

ABSTRACT
Bacteriophage (phage) therapy is an alternative to traditional antibiotic treatments that is particularly important for multidrug-resistant pathogens, such as Pseudomonas aeruginosa. Unfortunately, phage resistance commonly arises during treatment as bacteria evolve to survive phage predation. During in vitro phage treatment of a P. aeruginosa-type strain, we observed the emergence of phage-resistant mutants with brown pigmentation that was indicative of pyomelanin. As increased pyomelanin (due to hmgA gene mutation) was recently associated with enhanced resistance to hydrogen peroxide and persistence in experimental lung infection, we questioned if therapeutic phage applications could inadvertently select for hypervirulent populations. Pyomelanogenic phage-resistant mutants of P. aeruginosa PAO1 were selected for upon treatment with three distinct phages. Phage-resistant pyomelanogenic mutants did not possess increased survival of pyomelanogenic ΔhmgA in hydrogen peroxide. At the genomic level, large (~300 kb) deletions in the phage-resistant mutants resulted in the loss of ≥227 genes, many of which had roles in survival, virulence, and antibiotic resistance. Phage-resistant pyomelanogenic mutants were hypersusceptible to cationic peptides LL-37 and colistin and were more easily cleared in human whole blood, serum, and a murine infection model. Our findings suggest that hyperpigmented phage-resistant mutants that may arise during phage therapy are markedly less virulent than their predecessors due to large genomic deletions. Thus, their existence does not present a contraindication to using anti-pseudomonal phage therapy, especially considering that these mutants develop drug susceptibility to the familiar FDA-approved antibiotic, colistin.
KEYWORDS: bacteriophage therapy, bacteriophage resistance, Pseudomonas aeruginosa, pyomelanin, innate immunity
INTRODUCTION
Pseudomonas aeruginosa is a leading cause of human opportunistic infections, such as ventilator-associated pneumonia (1), chronic cystic fibrosis lung disease (2), burn sepsis (3), and infections following chemotherapy-induced neutropenia (4). Treatment of these infections is challenging due to intrinsic, adaptive, and acquired resistance to nearly all classes of antibiotics (5). Furthermore, the global spread of multidrug-resistant or extensively drug-resistant (MDR/XDR) P. aeruginosa strains presents an immediate public health threat (6). With the declining efficacy of traditional antibiotics, alternative treatment approaches for MDR P. aeruginosa infections are receiving increased attention, including the successful adjunctive use of bacteriophage (phage) therapy in several case reports (7–10) and a small clinical trial (11). As phage therapeutics increase the toolkit to combat MDR pathogens, natural selection of phage-resistant bacterial mutants (12) becomes another knowledge domain in which the infectious diseases clinician must gain familiarity of underlying mechanisms and clinical implications.
In our in vitro studies of candidate bacteriophages for P. aeruginosa treatment, we observed that three genetically distinct phages (AM.P2, Mat, and Kat) often selected for phage-resistant mutants in model strain PAO1 with a dark brown color indicative of hyperproduction of the pigment pyomelanin (13, 14), a phenomenon sometimes reported in other studies in which P. aeruginosa laboratory strains or clinical isolates were exposed to a phage (15–17). For several pathogenic microbes, pigments can function as virulence factors that interfere with host immune clearance mechanisms (18). Melanins in particular are highly effective free radical scavengers with electron transfer properties (19, 20), and their production by opportunistic pathogens such as Cryptococcus neoformans or Aspergillus spp. promotes resistance to reactive oxygen species (ROS) generated in the phagolysosome of host neutrophils and macrophages (21, 22). Indeed, a recent report demonstrated that increased production of pyomelanin by P. aeruginosa was linked to hydrogen peroxide resistance and in vivo persistence of the pathogen in a murine lung infection model (23).
The selection of hyper-melanogenic P. aeruginosa upon treatment with diverse bacteriophage types appears at first blush to be a concerning development for clinicians, as the resulting mutants would theoretically both resist phage infection and be better able to withstand host innate immune clearance. We applied whole-genome sequencing (WGS), antimicrobial and ROS sensitivity testing, bacterial cytological profiling, blood and serum survival assays, and a murine infection model to evaluate the phage-resistant pyomelanogenic P. aeruginosa mutants. We found that phage resistance arises in the context of large genomic deletions that increase pyomelanin production. At the same time, we found that these deletions impact lipopolysaccharide (LPS) and other key P. aeruginosa stress response pathways, cumulatively diminishing the pathogen’s innate immune resistance and virulence.
RESULTS
Bacteriophage-resistant P. aeruginosa mutants produce pyomelanin.
Bacteriophages AM.P2, Mat, and Kat were originally isolated and enriched using PAO1 as a host. The genome sequences of the three phages suggested a unique taxonomic lineage and genome size for each phage (Table 1). Additionally, there was little to no alignment between the three phage genomes (Fig. S1 and S2), indicating that these are indeed genetically distinct phages. Exposure to each phage for 24 h selected for mutants (AM.P2-X-1, Mat-X-1, and Kat-X-2) resistant to phage infection and also producing a dark brown pigment suggestive of pyomelanin (Fig. 1A). This phenotype represented approximately 15% of all phage-resistant mutants selected. Pyomelanin production in P. aeruginosa and other bacteria reflects an aberration of the tyrosine metabolism pathway through mutations in hmgA (23), leading to the overaccumulation and auto-oxidation of homogentisate, which polymerizes to produce the brown pigment (Fig. 1B). We used two strategies to confirm that the pigment of the phage-resistant mutants was indeed pyomelanin: (i) supplementation with 5 mM l-Tyrosine and (ii) cloning of functional hmgA from parent strain PAO1 into the phage-resistant mutants. An isogenic PAO1 hmgA transposon mutant (ΔhmgA) was used as a control. l-Tyrosine supplementation enhanced pigment production in phage-resistant mutants AM.P2-X-1, Mat-X-1, Kat-X-2, and ΔhmgA (Fig. 1C), while hmgA complementation of all strains rescued the hyperpigmentation phenotype (Fig. 1D), confirming that the phage-resistant mutants were indeed pyomelanogenic.
