

ABSTRACT
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the infectious agent that has caused the current coronavirus disease (COVID) pandemic. Viral infection relies on the viral S (spike) protein/cellular receptor ACE2 interaction. Disrupting this interaction would lead to early blockage of viral replication. To identify chemical tools to further study these functional interfaces, 139,146 compounds from different chemical libraries were screened through an S/ACE2 in silico virtual molecular model. The best compounds were selected for further characterization using both cellular and biochemical approaches, reiterating SARS-CoV-2 entry and the S/ACE2 interaction. We report here two selected hits, bis-indolyl pyridine AB-00011778 and triphenylamine AB-00047476. Both of these compounds can block the infectivity of lentiviral vectors pseudotyped with the SARS-CoV-2 S protein as well as wild-type and circulating variant SARS-CoV-2 strains in various human cell lines, including pulmonary cells naturally susceptible to infection. AlphaLISA and biolayer interferometry confirmed a direct inhibitory effect of these drugs on the S/ACE2 association. A specific study of the AB-00011778 inhibitory properties showed that this drug inhibits viral replication with a 50% effective concentration (EC50) between 0.1 and 0.5 μM depending on the cell lines. Molecular docking calculations of the interaction parameters of the molecules within the S/ACE2 complex from both wild-type and circulating variants of the virus showed that the molecules may target multiple sites within the S/ACE2 interface. Our work indicates that AB-00011778 constitutes a good tool for modulating this interface and a strong lead compound for further therapeutic purposes.
KEYWORDS: ACE2, COVID-19, inhibitor, SARS-CoV-2, spike, viral entry
INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a positive-strand RNA virus that causes severe respiratory syndrome (COVID-19) in humans. While several vaccines have been developed since the beginning of the pandemic, no efficient drugs have been identified as having a strong effect on viral replication for use in the clinic. Therefore, a better understanding of the viral replication processes and the search for new antiviral approaches remain of interest.
Viral entry into cells requires an interaction between the spike (S) protein of the virus and angiotensin-converting enzyme 2 (ACE2) at the surface of the cell membrane. ACE2 has been reported as a receptor for SARS-CoV-2 in the lung, as previously shown for SARS-CoV, which was responsible for the first SARS outbreak in 2003 (1, 2). ACE2 is expressed in human airway epithelia, lung parenchyma, vascular endothelia, kidney cells, and small intestine cells but is only weakly expressed in the brain (3, 4). Thus, converging data from the literature indicate that the S/ACE2 interaction remains the central entry pathway for SARS-CoV-2. More recently, the expression of the cellular protease TMPRSS2 has been shown to dictate the entry route used by the virus to infect cells (5). These entry routes, as well as the role of the S/ACE2 interaction in these processes, remain to be fully characterized. However, as the first contact point between the virus and the cell, the S/ACE2 complex constitutes an attractive target for blocking very early events of viral infection. Furthermore, in addition to being potential therapeutic agents, drugs targeting this association may constitute useful tools for dissecting the further processes of the entry step.
In this work, we used in silico, in vitro, and in cellulo models that allow the monitoring of the S/ACE2 complex (6) and select drugs that may target the S/ACE2 interface from 139,146 compounds. Using these systems, we selected approximatively 20 potential S/ACE2 inhibitors, and among them, we identified two novel chemical series capable of specifically blocking the SARS-CoV-2 entry step by impairing the ACE2/S interaction, leading to inhibition of viral SARS-CoV-2 replication in naturally susceptible cells. This work also provides molecular details on the inhibitory mechanism, paves the way for a potential new chemical strategy to interfere with the early stage of viral entry, and reports the first druggable protein/protein inhibitor (PPI) series able to disrupt the S/ACE2 interaction.
RESULTS
In silico virtual screening of drugs targeting the S/ACE2 interfaces.
