breast cancer | MR-Egger | 0.71 (0.41–1.24) | 2.38E-01 | 1.08E-01 | 62.39 | 2.30E-01 | 0.007 (–0.007–0.022) | 3.40E-01 |
| (ER–) | Inverse-variance weighted | 0.93 (0.81–1.07) | 3.00E-01 | 1.55E-01 | 63.43 | 2.31E-01 | ||
| Weighted median | 0.89 (0.72–1.09) | 2.54E-01 | 3.08E-01 | |||||
| MR-PRESSO | 0.90 (0.78–1.03) | 1.46E-01 | 1.53E-01 | |||||
| Lung cancer | MR-Egger | 1.21 (0.75–1.94) | 4.37E-01 | 1.37E-01 | 267.59 | 9.94E-04 | 0.006 (–0.009–0.106) | 9.12E-01 |
| Inverse-variance weighted | 1.24 (1.06–1.45) | 6.90E-03 | 1.56E-02* | 267.60 | 1.16E-03 | |||
| Weighted median | 1.21 (0.96–1.50) | 1.03E-01 | 1.92E-01 | |||||
| MR-PRESSO | 1.24 (1.07–1.45) | 4.81E-03 | 1.01E-02* | |||||
| Lung cancer | MR-Egger | 0.87 (0.44–1.76) | 7.07E-01 | 1.71E-01 | 253.86 | 8.85E-03 | 0.004 (–0.011–0.018) | 6.20E-01 |
| (Adenocarcinoma) | Inverse-variance weighted | 1.03 (0.82–1.30) | 7.79E-01 | 2.35E-01 | 254.17 | 9.71E-03 | ||
| Weighted median | 1.09 (0.78–1.51) | 6.11E-01 | 5.17E-01 | |||||
| MR-PRESSO | 1.09 (0.87–1.37) | 4.56E-01 | 2.84E-01 | |||||
| Lung cancer | MR-Egger | 1.62 (0.84–3.13) | 1.50E-01 | 9.59E-02 | 210.37 | 2.94E-01 | –0.002 (–0.016–0.012) | 7.59E-01 |
| (Squamous-cell carcinoma) | Inverse-variance weighted | 1.47 (1.20–1.82) | 3.22E-04 | 1.46E-03* | 210.47 | 3.09E-01 | ||
| Weighted median | 1.32(0.94–1.86) | 1.13E-01 | 1.92E-01 | |||||
| MR-PRESSO | 1.44 (1.17–1.76) | 6.50E-04 | 2.72E-03* | |||||
| Colorectal cancer | MR-Egger | 1.32 (0.76–2.30) | 3.21E-01 | 1.16E-01 | 223.75 | 2.45E-01 | –0.005 (–0.017–0.007) | 3.90E-01 |
| Inverse-variance weighted | 1.05 (0.88–1.26) | 5.78E-01 | 1.87E-01 | 224.54 | 2.49E-01 | |||
| Weighted median | 0.99 (0.74–1.33) | 9.47E-01 | 6.24E-01 | |||||
| Prostate cancer | MR-Egger | 0.85 (0.64–1.13) | 2.69E-01 | 1.12E-01 | 128.39 | 1.66E-03 | 0.004 (–0.004–0.122) | 2.89E-01 |
| Inverse-variance weighted | 0.99 (0.91–1.07) | 7.51E-01 | 2.28E-01 | 130.11 | 1.51E-03 | |||
| Weighted median | 0.99 (0.88–1.11) | 8.19E-01 | 4.85E-01 | |||||
| MR-PRESSO | 0.98 (0.90–1.07) | 6.61E-01 | 5.89E-01 |
Estimated by the false discovery rate (FDR) method for multiple testing correction.
q-value < 0.05.
ER, oestrogen receptor; NA, not applicable; Ph, P-value for heterogeneity; Pintercept, P-value for intercept of MR-Egger regression.
