

Abstract
Corticobasal syndrome (CBS) is a clinical syndrome characterised by progressive asymmetric limb rigidity and apraxia with dystonia, myoclonus, cortical sensory loss and alien limb phenomenon. Corticobasal degeneration (CBD) is one of the most common underlying pathologies of CBS, but other disorders, such as progressive supranuclear palsy (PSP), Alzheimer’s disease (AD) and frontotemporal lobar degeneration with TDP-43 inclusions, are also associated with this syndrome.
In this review, we describe common and rare neuropathological findings in CBS, including tauopathies, synucleinopathies, TDP-43 proteinopathies, fused in sarcoma proteinopathy, prion disease (Creutzfeldt-Jakob disease) and cerebrovascular disease, based on a narrative review of the literature and clinicopathological studies from two brain banks. Genetic mutations associated with CBS, including GRN and MAPT, are also reviewed. Clinicopathological studies on neurodegenerative disorders associated with CBS have shown that regardless of the underlying pathology, frontoparietal, as well as motor and premotor pathology is associated with CBS. Clinical features that can predict the underlying pathology of CBS remain unclear. Using AD-related biomarkers (ie, amyloid and tau positron emission tomography (PET) and fluid biomarkers), CBS caused by AD often can be differentiated from other causes of CBS. Tau PET may help distinguish AD from other tauopathies and non-tauopathies, but it remains challenging to differentiate non-AD tauopathies, especially PSP and CBD. Although the current clinical diagnostic criteria for CBS have suboptimal sensitivity and specificity, emerging biomarkers hold promise for future improvements in the diagnosis of underlying pathology in patients with CBS.
Keywords: corticobasal degeneration, alzheimer's disease, supranuclear palsy, neuropathology, genetics
Introduction
Corticobasal degeneration (CBD) is a progressive neurodegenerative tauopathy presenting with asymmetric rigidity, apraxia, dystonia, myoclonus, cortical sensory loss and dystonia, as well as behavioural and cognitive impairment such as aphasia.1 It has been increasingly recognised that other disorders, such as progressive supranuclear palsy (PSP) and Alzheimer’s disease (AD), can present with characteristic features that mimic CBD. Boeve et al investigated the postmortem neuropathology of 13 patients who were thought to have CBD based on clinical criteria and found that only 7 patients (54%) had neuropathological findings of CBD (figure 1).2 Other pathological diagnoses were AD in two patients, PSP, Pick’s disease, Creutzfeldt-Jakob disease (CJD) and non-specific degenerative changes. They considered that characteristic clinical features were explained by the topographical distribution of lesions, regardless of the underlying pathology. Subsequently, Cordato et al coined the term ‘corticobasal syndrome’ (CBS) to refer to the shared clinical features of six patients, four of whom at postmortem evaluation had CBD; the other two had PSP.3 Subsequently, Boeve et al proposed that CBD be limited to the neuropathological disorder, while CBS should be used to refer to the clinical syndrome of progressive asymmetric rigidity and apraxia.4
Figure 1.

Frequency of the common underlying pathologies associated with CBS. The pie charts (not drawn to scale) on the upper row are from review of the literature.2 11 12 The pie charts on the lower row are from unpublished data from the brain bank at the Institute for Medical Science of Aging, Aichi Medical University (1994–2020) and the Mayo Clinic brain bank (1998–2021). CBD is the most frequent underlying pathology, followed by PSP and AD. AD, Alzheimer’s disease; CBD, corticobasal degeneration; CBS, corticobasal syndrome; FTLD-TDP, frontotemporal lobar degeneration with TDP-43 inclusions; PSP, progressive supranuclear palsy.
Diagnostic criteria for CBS
Various diagnostic criteria for CBS have been proposed.4–8 Progressive, asymmetric akinetic rigid syndrome with apraxia is a feature in all criteria, but cognitive impairment, including aphasia, is included in some criteria (figure 2). The Mayo Clinic criteria include apraxia of speech/non-fluent aphasia as one of the core features, while frontal dysfunction was considered a supportive feature.4 The University of Toronto criteria do not include cognitive or language impairment.5 The modified Cambridge criteria have greater emphasis on cognitive and language dysfunction, including aphasia, executive dysfunction and visuospatial dysfunction.6 Criteria proposed by a group of international experts, including data from unpublished autopsy series, were reported by Armstrong et al.7 In these criteria, four clinical phenotypes of CBD are proposed: CBS, PSP syndrome, frontal behavioral-spatial syndrome and non-fluent/agrammatic variant of primary progressive aphasia. CBS is further classified as probable or possible. Probable CBS requires asymmetric motor signs, while possible CBS could have symmetric motor signs. Based on the combination of clinical phenotypes and other features, two degrees of certainty are defined in the Armstrong criteria—clinically probable CBD and clinically possible CBD. The former aimed to identify CBD without significant other pathology, while the latter would include cases that might have other (ie, mixed) pathology. Although it was recognised that some patients with autopsy-proven CBD can present with AD-type dementia,9 this clinical phenotype was excluded from the criteria. The significant advance in the Armstrong criteria is that it includes clinical phenotypes other than CBS. Subsequent clinicopathological studies have shown that the sensitivity and specificity of the Armstrong criteria are suboptimal.10
Figure 2.

Comparison of diagnostic criteria for CBS. Five commonly used criteria for CBS are compared. The motor symptoms and signs are in common in CBS, but the cognitive and language impairment are differently included between Mayo Clinic criteria, University of Toronto criteria and the modified Cambridge criteria.4–6 Armstrong criteria define four clinical presentations in CBD.7 The CBS of the modified Cambridge criteria roughly corresponds to CBS, PNFA and FBS of the Armstrong criteria. The MDS diagnostic criteria for PSP define PSP-CBS, which partially overlaps PSP syndrome of Armstrong criteria, although dystonia is not included in the criteria for PSP-CBS.8 Both Armstrong and MDS criteria list exclusion criteria, which suggest other underlying pathology. CBS, corticobasal syndrome; FBS, frontal behavioral-spatial syndrome; MDS, International Parkinson and Movement Disorder Society; PNFA, progressive non-fluent aphasia; PSPS, progressive supranuclear palsy syndrome.
More recently, the International Parkinson and Movement Disorder Society (MDS) proposed diagnostic criteria for PSP, which include eight clinical phenotypes for PSP, including CBS (possible PSP-CBS).8 This clinical phenotype is considered a probable 4-repeat (4R)-tauopathy, which has a high likelihood of underlying PSP or CBD pathology. The MDS criteria include AD-related biomarkers to exclude primary AD pathology from PSP-CBS, but they did not adopt tau positron emission tomography (PET) to detect PSP pathology. With development of molecular biomarkers for amyloid-β and tau, it will be necessary to incorporate these into diagnostic criteria to achieve optimal sensitivity and specificity.
Neuropathology of CBS
Heterogeneity of the underlying neuropathology associated with CBS has been reported in several autopsy series.2–4 11 12 Figure 1 summarises the frequency of the underlying pathology of CBS reported from the USA, UK and Japan, including unpublished data from two brain banks at the Institute for Medical Science of Aging, Aichi Medical University (1994–2020) and Mayo Clinic Florida (1998–2021). CBD accounted for less than half of patients with CBS, while PSP and AD were the most common non-CBD pathological diagnoses.11 12 Other neuropathological findings in patients with CBS were Lewy body disease (LBD),13 frontotemporal lobar degeneration (FTLD) with TDP-43 inclusions (FTLD-TDP),14 motor neuron disease,15 FTLD with fused-in-sarcoma pathology (FTLD-FUS),16 cerebrovascular disease,17 CJD18 and atypical multiple system atrophy (MSA).19
As of November 2021, the Mayo Clinic brain bank for neurodegenerative disorders has 345 brains from patients with an antemortem diagnosis of CBS. This clinical diagnosis is based on the final evaluation by clinicians who saw the patient later in the disease course. Rigorous clinical diagnostic criteria were not always applied, and not all patients were longitudinally followed by movement disorders specialists; therefore, the certainty of clinical diagnoses varies. The most common pathological diagnoses were CBD (32%), followed by PSP (31%), AD (20%) and others (17%), including LBD, FTLD-TDP, motor neuron disease, Pick’s disease, FTLD-FUS, CJD and cerebrovascular disease (figure 1).
In the following section, we will discuss clinicopathological features and genetic correlates of the disorders that can present with CBS, including tauopathies, synucleinopathies, TDP-43 proteinopathies, FUS proteinopathy, CJD and cerebrovascular disease (figure 3).20
Figure 3.

Genetic–pathological–clinical correlations in CBS. Font size reflects the frequency of each mutation or pathological diagnosis. Note that the majority of pathologies are sporadic; only a small subset of cases has gene mutations indicated. AD, Alzheimer’s disease; CBD, corticobasal degeneration; CBS, corticobasal syndrome; CJD, Creutzfeldt-Jakob disease; FTLD-FUS, frontotemporal lobar degeneration with fused-in-sarcoma pathology; FTLD-TDP, frontotemporal lobar degeneration with TDP-43 inclusions; LBD, Lewy body disease; PSP, progressive supranuclear palsy.
Tauopathies
Tauopathy, a collective term for neurodegenerative disorders characterised by accumulation of hyperphosphorylated tau proteins in neurons and glia,21 is the most common pathology associated with CBS. Tau protein, encoded by the MAPT gene, has six tau isoforms in adult brains. Alternative splicing of exon 10 in MAPT gene gives rise to 3-repeat (3R) or 4R tau.22 The tau isoforms in CBD, PSP and globular glial tauopathy (GGT) are primarily composed of 4R tau, hence these disorders are categorised as 4R tauopathies. Pick’s disease is a 3R tauopathy. All six tau isoforms are found in AD; thus, AD is a mixed 3R/4R tauopathy.
Corticobasal degeneration
CBD is characterised by tau aggregates in neurons and glia in both cortical and subcortical regions.23 Macroscopically, atrophy in the medial surface of the superior frontal gyrus, thinning of the corpus callosum, subcortical white matter atrophy and discolouration of the globus pallidus are typical findings in patients with CBS (figure 4A, B). Atrophy may be asymmetric, but the lack of marked asymmetry does not rule out CBD.24 The subthalamic nucleus may have atrophy, but it is less affected than in PSP. A variable degree of loss of pigment in the substantia nigra is observed. In contrast, loss of pigment in the locus coeruleus is less consistent, which differs from LBD and AD. On microscopic examination, spongiosis in the superficial layers and balloon neurons in the lower cortical layers are often seen in affected neocortices. Rarefaction of subcortical white matter, including U-fibres, is a characteristic feature of CBD. The pathognomic features of CBD are astrocytic plaques in grey matter (figure 4C) and numerous threads in both grey and white matter (figure 4D). Pretangles are more prominent than mature neurofibrillary tangles, which is different from tau pathology in AD and PSP (figure 4E). Astrocytic plaques are usually frequent in the neostriatum, although neuronal loss is relatively mild in this region compared with the globus pallidus and substantia nigra.