TABLE 1.
Bacteriophages used in this study
| Phage | AM.P2 | Mat | Kat |
|---|---|---|---|
| Family | Podoviridae | Myoviridae | Myoviridae |
| Genus | Luzseptimaviridae | Pbunaviridae | Pakpunaviridae |
| Genome size | 73 kb | 65 kb | 90 kb |
FIG 1.

Pyomelanin production and hydrogen peroxide-mediated killing of phage-resistant mutants. (A) Treatment of PAO1 with bacteriophages AM.P2, Mat, and Kat led to the generation of pyomelanogenic phage-resistant mutants AM.P2-X-1, Mat-X-1, and Kat-X-2, respectively. (B) Pyomelanin is produced by auto-oxidation and polymerization of excess homogentisate due to the loss of function of hmgA. (C) Visual changes in pigmentation of PAO1, ΔhmgA, and phage-resistant mutants (AM.P2-X-1, Mat-X-1, and Kat-X-2) upon supplementation of 5 mM Tyrosine to growth media and (D) quantified changes in pyomelanin production in the same strains when complemented with functional hmgA. Data are representative of the mean ± SEM of three independent experiments performed in triplicate. *, P < 0.05; ***, P < 0.0005; ****, P < 0.0001, or no statistical significance, ns, denoted based on one-way analyses of variance and Dunnett’s test.
Pyomelanin-mediated H2O2 resistance observed in ΔhmgA but not phage-resistant P. aeruginosa mutants.
Pyomelanin enhanced the survival of P. aeruginosa challenged with H2O2 (23) and could theoretically promote virulence and persistent infection by allowing the pathogen to resist ROS killing in the phagolysosome. We confirmed that ΔhmgA had significantly increased survival following 30 min H2O2 exposure compared to the parent strain PAO1 or the complemented mutant (ΔhmgA::hmgA) (Fig. 2A). However, enhanced H2O2 survival was not seen in the three phage-resistant mutants (Fig. 2B), suggesting additional factors present in these strains that counteract the protective effects of pyomelanin.
FIG 2.
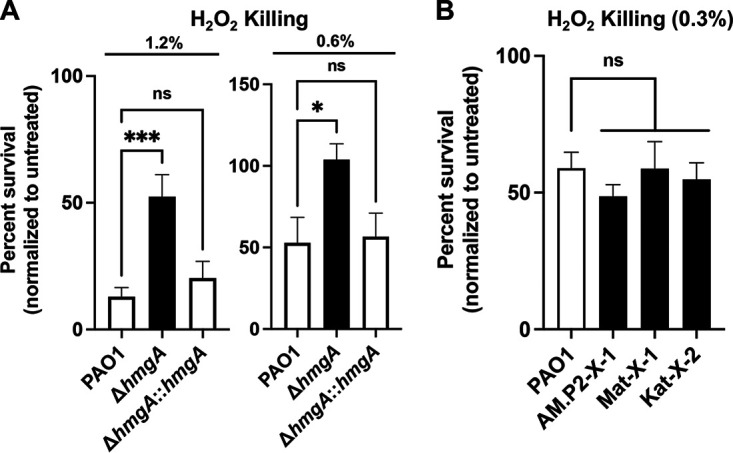
Surviving CFU/mL of (A) PAO1, ΔhmgA, and ΔhmgA::hmgA treated with 1.2% and 6% hydrogen peroxide and of (B) PAO1, AM.P2-X-1, Mat-X-1, and Kat-X-2 treated with 0.3% hydrogen peroxide. Data are representative of the mean ± SEM of three independent experiments performed in triplicate. *, P < 0.05; ***, P < 0.0005, or no statistical significance, ns, denoted based on one-way analyses of variance and Dunnett’s test.
Phage-resistant pyomelanogenic P. aeruginosa mutants have large genomic deletions.
Whole-genome sequencing of AM.P2-X-1, Mat-X-1, and Kat-X-2 indicated that exposure to phages AM.P2, Mat, and Kat independently resulted in the selection of mutants with approximately 300 kb deletions (~5% of the total PAO1 genome) in a region spanning the 2 to 2.5 Mb mark (Fig. 3A and B, Table S1). This corresponds to a common set of 227 coding regions deleted in each mutant strain. Notably, the deletions had unique start and stop sites: deletions spanned 2166285 to 2422315 bp in AM.P2-X-1, 2114437 to 2432633 bp in Mat-X-1, and 2091235 to 2445434 bp in Kat-X-2. No repeats or IS elements were found at these deletion start and stop sites. The deleted region includes hmgA, accounting for the hyperpigmentation phenotype, and galU, a gene required for complete LPS core synthesis (24). Similar galU mutations have been previously associated with phage resistance for a number of LPS-targeting phages (15–17). The ΔgalU mutant was resistant to Mat and Kat phage infection and had intermediate sensitivity to AM.P2, suggesting that these are LPS-targeting phages that do not depend on flagella, pili, or pyomelanin to aid in infection (Fig. 3C). Loss of galU results in a truncated LPS core and a “rough” LPS phenotype, reducing P. aeruginosa virulence in a corneal infection model and the pathogen’s potential for systemic spread (25). Also, among the 227 coding regions missing in the phage-resistant P. aeruginosa mutants are many genes essential for opportunistic survival, including aerobic ethanol oxidation, pyrroloquinoline quinone (PQQ) biosynthesis, amino acid (tyrosine, leucine, and cysteine) metabolism, carnitine and carbohydrates, copper resistance, and osmotic stress. Virulence determinants lost include genes associated with pili synthesis (important for biofilm formation), exoY toxin and hydrogen cyanide production, Pseudomonas quinolone signal (PQS) precursor synthesis via the kynurenine pathway (for quorum sensing), siderophore transporters (for iron sequestration from host), and multidrug efflux pump systems.