MR sensitivity analysis results
We conducted a series of sensitivity analyses to evaluate the heterogeneity and potential horizontal pleiotropy (Table 2). Cochran’s Q-test showed evidence (Ph < 0.05) for the presence of heterogeneity in the IVW results for high-serous ovarian cancer, endometrioid ovarian cancer, breast cancer and its subtype oestrogen-receptor-positive breast cancer, lung cancer and its subtype lung adenocarcinoma, and prostate cancer (Ph < 0.05). The MR-Egger intercept tests showed the presence of unbalanced horizontal pleiotropy (Pintercept < 0.05) for breast cancer and pancreatic cancer. MR-PRESSO and Radial MR did not identify any outlier SNPs for pancreatic cancer. The funnel plots showed a relatively symmetrical distribution of variant effects for pancreatic cancer and lung squamous-cell carcinoma, indicating an absence of directional pleiotropy (Figure 4). The leave-one-out analysis found that the MR estimates remained stable when sequentially dropping a single SNP out (Supplementary Figures S2–S7, available as Supplementary data at IJE online).
Figure 4.

Scatter plots and funnel plots for effects of visceral adipose tissue on pancreatic cancer (A, B) and lung squamous-cell carcinoma (C, D) VAT, visceral adipose tissue; MR, Mendelian randomization.
Multivariable MR results adjusted for BMI
Although the associations of VAT with pancreatic cancer (OR = 1.35, 95% CI = 0.63 to 2.93) and lung squamous-cell carcinoma (OR = 1.40, 95% CI = 0.97 to 2.01) were attenuated in multivariable MR with the adjustment for BMI, they still showed the same effect direction and overlapped confidence intervals with the IVW results. There was no evidence of a causal relationship between VAT and the risk of any other types of cancer (Figure 3).
Figure 3.

Comparison of the results between univariable and multivariable Mendelian randomization for the effect of visceral adipose tissue on cancer risks (outlier SNPs have been removed) ER, oestrogen receptor; VAT, visceral adipose tissue; IVW, inverse-variance weighted; MVMR, multivariable Mendelian randomization.
Discussion
In this study, we performed MR analyses to evaluate the causal relationship between VAT and the risk of six common cancers. We found that genetically increased VAT had a causal effect on the risk of pancreatic cancer and lung squamous-cell carcinoma. However, some of our findings were inconsistent with previous observational studies (Table 3 and Figure 3).
Table 3.
Observational studies on the associations between visceral obesity and cancer risks
| Cancer types | Visceral obesity | Study design | Population | Sample size | Age (years) | Findings | First author |
|---|---|---|---|---|---|---|---|
| Breast cancer | MRI-measured VAT | Nested case– control study | European American (19.3%) African American (16.2%) Native Hawaiian (11.2%) Japanese American (32.8%) Latino (20.5%) |
Case: 950 Control: 950 |
Case: 66.8 ± 7.9 Control: 67.0 ± 7.8 |
Risk factor OR (95% CI) by increasing tertiles: 1.00, 1.09 (0.86–1.39), 1.48 (1.16–1.89); Ptrend = 0.002 |
Le Marchand et al.31 |
| CT-measured VAT | Case–control study | East Asian | Case: 234 Control: 211 |
Case: 52.6 Control: 52.3 |
No significance Pre-menopause: Tertile3 vs Tertile1, OR = 0.98 (0.49–1.93); Post-menopause: Tertile3 vs Tertile1, OR = 1.84 (0.81–3.76) |
Kim et al.32 | |
| BIA-measured VAT | Case–control study | Southeast Asian | Case: 56 control: 56 |
Case: 47 ± 8 Control: 42 ± 9 |
No significance Per unit increase: Crude OR = 1.01(0.91–1.13) |
Zunura'in et al.33 | |
| Colorectal cancer | MRI-measured VAT | Nested case– control study | European American (14.2%) African American (21.7%) Native Hawaiian (6.6%) Japanese American (32.6%) Latino (24.9%) |
Case: 831 Control: 831 |
Case: 69.9 ± 7.8 Control: 70.5 ± 7.9 |