Figure 4.

Macroscopic and microscopic findings of patients with CBS and underlying pathology of CBD (A–E), PSP (F–J), and AD (K–O). Microscopic images show immunohistochemistry for phosphorylated-tau (C–E, H–J, N–O) and amyloid-β (panel M). In CBD, the characteristic findings include astrocytic plaques (C), numerous tau-positive threads in the white matter (D) and pretangles (E). In PSP, the characteristic findings are tufted astrocytes (H), oligodendroglial coiled bodies (I), and globose neurofibrillary tangles (J). In AD, the characteristic findings with tau immunohistochemistry are neuritic plaques (N) and neurofibrillary tangles (O), as well as amyloid-β positive amyloid plaques (M). Gross patterns of brain atrophy (A, B, F, G, K, L) are not specific, but show frontal lobe atrophy and enlargement of the frontal horn of the lateral ventricle in cases with CBS. AD, Alzheimer’s disease; CBD, corticobasal degeneration; PSP, progressive supranuclear palsy.
Pathologically, CBD can be divided into three subtypes on the basis of the distribution and severity of the lesions: typical CBD, ‘basal ganglia predominant CBD’ and ‘PSP-like CBD’.25 26 These subtypes correspond well to clinical phenotypes. Typical CBD is associated with CBS or frontal behavioral-spatial syndrome. ‘Basal ganglia predominant’ and ‘PSP-like’ CBD are associated with PSP syndrome. Another study reported that CBD-CBS had a greater tau burden in the frontal and parietal cortices, including the motor cortex, than CBD presenting as PSP (ie, Richardson syndrome—RS), CBD-RS.27 In contrast, tau burden in the cerebellum and medulla was greater in CBD-RS than in CBD-CBS. These findings suggest that the distribution of tau pathology differs between the two major clinical presentations of CBD (ie, CBS and RS). The presence of additional pathological processes may also affect clinical phenotypes. For example, TDP-43 pathology, which occurs commonly in elderly individuals,28 is more frequent in CBD-RS than CBD-CBS with the midbrain tegmentum being most commonly affected,29 although there was not a clear difference in age between the two groups. Another study demonstrated the correlation between the severity of TDP-43 pathology and neuronal loss in CBD, although the association with the clinical syndrome was not confirmed.30
Progressive supranuclear palsy
PSP is also a 4R tauopathy characterised by neuronal and glial tau aggregates predominantly in the subcortical regions. In typical PSP, macroscopic findings of the brain include atrophy of the subthalamic nucleus, midbrain, superior cerebellar peduncle and dentate nucleus. Most PSP patients had at least mild to moderate loss of pigment in substantia nigra. In PSP presenting as CBS (PSP-CBS), macroscopic findings are similar to those of CBD-CBS; namely, atrophy in the superior frontal gyrus, thinning of the corpus callosum and relative preservation of the subthalamic nucleus (figure 4F, G). In contrast to astrocytic plaques in CBD, the hallmark pathological glial lesion in PSP is the tufted astrocyte (figure 4H). Coiled bodies (figure 4I) and globose tangles (figure 4J) are also more frequent in PSP than in CBD.
Several clinical phenotypes of autopsy-confirmed PSP have been described. The most recent criteria proposed by the MDS define eight major clinical phenotypes.8 In addition to RS, which is the typical phenotype characterised by postural instability with early falls and supranuclear gaze palsy, seven atypical clinical phenotypes are proposed. Although PSP is the second most common underlying pathology of CBS, CBS is a relatively rare clinical presentation of PSP—in the range of 4%–9%.25 31 32
As in CBD, differences in clinical phenotypes in PSP can be explained by relative distributions of neurodegeneration and tau pathology. Compared with PSP-RS, PSP-CBS has greater tau burden in the middle frontal and inferior parietal cortices.33 A subsequent study validated this finding, showing increased tau burden in the frontal grey matter and parietal white matter as well as reduced tau burden in the caudate nucleus, subthalamic nucleus and cerebellar white matter in PSP-CBS.32 A recent study using cryogenic electron microscopy has revealed distinct ultrastructure characteristics of tau filaments in various tauopathies, such as PSP, CBD, GGT and Pick’s disease.34 Interestingly, however, tau filaments from different PSP subtypes (ie, PSP-RS, PSP-CBS, PSP with predominant parkinsonism and PSP with predominant frontal presentation) were identical, suggesting that distinct clinical phenotypes were not due to different tau filament structure, but rather distribution of the pathology.
Ling et al studied clinical features of CBS with various underlying pathologies and found that the mean duration between onset of vertical supranuclear gaze palsy and the first cardinal symptom was shorter in PSP-CBS than CBD-CBS (2.2 vs 5.9 years). Apraxia of eyelid opening was observed in 3 of 6 PSP-CBS patients, but in none of the patients with CBD-CBS.11 Lee et al also compared clinical features of CBS with various underlying pathologies, but they could not find any clinical features that were significantly different between the pathological groups.12
Both CBD and PSP share genetic risk factors, such as MAPT H1 haplotype. Interestingly, the frequency of homozygous MAPT H1c alleles, a risk subhaplotype of both CBD and PSP, was higher in PSP-CBS than in CBD-CBS.35 Another study, however, did not find strong associations between MAPT H1c and clinicopathological features of CBD.36
Alzheimer’s disease
Since genes that cause familial AD, such as amyloid precursor protein (APP)37 and presenilin (PSEN),38 drive amyloid deposits in the brain, not tau pathology, AD is considered to be a secondary tauopathy.39 AD can be divided into at least three neuropathological subtypes based on the relative distribution of neurofibrillary tangles in the hippocampus and neocortices: typical, hippocampal-sparing and limbic-predominant types.40 Using tau PET imaging, a fourth subtype—asymmetrical AD—has recently been reported.41 Hippocampal sparing AD is associated with young age of onset and high frequency of non-amnestic clinical syndrome, such as logopenic variant of primary progressive aphasia, posterior cortical syndrome and CBS.40 Macroscopic findings of AD associated with CBS are characterised by focal atrophy in the motor cortex and superior frontal gyrus (figure 4K, L). Pathological hallmarks of AD are senile plaques composed of amyloid-β and neurofibrillary tangles composed of hyperphosphorylated tau protein (figure 4M–O). One study reported that 3.5% of AD patients presented as CBS (AD-CBS).42 AD-CBS had greater atrophy in the motor cortex and more neuronal loss in the substantia nigra compared with amnestic type AD. Tau burden in the motor cortex was greater in AD-CBS than in amnestic type AD.
Several studies have investigated clinical features that differentiate CBD from AD by comparing clinical features of AD-CBS and CBD-CBS.12 42–44 The age of onset tends to be younger in AD-CBS than CBD-CBS.43 Data from the Mayo Clinic brain bank supports this finding; the age at onset is significantly younger in AD-CBS than CBD-CBS or PSP-CBS (59 vs 63 vs 70 years; p<0.001). Myoclonus was more frequent in AD-CBS than CBD-CBS in one study,43 but others did not replicate this finding.12 42 44 Tremor was more frequent in CBD-CBS than in AD-CBS in one study, but another study did not show a significant difference. Only one study compared the frequency of visual neglect, which was more common in AD-CBS.12 It is challenging to predict the underlying pathology of CBS based solely on clinical features, but molecular biomarkers may help differentiate AD from other aetiologies (see below). In rare cases, familial CBS is caused by mutations associated with early-onset AD, such as PSEN1 and APP.45–48
Pick’s disease
Pick’s disease is a 3R tauopathy often characterised by severe circumscribed atrophy (‘knife-edge’ atrophy) in the frontal and temporal lobes.49 Prominent neuronal loss with gliosis is observed in affected cortices, and some of the remaining neurons are swollen and chromatolytic, so-called Pick cells. Immunohistochemistry for phosphorylated-tau or 3R-tau shows highly characteristic round neuronal cytoplasmic inclusions (Pick bodies). Pick bodies are often numerous in the dentate gyrus of the hippocampus, as well as the amygdala, frontal and temporal neocortices, and subcortical nuclei, especially the corpus striatum. The most common clinical presentation of Pick’s disease is behavioural variant frontotemporal dementia (bvFTD), but progressive non-fluent aphasia and semantic dementia are also reported.50 51 A few case reports have described patients presenting as CBS with underlying pathology of Pick’s disease with52 or without53–55 concomitant AD pathology. One of these patients showed severe focal atrophy of the frontal and parietal lobes, especially around the left central sulcus, while anterior temporal cortices showed relatively mild atrophy.54 In a recent study, 5 of 21 patients with Pick’s disease (24%) had a clinical diagnosis of CBS, suggesting that CBS may not be a rare presentation of Pick’s disease.56 Further clinicopathological studies are warranted to determine whether sparing of temporal cortices is a pattern that correlates with the clinical presentation of CBS.
Globular glial tauopathy
GGT is an extremely rare 4R tauopathy characterised by 4R-tau-positive globular inclusions in oligodendrocytes and astrocytes.57 The distribution of tau pathology divides GGT into three pathological subtypes. Type I involves the prefrontal and temporal cortices; type II involves both the motor cortex and the corticospinal tracts; and type III involves the prefrontal, temporal, and motor cortices, and the corticospinal tracts. In a clinicopathological study of 11 GGT patients, the most common clinical diagnosis was bvFTD.58 Only one patient had CBS with no obvious pyramidal signs.58 Clinical diagnostic criteria for GGT are not established; therefore, the diagnosis of GGT can be made only by pathological assessment. In this sense, it is currently impossible to predict GGT pathology in patients with CBS.
MAPT mutations
To date, more than 70 MAPT mutations or variants have been reported to cause various types of tau pathologies, including CBD, Pick’s disease, GGT and ‘unclassifiable’ tauopathies.59 60 Each tauopathy is heterogeneous in clinical presentations and can present as CBS. Common clinical presentations in MAPT mutations are parkinsonism and dementia; hence, the name FTD and parkinsonism linked to chromosome 17.61 Many cases are familial, but seemingly sporadic patients have been described with MAPT mutations.