To select potential drugs targeting the interaction between the spike protein and its cellular receptor, ACE2, we used the 100-ns molecular dynamics (MD) simulation previously performed (6) with the AMBER force field and TIP3P water box with the published S/ACE2 complex (7). The identified transient interfaces within the S/ACE2 complex were used after incorporating the dynamic nature of the interface to determine the pockets to be explored during the virtual screening campaigns. This simulation allowed us to identify druggable sites within the S/ACE2 complex, which are reported in Fig. 1A and B. These druggable sites were then used for the virtual drug screening steps. To this purpose, the Mu.Ta.Lig. Virtual Chemotheca (approximatively 60,000 molecules; http://chemotheca.unicz.it), the ZINC FDA Approved and ZINC In-Trials Database (1,379 and 5,811 molecules for evaluation for drug repurposing evaluation; https://zinc.docking.org), the French National Chemical Library (Chimiothèque Nationale [CN], 70,000 molecules; https://chembiofrance.cn.cnrs.fr/fr/composante/chimiotheque), and the Inhibitors of Protein-Protein Interactions Database (1,956 molecules; https://ippidb.pasteur.fr) were used. Among the 139,146 selected compounds, 200 molecules were first selected and classified based on their Vina and PLP scores when tested either on the full S/ACE2 complex (Fig. 1C) or the S protein alone (Fig. 1D). The best compounds from each library were then extracted, allowing us to select approximatively 20 hits for further analyses (see the list of selected compounds in Table S1 and their structure in Fig. S2, both in the supplemental material). Based on their originality and availability, we focused our present work on the molecules selected from the CN.
FIG 1.
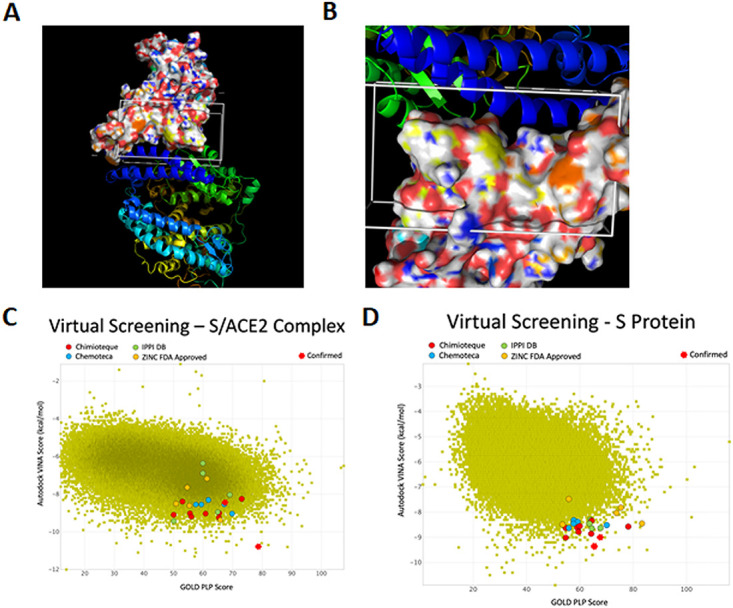
Selection of drugs targeting the S/ACE2 interaction by virtual screening studies. (A and B) The virtual screenings targeting the reported region in the S/ACE2 complex and in S alone were performed by molecular dynamics calculations. The surface representation corresponds to S, while the ribbon representations represent ACE2. (C and D) Results of the virtual drug screening.
In cellulo screening for SARS-CoV-2 S/ACE2-mediated entry.
The 10 best molecules from the CN (see their structure in Fig. S2) were selected and first tested on the lentiviral vector-based cellular approach previously set up in the laboratory (6) using lentiviral vectors pseudotyped with the SARS-CoV-2 spike protein (CoV-2 LV) and HEK293T-ACE2 cells containing 9 copies of the ACE2 gene (see reference 6 and Materials and Methods below for a full description of the procedures). As reported in Fig. S3A and B, the CoV-2 LVs were shown to efficiently transduce only the 293T-ACE2 cells, in contrast to the original 293T cells, leading to the integration and expression of the enhanced green fluorescent protein (eGFP) gene carried by the LV. This confirmed that the transduction efficiency indicated by the percentage of eGFP-positive cells relied on the early S/ACE2 interaction. In contrast, typical vesicular stomatitis virus G protein (vesicular stomatitis virus [VSV]-g) pseudotyped LVs able to transduce both 293T and 293T-ACE2 cells served as a specificity control for the next selection of drugs specific for S/ACE2 entry. Competition experiments using the soluble ACE2 protein fully validated the S/ACE2 dependency of LV CoV-2 infectivity, while LV VSV-g was not affected by the protein (Fig. S3C and reference 6).