No significance (P = 0.84) OR (95% CI) by increasing tertiles: 1.00, 0.98 (0.68–1.39), 1.24 (0.88–1.76); Ptrend = 0.08 |
Le Marchand et al.31 |
| CT-measured VAT | Cross-sectional study | East Asian | 200 | 50.9 ± 8.5 | Risk factor OR 4.07 (95% CI: 1.01–16.43, P = 0.03) for those with VAT over 136.61 cm2 relative to those with VAT under 67.23 cm2 |
Oh et al.34 | |
| CT-measured VAT | Case–control study | East Asian | Case: 22 Control: 66 |
Case: 53.8 ± 7.9 Control: 53.8 ± 7.7 |
OR (95% CI) for the lowest to highest tertile of visceral fat area of 1 (reference), 2.17 (0.45–10.46) and 5.92 (1.22–28.65), respectively (Ptrend = 0.02) | Yamamoto et al.35 | |
| CT-measured VAT | Cross-sectional study | East Asian post-menopausal women |
398 | 60.73 ± 8.55 | Highest vs the lowest visceral fat tertiles were 2.96 (1.38–6.33) | Lee et al.36 | |
| CT-measured VAT | Case–control study | European | Case: 23 Control: 50 |
Case: 57 ± 9.7 Control: 59 ± 9.2 |
VFA was not different in the colorectal carcinoma groups than controls (P = 0.156) | Erarslan et al.37 | |
| Prostate cancer | CT-measured VAT | Case–control study | European | Case: 63 Control: 63 |
Case: 71.0 ± 7.3 Control: 68.9 ± 10.5 |
Risk factor OR (95% CI), 4.6 (2.6–8.2) per SD increased visceral fat |
von Hafe et al.38 |
| CT-measured VAT | Prospective cohort studies | European | 1832 | NA | No association between VAT and the risk of total prostate cancer: HR 1.02 (0.88–1.19) | Dickerman et al.39 | |
| CT-measured VAT | Cross-sectional analysis | African American (62.7%) | 308 | Non-Black: 65.4 ± 6.4 Black: 63.4 ± 6.5 |
Risk factor Tertile 3 vs Tertile 1: OR = 2.12 (1.07–4.22) |
Allott et al.40 |
VAT, visceral adipose tissue; CT, computed tomography; MRI, magnetic resonance imaging; BIA, bioelectrical impedance analysis.
Few observational studies have specifically investigated the association between VAT and ovarian cancer, and most published studies have only focused on BMI or weight circumference (WC) as the exposure.41,42 It has been reported that the adipocytes in the tumour microenvironment may result in the metastasis, growth and angiogenesis of ovarian cancer.43 However, we found that ovarian cancer and its subtypes were not causally affected by VAT in our MR analysis. For lung cancer, we failed to retrieve any publications describing the association of VAT with lung cancer and its subtypes. A meta-analysis of prospective studies suggests that abdominal obesity, measured by WC, may play a critical role in the development of lung cancer.44 We observed a causal relationship between VAT and a higher risk of lung squamous-cell carcinoma, other than lung adenocarcinoma.
Notably, lung squamous-cell carcinoma has been demonstrated to be remarkably distinct from the other subtype. The underlying mechanisms may be attributable to the following two aspects. First, different cell types differ in their ability to repair DNA damage, which is associated with chronic inflammation caused by obesity.45 Compared with subcutaneous adiposity, visceral adiposity is more metabolically active and may be more strongly linked with chronic inflammation.46 Then more cytokines and adipokines are released, which promote DNA damage and dysregulation of DNA repair pathways, increasing the mutation rate and leading to the transformation of healthy tissues into cancer,47 especially for repair-deficient cells. Second, different cancer types may have different susceptibility to environmental influences. For instance, lung squamous-cell carcinoma originates from squamous metaplasia of bronchial epithelium, which is more vulnerable to environmental factors.48–50 Further observational studies focusing on the association between VAT and ovarian and lung cancer subtypes and tissue-specific basic research are needed to reveal the possible mechanisms.