MAPT p.P301S mutation was first reported in a family with early-onset FTD with parkinsonism,62 but later reported to cause early-onset CBS (ie, 20s and 30s years old) in several patients.63 64 None of the patients with CBS due to this mutation had autopsy confirmation. MAPT p.P301L mutation was also initially reported in several families with FTD with parkinsonism.65 66 One study reported a patient with CBS, but lacked autopsy confirmation.67 MAPT p.V363I mutation has been reported to cause bvFTD, non-fluent PPA, posterior cortical atrophy and rarely CBS.68–70 Two patients with MAPT p.V363I mutation were identified by screening of 173 CBS patients for tau mutaitons.71 Pathological assessment was performed in one patient, which confirmed CBD-like pathology.71 MAPT p.N410H mutation was reported in a patient with a mixture of CBS and PSP syndrome, whose pathology was CBD.72 A patient with MAPT p.G389R presented with progressive aphasia at 38 years of age and later developed CBS. This patient had severe frontotemporal atrophy with Pick body-like inclusions in the cortices and tau-positive threads in the white matter, particularly numerous in the temporal lobe.73 A recent study reported five patients with MAPT p.P301T mutation from two pedigrees: the clinical syndrome was CBS in three patients with age of onset in the 40s in two, FTD in one patient, and primary lateral sclerosis in one patient.74 All but one CBS patient had autopsies and confirmation of neuropathological features consistent with GGT type II.
Anti-IgLON5 disease
Anti-IgLON5 disease is a recently proposed tauopathy characterised by autoantibodies against the neural cell adhesion protein IgLON5 and deposition of both 3R and 4R tau in neurons in the hypothalamus and the tegmental nuclei of the brainstem.75 The cardinal clinical features are non-REM sleep parasomnias, bulbar dysfunction, movement disorders, oculomotor abnormalities and cognitive impairment.75 76 Since this is thought to be an immune-mediated secondary tauopathy, it may be a treatable form of CBS using immunotherapeutic approaches.77
TDP-43 proteinopathy
TDP-43 is a pathognomonic protein in amyotrophic lateral sclerosis and a subset of FTLD (ie, FTLD-TDP).78 79 TDP-43 is a DNA/RNA binding protein located in the nucleus in the physiological state, but forming aggregates of hyper-phosphorylated and ubiquitinated TDP-43 protein in the pathological state. Based on the topographical and cellular distribution of TDP-43 inclusions, FTLD-TDP can be divided into five pathological subtypes.80 81 Of those, types A, B and C are frequent. Type A is characterised by numerous neuronal cytoplasmic inclusions and short dystrophic neurites, predominantly in the upper layers of the cortex. The common clinical phenotypes of Type A are bvFTD and progressive non-fluent aphasia. GRN mutations are most often associated with this subtype. Type B is characterised by neuronal cytoplasmic inclusions and sparse dystrophic neurites in all layers in the neocortex. Common clinical phenotypes are bvFTD and motor neuron disease with FTD, and the most common genetic cause is C9orf72 hexanucleotide repeat expansion.82 83 Type C is characterised by many long dystrophic neurites and few neuronal cytoplasmic inclusions predominantly in layer 2 of the neocortex, as well as Pick body-like inclusions in the dentate gyrus and neostriatum.84 Semantic dementia and bvFTD are common clinical presentations of Type C. No genetic association is known for this subtype.
CBS has been reported as a phenotype of FTLD-TDP.11 12 14 85 86 In an early study, Masellis et al reported a GRN mutation in familial CBS patients.87 This study predated the discovery of TDP-43 protein as the major constituent of neuronal cytoplasmic inclusions in non-tau FTLD. Benussi et al investigated the frequency of GRN mutations in FTLD and CBS, and found GRN mutations in three of four (75%) familial CBS and one of nine (11%) sporadic CBS patients.88 Le Ber et al reported that 3% (1/30) of CBS patients had GRN mutations.89 Although these studies lacked pathological assessment, it is reasonable to assume that TDP-43 pathology is the underlying pathology in these patients. Autopsy findings of patients with CBS due to GRN mutations have also been reported. Both p.A9D and p.Serine301CysfsX61 mutations had TDP-43 pathology in the frontal cortex consistent with FTLD-TDP type A.90 91 A neuroimaging study by Whitwell et al included five sporadic patients with CBS and FTLD-TDP type A; one of them had GRN mutation.86 Several studies reported FTLD-TDP due to C9orf72 hexanucleotide repeat expansion is rarely associated with CBS.92–94 To date, GRN mutations are the most common cause of familial CBS.95 Of three patients with familial CBS with known genetic mutations in the Mayo Clinic brain bank, two had GRN mutations and one had a PSEN1 mutation.
Sporadic CBS associated with FTLD-TDP type A has also been reported.14 In a cohort of 38 patients, 3 of 30 patients with FTLD-TDP type A had CBS, while none of 8 patients with FTLD-TDP type C had CBS.85 A study comparing brain atrophy on MRI across patients with CBS, FTLD-TDP showed more significant volume loss in the prefrontal cortex and posterior temporal lobe in the dominant hemisphere than all the other pathologies (ie, AD, CBD and PSP).86
FUS proteinopathy
FTLD-FUS is a rare subtype of FTLD compared with both FTLD-TDP and FTLD-tau.96 Like TDP-43, FUS exists in the nucleus under the physiological conditions and relocates to the cytoplasm and forms insoluble aggregates under the pathological conditions.97 FTLD-FUS can be classified into three clinicopathological subtypes: neuronal intermediate filament inclusion body disease (NIFID), basophilic inclusion body disease (BIBD) and atypical FTLD with ubiquitin-immunoreactive inclusions (aFTLD-U). NIFID is most commonly associated with early-onset bvFTD and less commonly CBS.98 99 BIBD is often associated with bvFTD and motor neuron disease,100–102 but CBS in patients with BIBD has also been reported.102 103 A recent study of NIFID and aFTLD-U from the Mayo Clinic brain bank reported that one of seven NIFID and one of eight aFTLD-U patients were clinically diagnosed as CBS.16
Synucleinopathy
Lewy body disease
LBD is an umbrella term for neurodegenerative disorders characterised by pathological inclusions composed of hyperphosphorylated α-synuclein in neuronal perikarya in the form of Lewy bodies and in neuronal processes as Lewy neurites.104 105 Common clinical presentations are Parkinson’s disease and dementia with Lewy bodies, but LBD can also present with focal cortical syndromes, including CBS.13 106 Although pathological findings of a few LBD patients presenting with CBS (LBD-CBS) have been reported,11 107 108 it is difficult to demonstrate a correlation between LBD pathology and CBS because most cases also had a variable degree of AD pathology. A study from Mayo Clinic reported that 11 of 532 (2%) patients with diffuse LBD (DLBD) had clinical presentation of probable CBS (DLBD-CBS).13 Of these cases, four patients had a primary pathological diagnosis of PSP, four had a primary pathological diagnosis of AD, and the remaining three patients had DLBD with mild-to-moderate Alzheimer’s type pathology consistent with intermediate likelihood AD; therefore, these concomitant pathologies are the likely pathological correlate of CBS.13 In neuropathological assessments, DLBD-CBS patients had greater atrophy in premotor and motor cortices, as well as corpus callosum than typical DLBD. The number of Lewy bodies and the severity of spongiform changes in the motor cortex were greater in DLBD-CBS than in typical DLBD. These findings indicate that the distribution of pathological lesions is associated with clinical phenotypes, similar to that observed in AD and tauopathies. Recently, Ichinose et al reported a patient with ‘pure’ DLBD who presented as CBS.109 This patient did not have amyloid-β, tau or TDP-43 pathologies; therefore, this case illustrates that the pure LBD can cause CBS.109 Autonomic dysfunction and responsiveness to L-DOPA may help distinguish LBD-CBS from other aetiologies of CBS.109
Multiple system atrophy
MSA is a synucleinopathy characterised by glial cytoplasmic inclusions in oligodendrocytes, composed of hyperphosphorylated α-synuclein, along with neurodegeneration in the striatonigral and olivopontocerebellar systems.110 111 Typical clinical presentations of MSA include variable combinations of autonomic dysfunction, parkinsonism and cerebellar ataxia.112 In a case series of atypical MSA presenting as FTLD, two patients presented as CBS with a disease duration of only 3 years.19 Neither patients had documented autonomic dysfunction that might have suggested clinically probable MSA. In addition to the widespread glial cytoplasmic inclusions with striatonigral degeneration, severe frontotemporal atrophy and abundant α-synuclein-positive neuronal cytoplasmic inclusions, including Pick body-like inclusions, were observed in limbic structures and the neocortex. A caveat of this report is that one of the patients also had moderate Alzheimer’s type pathology, which may have contributed to the CBS clinical presentation. The other patient, however, had only mild medial temporal neurofibrillary tangles without amyloid-β plaques consistent with primary age-related tauopathy,113 which has no known clinical significance. As with cases of LBD, concomitant AD pathology in MSA makes it difficult to demonstrate a causal relationship between MSA pathology and CBS.
Prion disease
There have been multiple reports of CJD presenting with CBS.2 18 114–116 In the Australian National CJD Registry, 7 of 387 sporadic CJD patients (1.8%) presented as CBS.18 As expected, the disease duration of CJD-CBS was significantly shorter than that of CBD-CBS (5 vs 68 months). Abnormal hyperintensity on diffusion-weighted images, the presence of 14-3-3 protein in cerebrospinal fluid (CSF), and periodic sharp wave complexes on electroencephalogram also suggested CJD. Detection of pathogenic seeding of prions by RT-QuIC using CSF is currently used to diagnose CJD.117 Based on these characteristic clinical and laboratory findings, CJD can be distinguished from other pathologies causing CBS. In rare cases, carriers of mutations in PRNP gene, encoding the prion protein, present as CBS.115 116
Cerebrovascular disease
Cerebrovascular disease can be associated with parkinsonism, and this syndrome is referred to as vascular parkinsonism.118 In rare cases, vascular parkinsonism can mimic atypical parkinsonian disorders, such as PSP or CBS.17 119 Several case reports and autopsy series have described patients with vascular CBS. In most cases, multiple infarcts in the neocortex and basal ganglia are detected with antemortem imaging studies, but a specific pattern of vascular pathology for CBS has not been defined.120–122 Furthermore, most of these studies lack pathological confirmation; thus, the presence of comorbid neurodegenerative changes cannot be excluded. A recent study from the Mayo Clinic brain bank identified 3 of 217 CBS patients with cerebrovascular pathology as a possible correlate of CBS.17 All three patients had small vessel disease in watershed regions, primary motor cortex, deep white matter, thalamus and basal ganglia, as well as secondary corticospinal tract degeneration without evidence of neurodegenerative pathology.