FIG 3.

(A) Comparative genomics of AM.P2-X-1 (black), Mat-X-1 (dark gray), and Kat-X-2 (light gray) to PAO1. (B) Common region of the large genomic deletions (2166553 to 2422738 bp) between AM.P2-X-1, Mat-X-1, and Kat-X-2. (C) PAO1, ΔfliC, ΔpilA, ΔgalU, and ΔhmgA sensitivity to AM.P2, Mat, and Kat phages.
Phage-resistant pyomelanogenic P. aeruginosa mutants are hypersusceptible to cationic peptide antimicrobials.
Antibiotic susceptibility testing shows that phage-resistant pyomelanogenic mutants had changes in antibiotic susceptibility compared to wild-type PAO1 (Table S2). Human cathelicidin LL-37 is a cationic antimicrobial peptide produced by leukocytes and epithelial cells that serves a frontline role in innate immunity to bacterial pathogens (26). Colistin, a last resort antibiotic for MDR Gram-negative pathogens, including P. aeruginosa, is likewise a cationic peptide antibiotic. These amphipathic antimicrobials can bind LPS and destabilize negatively charged bacterial cell membranes, resulting in membrane permeabilization and cell lysis. Compared to parent strain PAO1, all three phage-resistant pyomelanogenic P. aeruginosa mutants displayed 16-fold and 4-fold decreases in MICs for LL-37 and colistin, respectively (Fig. 4A), and were more rapidly killed by the peptides in kinetic assays (Fig. 4B). Increased cationic peptide susceptibility was decoupled from hyperpigmentation, since the isogenic PAO1 ΔhmgA LL-37 and colistin MICs matched the PAO1 and hmgA complemented mutant. Bacterial cytological profiling was used for the visualization and quantification of LL-37 and colistin-induced P. aeruginosa cell death, showing that the treated AM.P2-X-1, Mat-X-1, and Kat-X-2 mutants had significantly increased SYTOX Green staining compared to PAO1 (Fig. 4C to F). AM.P2-X-1, Mat-X-1, and Kat-X-2 all bound greater amounts of the cationic test molecule, cytochrome c, compared to PAO1 (Fig. 4G), indicating that increased negative surface charge accompanied the genomic deletions and likely underlies the increased antimicrobial peptide susceptibility.
FIG 4.

Susceptibility to cationic human antimicrobial peptide LL-37 and colistin. MIC90 concentrations for LL-37 and colistin against PAO1, ΔhmgA, ΔhmgA::hmgA, AM.P2-X-1, Mat-X-1, and Kat-X-2 (A). Time-kill curves for PAO1, AM.P2-X-1, Mat-X-1, and Kat-X-2 treated with 1x MIC and 2x MIC concentrations of LL-37 and colistin over 2 η (B). Cytological profiles of PAO1, AM.P2-X-1, Mat-X-1, and Kat-X-2 treated with LL-37 (4 μM) for 1 h (C) and colistin (2 μg/mL) for 30 min (D). Images were taken after staining the cells with FM4-64 (red), DAPI (blue), and SYTOX Green (green) and show the respective percentages of bacteria with SYTOX Green (E and F). Scale bars represent 1 μm. Relative bacterial surface charge of PAO1, AM.P2-X-1, Mat-X-1, and Kat-X-2, represented as percentage of unbound cytochrome c (G). Data are representative of the mean ± SEM of three independent experiments performed in triplicate. *, P < 0.05; **, P < 0.005; ***, P < 0.0005; ****, P < 0.0001, denoted based on one-way or two-way analyses of variance and Dunnett’s test. LOD, limit of detection.
Phage-resistant pyomelanogenic P. aeruginosa mutants are more rapidly cleared in human whole blood, serum, and a murine pneumonia model.
Increased susceptibility to host defense peptide LL-37 led us to further explore virulence phenotypes of the phage-resistant P. aeruginosa mutants. AM.P2-X-1, Mat-X-1, and Kat-X-2 were killed markedly more rapidly than original PAO1 in freshly isolated human whole blood (Fig. 5A) or serum (Fig. 5B), with multilog-fold fewer bacteria surviving within the first 30 to 60 min of incubation. These results suggest greater susceptibility to neutrophil- or platelet-derived antimicrobial factors and/or the terminal membrane attack complex of serum complement cascade. Moving to an in vivo model, we challenged mice by intratracheal infection with WT PAO1, ΔhmgA (pyomelanogenic), or AM.P2-X-1 (pyomelanogenic and phage-resistant) and then humanely sacrificed the mice 24 h later for enumeration of lung CFU (Fig. 5C). AM.P2-X-1 was selected as the representative since it harbors the shortest genomic deletion of the three phage-resistant mutants. We found that ΔhmgA showed a ~10-fold increase in surviving bacteria in the lungs compared to PAO1 (Fig. 5D), indicating that increased pyomelanin per se inhibited host clearance of infection. However, phage-resistant pyomelanogenic mutant AM.P2-X-1 had a ~10-fold decrease in survival in the lungs, relative to PAO1 (Fig. 5D), indicating that the overall loss of virulence factors in its large genomic deletion outweighed the survival benefit of pyomelanin, rendering the strain hypersusceptible to host-mediated clearance in the lung.
FIG 5.

Susceptibility to host innate immunity. Human whole blood (A) and serum (B) killing of PAO1, AM.P2-X-1, Mat-X-1, and Kat-X-2 in active (solid line) and heat inactivated blood/serum (dotted line). For the murine lung infection model, mice were intratracheally infected with PAO1, ΔhmgA, and AM.P2-X-1, and lungs were harvested after 24 h to determine bacterial survival in the host environment (C and D). *, P < 0.05; **, P < 0.005; ****, P < 0.0001, denoted based on one-way or two-way analyses of variance and Dunnett’s test. LOD, limit of detection.