Since VAT is in close proximity to the pancreas, they may directly interact with each other. For example, increased VAT leads to fatty infiltration in the pancreas and is correlated with pancreatic intraepithelial neoplasia (PanIN), which has a high risk of conversion to pancreatic ductal adenocarcinoma (PDAC).51 Similarly, our MR results showed a causal relationship between genetically determined VAT and pancreatic cancer, which was supported by further sensitivity analysis.
We did not find evidence of a causal effect of VAT on breast cancer and its two subtypes in either univariable or multivariable MR analysis. In contrast, most observational studies have reported a positive association between VAT and breast cancer risk.31,52 As VAT is more metabolically active than subcutaneous adipose tissue, the increased levels of adipokines such as IL-6, IL-1β and leptin contribute to insulin resistance,53,54 which is in turn associated with an increased risk of breast cancer.55,56 Moreover, hyposecretion of adiponectin due to VAT accumulation has been associated with increased proliferation of tumour cells in breast cancer.54,56 It has been reported that the association between BMI and breast cancer is complicated by different menopausal statuses. More specifically, the inverse association between adult BMI and pre-menopausal breast cancer is consistently supported by previous studies, whereas MR results for post-menopausal breast cancer are in contrast with conventional observational studies in favour of a positive association. The discrepancy may be partly attributed to early-life body shape and post-menopausal weight gain.27,28
There was no evidence supporting VAT as a causal factor on the risk of colorectal cancer or prostate cancer in our study. Although meta-analysis and observational studies have found that increased VAT measured by CT is linked to the aetiology of colorectal adenoma and colorectal cancer,34,57–60 these studies were all based on Asian populations, which may not be generalizable to other ethnic groups. The evidence of European populations came from a small case–control study, which did not show different volumes of visceral fat between cases and controls (P = 0.156).37 On the other hand, CT-measured VAT has been shown as a risk factor (OR = 4.6, 95% CI = 2.6 to 8.2) for prostate cancer in a case–control study.38 No association between VAT and the risk of total prostate cancer (OR = 1.02, 95% CI = 0.88 to 1.19) was found in another prospective study including 1832 participants.39 These observational studies, nonetheless, might have suffered from issues such as small sample sizes, reverse causality and residual confounding.
Strength and limitations
To the best of our knowledge, this study is the first to systematically assess the causal effect of VAT on multiple cancer risks using MR. We applied a series of sensitivity analyses and multivariable MR to test the assumptions of MR and minimize the influence of potential confounders and horizontal pleiotropy. Given that the profound differences of male/female proportions between the exposure and outcome populations could be a potential confounder, which might substantially influence the direction or magnitude of causal relationships between VAT and sex-specific cancers, we conducted sex-specific MR analyses to reduce the bias of causal effect estimation and make our MR results more reliable.
Notably, there are also four major limitations in our study. First, as the training models for the VAT prediction was established on a relatively small subset of data, GWAS results for predicted VAT may not reflect genetic associations with the true volume of VAT, and the IVs selected from these GWAS results was likely to introduce biases. Second, these IVs could only explain a small part of the variation in VAT, resulting in limited statistical power and imprecision of MR estimates. Third, to ensure the consistency of genetic background, only European-ancestry participants were included in our MR analysis, limiting the generalizability of the conclusions to other ethnic groups. Fourth, we could not rule out the possibility that the association between VAT and cancer risks may be non-linear. Current MR methods based on summary-level data assume that the exposure–outcome relationship is linear when estimating causal effects. Therefore, this possible non-linear relation should be investigated using individual-level data in future research.