Mixed pathologies
Since ageing is the most important risk factor for neurodegenerative disorders, multiple neuropathological processes, such as AD neuropathological change, argyrophilic grain disease, TDP-43 pathology, Lewy body pathology and cerebrovascular disease occur in elderly individuals.35 123 124 AD neuropathological change (ie, amyloid plaques, neurofibrillary tangles and neuritic plaques)125 is the most important coexisting neuropathological process in CBS. For example, concurrent AD in PSP may modify clinical presentations, leading to a clinical diagnosis of CBS or dementia, rather than RS.126
Recent advances in molecular biomarkers have greatly improved the accuracy of the clinical diagnosis of AD (see below). It must be noted, however, that a clinical diagnosis of AD does not rule out other disease processes, such as CBD and PSP. A recent study from the Mayo Clinic brain bank reported that 86% of CBD and 89% of PSP had at least minimal AD neuropathological change (ie, Braak neurofibrillary tangle stage ≥0), and 6% of CBD (11/199) and 10% of PSP (97/1020) met the criteria of neuropathological diagnosis of AD (ie, high AD neuropathological change).126 Although a small subset, it should be recognised that high AD neuropathological change can coexist with CBD and PSP, particularly in elderly individuals.
In addition, one should be cautious in drawing correlations between primary pathological diagnoses and clinical presentations. LBD, for instance, almost always has some degree of AD neuropathological change127; therefore, it is difficult to weigh the relative contribution of LBD pathology and AD pathology to the clinical syndrome.13
Fluid and imaging biomarkers for CBS
As discussed above, various neurodegenerative disorders are associated with CBS, but clinical features are not sufficient to predict the underlying pathologies.44 Molecular biomarkers that can detect specific pathological proteins hold promise for improved antemortem diagnoses. Since several reliable biomarkers for AD have been established, a practical approach is first to differentiate AD from other disorders. Representative imaging of AD-CBS and CBD-CBS are provided in figure 5. In this section, CSF and blood biomarkers and PET using tracers for amyloid β, tau and α-synuclein are discussed.
Figure 5.
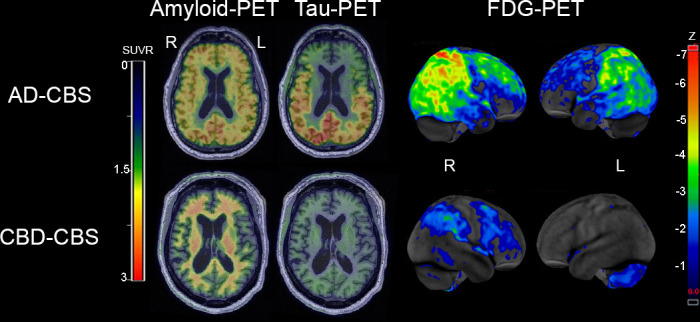
Representative amyloid PET with [11C]PiB, tau PET with [18F]flortaucipir and glucose PET with [18F]fluoro-2-deoxyglucose (FDG) in AD-CBS and CBD-CBS. The amyloid PET and tau PET are shown on representative axial slices on the same colour scale, while the FDG-PET highlights regional metabolism with z-scores between −1 and −7 SD from controls using CortexID suite software. Upper images are from a patient with AD-CBS. Amyloid PET shows diffuse ligand uptake in cortical grey matter, and tau PET reveals increased signal in the temporo-parieto-occipital lobes. FDG-PET reveals hypometabolism in bilateral parietal cortex extending into occipital lobes (right >left) and right >left superior frontal cortex. Lower images are from a patient with CBD-CBS. There is no evidence of increase amyloid PET uptake in the cortical grey matter and there is no increase tau PET uptake. FDG-PET reveals mild hypometabolism in the right premotor cortex and postcentral gyrus in CBD-CBS. AD-CBS, Alzheimer’s disease presenting as corticobasal syndrome; CBD-CBS, corticobasal degeneration presenting as corticobasal syndrome; FDG, fluorodeoxyglucose; L, left; PET, positron emission tomography; PIB, Pittsburgh compound-B; R, right; SUVR, standardised uptake value ratio.
CSF and blood biomarkers
Decreased Aβ42, elevated phosphorylated tau and elevated total tau in CSF have been established as AD-specific patterns, which are useful in the antemortem diagnosis of AD.128 Toledo et al analysed antemortem AD-related CSF biomarkers in 142 autopsy cases, including PSP and CBD and reported that the group with AD pathology could be differentiated from the group without AD pathology with a sensitivity of 96% and specificity of 87%.129 Therefore, the presence of AD pathology in CBS patients can be predicted by measuring AD-associated CSF biomarkers. AD can be ruled out if the biomarker profile is not consistent with AD. Even if the CSF biomarkers show AD pattern, interpretation needs to be made with caution; while AD pathology may be present, it does not exclude other pathologies.
The presence of phosphorylated tau and total tau in CSF have been investigated as tools to differentiate CBS from Parkinson’s disease and PSP. Both phosphorylated and total tau are elevated in CBS compared with PSP and Parkinson’s disease, but CSF tau has not been shown to differentiate CBS from PSP.130
Neurofilament light chain (NfL) has also been investigated as a potential CSF biomarker for neurodegenerative disorders. NfL is a low molecular weight constituent of neuronal intermediate filaments, and NfL in CSF is considered a marker for axonal damage that lacks disease specificity.131 CSF NfL levels have been reported to be elevated in CBS and PSP clinical syndromes compared with controls, but the levels do not differentiate PSP from CBS.132 Hansson et al used single molecule array, a technology that enables ultrasensitive protein detection in blood, to quantify plasma NfL.133 Plasma NfL levels were more elevated in MSA, PSP and CBS than Parkinson’s disease or controls, but the levels could not differentiate MSA, PSP and CBS. Another study also reported that serum NfL level was able to accurately distinguish Parkinson’s disease from a combined group of PSP and CBS.134 Taken together, NfL in CSF or blood may be a promising biomarker to differentiate atypical parkinsonian disorders from Parkinson’s disease, but it has not been proven to be useful in distinguishing CBD from PSP.
Amyloid PET
Amyloid PET has been developed as an imaging biomarker to visualise amyloid accumulation in vivo. Together with CSF biomarkers, its efficacy in the diagnosis of AD has been established.135 136 A meta-analysis of amyloid PET studies reported 23 of 61 (38%) patients with CBS showed amyloid positivity on PET.137 Interestingly, the frequency of amyloid positivity decreases with age. This finding may be explained by the fact that CBS is strongly associated with hippocampal sparing AD, which usually affects younger individuals compared with other subtypes of AD.40 AD may contribute to the underlying pathology in young CBS patients, whereas tauopathies and other aetiologies may predominate with increasing age.
As with the interpretation of CSF biomarkers, the possibility of coexisting pathologies other than AD (eg, CBD and PSP) cannot be excluded even in cases where amyloid PET is positive. Therefore, it is not possible to confirm AD as the primary pathology in cases with positive amyloid PET, and it may be necessary to make a diagnosis in combination with other biomarkers such as tau PET. On the other hand, if the amyloid PET is negative, the presence of AD pathology can be ruled out. Other underlying pathologies, especially CBD and PSP, would then be more likely.
Tau PET
Following the success of amyloid PET, tau protein became a target for in vivo molecular diagnosis. Several tau PET tracers have been developed in the last decade. Tau PET may help distinguish tauopathy-CBS from non-tauopathy-CBS and AD-CBS from non-AD tauopathies138 139; however, it remains challenging to differentiate non-AD-tauopathies, particularly CBD and PSP.
[18F]flortaucipir, formerly known as [18F]T807 and [18F]AV-1451,140 141 is one of the most widely used tau PET ligands. This tracer has excellent correlation between [18F]flortaucipir retention and distribution of neurofibrillary tangles in AD,142 143 but there have been inconsistent reports on its utility for non-AD tauopathies. One study assessed regional patterns of uptake on [18F]flortaucipir in 14 patients with CBS. Three of six amyloid PET-positive patients showed elevated [18F]flortaucipir uptake across many cortical regions, consistent with AD-CBS. Interestingly, amyloid PET-negative patients who initially presented with apraxia of speech showed elevated [18F]flortaucipir retention in the supplementary motor area and precentral cortex; however, patients who had CBS without apraxia of speech did not show significant retention.144 Another study investigated the retention of [18F]flortaucipir in eight patients with CBS; two had bilateral temporoparietal uptake consistent with AD, and six had retention in the motor cortex and subcortical white matter contralateral to the side of symptoms, presumably due to CBD. Again, the lack of autopsy confirmation is a limitation of this study. PSP-CBS may have similar retention patterns; therefore, the underlying pathology of these six patients remains unclear. In vitro autoradiography using brains from non-AD tauopathies has revealed less binding affinity of [F18F]AV-1451 to 4R tauopathies than to AD145 146; therefore, [18F]flortaucipir PET may not reliably detect non-AD tauopathies.147 148
[18F]PI-2620, an analogue of [18F]flortaucipir, has been developed as a ‘second generation’ tau PET tracer. It has a high affinity for pathological tau aggregates with low off-target binding towards amyloid, monoamine oxidase (MAO)-A and MAO-B.149 [18F]PI-2620 PET could differentiate patients with CBS from cognitively normal individuals without motor symptoms with high sensitivity and specificity. Positive [18F]PI-2620 retention was observed in 91% of amyloid-β-positive CBS and 65% of amyloid-β-negative CBS, while only 7% of control individuals showed positive [18F]PI-2620 retention (93% specificity).138 The putamen and external segment of the pallidum were the most common regions of retention in the CBS cohort.138 Cortical retention was highest and most frequent in amyloid-β-positive CBS compared with amyloid-β-negative CBS, with the dorsolateral prefrontal cortex being the most frequent cortical area with retention. This tracer also showed promising characteristics as an imaging agent for PSP in a non-clinical study.150 Patients with PSP-RS had the highest retention in the internal part of globus pallidus and had a more frequent elevation in the subthalamic nucleus and substantia nigra than controls.150 A recent study using autopsy-confirmed AD and non-AD tauopathies, however, revealed that [18F]PI-2620 retention was not correlated with tau burden at postmortem pathological assessment.151
[18F]PM-PBB3 is another second-generation tau PET tracer, which has great promise to visualise pathological tau aggregates in AD and non-AD tauopathies.139 The retention of [18F]PM-PBB3 was increased in the primary motor cortex, basal ganglia, brainstem and middle frontal gyrus in a patient with CBD confirmed by brain biopsy. In a patient with autopsy-confirmed PSP, [18F]PM-PBB3 retention was increased in the subthalamic nucleus and midbrain. Furthermore, increased retention of [18F]PM-PBB3 was observed in frontal and temporal cortices in a patient with autopsy-confirmed Pick’s disease. Although only one patient from each tauopathy was studied, retention of [18F]PM-PBB3 strongly suggests the possibility of imaging-pathology relationships at a single subject level.