DISCUSSION
Pyomelanin production by P. aeruginosa has been reported in numerous clinical scenarios, including lung infections in cystic fibrosis patients and urinary tract infections (27, 28). Pyomelanin production can arise through various mechanisms, including a loss-of-function mutation or deletion of hmgA, the gene that encodes the enzyme responsible for the conversion of homogentisate to maleylacetoacetate (23), changes in hmgA expression by hmgR or sawR mediated regulation (29), or variations in tyrosine substrate concentrations. In our study, we observed that pyomelanin production was instead attributed to large genomic deletions that were selected for via bacteriophage treatment. Phage-resistant mutants harbored deletion of hmgA (resulting in pyomelanin production), galU (required for complete LPS core and O-antigen), and over 200 other additional genes, several of which play established roles in P. aeruginosa infection. These large deletions are similar to those seen by other groups that have sequenced pyomelanogenic clinical isolates (27, 30), phage-resistant mutants (15–17, 31–33), and ceftazidime resistant mutants (34), suggesting that the excision of this region of the genome may be a conserved mechanism for surviving a variety of stresses initiated at the bacteria cell membrane. Although phage-resistant mutants produced pyomelanin, isogenic phage selection for hmgA mutants seems unlikely, as ΔhmgA are susceptible to phage infection, suggesting that pyomelanin does not play a protective role against phage predation.
Exposure of P. aeruginosa to candidate therapeutic bacteriophages AM.P2, Mat, and Kat resulted in the selection of phage resistance coupled with pyomelanin production. Resistance to a treatment modality would generally be a concerning clinical development, but in this case, as the bacteria modifies its surface to evade the immediate threat of phage predation, it sacrifices its ability to persist in the host. The effects of pyomelanin, a virulence factor that by itself protects P. aeruginosa from reactive oxygen species (ROS) and promotes lung persistence, are overcome by the cumulative effects of the deletion, which render the phage-resistant bacteria highly susceptible to cathelicidin antimicrobial peptide LL-37, serum, and whole blood killing. Furthermore, hypersusceptibility to colistin suggests a potential antibiotic approach that can be delivered systemically or by aerosol as a companion to phage therapy. Evolutionary tradeoffs like these are a common theme in bacterial survival, bolstering the concept of “phage steering”, i.e., using phages to “steer” a bacterial population to less virulent or more antibiotic-susceptible phenotypes, thereby achieving immuno-phage-antibiotic synergy (35, 36).
MATERIALS AND METHODS
Bacteriophage isolation.
The isolation of AM.P2 was described previously (37). Phages Mat and Kat were independently isolated using the double-agar layer technique on a fluorescent PAO1 derivative (ATCC 15692GFP). Local wastewater was combined with Luria-Bertani (LB) broth, incubated 37°C overnight with 250 rpm shaking, and centrifuged at 5000 × g for 15 min. The supernatant was filtered through a 0.45 μm filter. 10-fold dilutions of filtrate were combined with log-phase P. aeruginosa and soft LB with 0.3% agar, and the mixtures were poured onto a bottom LB agar layer, then incubated overnight at 37°C after solidification. Individual plaques were suspended in 100 μL sterile PBS, 10-fold serial dilutions were created, and the double-agar layer technique was repeated for three additional rounds. Purified phage plaques were selected using a pipette tip, suspended in 100 μL of PBS, combined with 10 mL of P. aeruginosa at OD600 = 0.2 in LB broth, and incubated overnight at 37°C/250 rpm. Lysates were prepared as described above, and the final phage titer was quantified via the double-agar layer method. Mat and Kat phage lysates (with titer of 108 PFU/mL confirmed on P. aeruginosa PAO1) were stored at 4°C.
Bacterial strains and culturing conditions.
All bacteria used in this study are listed in Table 2. MPAO1 (PAO1) and transposon mutants were obtained from the University of Washington Manoil Lab P. aeruginosa PAO1 Two-Allele Transposon Mutant Library (38, 39). Phage-resistant mutants AM.P2-X-1, Mat-X-1, and Kat-X-2 were generated by co-incubating PAO1 with the respective phages at MOI = 0.1 for 24 h, followed by serial dilution and plating (37). Single brown pigmented colonies that survived phage treatment were restreaked to purity onto LB plates twice, their resistance was confirmed via the double-agar layer method on PAO1, and the glycerol stocks were stored at −80°C. The complementation of functional hmgA used a published protocol (40). MPAO1 genomic DNA was purified by the DNEasy Blood and Tissue Kit (Qiagen) and hmgA + 139 bp upstream sequence cloned into pUC18T-mini-Tn7T-hph using HindIII-HF and BamHI-HF (New England Biosciences). The final vector was electroporated into ΔhmgA, AM.P2-X-1, Mat-X-1, and Kat-X-2, and transformants were selected on 100 μg/mL hygromycin and confirmed by PCR. All primers used are detailed in Table 3. Bacteria were routinely propagated on LB agar/broth at 37°C to mid-log-phase (OD600 = 0.4) at 37°C/200 rpm, then diluted to assay concentrations. For hydrogen peroxide killing and pigment quantification, bacteria were grown in RPMI 1640 medium without phenol red (Gibco) + 10% LB broth ± 5 mM l-Tyrosine (Alfa Aesar). For other studies, bacteria were grown in LB broth.
TABLE 2.