The selected drugs were tested for both VSV-g and SARS-CoV-2 LV infectivity to determine their possible effects on the S/ACE2-mediated entry pathway. The first screening, using a 10 μM concentration of each compound, showed a significant inhibitory effect for certain molecules on CoV-2 LVs without any effect on VSV-g LVs (Fig. S4). Among the selected drugs, AB-00074850, AB-00054798, AB-00047476, and AB-00011778 were found to be the most efficient by inducing a 40% to 80% decrease in CoV-2 LV viral infectivity without any effect on VSV-g LV. Further accurate analysis of the inhibitory effects of the molecules using increasing concentrations showed that AB-00074860 and AB-00054798 exhibited only a modest inhibition of CoV-2 LV infectivity at concentrations below 10 μM while AB-00047476 and AB-00011778 conserved a significant inhibition capability without any effect on VSVg LV (Fig. 2). We thus decided to focus on these two molecules, AB-00047476 and AB-00011778. The cytotoxicity of the two compounds was then determined in different cell lines, including HEK293T, A549, Calu3, and Caco2, using the MTT 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide assay. No cytotoxicity was shown within the 0 to 30 μM range, while some slight decrease in cell viability could be observed at higher concentrations (Fig. S5).
FIG 2.

Effect of AB-00047476 (A) and AB-00011778 (B) on SARS-CoV-2 and VSV-g lentivirus infectivity. AB-00047476 and AB-00011778 were selected from 10 μM concentrations from a previous screen for further characterization using increasing concentrations of drug. Data are shown as the means of results of at least three independent experiments ± standard deviations. ***, P < 0.001; **, P < 0.01 (Student's t test).
Taken together, our data strongly suggest that both AB-00047476 and AB-00011778 interfered with the early S/ACE2-mediated SARS-CoV-2 entry process. To confirm this, we next investigated using molecular docking and biochemical approaches whether this inhibitory effect could be due to a direct effect of the drugs on the physical interaction between S and ACE2.
Molecular docking analysis of the drug interactions with S and ACE2.
To better understand the binding of AB-00011778 and AB-00047476 to the target protein, molecular docking studies were performed. Figure 3 displays the results from the predicted binding poses to the interface between the minimal receptor binding domain from SARS-CoV-2 spike protein (S-RBD) and ACE2 and to the S-RBD alone, highlighting the interacting amino acid residues.
FIG 3.

Molecular docking of AB-00011778 and AB-00047476 to the spike/ACE2 complex and to the spike protein alone. Data were obtained from the predicted binding poses of the compounds to the S-RBD and to the S-RBD/ACE2 interface. The interacting amino acid residues are highlighted. (A) AB-00011778 bound to the spike/ACE2 complex; (B) AB-00047476 bound to the spike/ACE2 complex; (C) AB-00011778 bound to the spike protein alone; (D) AB-00047476 bound to the spike protein alone.
The docking of AB-00011778 to the S-RDB/ACE2 interface (Fig. 3A) resulted in a score of 92.05. S-RDB residues Arg403, Gln409, and Lys417 establish strong hydrogen bonds with this molecule. Arg403 interacts with one oxygen from one of the sulfonyl groups with a distance of 3.00 Å. Gln409 and Lys417 interact with the nitrogen from the central pyridine with distances of 3.78 Å and 3.55 Å, respectively. In addition, the ACE2 His34 residue interacts with the indole group via π-π stacking interactions. ACE2 residues Lys26, Leu29, and Asp38 and S-RDB residues Lys417 and Gln493 are also involved in hydrophobic interactions with AB-00011778. The interactions of AB-00011778 with the amino acid residue Asp38 of ACE2 and with S-RDB Lys417 and Gln493 are of particular relevance. In fact, previous molecular docking studies of the S-RDB/ACE2 interface (6) have shown that Asp38 from ACE2 establishes important hydrogen bonds with S-RDB Tyr449 and Gln498, that S-RDB Lys417 was shown to be involved in very stable hydrogen bonds and charge interactions with Asp30 from ACE2, and that S-RDB Gln493 formed important hydrogen bonds with ACE2 Glu35. The disruption of these interactions caused by AB-00011778 binding should lead to a blockade of the S/ACE2 interaction and could be one of the reasons why this molecule is a successful inhibitor of this system.