A head-to-head comparison of [18F]PM-PBB3 and [18F]PI-2620 in a patient with PSP-CBS has been reported.151 Retention of [18F]PM-PBB3 was increased in the midbrain, subthalamic nucleus, thalamus, globus pallidus and precentral gyrus, consistent with the expected topographical distribution of tau pathology in PSP. In contrast, retention of [18F]PI-2620 showed a different pattern from that of [18F]PM-PBB3, with increased in retention in the globus pallidus and substantia nigra, but weak retention in the thalamus, frontal gyrus and subthalamic nucleus.151
THK-5351 PET visualises both tau deposits and MAO-B in vivo. Increased THK-5351 retention was greater in the pre-and post-central gyri and globus pallidus in patients with CBS than in AD and healthy individuals.152 The THK-5351 retention pattern was different among AD, PSP and CBS, but none of the patients had autopsy confirmation.153 The underlying pathology of CBS patients remained unclear; therefore, the usefulness of THK-5351 in differentiating the pathology of CBS, particularly CBD or PSP, needs to be elucidated.
α-Synuclein PET
PET tracers for α-synuclein are currently under development. Both in vitro and in vivo studies suggest that [11C]PBB3, a first-generation tau PET ligand,154 has some degree of binding affinity to α-synuclein.155 156 C05-01 is the first PBB3 analogue developed as a potential α-synuclein PET tracer. In vitro autoradiography using [3H]C05-01 showed specific binding to α-synuclein in Parkinson’s disease and MSA, but also to amyloid-β and tau pathology.157 More recently, another PBB3 analogue, C05–05, another promising α-synuclein PET tracer, detected α-synuclein in mouse and monkey models.158 Although the results of clinical studies in humans have not been reported, this modality will be useful for the early diagnosis of synucleinopathies, which account for a small subset of the underlying pathology of CBS.
Conclusions
CBS is a clinical syndrome caused by various underlying pathologies, with the most frequent pathologies at autopsy being CBD and PSP. At present, pathological assessment is required for definitive diagnosis, but in the future, it may be possible to predict the underlying pathology during life based on a combination of clinical symptoms and signs with supporting information from various biomarkers. Biomarker research is most reliable when it is based on neuropathologically confirmed cases. Accumulation of autopsy-confirmed cases is time-consuming, but extremely important. Clarification of the abnormal proteins that cause the disease is expected to lead to the development of disease-modifying therapies that target those proteins for treatment.20 For this purpose, it is necessary to conduct prospective cohort studies with pathological diagnosis to gather knowledge on the correlations between clinical symptoms/signs, biomarkers and pathological diagnosis.
Acknowledgments
We would like to thank the patients and their families who donated brains to Mayo Clinic brain bank to help further the scientific understanding of neurodegeneration.
Footnotes
Twitter: @shunsuke_koga, @dennis_w_d
Contributors: SK searched the literature, prepared tables and figures, and wrote the first draft. KAJ, MY and DWD provided the data. KAJ, IA, MY and DWD reviewed and edited the manuscript. All authors read and approved the final manuscript.
Funding: The Mayo Clinic brain bank is supported by Rainwater Charitable Foundation. SK receives funding from the Rainwater Charitable Foundation and CurePSP (672-2020-12). KAJ (RF1 NA112153 and R01 NS089757) and DWD (UG3 NS104095) receive funding from the National Institute of Neurological Disorders and Stroke.
Competing interests: None declared.
Provenance and peer review: Commissioned; externally peer reviewed.
Ethics statements
Patient consent for publication
Not applicable.
References
- 1. Rebeiz JJ, Kolodny EH, Richardson EP. Corticodentatonigral degeneration with neuronal achromasia. Arch Neurol 1968;18:20–33. 10.1001/archneur.1968.00470310034003 [DOI] [PubMed] [Google Scholar]
- 2. Boeve BF, Maraganore DM, Parisi JE, et al. Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology 1999;53:795–800. 10.1212/wnl.53.4.795 [DOI] [PubMed] [Google Scholar]
- 3. Cordato NJ, Halliday GM, McCann H, et al. Corticobasal syndrome with tau pathology. Mov Disord 2001;16:656–67. 10.1002/mds.1124 [DOI] [PubMed] [Google Scholar]
- 4. Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol 2003;54 Suppl 5:S15–19. 10.1002/ana.10570 [DOI] [PubMed] [Google Scholar]
- 5. Lang A, Riley DE, Bergeron C. Cortical-Basal ganglionic degeneration. neurodegenerative diseases. Philadelphia: WB Saunders, 1994: 877–94. [Google Scholar]
- 6. Mathew R, Bak TH, Hodges JR. Diagnostic criteria for corticobasal syndrome: a comparative study. J Neurol Neurosurg Psychiatry 2012;83:405–10. 10.1136/jnnp-2011-300875 [DOI] [PubMed] [Google Scholar]
- 7. Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013;80:496–503. 10.1212/WNL.0b013e31827f0fd1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the movement disorder Society criteria. Mov Disord 2017;32:853–64. 10.1002/mds.26987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Day GS, Lim TS, Hassenstab J, et al. Differentiating cognitive impairment due to corticobasal degeneration and Alzheimer disease. Neurology 2017;88:1273–81. 10.1212/WNL.0000000000003770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alexander SK, Rittman T, Xuereb JH, et al. Validation of the new consensus criteria for the diagnosis of corticobasal degeneration. J Neurol Neurosurg Psychiatry 2014;85:925–9. 10.1136/jnnp-2013-307035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ling H, O'Sullivan SS, Holton JL, et al. Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain 2010;133:2045–57. 10.1093/brain/awq123 [DOI] [PubMed] [Google Scholar]
- 12. Lee SE, Rabinovici GD, Mayo MC, et al. Clinicopathological correlations in corticobasal degeneration. Ann Neurol 2011;70:327–40. 10.1002/ana.22424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kasanuki K, Josephs KA, Ferman TJ, et al. Diffuse Lewy body disease manifesting as corticobasal syndrome: a rare form of Lewy body disease. Neurology 2018;91:e268–79. 10.1212/WNL.0000000000005828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tartaglia MC, Sidhu M, Laluz V, et al. Sporadic corticobasal syndrome due to FTLD-TDP. Acta Neuropathol 2010;119:365–74. 10.1007/s00401-009-0605-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Murakami A, Koga S, Dickson DW. Asymmetrical primary lateral sclerosis presenting as corticobasal syndrome. J Neuropathol Exp Neurol 2022;81:154–6. 10.1093/jnen/nlab104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bieniek KF, Josephs KA, Lin W-L, et al. Neuronal intermediate filament inclusion disease may be incorrectly classified as a subtype of FTLD-FUS. Free Neuropathol 2020;1:9. 10.17879/freeneuropathology-2020-2639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Koga S, Roemer SF, Kasanuki K, et al. Cerebrovascular pathology presenting as corticobasal syndrome: An autopsy case series of "vascular CBS". Parkinsonism Relat Disord 2019;68:79–84. 10.1016/j.parkreldis.2019.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee W, Simpson M, Ling H, et al. Characterising the uncommon corticobasal syndrome presentation of sporadic Creutzfeldt-Jakob disease. Parkinsonism Relat Disord 2013;19:81–5. 10.1016/j.parkreldis.2012.07.010 [DOI] [PubMed] [Google Scholar]
- 19. Aoki N, Boyer PJ, Lund C, et al. Atypical multiple system atrophy is a new subtype of frontotemporal lobar degeneration: frontotemporal lobar degeneration associated with α-synuclein. Acta Neuropathol 2015;130:93–105. 10.1007/s00401-015-1442-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aiba I. [Corticobasal syndrome: recent advances and future directions]. Brain Nerve 2012;64:462–73. [PubMed] [Google Scholar]
- 21. Spillantini MG, Goedert M. Tau protein pathology in neurodegenerative diseases. Trends Neurosci 1998;21:428–33. 10.1016/s0166-2236(98)01337-x [DOI] [PubMed] [Google Scholar]
- 22. Goedert M, Spillantini MG, Jakes R, et al. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron 1989;3:519–26. 10.1016/0896-6273(89)90210-9 [DOI] [PubMed] [Google Scholar]