Bacteria used in this study
| Strain | Additional information | Source |
|---|---|---|
| MPAO1 | Wild-type PAO1 | Manoil Lab (38, 39) |
| △hmgA | PW4489; genotype hmgA(PA2009)-C03::ISphoA/hah | Manoil Lab (38, 39) |
| △galU | PW4507; genotype galU(PA2023)-E09::ISphoA/hah | Manoil Lab (38, 39) |
| △fliC | PW2971; genotype fliC(PA1092)-G10::ISphoA/hah | Manoil Lab (38, 39) |
| △pilA | PW8621; genotype pilA(PA4525)-E01::ISphoA/hah | Manoil Lab (38, 39) |
| AM.P2-X-1 | AM.P2 phage-resistant PAO1 mutant | This study |
| Mat-X-1 | Mat phage-resistant PAO1 mutant | This study |
| Kat-X-2 | Kat phage-resistant PAO1 mutant | This study |
| △hmgA::hmgA | △hmgA complemented with functional hmgA | This study |
| AM.P2-X-1::hmgA | AM.P2-X-1 complemented with functional hmgA | This study |
| Mat-X-1::hmgA | Mat-X-1 complemented with functional hmgA | This study |
| Kat-X-2::hmgA | Kat-X-2 complemented with functional hmgA | This study |
TABLE 3.
Primers used in this study
| Name | Direction | Sequence (5′–3′) | Purpose | Reference |
|---|---|---|---|---|
| hmgA-cloning | F | GTACTGAAGCTTGGGCCGACGTTATAGCTTGACG | For cloning hmgA + 139 bp upstream into puc18T-mini-Tn7T-hph | This study |
| R | GTATAAGGATCCTTATCTCCGTTGCGGGTTGAAGG | |||
| hmgA-PCR | F | ATGAACCTCGACTCCACTGCC | For transposition confirmation of hmgA | This study |
| R | TTATCTCCGTTGCGGGTTGAAGG | |||
| PTn7R-PglmS-down | PTn7R | CACAGCATAACTGGACTGATTTC | For transposition confirmation at attTn7 site | (40) |
| PglmS-down | GCACATCGGCGACGTGCTCTC |
Fluctuation assay.
Assays were performed as previously described with few adjustments (37). Briefly, culture was mixed with phage at an MOI of 10, incubated at 37°C for 10 min to allow for phage adsorption, plated using the double-agar overlay method, and incubated at 37°C for 24 to 48 h. The number of phage-resistant mutants was enumerated after 24 to 48 h. Based on the coloration of resistant colonies, the percentage of total phage-resistant mutants with hyperpigmentation was determined.
Pyomelanin production and quantification.
Overnight cultures were diluted 1:100 in either RPMI + 10%LB ± 5 mM Tyrosine, and 100 μL were added to the wells of a round bottomed 96-well plate and incubated, shaking at 200 rpm at 37°C for 16 to 20 h. For quantifying pyomelanin, 96-well plate was centrifuged at 3200 × g for 10 min, and 50 μL of the supernatant were transferred to a flat bottomed 96-well plate. Absorbance at OD405 was measured using an Enspire Alpha multimode plate reader (PerkinElmer). Absorbance values were normalized to the average absorbance for PAO1.
Reagents, antibiotics, and antimicrobial peptides.
Single-use aliquots of colistin sulfate (Sigma) 1 mg/mL and cytochrome c (Sigma) 10 mg/mL stocks were prepared in molecular biology-grade water and stored at −20°C. Single-use aliquots in DPBS of hygromycin B (Invitrogen) 50 mg/mL and resazurin sodium salt (Sigma) 25 mM were stored at 4°C, and 640 μM stocks of LL-37 (Bachem) in DPBS were stored at −80°C.
Whole-genome sequencing.
P. aeruginosa and phage DNA was extracted by the DNeasy Blood and Tissue Kit (Qiagen) and quantified by dsDNA HS assay (Qubit). DNA preparation and whole-genome sequencing was done using the Illumina Nextera DNA FlexKit and Adapter Indexes and the MiSeq platform. Sequencing reads were downloaded from Illumina Basespace, trimmed for length and quality, and assembled de novo using CLC Genomics Workbench 9. Bacterial genomes were aligned to PAO1 (NC_002516.2), and progressiveMauve alignment was performed using both the Mauve module in Geneious Prime and the Mauve desktop application and was visualized using PATRIC v.3.6.12. The taxonomic classification of phages was determined using Kraken2 on the PATRIC platform.
Hydrogen peroxide killing.
Overnight cultures were diluted 1:100 in 1.5 mL RPMI + 10%LB + 5 mM l-Tyrosine and incubated at 37°C/200 rpm for 16 to 20 h, and 45 μL were added to 5 μL of 12%, 6%, or 3% H2O2 (Fischer Chemical) in a 96-well plate for 30 min at 37°C/200 rpm prior to dilution plating on LB agar overnight at 37°C for CFU determination.
MIC (MIC90) and kinetic killing assays.
Mid-log-phase P. aeruginosa were diluted 1:100 in RPMI + 10%LB to ~2 × 106 CFU/mL, 180 μL were added to wells of a 96-well plate, and 20 μL of antibiotic/antimicrobial peptide or vehicle were added to achieve desired final concentrations and incubated at 37°C/200 rpm for 16 to 20 h following OD600 with an Enspire Alpha multimode plate reader (PerkinElmer). For LL-37 assays, bacterial growth was further assessed by adding 10 μL of 200 μM resazurin at 37°C for an additional 2 h. MIC90 was defined as the minimum concentration of antimicrobial inhibiting bacterial growth ≥90% versus vehicle-treated controls. For colistin and LL-37 kinetic killing studies at 1x and 2x MIC, plates were set up as above, and aliquots were collected at T = 0, 30, 60, and 120 min for CFU enumeration.
Cytochrome c binding.
P. aeruginosa was grown to mid-log-phase and concentrated to ~3 × 109 CFU/mL, and 190 μL were added to 10 μL cytochrome c (final concentration 0.5 mg/mL) in 96-well format, incubated at room temperature for 10 min, and centrifuged at 3200 × g for 10 min. The OD530 was determined to assess the surface charge, relative to that of PAO1.
Bacterial cytological profiling.