Conclusion
In summary, this MR study suggests that lifelong exposure to elevated volumes of VAT might increase the risk of pancreatic cancer and lung squamous-cell carcinoma. Further studies are needed to determine the reliability of VAT as a predictor of cancer risks, evaluate the mediating mechanisms for potential intervention targets and explore the possible non-linear relationship using individual-level data.
Ethics approval
The UK Biobank study has ethical approval from the North West Multicentre Research Ethics Committee (MREC). For cancer-related consortia, all participating studies were approved by their appropriate ethics review board and all participants provided informed written consent.
Supplementary Material
Contributor Information
Yao Lu, Health Management Center, The Third Xiangya Hospital, Central South University, Changsha, China; Schools of Life Course Sciences, King’s College London, London, UK; Center of Clinical Pharmacology, The Third Xiangya Hospital, Central South University, Changsha, China.
Haibo Tang, Department of Metabolic and Bariatric Surgery, The Third Xiangya Hospital, Central South University, Changsha, China.
Peiyuan Huang, MRC Integrative Epidemiology Unit (IEU), Bristol Medical School, University of Bristol, Bristol, UK.
Jie Wang, Center of Clinical Pharmacology, The Third Xiangya Hospital, Central South University, Changsha, China.
Peizhi Deng, Center of Clinical Pharmacology, The Third Xiangya Hospital, Central South University, Changsha, China.
Yalan Li, Center of Clinical Pharmacology, The Third Xiangya Hospital, Central South University, Changsha, China.
Jie Zheng, MRC Integrative Epidemiology Unit (IEU), Bristol Medical School, University of Bristol, Bristol, UK; Department of Endocrine and Metabolic Diseases, Shanghai Institute of Endocrine and Metabolic Diseases, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China; Shanghai National Clinical Research Center for Metabolic Diseases, Key Laboratory for Endocrine and Metabolic Diseases of the National Health Commission of the PR China, Shanghai National Center for Translational Medicine, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China.
Liang Weng, Department of Pathology, Xiangya Hospital, Central South University, Changsha, China; Xiangya Cancer Center, Xiangya Hospital, Central South University, Changsha, China.
Data Availability
The data sets were derived from sources in the public domain: GWAS Catalog (https://www.ebi.ac.uk/gwas/home) and MR-Base (https://www.mrbase.org/).
Supplementary data
Supplementary data are available at IJE online.
Author contributions
Y.L., H.B.T. and P.Y.H.: formal analysis; statistic analysis; writing original draft. J.W., P.Z.D. and Y.L.L.: data collection. J.Z. and L.W.: methodology; writing review and editing; supervision.
Funding
This work was supported by the National Natural Science Foundations of China (grant number 81974465, 81900199); Hunan province natural science funds for Excellent Young Scholars (grant number 2019JJ30043); Outstanding Young Investigator of Hunan province (grant number 2020JJ2056); the Hunan Youth Talent Project (grant number 2019RS2014); UK Medical Research Council Integrative Epidemiology Unit (grant number MC_UU_00011/1, MC_UU_00011/4 to J.Z.); Academy of Medical Sciences (AMS) Springboard Award, the Wellcome Trust, the Government Department of Business, Energy and Industrial Strategy (BEIS), the British Heart Foundation and Diabetes UK (grant number SBF006\1117 to J.Z.); J.Z. is funded by the Vice-Chancellor Fellowship from the University of Bristol.
Conflict of interest
None declared.