The docking of AB-00011778 to the S-RBD (Fig. 3C) in the absence of ACE2 led to a score of 89.12. The obtained docking pose shows the formation of a strong hydrogen bond between Gly502 and one of the AB-0001778 ether groups, with a length of 1.90 Å. There is also the formation of π-π stacking interactions between Tyr505 of the indole group and two of the ligand’s phenyl groups. Finally, Lys417, Leu455, Asn501, and Tyr505 are involved in hydrophobic interactions with AB-00011778. Gly502 and Tyr505 have been implicated from molecular docking studies of the S-RDB/ACE2 interface to participate in very stable hydrogen bonds with ACE2, with Gly502 interacting with Lys353 and Tyr505 interacting with ACE2 Glu37, during 57% and 56% of the simulation times, respectively (6). Hence, the interactions of AB-00011778 with these two residues are likely to significantly interfere with the S-RDB/ACE2 association.
The docking of AB-00047476 to the S-RDB/ACE2 interface (Fig. 3B) resulted in a score of 100.71 (GOLD/PLP). The most favored pose suggests that this molecule forms one hydrogen bond through the nitrogen of one of its three benzothiazole groups and S-RDB Arg403, with a distance of 2.14 Å. In addition, AB-00047476 forms hydrophobic interactions with ACE2 Asn33, Thr92, and Pro389 and with S-RDB Lys417 and Tyr495.
The docking of AB-00047476 to the S-RDB protein alone (Fig. 3D) resulted in a score of 89.58. The most favored pose of this molecule reveals two hydrogen bonds: one between Arg403 and the nitrogen from one of the benzothiazole groups and another between Gly502 and the nitrogen from a second benzothiazole group, with lengths of 2.28 Å and 2.26 Å, respectively. There is also the formation of π-π stacking interactions between Tyr505 and one of the phenyl groups of the triphenylamine core in the center of the molecule. Residues Arg403, Lys417, Tyr449, and Tyr505 from the S-RDB are also involved in hydrophobic interactions with the molecule. The interactions of AB-00047476 with Tyr449, Gly502, and Tyr505 from the S-RDB are of particular relevance, as these residues have been shown to be important for the formation of the S-RDB/ACE2 complex. A previous molecular docking simulation (6) of the S-RDB/ACE2 complex showed that these residues of the spike protein are involved in important hydrogen bonds with ACE2. Tyr449 interacts with ACE2 Asp38, Gly502 interacts with ACE2 Lys353, and Tyr505 interacts with ACE2 Glu37. All of these are major interactions, being present during 54%, 57%, and 56% of the simulation times, respectively. Therefore, the results suggest that AB-00047476 binding to S-RDB would prevent the normal S-RDB/ACE2 association.
Additionally, the abilities of AB-00011778 and AB-00047476 to competitively bind to ACE2 with the spike protein was evaluated. Both molecules were docked into the ACE2 receptor, defining a potential binding region on the surface of ACE2 that is known to interact and recognize the spike protein. The results are presented in Fig. S6, showing that AB-00047476 and AB-00011778 can bind ACE2 with high binding scores that are comparable to those observed when these molecules were docked to the S-RDB/ACE2 interface and to S-RDB alone.
The docking of AB-00011778 to ACE2 alone resulted in a score of 81.41 (GOLD/PLP). The most stable pose reveals the formation of multiple hydrogen bonds between ACE2 and AB-00011778: (i) between Tyr41 and the nitrogen from the central pyridine group, with a length of 1.66 Å; (ii) between Gln325 and Gly326 interacting with the oxygen from one of the ligand’s ether groups, with lengths of 2.04 Å and 3.09 Å, respectively; and (iii) between Asn330 and one oxygen from one of the sulfonyl groups, with a distance of 2.10 Å. One can also observe the formation of one π-cation interaction between Lys353 and the phenyl ring from one of the indole groups. Glu37, Tyr41, Thr324, Gln325, and Lys353 are involved in hydrophobic interactions. Glu37, Tyr41, and Lys335 have also been previously implicated (6) to participate in important hydrogen bonds to the spike protein, also suggesting that the interaction of AB-00011778 with these residues could interfere with the ability of ACE2 to bind the spike protein.