High-resolution fluorescence microscopy and bacterial cytological profiling (BCP) were performed as described (41). Briefly, P. aeruginosa cultures were diluted 1:100 in RPMI 1640 + 10% LB, incubated with rolling at 37°C to OD600 = 0.12 to 0.15, and 300 μL of bacterial cells were treated with compounds while rolling at 37°C. Microscopy images were captured at 30 min (2 μg/mL colistin) and 1 h (4 μM LL-37), and staining was performed with a dye mixture of FM4-64 (100 μg/mL), DAPI (49,69-diamidino-2-phenylindole) (100 μg/mL), and SYTOX Green (10 μM). The percentage of bacterial cells with SYTOX Green permeability was calculated, counting 100 to 200 cells in at least three frames for all samples and correcting for the baseline SYTOX Green permeability of untreated samples for each strain.
Ethics statement.
Blood was collected via venipuncture from healthy volunteers under written informed consent approved by the UC San Diego Human Research Protection Program (no.131002). Mouse studies were conducted in accord with protocols approved by the UC San Diego IACUC (no. S00227M) and optimized to minimize animal numbers and suffering.
Whole blood and serum bactericidal activity.
Blood was collected from healthy volunteers in heparinized tubes or serum separation tubes (SST, Becton Dickenson). After 30 min clot formation, SST were centrifuged at 1000 × g for 10 min to collect serum. Mid-log-phase P. aeruginosa were diluted 1:100 in RPMI 1640 and either (i) 15 μL (~3 × 104 CFU) were added to 135 μL of either active or heat-inactivated blood, or (ii) 10 μL (~w × 104 CFU) were added to 90 μL of either active or heat-inactivated serum in 96-well plate format, incubated rotating at 37°C. Aliquots were plated at the indicated time points for CFU determination.
Murine lung infection.
8-week-old CD1 mice (Charles River) were anesthetized with 10 mg/mL ketamine and 1 mg/mL xylazine and were intratracheally infected with mid-log-phase P. aeruginosa, washed and resuspended in DPBS to ~1 × 107 CFU in 30 μL or DPBS only control. 24 h postinfection, mice were humanely euthanized by CO2 asphyxiation and cervical dislocation, lungs immediately harvested, homogenized in 1 mL of DPBS, and 20 μL of the suspension serially diluted 1:10 in on LB plates to determine CFU/g of lung tissue.
Statistical analysis.
Statistical analyses were performed with GraphPad Prism 9 software. Raw CFU/mL or CFU/g counts were log-transformed prior to statistical analysis. Data are expressed as the mean ± SEM from triplicate readings from three independent experiments, except for animal experiments, which are expressed as individual CFU/g counts from each mouse and the corresponding median value. Means were compared using one-way or two-way analyses of variance with Dunnett’s post-test, as appropriate; P < 0.05 was considered statistically significant.
Data availability.
Sequences are available under the following genome accession numbers in NCBI GenBank: MT416090 (vB_Pae_AM.P2), ON361134 (vB_Pae_Mat), ON361135 (vB_Pae_Kat), JAMKNC000000000 (AM.P2-X-1), CP096665 (Mat-X-1), and CP096664 (Kat-X-2).
ACKNOWLEDGMENTS
This work was supported by NIH grants R01-AI145310 and R37-AI052453 to V.N., the University Grants Commission of India (grant number F.16-6 [DEC.2016]/2017 [NET]) to N.D.M., the UC San Diego Infectious Diseases Training Program T32-AI007036 to S.P., and the San Diego IRACDA Postdoctoral Fellowship Program K12-GM068524 to E.M. We thank Robert Schooley for his valuable insights into this line of investigation.
N.D.M. and V.N. conceived and designed the experiments for the study. Experiments were done by N.D.M., S.P., E.M., and R.Z. Analysis was done by N.D.M., S.P., E.M., D.P., B.N., G.B.K., and V.N. N.D.M., S.P., E.M., and V.N. drafted the manuscript. All authors reviewed the final manuscript.
Footnotes
Supplemental material is available online only.
Contributor Information
Geetha B. Kumar, Email: gkumar@am.amrita.edu.
Victor Nizet, Email: vnizet@health.ucsd.edu.
REFERENCES
- 1.Jones RN. 2010. Microbial etiologies of hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia. Clin Infect Dis 51 Suppl 1:S81–7. 10.1086/653053. [DOI] [PubMed] [Google Scholar]
- 2.Crull MR, Somayaji R, Ramos KJ, Caldwell E, Mayer-Hamblett N, Aitken ML, Nichols DP, Rowhani-Rahbar A, Goss CH. 2018. Changing rates of chronic Pseudomonas aeruginosa infections in cystic fibrosis: a population-based cohort study. Clin Infect Dis 67:1089–1095. 10.1093/cid/ciy215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lachiewicz AM, Hauck CG, Weber DJ, Cairns BA, van Duin D. 2017. Bacterial infections after burn injuries: impact of multidrug resistance. Clin Infect Dis 65:2130–2136. 10.1093/cid/cix682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perez F, Adachi J, Bonomo RA. 2014. Antibiotic-resistant Gram-negative bacterial infections in patients with cancer. Clin Infect Dis 59 Suppl 5:S335–9. 10.1093/cid/ciu612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Langendonk RF, Neill DR, Fothergill JL. 2021. The building blocks of antimicrobial resistance in Pseudomonas aeruginosa: implications for current resistance-breaking therapies. Front Cell Infect Microbiol 11:665759. 