- 23. Dickson DW, Bergeron C, Chin SS, et al. Office of rare diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol 2002;61:935–46. 10.1093/jnen/61.11.935 [DOI] [PubMed] [Google Scholar]
- 24. Hassan A, Whitwell JL, Boeve BF, et al. Symmetric corticobasal degeneration (S-CBD). Parkinsonism Relat Disord 2010;16:208–14. 10.1016/j.parkreldis.2009.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yoshida M. Astrocytic inclusions in progressive supranuclear palsy and corticobasal degeneration. Neuropathology 2014;34:555–70. 10.1111/neup.12143 [DOI] [PubMed] [Google Scholar]
- 26. Mimuro M, Yoshida M. Chameleons and mimics: progressive supranuclear palsy and corticobasal degeneration. Neuropathology 2020;40:57–67. 10.1111/neup.12590 [DOI] [PubMed] [Google Scholar]
- 27. Kouri N, Murray ME, Hassan A, et al. Neuropathological features of corticobasal degeneration presenting as corticobasal syndrome or Richardson syndrome. Brain 2011;134:3264–75. 10.1093/brain/awr234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nelson PT, Dickson DW, Trojanowski JQ, et al. Limbic-predominant age-related TDP-43 encephalopathy (late): consensus Working Group report. Brain 2019;142:1503–27. 10.1093/brain/awz099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Koga S, Kouri N, Walton RL, et al. Corticobasal degeneration with TDP-43 pathology presenting with progressive supranuclear palsy syndrome: a distinct clinicopathologic subtype. Acta Neuropathol 2018;136:389–404. 10.1007/s00401-018-1878-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sainouchi M, Tada M, Fitrah YA, et al. Brain TDP-43 pathology in corticobasal degeneration: topographical correlation with neuronal loss. Neuropathol Appl Neurobiol 2022;48:e12786. 10.1111/nan.12786 [DOI] [PubMed] [Google Scholar]
- 31. Respondek G, Stamelou M, Kurz C, et al. The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Mov Disord 2014;29:1758–66. 10.1002/mds.26054 [DOI] [PubMed] [Google Scholar]
- 32. Ling H, Ling H, de Silva R, et al. Characteristics of progressive supranuclear palsy presenting with corticobasal syndrome: a cortical variant. Neuropathol Appl Neurobiol 2014;40:149–63. 10.1111/nan.12037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tsuboi Y, Josephs KA, Boeve BF, et al. Increased tau burden in the cortices of progressive supranuclear palsy presenting with corticobasal syndrome. Mov Disord 2005;20:982–8. 10.1002/mds.20478 [DOI] [PubMed] [Google Scholar]
- 34. Shi Y, Zhang W, Yang Y, et al. Structure-Based classification of tauopathies. Nature 2021;598:359–63. 10.1038/s41586-021-03911-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Robinson JL, Yan N, Caswell C, et al. Primary tau pathology, not Copathology, correlates with clinical symptoms in PSP and CBD. J Neuropathol Exp Neurol 2020;79:296–304. 10.1093/jnen/nlz141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Valentino RR, Koga S, Walton RL, et al. Mapt subhaplotypes in corticobasal degeneration: assessing associations with disease risk, severity of tau pathology, and clinical features. Acta Neuropathol Commun 2020;8:218. 10.1186/s40478-020-01097-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kang J, Lemaire HG, Unterbeck A, et al. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987;325:733–6. 10.1038/325733a0 [DOI] [PubMed] [Google Scholar]
- 38. Sherrington R, Rogaev EI, Liang Y, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature 1995;375:754–60. 10.1038/375754a0 [DOI] [PubMed] [Google Scholar]
- 39. DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer's disease. Mol Neurodegener 2019;14:32. 10.1186/s13024-019-0333-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Murray ME, Graff-Radford NR, Ross OA, et al. Neuropathologically defined subtypes of Alzheimer's disease with distinct clinical characteristics: a retrospective study. Lancet Neurol 2011;10:785–96. 10.1016/S1474-4422(11)70156-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ossenkoppele R, Lyoo CH, Sudre CH, et al. Distinct tau PET patterns in atrophy-defined subtypes of Alzheimer's disease. Alzheimers Dement 2020;16:335–44. 10.1016/j.jalz.2019.08.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sakae N, Josephs KA, Litvan I, et al. Clinicopathologic subtype of Alzheimer's disease presenting as corticobasal syndrome. Alzheimers Dement 2019;15:1218–28. 10.1016/j.jalz.2019.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hu WT, Rippon GW, Boeve BF, et al. Alzheimer's disease and corticobasal degeneration presenting as corticobasal syndrome. Mov Disord 2009;24:1375–9. 10.1002/mds.22574 [DOI] [PubMed] [Google Scholar]
- 44. Shelley BP, Hodges JR, Kipps CM, et al. Is the pathology of corticobasal syndrome predictable in life? Mov Disord 2009;24:1593–9. 10.1002/mds.22558 [DOI] [PubMed] [Google Scholar]
- 45. Navarro E, De Andrés C, Guerrero C, et al. Corticobasal syndrome in a family with early-onset Alzheimer's disease linked to a presenilin-1 gene mutation. Mov Disord Clin Pract 2015;2:388–94. 10.1002/mdc3.12212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lam B, Khan A, Keith J, et al. Characterizing familial corticobasal syndrome due to Alzheimer's disease pathology and PSEN1 mutations. Alzheimers Dement 2017;13:520–30. 10.1016/j.jalz.2016.08.014 [DOI] [PubMed] [Google Scholar]
- 47. López-García S, Jiménez-Bonilla J, López Delgado A, et al. A rare PSEN1 (Leu85Pro) mutation causing Alzheimer's disease in a 29-year-old woman presenting as corticobasal syndrome. J Alzheimers Dis 2019;70:655–8. 10.3233/JAD-190107 [DOI] [PubMed] [Google Scholar]
- 48. Abate F, Dati G, Ginevrino M, et al. APP-Related Corticobasal Syndrome: Expanding the List of Corticobasal Degeneration Look Alikes. Mov Disord Clin Pract 2020;7:849–51. 10.1002/mdc3.13037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McKhann GM, Albert MS, Grossman M, et al. Clinical and pathological diagnosis of frontotemporal dementia: report of the work group on frontotemporal dementia and Pick's disease. Arch Neurol 2001;58:1803–9. 10.1001/archneur.58.11.1803 [DOI] [PubMed] [Google Scholar]
- 50. Hodges JR. Frontotemporal dementia (Pick's disease): clinical features and assessment. Neurology 2001;56:S6–10. 10.1212/WNL.56.suppl_4.S6 [DOI] [PubMed] [Google Scholar]
- 51. Graff-Radford NR, Damasio AR, Hyman BT, et al. Progressive aphasia in a patient with Pick's disease: a neuropsychological, radiologic, and anatomic study. Neurology 1990;40:620–6. 10.1212/wnl.40.4.620 [DOI] [PubMed] [Google Scholar]
- 52. Rusina R, Pazdera L, Kulišťák P, et al. Pick and Alzheimer diseases: a rare comorbidity presenting as corticobasal syndrome. Cogn Behav Neurol 2013;26:189–94. 10.1097/WNN.0000000000000011 [DOI] [PubMed] [Google Scholar]
- 53. Fukui T, Sugita K, Kawamura M, et al. Primary progressive apraxia in Pick's disease: a clinicopathologic study. Neurology 1996;47:467–73. 10.1212/wnl.47.2.467 [DOI] [PubMed] [Google Scholar]
- 54. Uchida Y, Yoshida M, Takada K, et al. Corticobasal syndrome-Pick's disease: a clinicopathological study. J Neurol Sci 2020;412:116752. 10.1016/j.jns.2020.116752 [DOI] [PubMed] [Google Scholar]
- 55. Lang AE, Bergeron C, Pollanen MS, et al. Parietal Pick's disease mimicking cortical-basal ganglionic degeneration. Neurology 1994;44:1436–40. 10.1212/wnl.44.8.1436 [DOI] [PubMed] [Google Scholar]
- 56. Koga S, Ikeda A, Dickson DW. Deep learning-based model for diagnosing Alzheimer's disease and tauopathies. Neuropathol Appl Neurobiol 2022;48:e12759. 10.1111/nan.12759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ahmed Z, Bigio EH, Budka H, et al. Globular glial tauopathies (GGT): consensus recommendations. Acta Neuropathol 2013;126:537–44. 10.1007/s00401-013-1171-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Burrell JR, Forrest S, Bak TH, et al. Expanding the phenotypic associations of globular glial tau subtypes. Alzheimers Dement 2016;4:6–13. 10.1016/j.dadm.2016.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Moore KM, Nicholas J, Grossman M, et al. Age at symptom onset and death and disease duration in genetic frontotemporal dementia: an international retrospective cohort study. Lancet Neurol 2020;19:145–56. 10.1016/S1474-4422(19)30394-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. FTDtalk . Mapt mutations. Available: https://www.ftdtalk.org/what-is-ftd/genetics/mapt-mutations/ [Accessed 06 Mar 2022].