References
- 1. Sung H, Siegel RL, Torre LA. et al. Global patterns in excess body weight and the associated cancer burden. CA Cancer J Clin 2019;69:88–112. [DOI] [PubMed] [Google Scholar]
- 2. Fox CS, Massaro JM, Hoffmann U. et al. Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation 2007;116:39–48. [DOI] [PubMed] [Google Scholar]
- 3. Doyle SL, Donohoe CL, Lysaght J, Reynolds JV.. Visceral obesity, metabolic syndrome, insulin resistance and cancer. Proc Nutr Soc 2012;71:181–89. [DOI] [PubMed] [Google Scholar]
- 4. Barberio AM, Alareeki A, Viner B. et al. Central body fatness is a stronger predictor of cancer risk than overall body size. Nat Commun 2019;10:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Silveira EA, Kliemann N, Noll M, Sarrafzadegan N, de Oliveira C.. Visceral obesity and incident cancer and cardiovascular disease: an integrative review of the epidemiological evidence. Obes Rev 2021;22:e13088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Riaz H, Khan MS, Siddiqi TJ. et al. Association between obesity and cardiovascular outcomes: a systematic review and meta-analysis of Mendelian randomization studies. JAMA Netw Open 2018;1:e183788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thanassoulis G, O'Donnell CJ.. Mendelian randomization: nature's randomized trial in the post-genome era. JAMA 2009;301:2386–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Smith GD, Ebrahim S.. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol 2003;32:1–22. [DOI] [PubMed] [Google Scholar]
- 9. Karlsson T, Rask-Andersen M, Pan G. et al. Contribution of genetics to visceral adiposity and its relation to cardiovascular and metabolic disease. Nat Med 2019;25:1390–95. [DOI] [PubMed] [Google Scholar]
- 10. Burgess S, Thompson SG, CRP CHD Genetics Collaboration. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol 2011;40:755–64. [DOI] [PubMed] [Google Scholar]
- 11. Shungin D, Winkler TW, Croteau-Chonka DC. et al. ; The ADIPOGen Consortium. New genetic loci link adipose and insulin biology to body fat distribution. Nature 2015;518:187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Locke AE, Kahali B, Berndt SI. et al. ; The LifeLines Cohort Study. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015;518:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Michailidou K, Lindström S, Dennis J. et al. ; ConFab/AOCS Investigators. Association analysis identifies 65 new breast cancer risk loci. Nature 2017;551:92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Y, McKay JD, Rafnar T. et al. Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nat Genet 2014;46:736–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rashkin SR, Graff RE, Kachuri L. et al. Pan-cancer study detects genetic risk variants and shared genetic basis in two large cohorts. Nat Commun 2020;11:4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Phelan CM, Kuchenbaecker KB, Tyrer JP. et al. ; OPAL study group. Identification of 12 new susceptibility loci for different histotypes of epithelial ovarian cancer. Nat Genet 2017;49:680–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Amundadottir L, Kraft P, Stolzenberg-Solomon RZ. et al. Genome-wide association study identifies variants in the ABO locus associated with susceptibility to pancreatic cancer. Nat Genet 2009;41:986–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hemani G, Zheng J, Elsworth B. et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zheng J, Baird D, Borges MC. et al. Recent developments in Mendelian randomization studies. Curr Epidemiol Rep 2017;4:330–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Burgess S, Scott RA, Timpson NJ, Davey Smith G, Thompson SG, EPIC- InterAct Consortium. Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol 2015;30:543–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Johnson T, Uk S, Efficient calculation for multi-SNP genetic risk scores, 2012. http://cran.r-project.org/web/packages/gtx (12 June 2021, date last accessed).