10.3389/fcimb.2021.665759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horcajada JP, Montero M, Oliver A, Sorlí L, Luque S, Gómez-Zorrilla S, Benito N, Grau S. 2019. Epidemiology and treatment of multidrug-resistant and extensively drug-resistant Pseudomonas aeruginosa infections. Clin Microbiol Rev 32:e00031-19. 10.1128/CMR.00031-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jennes S, Merabishvili M, Soentjens P, Pang KW, Rose T, Keersebilck E, Soete O, François P-M, Teodorescu S, Verween G, Verbeken G, De Vos D, Pirnay J-P. 2017. Use of bacteriophages in the treatment of colistin-only-sensitive Pseudomonas aeruginosa septicaemia in a patient with acute kidney injury-a case report. Crit Care 21:129. 10.1186/s13054-017-1709-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan BK, Turner PE, Kim S, Mojibian HR, Elefteriades JA, Narayan D. 2018. Phage treatment of an aortic graft infected with Pseudomonas aeruginosa. Evol Med Public Health 2018:60–66. 10.1093/emph/eoy005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Law N, Logan C, Yung G, Furr C-LL, Lehman SM, Morales S, Rosas F, Gaidamaka A, Bilinsky I, Grint P, Schooley RT, Aslam S. 2019. Successful adjunctive use of bacteriophage therapy for treatment of multidrug-resistant Pseudomonas aeruginosa infection in a cystic fibrosis patient. Infection 47:665–668. 10.1007/s15010-019-01319-0. [DOI] [PubMed] [Google Scholar]
- 10.Aslam S, Courtwright AM, Koval C, Lehman SM, Morales S, Furr C-LL, Rosas F, Brownstein MJ, Fackler JR, Sisson BM, Biswas B, Henry M, Luu T, Bivens BN, Hamilton T, Duplessis C, Logan C, Law N, Yung G, Turowski J, Anesi J, Strathdee SA, Schooley RT. 2019. Early clinical experience of bacteriophage therapy in 3 lung transplant recipients. Am J Transplant 19:2631–2639. 10.1111/ajt.15503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jault P, Leclerc T, Jennes S, Pirnay JP, Que Y-A, Resch G, Rousseau AF, Ravat F, Carsin H, Le Floch R, Schaal JV, Soler C, Fevre C, Arnaud I, Bretaudeau L, Gabard J. 2019. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): a randomised, controlled, double-blind phase 1/2 trial. Lancet Infect Dis 19:35–45. 10.1016/S1473-3099(18)30482-1. [DOI] [PubMed] [Google Scholar]
- 12.Broniewski JM, Meaden S, Paterson S, Buckling A, Westra ER. 2020. The effect of phage genetic diversity on bacterial resistance evolution. ISME J 14:828–836. 10.1038/s41396-019-0577-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elston HR. 1968. Melanogenic strains of Pseudomonas aeruginosa in biological specimens. Am J Med Technol 34:189–194. [PubMed] [Google Scholar]
- 14.Ogunnariwo J, Hamilton-Miller JM. 1975. Brown- and red-pigmented Pseudomonas aeruginosa: differentiation between melanin and pyorubrin. J Med Microbiol 8:199–203. 10.1099/00222615-8-1-199. [DOI] [PubMed] [Google Scholar]
- 15.Latino L, Midoux C, Vergnaud G, Pourcel C. 2019. Investigation of Pseudomonas aeruginosa strain PcyII-10 variants resisting infection by N4-like phage Ab09 in search for genes involved in phage adsorption. PLoS One 14:e0215456. 10.1371/journal.pone.0215456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen M, Zhang H, Shen W, Zou Z, Lu S, Li G, He X, Agnello M, Shi W, Hu F, Le S. 2018. Pseudomonas aeruginosa MutL promotes large chromosomal deletions through non-homologous end joining to prevent bacteriophage predation. Nucleic Acids Res 46:4505–4514. 10.1093/nar/gky160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le S, Yao X, Lu S, Tan Y, Rao X, Li M, Jin X, Wang J, Zhao Y, Wu NC, Lux R, He X, Shi W, Hu F. 2014. Chromosomal DNA deletion confers phage resistance to Pseudomonas aeruginosa. Sci Rep 4:4738. 10.1038/srep04738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu GY, Nizet V. 2009. Color me bad: microbial pigments as virulence factors. Trends Microbiol 17:406–413. 10.1016/j.tim.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sichel G, Corsaro C, Scalia M, Di Bilio AJ, Bonomo RP. 1991. In vitro scavenger activity of some flavonoids and melanins against O2−dot. Free Radic Biol Med 11:1–8. 10.1016/0891-5849(91)90181-2. [DOI] [PubMed] [Google Scholar]
- 20.Gan EV, Haberman HF, Menon IA. 1976. Electron transfer properties of melanin. Arch Biochem Biophys 173:666–672. 10.1016/0003-9861(76)90304-0. [DOI] [PubMed] [Google Scholar]
- 21.Williamson PR. 1997. Laccase and melanin in the pathogenesis of Cryptococcus neoformans. Front Biosci 2:e99–e107. 10.2741/a231. [DOI] [PubMed] [Google Scholar]
- 22.de Cássia R Goncalves R, Pombeiro-Sponchiado SR. 2005. Antioxidant activity of the melanin pigment extracted from Aspergillus nidulans. Biol Pharm Bull 28:1129–1131. 10.1248/bpb.28.1129. [DOI] [PubMed] [Google Scholar]
- 23.Rodríguez-Rojas A, Mena A, Martín S, Borrell N, Oliver A, Blázquez J. 2009. Inactivation of the hmgA gene of Pseudomonas aeruginosa leads to pyomelanin hyperproduction, stress resistance and increased persistence in chronic lung infection. Microbiology (Reading) 155:1050–1057. 10.1099/mic.0.024745-0. [DOI] [PubMed] [Google Scholar]
- 24.Dean CR, Goldberg JB. 2002. Pseudomonas aeruginosa galU is required for a complete lipopolysaccharide core and repairs a secondary mutation in a PA103 (serogroup O11) wbpM mutant. FEMS Microbiol Lett 210:277–283. 10.1111/j.1574-6968.2002.tb11193.x. [DOI] [PubMed] [Google Scholar]