- 61. Foster NL, Wilhelmsen K, Sima AA, et al. Frontotemporal dementia and parkinsonism linked to chromosome 17: a consensus conference. conference participants. Ann Neurol 1997;41:706–15. 10.1002/ana.410410606 [DOI] [PubMed] [Google Scholar]
- 62. Sperfeld AD, Collatz MB, Baier H, et al. FTDP-17: an early-onset phenotype with parkinsonism and epileptic seizures caused by a novel mutation. Ann Neurol 1999;46:708–15. 10.1002/1531-8249(199911)46:5<708::aid-ana5>3.0.co;2-k [DOI] [PubMed] [Google Scholar]
- 63. Bugiani O, Murrell JR, Giaccone G, et al. Frontotemporal dementia and corticobasal degeneration in a family with a P301S mutation in tau. J Neuropathol Exp Neurol 1999;58:667–77. 10.1097/00005072-199906000-00011 [DOI] [PubMed] [Google Scholar]
- 64. Casseron W, Azulay JP, Guedj E, et al. Familial autosomal dominant cortico-basal degeneration with the P301S mutation in the tau gene: an example of phenotype variability. J Neurol 2005;252:1546–8. 10.1007/s00415-005-0880-2 [DOI] [PubMed] [Google Scholar]
- 65. Dumanchin C, Camuzat A, Campion D, et al. Segregation of a missense mutation in the microtubule-associated protein tau gene with familial frontotemporal dementia and parkinsonism. Hum Mol Genet 1998;7:1825–9. 10.1093/hmg/7.11.1825 [DOI] [PubMed] [Google Scholar]
- 66. Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998;393:702–5. 10.1038/31508 [DOI] [PubMed] [Google Scholar]
- 67. Gatto EM, Allegri RF, Da Prat G, et al. Intrafamilial variable phenotype including corticobasal syndrome in a family with p.P301L mutation in the MAPT gene: first report in South America. Neurobiol Aging 2017;53:195.e11–195.e17. 10.1016/j.neurobiolaging.2017.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Munoz DG, Ros R, Fatas M, et al. Progressive nonfluent aphasia associated with a new mutation V363I in tau gene. Am J Alzheimers Dis Other Demen 2007;22:294–9. 10.1177/1533317507302320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Anfossi M, Bernardi L, Gallo M, et al. Mapt V363I variation in a sporadic case of frontotemporal dementia: variable penetrant mutation or rare polymorphism? Alzheimer Dis Assoc Disord 2011;25:96–9. 10.1097/WAD.0b013e3181eff860 [DOI] [PubMed] [Google Scholar]
- 70. Rossi G, Bastone A, Piccoli E, et al. Different mutations at V363 MAPT codon are associated with atypical clinical phenotypes and show unusual structural and functional features. Neurobiol Aging 2014;35:408–17. 10.1016/j.neurobiolaging.2013.08.004 [DOI] [PubMed] [Google Scholar]
- 71. Ahmed S, Fairen MD, Sabir MS, et al. MAPT p.V363I mutation: A rare cause of corticobasal degeneration. Neurol Genet 2019;5:e347. 10.1212/NXG.0000000000000347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kouri N, Carlomagno Y, Baker M, et al. Novel mutation in MAPT exon 13 (p.N410H) causes corticobasal degeneration. Acta Neuropathol 2014;127:271–82. 10.1007/s00401-013-1193-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Murrell JR, Spillantini MG, Zolo P, et al. Tau gene mutation G389R causes a tauopathy with abundant Pick body-like inclusions and axonal deposits. J Neuropathol Exp Neurol 1999;58:1207–26. 10.1097/00005072-199912000-00002 [DOI] [PubMed] [Google Scholar]
- 74. Erro ME, Zelaya MV, Mendioroz M, et al. Globular glial tauopathy caused by MAPT P301T mutation: clinical and neuropathological findings. J Neurol 2019;266:2396–405. 10.1007/s00415-019-09414-w [DOI] [PubMed] [Google Scholar]
- 75. Gelpi E, Höftberger R, Graus F, et al. Neuropathological criteria of anti-IgLON5-related tauopathy. Acta Neuropathol 2016;132:531–43. 10.1007/s00401-016-1591-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sabater L, Gaig C, Gelpi E, et al. A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: a case series, characterisation of the antigen, and post-mortem study. Lancet Neurol 2014;13:575–86. 10.1016/S1474-4422(14)70051-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fuseya K, Kimura A, Yoshikura N, et al. Corticobasal syndrome in a patient with anti-IgLON5 antibodies. Mov Disord Clin Pract 2020;7:557–9. 10.1002/mdc3.12957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Arai T, Hasegawa M, Akiyama H, et al. Tdp-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 2006;351:602–11. 10.1016/j.bbrc.2006.10.093 [DOI] [PubMed] [Google Scholar]
- 79. Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006;314:130–3. 10.1126/science.1134108 [DOI] [PubMed] [Google Scholar]
- 80. Mackenzie IRA, Neumann M, Baborie A, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 2011;122:111–3. 10.1007/s00401-011-0845-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lee EB, Porta S, Michael Baer G, et al. Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol 2017;134:65–78. 10.1007/s00401-017-1679-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9orf72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72:245–56. 10.1016/j.neuron.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9orf72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72:257–68. 10.1016/j.neuron.2011.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Mackenzie IR, Neumann M. Subcortical TDP-43 pathology patterns validate cortical FTLD-TDP subtypes and demonstrate unique aspects of C9orf72 mutation cases. Acta Neuropathol 2020;139:83–98. 10.1007/s00401-019-02070-4 [DOI] [PubMed] [Google Scholar]
- 85. Gefen T, Ahmadian SS, Mao Q, et al. Combined pathologies in FTLD-TDP types A and C. J Neuropathol Exp Neurol 2018;77:405–12. 10.1093/jnen/nly018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Whitwell JL, Jack CR, Boeve BF, et al. Imaging correlates of pathology in corticobasal syndrome. Neurology 2010;75:1879–87. 10.1212/WNL.0b013e3181feb2e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Masellis M, Momeni P, Meschino W, et al. Novel splicing mutation in the progranulin gene causing familial corticobasal syndrome. Brain 2006;129:3115–23. 10.1093/brain/awl276 [DOI] [PubMed] [Google Scholar]
- 88. Benussi L, Binetti G, Sina E, et al. A novel deletion in progranulin gene is associated with FTDP-17 and CBS. Neurobiol Aging 2008;29:427–35. 10.1016/j.neurobiolaging.2006.10.028 [DOI] [PubMed] [Google Scholar]
- 89. Le Ber I, Camuzat A, Hannequin D, et al. Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain 2008;131:732–46. 10.1093/brain/awn012 [DOI] [PubMed] [Google Scholar]
- 90. Spina S, Murrell JR, Huey ED, et al. Corticobasal syndrome associated with the A9D progranulin mutation. J Neuropathol Exp Neurol 2007;66:892–900. 10.1097/nen.0b013e3181567873 [DOI] [PubMed] [Google Scholar]
- 91. Guerreiro RJ, Santana I, Bras JM, et al. Novel progranulin mutation: screening for PGRN mutations in a Portuguese series of FTD/CBS cases. Mov Disord 2008;23:1269–73. 10.1002/mds.22078 [DOI] [PubMed] [Google Scholar]
- 92. Lindquist SG, Duno M, Batbayli M, et al. Corticobasal and ataxia syndromes widen the spectrum of C9orf72 hexanucleotide expansion disease. Clin Genet 2013;83:279–83. 10.1111/j.1399-0004.2012.01903.x [DOI] [PubMed] [Google Scholar]
- 93. Anor CJ, Xi Z, Zhang M, et al. Mutation analysis of C9orf72 in patients with corticobasal syndrome. Neurobiol Aging 2015;36:2905 e2901–5. 10.1016/j.neurobiolaging.2015.06.008 [DOI] [PubMed] [Google Scholar]
- 94. Hokelekli FO, Whitwell JL, Machulda MM, et al. Underlying pathology identified after 20 years of disease course in two cases of slowly progressive frontotemporal dementia syndromes. Neurocase 2021;27:212–22. 10.1080/13554794.2021.1918723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Arienti F, Lazzeri G, Vizziello M, et al. Unravelling genetic factors underlying corticobasal syndrome: a systematic review. Cells 2021;10:171. 10.3390/cells10010171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Neumann M, Rademakers R, Roeber S, et al. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 2009;132:2922–31. 10.1093/brain/awp214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kwiatkowski TJ, Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009;323:1205–8. 10.1126/science.1166066 [DOI] [PubMed] [Google Scholar]
- 98. Josephs KA, Holton JL, Rossor MN, et al. Neurofilament inclusion body disease: a new proteinopathy? Brain 2003;126:2291–303. 10.1093/brain/awg231 [DOI] [PubMed] [Google Scholar]
- 99. Cairns NJ, Grossman M, Arnold SE, et al. Clinical and neuropathologic variation in neuronal intermediate filament inclusion disease. Neurology 2004;63:1376–84. 10.1212/01.wnl.0000139809.16817.dd [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Yokota O, Tsuchiya K, Terada S, et al. Basophilic inclusion body disease and neuronal intermediate filament inclusion disease: a comparative clinicopathological study. Acta Neuropathol 2008;115:561–75. 10.1007/s00401-007-0329-z [DOI] [PubMed] [Google Scholar]
- 101. Munoz DG, Neumann M, Kusaka H, et al. FUS pathology in basophilic inclusion body disease. Acta Neuropathol 2009;118:617–27. 10.1007/s00401-009-0598-9 [DOI] [PubMed] [Google Scholar]
- 102. Josephs KA, Lin W-L, Ahmed Z, et al. Frontotemporal lobar degeneration with ubiquitin-positive, but TDP-43-negative inclusions. Acta Neuropathol 2008;116:159–67. 10.1007/s00401-008-0397-8 [DOI] [PubMed] [Google Scholar]
- 103. Matsumoto A, Suzuki H, Fukatsu R, et al. An autopsy case of frontotemporal lobar degeneration with the appearance of fused in sarcoma inclusions (basophilic inclusion body disease) clinically presenting corticobasal syndrome. Neuropathology 2016;36:77–87. 10.1111/neup.12232 [DOI] [PubMed] [Google Scholar]
- 104. Kosaka K, Matsushita M, Oyanagi S, et al. [A cliniconeurophathological study of the "Lewy body disease"]. Seishin Shinkeigaku Zasshi 1980;82:292–311. [PubMed] [Google Scholar]
- 105. Spillantini MG, Schmidt ML, Lee VM, et al. Alpha-Synuclein in Lewy bodies. Nature 1997;388:839–40. 10.1038/42166 [DOI] [PubMed] [Google Scholar]
- 106. Koga S, Sekiya H, Kondru N, et al. Neuropathology and molecular diagnosis of synucleinopathies. Mol Neurodegener 2021;16:83. 10.1186/s13024-021-00501-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Horoupian DS, Wasserstein PH. Alzheimer's disease pathology in motor cortex in dementia with Lewy bodies clinically mimicking corticobasal degeneration. Acta Neuropathol 1999;98:317–22. 10.1007/s004010051087 [DOI] [PubMed] [Google Scholar]
- 108. Haug A, Boyer P, Kluger B. Diffuse Lewy body disease presenting as corticobasal syndrome and progressive supranuclear palsy syndrome. Mov Disord 2013;28:1153–5. 10.1002/mds.25368 [DOI] [PubMed] [Google Scholar]
- 109. Ichinose K, Watanabe M, Mizutani S, et al. An autopsy case of corticobasal syndrome with pure diffuse Lewy body disease. Neurocase 2021;27:231–7. 10.1080/13554794.2021.1921220 [DOI] [PubMed] [Google Scholar]
- 110. Trojanowski JQ, Revesz T, Neuropathology Working Group on MSA . Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol Appl Neurobiol 2007;33:615–20. 10.1111/j.1365-2990.2007.00907.x [DOI] [PubMed] [Google Scholar]