The docking of AB-00047476 to ACE2 alone yielded a score of 78.19. The most stable pose suggests the formation of one hydrogen bond with a length of 2.39 Å between Lys353 and the nitrogen from one of the benzothiazole groups. AB-00047476 and ACE2 also interact via π-π stacking interactions. These interactions are observed between His34 of ACE2 and a second benzothiazole group and between Phe72 and a third benzothiazole group, with lengths of 3.47 and 4.57 Å, respectively. Hydrophobic interactions between AB-00047476 and ACE2 Glu37, Leu39, and Phe72 are also well defined. For most of the docking results shown here, there are some interactions featuring residues from ACE2 and S-RBD that have been reported to form hydrogen bonds. In this case, the interactions between AB-00047476 and ACE2 Glu37 and Lys353 are particularly noteworthy. During the MD simulations of the S-RDB/ACE interface (6), Glu37 was shown to form a hydrogen bond with S-RDB Tyr505 for 56% of the simulation times, while Lys353 formed a hydrogen bond with Gly502 for 57% of the simulation times. Disruption of these interactions on the side of ACE2 could further help explain the inhibitory capabilities of this molecule.
Effects on the S-RBD/ACE2 association in vitro.
To confirm that the selected drugs may physically prevent the formation of the S/ACE2 complex, we performed an AlphaLISA assay using the recombinant S minimal binding domain (RBD) fused to a 6×His tag [S-RBD(His)6] and human ACE2 protein (ACE2-Biot). For this purpose, the AlphaLISA assay previously set up (6) and described in the legend to Fig. S7 was adapted to 384-well microplates in a final reaction volume of 20 μL. As shown in Fig. S7 and Fig. 4A, the AlphaLISA data confirmed that both AB-00011778 and AB-00047476 could similarly interfere with the formation of the RBD/ACE2 complex, reaching 50% inhibition at 25 μM drug in both cases. A TruHits counterassay experiment did not show any significant effects from the compounds on the AslpaLISA (AS) signal, confirming the specific inhibition of the S-RBD/ACE2 interaction (Fig. S7). To obtain additional information regarding the effects of the drugs on the kinetics of the S-RBD/ACE2 interaction, we used the biolayer interferometry (BLI) approach set up previously (6). In these competition binding assays, biotinylated ACE2 was loaded onto streptavidin biosensors and binding kinetics were carried out with 50 nM S-RBD(His)6 preincubated with increasing concentrations of compound AB-00047476 or AB-00011778 as described in Materials and Methods. Strong binding of S-RBD(His)6 to ACE2 was observed, as indicated by the wavelength shift in the BLI sensorgrams (Fig. 4B and C). The kinetic binding parameters (absorption rate constant [ka] and dissociation constant [Kd]) and equilibrium dissociation constant (KD) were determined after local fitting of the association and dissociation curves using a 1:1 binding model with ForteBio BLItz Pro 1.1 software. The KD was found to be 3.42 ± 0.49 nM, confirming previous data (6, 7). The addition of either AB-00011778 or AB-00047476 inhibited the interaction, leading to a strong increase in the KD to 111.0 ± 16.0 nM and 68.8 ± 5.1 nM, respectively (Fig. 4D). Determination of the kinetic constants in the absence or presence of drug showed that the compounds interfered with both the S-RBD/ACE2 association and dissociation steps, indicating that they are able to not only prevent the formation of the S-RBD/ACE2 complex but also decrease its stability, confirming the docking analyses.
FIG 4.