- 22. Bowden J, Davey Smith G, Burgess S.. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 2015;44:512–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bowden J, Davey Smith G, Haycock PC, Burgess S.. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 2016;40:304–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Verbanck M, Chen C-Y, Neale B, Do R.. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet 2018;50:693–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bowden J, Spiller W, Del Greco M F. et al. Improving the visualization, interpretation and analysis of two-sample summary data Mendelian randomization via the Radial plot and Radial regression. Int J Epidemiol 2018;47:1264–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG.. Sensitivity analyses for robust causal inference from Mendelian randomization analyses with multiple genetic variants. Epidemiology 2017;28:30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mariosa D, Carreras-Torres R, Martin RM, Johansson M, Brennan P.. Commentary: What can Mendelian randomization tell us about causes of cancer? Int J Epidemiol 2019;48:816–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fang Z, Song M, Lee D, Giovannucci EL.. The role of Mendelian randomization studies in deciphering the effect of obesity on cancer. J Natl Cancer Inst 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carreras-Torres R, Johansson M, Gaborieau V. et al. The role of obesity, type 2 diabetes, and metabolic factors in pancreatic cancer: a Mendelian randomization study. J Natl Cancer Inst 2017;109:495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sanderson E, Davey Smith G, Windmeijer F, Bowden J.. An examination of multivariable Mendelian randomization in the single-sample and two-sample summary data settings. Int J Epidemiol 2019;48:713–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Le Marchand L, Wilkens LR, Castelfranco AM. et al. Circulating biomarker score for visceral fat and risks of incident colorectal and postmenopausal breast cancer: the multiethnic cohort adiposity phenotype study. Cancer Epidemiol Biomarkers Prev 2020;29:966–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim MS, Choi YJ, Lee YH.. Visceral fat measured by computed tomography and the risk of breast cancer. Transl Cancer Res 2019;8:1939–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zunura'in Z, Almardhiyah AR, Gan SH. et al. Anthropometric and reproductive factors among newly-diagnosed breast cancer patients and healthy women: a case-control study. Asian Pac J Cancer Prev 2016;17:4439–44. [PubMed] [Google Scholar]
- 34. Oh TH, Byeon JS, Myung SJ. et al. Visceral obesity as a risk factor for colorectal neoplasm. J Gastroenterol Hepatol 2008;23:411–17. [DOI] [PubMed] [Google Scholar]
- 35. Yamamoto S, Nakagawa T, Matsushita Y. et al. Visceral fat area and markers of insulin resistance in relation to colorectal neoplasia. Diabetes Care 2010;33:184–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee JY, Lee HS, Lee DC. et al. Visceral fat accumulation is associated with colorectal cancer in postmenopausal women. PLoS One 2014;9:e110587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Erarslan E, Turkay C, Koktener A, Koca C, Uz B, Bavbek N.. Association of visceral fat accumulation and adiponectin levels with colorectal neoplasia. Dig Dis Sci 2009;54:862–68. [DOI] [PubMed] [Google Scholar]
- 38. von Hafe P, Pina F, Pérez A, Tavares M, Barros H.. Visceral fat accumulation as a risk factor for prostate cancer. Obes Res 2004;12:1930–35. [DOI] [PubMed] [Google Scholar]
- 39. Dickerman BA, Torfadottir JE, Valdimarsdottir UA. et al. Body fat distribution on computed tomography imaging and prostate cancer risk and mortality in the AGES-Reykjavik study. Cancer 2019;125:2877–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Allott EH, Howard LE, Song HJ. et al. Racial differences in adipose tissue distribution and risk of aggressive prostate cancer among men undergoing radiotherapy. Cancer Epidemiol Biomarkers Prev 2014;23:2404–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Olsen CM, Green AC, Whiteman DC, Sadeghi S, Kolahdooz F, Webb PM.. Obesity and the risk of epithelial ovarian cancer: a systematic review and meta-analysis. Eur J Cancer 2007;43:690–709. [DOI] [PubMed] [Google Scholar]