- 111. Koga S, Dickson DW. Recent advances in neuropathology, biomarkers and therapeutic approach of multiple system atrophy. J Neurol Neurosurg Psychiatry 2018;89:175–84. 10.1136/jnnp-2017-315813 [DOI] [PubMed] [Google Scholar]
- 112. Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670–6. 10.1212/01.wnl.0000324625.00404.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age-related tauopathy (part): a common pathology associated with human aging. Acta Neuropathol 2014;128:755–66. 10.1007/s00401-014-1349-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Arnao V, Gangitano M, Giacalone F, et al. Corticobasal syndrome-like variant of Creutzfeldt-Jakob disease: clinical description of two cases. Neurol Sci 2015;36:1303–5. 10.1007/s10072-014-2043-7 [DOI] [PubMed] [Google Scholar]
- 115. Lim JG, Oh E, Park S, et al. Familial Creutzfeldt-Jakob disease with M232R mutation presented with corticobasal syndrome. Neurol Sci 2015;36:1291–3. 10.1007/s10072-014-2038-4 [DOI] [PubMed] [Google Scholar]
- 116. Necpál J, Stelzer M, Koščová S, et al. A corticobasal syndrome variant of familial Creutzfeldt-Jakob disease with stroke-like onset. Case Rep Neurol Med 2016;2016:4167391. 10.1155/2016/4167391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Hermann P, Laux M, Glatzel M, et al. Validation and utilization of amended diagnostic criteria in Creutzfeldt-Jakob disease surveillance. Neurology 2018;91:e331–8. 10.1212/WNL.0000000000005860 [DOI] [PubMed] [Google Scholar]
- 118. Thanvi B, Lo N, Robinson T. Vascular parkinsonism--an important cause of parkinsonism in older people. Age Ageing 2005;34:114–9. 10.1093/ageing/afi025 [DOI] [PubMed] [Google Scholar]
- 119. Josephs KA, Ishizawa T, Tsuboi Y, et al. A clinicopathological study of vascular progressive supranuclear palsy: a multi-infarct disorder presenting as progressive supranuclear palsy. Arch Neurol 2002;59:1597–601. 10.1001/archneur.59.10.1597 [DOI] [PubMed] [Google Scholar]
- 120. Kreisler A, Mastain B, Tison F, et al. [Multi-infarct disorder presenting as corticobasal degeneration (DCB): vascular pseudo-corticobasal degeneration?]. Rev Neurol 2007;163:1191–9. 10.1016/S0035-3787(07)78403-5 [DOI] [PubMed] [Google Scholar]
- 121. Kim Y-D, Kim J-S, Lee E-S, et al. Progressive "vascular" corticobasal syndrome due to bilateral ischemic hemispheric lesions. Intern Med 2009;48:1699–702. 10.2169/internalmedicine.48.2415 [DOI] [PubMed] [Google Scholar]
- 122. Miyaji Y, Koyama K, Kurokawa T, et al. Vascular corticobasal syndrome caused by unilateral internal carotid artery occlusion. J Stroke Cerebrovasc Dis 2013;22:1193–5. 10.1016/j.jstrokecerebrovasdis.2012.07.005 [DOI] [PubMed] [Google Scholar]
- 123. Kovacs GG. Are comorbidities compatible with a molecular pathological classification of neurodegenerative diseases? Curr Opin Neurol 2019;32:279–91. 10.1097/WCO.0000000000000664 [DOI] [PubMed] [Google Scholar]
- 124. Cornblath EJ, Robinson JL, Irwin DJ, et al. Defining and predicting transdiagnostic categories of neurodegenerative disease. Nat Biomed Eng 2020;4:787–800. 10.1038/s41551-020-0593-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement 2012;8:1–13. 10.1016/j.jalz.2011.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Koga S, Zhou X, Dickson DW. Machine learning-based decision tree classifier for the diagnosis of progressive supranuclear palsy and corticobasal degeneration. Neuropathol Appl Neurobiol 2021;47:931–41. 10.1111/nan.12710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology 2017;89:88–100. 10.1212/WNL.0000000000004058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Blennow K, Hampel H, Weiner M, et al. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol 2010;6:131–44. 10.1038/nrneurol.2010.4 [DOI] [PubMed] [Google Scholar]
- 129. Toledo JB, Brettschneider J, Grossman M, et al. Csf biomarkers cutoffs: the importance of coincident neuropathological diseases. Acta Neuropathol 2012;124:23–35. 10.1007/s00401-012-0983-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Aerts MB, Esselink RAJ, Bloem BR, et al. Cerebrospinal fluid tau and phosphorylated tau protein are elevated in corticobasal syndrome. Mov Disord 2011;26:169–73. 10.1002/mds.23341 [DOI] [PubMed] [Google Scholar]
- 131. Rosengren LE, Karlsson JE, Karlsson JO, et al. Patients with amyotrophic lateral sclerosis and other neurodegenerative diseases have increased levels of neurofilament protein in CSF. J Neurochem 1996;67:2013–8. 10.1046/j.1471-4159.1996.67052013.x [DOI] [PubMed] [Google Scholar]
- 132. Olsson B, Portelius E, Cullen NC, et al. Association of cerebrospinal fluid neurofilament light protein levels with cognition in patients with dementia, motor neuron disease, and movement disorders. JAMA Neurol 2019;76:318–25. 10.1001/jamaneurol.2018.3746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Hansson O, Janelidze S, Hall S, et al. Blood-Based NFL: a biomarker for differential diagnosis of parkinsonian disorder. Neurology 2017;88:930–7. 10.1212/WNL.0000000000003680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Jabbari E, Holland N, Chelban V, et al. Diagnosis across the spectrum of progressive supranuclear palsy and corticobasal syndrome. JAMA Neurol 2020;77:377–87. 10.1001/jamaneurol.2019.4347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Morris E, Chalkidou A, Hammers A, et al. Diagnostic accuracy of (18)F amyloid PET tracers for the diagnosis of Alzheimer's disease: a systematic review and meta-analysis. Eur J Nucl Med Mol Imaging 2016;43:374–85. 10.1007/s00259-015-3228-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Chételat G, Arbizu J, Barthel H, et al. Amyloid-PET and 18F-FDG-PET in the diagnostic investigation of Alzheimer's disease and other dementias. Lancet Neurol 2020;19:951–62. 10.1016/S1474-4422(20)30314-8 [DOI] [PubMed] [Google Scholar]
- 137. Ossenkoppele R, Jansen WJ, Rabinovici GD, et al. Prevalence of amyloid PET positivity in dementia syndromes: a meta-analysis. JAMA 2015;313:1939–49. 10.1001/jama.2015.4669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Palleis C, Brendel M, Finze A, et al. Cortical [18 F]PI-2620 Binding Differentiates Corticobasal Syndrome Subtypes. Mov Disord 2021;36:2104–15. 10.1002/mds.28624 [DOI] [PubMed] [Google Scholar]
- 139. Tagai K, Ono M, Kubota M, et al. High-Contrast In Vivo Imaging of Tau Pathologies in Alzheimer's and Non-Alzheimer's Disease Tauopathies. Neuron 2021;109:42–58. 10.1016/j.neuron.2020.09.042 [DOI] [PubMed] [Google Scholar]
- 140. Marquié M, Normandin MD, Vanderburg CR, et al. Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol 2015;78:787–800. 10.1002/ana.24517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Nasrallah IM, Chen YJ, Hsieh M-K, et al. 18F-Flortaucipir PET/MRI Correlations in Nonamnestic and Amnestic Variants of Alzheimer Disease. J Nucl Med 2018;59:299–306. 10.2967/jnumed.117.194282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Schwarz AJ, Yu P, Miller BB, et al. Regional profiles of the candidate tau PET ligand 18F-AV-1451 recapitulate key features of Braak histopathological stages. Brain 2016;139:1539–50. 10.1093/brain/aww023 [DOI] [PubMed] [Google Scholar]
- 143. Marquié M, Siao Tick Chong M, Antón-Fernández A, et al. [F-18]-AV-1451 binding correlates with postmortem neurofibrillary tangle Braak staging. Acta Neuropathol 2017;134:619–28. 10.1007/s00401-017-1740-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Ali F, Whitwell JL, Martin PR, et al. [18F] AV-1451 uptake in corticobasal syndrome: the influence of beta-amyloid and clinical presentation. J Neurol 2018;265:1079–88. 10.1007/s00415-018-8815-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Marquié M, Normandin MD, Meltzer AC, et al. Pathological correlations of [F-18]-AV-1451 imaging in non-alzheimer tauopathies. Ann Neurol 2017;81:117–28. 10.1002/ana.24844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Ono M, Sahara N, Kumata K, et al. Distinct binding of PET ligands PBB3 and AV-1451 to tau fibril strains in neurodegenerative tauopathies. Brain 2017;140:764–80. 10.1093/brain/aww339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Soleimani-Meigooni DN, Iaccarino L, La Joie R, et al. 18F-flortaucipir PET to autopsy comparisons in Alzheimer's disease and other neurodegenerative diseases. Brain 2020;143:3477–94. 10.1093/brain/awaa276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Marquié M, Verwer EE, Meltzer AC, et al. Lessons learned about [F-18]-AV-1451 off-target binding from an autopsy-confirmed Parkinson's case. Acta Neuropathol Commun 2017;5:75. 10.1186/s40478-017-0482-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Kroth H, Oden F, Molette J, et al. Discovery and preclinical characterization of [18F]PI-2620, a next-generation tau PET tracer for the assessment of tau pathology in Alzheimer's disease and other tauopathies. Eur J Nucl Med Mol Imaging 2019;46:2178–89. 10.1007/s00259-019-04397-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Brendel M, Barthel H, van Eimeren T, et al. Assessment of 18F-PI-2620 as a biomarker in progressive supranuclear palsy. JAMA Neurol 2020;77:1408–19. 10.1001/jamaneurol.2020.2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Tezuka T, Takahata K, Seki M, et al. Evaluation of [18F]PI-2620, a second-generation selective tau tracer, for assessing four-repeat tauopathies. Brain Commun 2021;3:fcab190. 10.1093/braincomms/fcab190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Kikuchi A, Okamura N, Hasegawa T, et al. In vivo visualization of tau deposits in corticobasal syndrome by 18F-THK5351 PET. Neurology 2016;87:2309–16. 10.1212/WNL.0000000000003375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Ezura M, Kikuchi A, Okamura N, et al. 18F-THK5351 Positron Emission Tomography Imaging in Neurodegenerative Tauopathies. Front Aging Neurosci 2021;13:761010. 10.3389/fnagi.2021.761010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Maruyama M, Shimada H, Suhara T, et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron 2013;79:1094–108. 10.1016/j.neuron.2013.07.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Koga S, Ono M, Sahara N, et al. Fluorescence and autoradiographic evaluation of tau PET ligand PBB3 to α-synuclein pathology. Mov Disord 2017;32:884–92. 10.1002/mds.27013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Perez-Soriano A, Arena JE, Dinelle K, et al. PBB3 imaging in parkinsonian disorders: evidence for binding to tau and other proteins. Mov Disord 2017;32:1016–24. 10.1002/mds.27029 [DOI] [PubMed] [Google Scholar]
- 157. Miranda-Azpiazu P, Svedberg M, Higuchi M, et al. Identification and in vitro characterization of C05-01, a PBB3 derivative with improved affinity for alpha-synuclein. Brain Res 2020;1749:147131. 10.1016/j.brainres.2020.147131 [DOI] [PubMed] [Google Scholar]
- 158. Ono M, Takahashi M, Shimozawa A. Visualization of propagating α-synuclein pathologies in mouse and marmoset models by a bimodal imaging probe, C05-05. bioRxiv 2021:2020.2010.2023.349860. [Google Scholar]