Effects of AB-00011778 and AB-00047476 on the in vitro S-RBD/ACE2 interaction using AlphaLISA and biolayer interferometry (BLI) technologies. (A) SARS-CoV-2 S-RBD(His)6 and human ACE2-Biot interactions were first monitored by AlphaLISA using a 3 nM concentration of each protein. Increasing concentrations of AB-00011778 and AB-00047476 were incubated for 30 min with S-RBD(His)6 before being mixed with ACE2-Biot for 2 h. The microplate results were read after 2 h of incubation with the anti-6×His acceptor and streptavidin donor beads. Data obtained with the compounds were compared to the DMSO control and are reported as the mean percentage of binding inhibition from two or three independent experiments in duplicate ± standard deviation. (B and C) The effect of the molecules on the binding kinetics of S-RBD to biotinylated ACE2 immobilized on a streptavidin biosensor was also determined using BLI experiments. Baseline reaction buffer was measured for 60 s. For the association step (300 s), each loaded biosensor was dipped into 50 nM S-RBD(His)6 preincubated for 15 min with increasing concentrations of AB-00011778 or AB-00047476. The dissociation step was subsequently measured for 300 s. Sensorgram curves shown in panels B and C were plotted using Prism 5.0 software (GraphPad Software, La Jolla, CA). The association and dissociation experimental curves were locally fitted using a 1:1 binding model with BLItz Pro 1.1 software. (D) Kinetic binding parameters (kon, koff) and the equilibrium dissociation constant (KD) were determined as the means of results of two to three independent experiments. Data are shown as the means of results of two to three independent experiments ± standard deviations.
Effects on the S-RBD/ACE2 association in cellulo.
In a cellular context, both ACE2 and S are embedded in the membranous environment constituted by the viral envelope and the cellular membrane. Thus, we next investigated whether these compounds could also prevent the S/ACE2 interaction in a more physiological context. For this purpose, we used the cellular S-RBD/ACE2 interaction model developed previously (6). This model allows the association between recombinant S-RBD protein and the cellular membranous ACE2 protein to be monitored at the HEK293T-ACE2 cell surface by anti-S immunofluorescence (Fig. 5A). Flow cytometry analysis allowed the quantification of the global S-RBD association with the ACE2 protein (Fig. 5B), and competition with the soluble ACE2 protein confirmed the specificity of the interaction and validated the assay to test potential S/ACE2 inhibitors (Fig. 5C). The addition of AB-00014778 or AB-00047476 significantly decreased the efficiency of the S-RBD/ACE2 association at the surface of the cell (Fig. 5D). AB-00014778 was found to be more efficient than AB-00047476, which is in agreement with data obtained from the previous infectivity and BLI assays reported in Fig. 2 and 4.
FIG 5.

Effects of AB-00014778 and AB-00047476 on the S-RBD/ACE2 interaction in a cellular context. The SARS-CoV-2 S-RBD fragment was added to either HEK293T or HEK293-ACE2 cells for 0 to 60 min, and immunofluorescence staining was performed using an anti-6×His antibody and secondary antibody coupled to Alexa Fluor 488. (A) Cells were observed by epifluorescence microscopy. (B) Cells were also analyzed using flow cytometry to detect the percentage of FITC-positive cells over time. (C and D) Increasing concentrations of either soluble ACE2 (C) or drugs (D) were added to the RBD. Data are reported as the mean percentage of positive cells from three to five independent experiments ± standard deviation. **, P < 0.05; ***, P < 0.005 (Student's t test).
Together, the data obtained demonstrate that the two selected drugs block the early interaction between S and ACE2, supporting, in concert with the data obtained with pseudoviruses, their effect on the early entry pathway mediated by this interaction.
Effect of the drugs on wild-type SARS-CoV-2 infectivity in naturally susceptible human cells.
To complete the study, and prove the potency of the series, we next investigated whether this S/ACE2 inhibitory effect could sufficiently block the replication of wild-type circulating SARS-CoV-2 strains in different human cell lines naturally susceptible to infection.
The two best hits were tested in different pulmonary cell lines overexpressing, or not, the human ACE2 viral receptor after infection with the SARS-CoV-2 Wuhan reference strain. As shown in Fig. 6A, both molecules induced inhibition of viral replication in A549-ACE2 cells, with 50% effective concentration (EC50) values for AB-00011778 and AB-00047476 of 250 nM and 2.5 μM, respectively. The two molecules were then further tested in infection assays using the Calu3 human lung cancer cell line that naturally expresses the ACE2 receptor. As shown in Fig. 6B, an inhibitory effect on viral replication was confirmed for both compounds, leading to 50% inhibition of viral replication at a concentration of approximatively 1 μM.