- 42. Aune D, Navarro Rosenblatt DA, Chan DS. et al. Anthropometric factors and ovarian cancer risk: a systematic review and nonlinear dose-response meta-analysis of prospective studies. Int J Cancer 2015;136:1888–98. [DOI] [PubMed] [Google Scholar]
- 43. Dai L, Song K, Di W.. Adipocytes: active facilitators in epithelial ovarian cancer progression? J Ovarian Res 2020;13:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hidayat K, Du X, Chen G, Shi M, Shi B.. Abdominal obesity and lung cancer risk: systematic review and meta-analysis of prospective studies. Nutrients 2016;8:810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Włodarczyk M, Nowicka G.. Obesity, DNA damage, and development of obesity-related diseases. IJMS 2019;20:1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Després JP, Lemieux I, Bergeron J. et al. Abdominal obesity and the metabolic syndrome: contribution to global cardiometabolic risk. Arterioscler Thromb Vasc Biol 2008;28:1039–49. [DOI] [PubMed] [Google Scholar]
- 47. Turgeon MO, Perry NJS, Poulogiannis G.. DNA damage, repair, and cancer metabolism. Front Oncol 2018;8:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chang CH, Hsiao CF, Chang GC. et al. Interactive effect of cigarette smoking with human 8-oxoguanine DNA N-glycosylase 1 (hOGG1) polymorphisms on the risk of lung cancer: a case-control study in Taiwan. Am J Epidemiol 2009;170:695–702. [DOI] [PubMed] [Google Scholar]
- 49. Yang Y, Wang M, Liu B.. Exploring and comparing of the gene expression and methylation differences between lung adenocarcinoma and squamous cell carcinoma. J Cell Physiol 2019;234:4454–59. [DOI] [PubMed] [Google Scholar]
- 50. Taylor AE, Richmond RC, Palviainen T. et al. The effect of body mass index on smoking behaviour and nicotine metabolism: a Mendelian randomization study. Hum Mol Genet 2019;28:1322–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rebours V, Gaujoux S, d'Assignies G. et al. Obesity and fatty pancreatic infiltration are risk factors for Pancreatic Precancerous Lesions (PanIN). Clin Cancer Res 2015;21:3522–28. [DOI] [PubMed] [Google Scholar]
- 52. Schapira DV, Clark RA, Wolff PA, Jarrett AR, Kumar NB, Aziz NM.. Visceral obesity and breast cancer risk. Cancer 1994;74:632–39. [DOI] [PubMed] [Google Scholar]
- 53. Fasshauer M, Paschke R.. Regulation of adipocytokines and insulin resistance. Diabetologia 2003;46:1594–603. [DOI] [PubMed] [Google Scholar]
- 54. Andò S, Gelsomino L, Panza S. et al. Obesity, leptin and breast cancer: epidemiological evidence and proposed mechanisms. Cancers (Basel) 2019;11:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bruning PF, Bonfrèr JM, van Noord PA, Hart AA, de Jong-Bakker M, Nooijen WJ.. Insulin resistance and breast-cancer risk. Int J Cancer 1992;52:511–16. [DOI] [PubMed] [Google Scholar]
- 56. Pan K, Chlebowski RT, Mortimer JE. et al. Insulin resistance and breast cancer incidence and mortality in postmenopausal women in the Women's Health Initiative. Cancer 2020;126:3638–47. [DOI] [PubMed] [Google Scholar]
- 57. Kang HW, Kim D, Kim HJ. et al. Visceral obesity and insulin resistance as risk factors for colorectal adenoma: a cross-sectional, case-control study. Am J Gastroenterol 2010;105:178–87. [DOI] [PubMed] [Google Scholar]
- 58. Yamaji T, Iwasaki M, Sasazuki S. et al. Visceral fat volume and the prevalence of colorectal adenoma. Am J Epidemiol 2009;170:1502–11. [DOI] [PubMed] [Google Scholar]
- 59. Im JP, Kim D, Chung SJ. et al. Visceral obesity as a risk factor for colorectal adenoma occurrence in surveillance colonoscopy. Gastrointest Endosc 2018;88:119–27. [DOI] [PubMed] [Google Scholar]
- 60. Keum N, Lee DH, Kim R, Greenwood DC, Giovannucci EL.. Visceral adiposity and colorectal adenomas: dose-response meta-analysis of observational studies. Ann Oncol 2015;26:1101–109. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets were derived from sources in the public domain: GWAS Catalog (https://www.ebi.ac.uk/gwas/home) and MR-Base (https://www.mrbase.org/).