- 25.Priebe GP, Dean CR, Zaidi T, Meluleni GJ, Coleman FT, Coutinho YS, Noto MJ, Urban TA, Pier GB, Goldberg JB. 2004. The galU gene of Pseudomonas aeruginosa is required for corneal infection and efficient systemic spread following pneumonia but not for infection confined to the lung. Infect Immun 72:4224–4232. 10.1128/IAI.72.7.4224-4232.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kai-Larsen Y, Agerberth B. 2008. The role of the multifunctional peptide LL-37 in host defense. Front Biosci 13:3760–3767. 10.2741/2964. [DOI] [PubMed] [Google Scholar]
- 27.Hocquet D, Petitjean M, Rohmer L, Valot B, Kulasekara HD, Bedel E, Bertrand X, Plésiat P, Köhler T, Pantel A, Jacobs MA, Hoffman LR, Miller SI. 2016. Pyomelanin-producing Pseudomonas aeruginosa selected during chronic infections have a large chromosomal deletion which confers resistance to pyocins. Environ Microbiol 18:3482–3493. 10.1111/1462-2920.13336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yabuuchi E, Ohyama A. 1972. Characterization of “pyomelanin”-producing strains of Pseudomonas aeruginosa. Int J Syst Bacteriol 22:53–64. 10.1099/00207713-22-2-53. [DOI] [Google Scholar]
- 29.Ben-David Y, Zlotnik E, Zander I, Yerushalmi G, Shoshani S, Banin E. 2018. SawR a new regulator controlling pyomelanin synthesis in Pseudomonas aeruginosa. Microbiol Res 206:91–98. 10.1016/j.micres.2017.10.004. [DOI] [PubMed] [Google Scholar]
- 30.Ernst RK, D'Argenio DA, Ichikawa JK, Bangera MG, Selgrade S, Burns JL, Hiatt P, McCoy K, Brittnacher M, Kas A, Spencer DH, Olson MV, Ramsey BW, Lory S, Miller SI. 2003. Genome mosaicism is conserved but not unique in Pseudomonas aeruginosa isolates from the airways of young children with cystic fibrosis. Environ Microbiol 5:1341–1349. 10.1111/j.1462-2920.2003.00518.x. [DOI] [PubMed] [Google Scholar]
- 31.Hosseinidoust Z, Tufenkji N, van de Ven TGM. 2013. Predation in homogeneous and heterogeneous phage environments affects virulence determinants of Pseudomonas aeruginosa. Appl Environ Microbiol 79:2862–2871. 10.1128/AEM.03817-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pires DP, Dötsch A, Anderson EM, Hao Y, Khursigara CM, Lam JS, Sillankorva S, Azeredo J. 2017. A genotypic analysis of five P aeruginosa strains after biofilm infection by phages targeting different cell surface receptors. Front Microbiol 8:1229. 10.3389/fmicb.2017.01229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Markwitz P, Olszak T, Gula G, Kowalska M, Arabski M, Drulis-Kawa Z. 2021. Emerging phage resistance in Pseudomonas aeruginosa PAO1 is accompanied by an enhanced heterogeneity and reduced virulence. Viruses 13:1332. 10.3390/v13071332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanz-García F, Hernando-Amado S, Martínez JL. 2018. Mutation-Driven Evolution of Pseudomonas aeruginosa in the presence of either ceftazidime or ceftazidime-avibactam. Antimicrob Agents Chemother 62:e01379-18. 10.1128/AAC.01379-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gurney J, Pradier L, Griffin JS, Gougat-Barbera C, Chan BK, Turner PE, Kaltz O, Hochberg ME. 2020. Phage steering of antibiotic-resistance evolution in the bacterial pathogen, Pseudomonas aeruginosa. Evol Med Public Health 2020:148–157. 10.1093/emph/eoaa026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gordillo Altamirano FL, Barr JJ. 2019. Phage therapy in the postantibiotic era. Clin Microbiol Rev 32:e00066-18. 10.1128/CMR.00066-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Menon ND, Kumar MS, Satheesh Babu TG, Bose S, Vijayakumar G, Baswe M, Chatterjee M, D’Silva JR, Shetty K, Haripriyan J, Kumar A, Nair S, Somanath P, Nair BG, Nizet V, Kumar GB. 2021. A novel N4-like bacteriophage isolated from a wastewater source in South India with activity against several multidrug-resistant clinical Pseudomonas aeruginosa isolates. mSphere 6:e01215-20. 10.1128/mSphere.01215-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Held K, Ramage E, Jacobs M, Gallagher L, Manoil C. 2012. Sequence-verified two-allele transposon mutant library for Pseudomonas aeruginosa PAO1. J Bacteriol 194:6387–6389. 10.1128/JB.01479-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jacobs MA, Alwood A, Thaipisuttikul I, Spencer D, Haugen E, Ernst S, Will O, Kaul R, Raymond C, Levy R, Chun-Rong L, Guenthner D, Bovee D, Olson MV, Manoil C. 2003. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc Natl Acad Sci USA 100:14339–14344. 10.1073/pnas.2036282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi K-H, Schweizer HP. 2006. mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nat Protoc 1:153–161. 10.1038/nprot.2006.24. [DOI] [PubMed] [Google Scholar]
- 41.Nonejuie P, Burkart M, Pogliano K, Pogliano J. 2013. Bacterial cytological profiling rapidly identifies the cellular pathways targeted by antibacterial molecules. Proc Natl Acad Sci USA 110:16169–16174. 10.1073/pnas.1311066110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 and S2 and Tables S1 and S2. Download aac.00239-22-s0001.pdf, PDF file, 0.4 MB (417KB, pdf)
Data Availability Statement
Sequences are available under the following genome accession numbers in NCBI GenBank: MT416090 (vB_Pae_AM.P2), ON361134 (vB_Pae_Mat), ON361135 (vB_Pae_Kat), JAMKNC000000000 (AM.P2-X-1), CP096665 (Mat-X-1), and CP096664 (Kat-X-2).