FIG 6.

Effects of AB-00011778 and AB-00047476 on SARS-CoV-2 replication in human pulmonary cells. A549-ACE2 (A) or Calu3 (B) cells were incubated with the SARS-CoV-2 reference strain (MOI = 1) and increasing concentrations of the tested compounds. Replication was evaluated by quantifying the viral genome copies after 24 h. Data are reported as the means of results from five independent experiments and are expressed as a percentage of the control without molecule ± standard deviation. ***, P < 0.001; **, P < 0.01; *, P < 0.05 (Student's t test).
As reported in Table 1, based on the apparent 50% inhibitory concentration (IC50) and 50% cytotoxic concentration (CC50) values reported in the different cell lines, both compounds gave selectivity index (SI) values greater than 30, making them good candidates and anti-SARS-CoV-2-selective PPIs. AB-00011778 was found to be the most promising molecule, reaching an SI of >120 in A549-ACE2 cells.
TABLE 1.
IC50, CC50, and SI values for AB-00047476 and AB-00011778a
| Parameter | Value for: |
|||||
|---|---|---|---|---|---|---|
| AB-00047476 |
AB-00011778 |
|||||
| 293T-ACE2 | A549-ACE2 | Calu3 | 293T-ACE2 | A549-ACE2 | Calu3 | |
| IC50 | 1 | 2.5 | 5 | 1 | 0.25 | 1 |
| CC50 | >30 | >30 | >30 | >50 | >30 | >30 |
| SI | >30 | >12 | >6 | >50 | >120 | 30 |
CC50 was calculated using four-parameter variable slope sigmoidal dose-response models based on MTT assays.
Effects of the selected drugs on SARS-CoV-2 circulating variant forms.
To determine whether the selected drugs could be effective against the current major SARS-CoV-2 variants, we first performed in silico modeling and molecular dynamics simulations of the SARS-CoV-2 S/ACE2 complex with alpha, beta, gamma, and delta variants.
Models for the S-RDB/ACE2 complex formed with the different SARS-CoV-2 mutants were prepared starting from the X-ray structure 6M0J (resolution, 2.45 Å) (7), which represents the original SARS-CoV-2 spike protein receptor binding domain complexed with ACE2. Mutant variants were prepared by modeling the interface mutations using the mutagenesis feature in PyMOL 1.7.2.1 software, using the rotamer libraries available from Dunbrack (8). Selection of the initial amino acid conformation of each mutant was based on the dominant conformer predicted, excluding clashes with other residues. This computational mutagenesis protocol has been previously used with success in the study of other enzymes (9, 10). The mutations modeled are reported in Table S8.
All four systems were subjected to the same molecular mechanics minimization and molecular dynamics procedure previously described, employing AMBER and the ff14sb force field. Molecular dynamics simulations were run for 400 ns for each mutant. Final trajectories were analyzed in terms of backbone root mean square deviation (RMSD), hydrogen bonds formed, and cluster analysis. From the cluster analysis performed on each mutant, a representative structure from the dominant cluster of conformations obtained for each mutant was selected using cpptraj from AmberTools and prepared for the molecular docking stage.
Protein-protein interaction inhibitors AB-00011778 and AB-00047476 were docked into the structures of each of the mutants using the PLP scoring function in GOLD with 500 independent genetic algorithm runs. Molecules were docked into (i) the S-RDB/ACE2 interface and (ii) S-RDB alone. Table 2 presents the best scores obtained when docking the two molecules to the different variants, compared with the values obtained against the initial nonmutated S-RDB/ACE2 interface. For the omicron variant, the cluster analysis was performed on the 500-ns MD simulation trajectory performed by Lupala et al. (11).
TABLE 2.
GOLD/PLP docking scores of AB-00011778 and AB-00047476 for the structures of the different SARS-CoV-2 mutants
| SARS-CoV-2 | Docking score of drug to indicated structure |
|||
|---|---|---|---|---|
| AB-00011778 |
AB-00047476 |
|||
| S-RDB/ACE2 | S-RDB | S-RDB/ACE2 | S-RDB | |
| Initial | 92.05 | 89.12 | 100.71 | 89.58 |
| Alpha | 89.75 | 92.99 | 102.11 | 85.04 |
| Beta | 78.14 | 92.67 | 79.48 | 89.02 |
| Gamma | 91.97 | 93.96 | 90.71 | 92.70 |
| Delta | 81.57 | 87.30 | 93.84 | 93.01 |
| Omicron | 91.95 | 92.81 | 100.12 | 88.57 |
| Avg | 87.6 ± 6.2 | 91.6 ± 2.7 | 94.5 ± 7.1 | 89.7 ± 2.7 |
The results presented in Table 2 show that both molecules consistently bind with high docking scores to the different mutants, reinforcing the idea that these molecules can be equally effective in impairing the S/ACE2 association in the different mutants.
To confirm this, we tested the most promising drug (AB-00011778) on the four major circulating forms of SARS-CoV-2, namely, the original Wuhan strain and the alpha, delta, and omicron viruses in pulmonary A549-ACE2 cells. This compound blocked the viral replication of all variants with efficiency very similar to that of the Wuhan strain, leading to an IC50 of 0.5 to 1 μM (Fig. 7).
FIG 7.

Effects of AB-00011778 on SARS-CoV-2 Wuhan, alpha, delta, and omicron replication in human pulmonary cells. A549-ACE2-TMPRSS2 cells were incubated with the SARS-CoV-2 Wuhan, alpha, or delta strain (MOI = 1) and increasing concentrations of drugs. (A and B) Replication was evaluated by quantifying the viral gene E (A) and S (B) genome copies normalized to actin RNA after 24 h (results of a representative experiment are shown here). (C) Means of results from two to three independent experiments expressed as a percentage of the control without molecule ± standard deviation.
Taken together, these data indicate that AB-00011778 is efficient against the main current SARS-CoV-2 strains and show an original entry inhibition mechanism by possibly acting simultaneously on distinct S/ACE2 interfaces. This compound thus constitutes a strong candidate for further evaluation in ACE2-humanized animal models and future clinical trials as a new SARS-CoV-2 entry inhibitor.
DISCUSSION
SARS-CoV-2 S/ACE2-mediated entry remains a major antiviral target not yet exploited in therapy, while the molecular mechanisms underlying this process are poorly characterized. By using in silico screening, we selected 10 hits from the French National Chemical Library based on their GOLD/PLP and AutoDock Vina scores. Among these 10 compounds, we identified two compounds showing potent entry inhibition in lentivirus-based pseudovirus infectivity assays. The specific inhibition of SARS-CoV-2 LV infectivity in comparison with VSV-g LVs suggests that the selected drugs target the early S/ACE2-dependent entry process reiterated in this model. The simplest explanation for the inhibition mechanism is blockage of the S/ACE2 association due to binding of the drug to the S/ACE2 interface. Biochemical approaches, including AlphaLISA and BLI, confirmed a direct effect of the drugs to block the S-RBD/ACE2 interaction by acting to prevent association and to dissociate the complex. Detailed analysis of the effects of the drugs on the kinetic parameters of S/ACE2 complex formation indicated that they could not only prevent the association between the two partners but also decrease the stability of the complex. These data are in full agreement with the molecular docking calculations performed on the S/ACE2 complex structure and on the S-RDB and ACE2 proteins alone, indicating that both drugs could make contact with the viral and cellular proteins. Interestingly, the molecular modeling studies reported in this work suggest that the selected drugs may interact with both the S and ACE2 proteins. This fully agrees with the BLI data indicating that the molecules could interfere with the formation of the S/ACE2 complex and decrease the stability of the formed complex. This may constitute an original inhibition mechanism of simultaneous blocking of various S/ACE2 interfaces, increasing the inhibition potency in addition to decreasing the emergence of resistance. This may also provide new tools to further study the functional implication of these interfaces during the viral entry stage.
The two chosen compounds were also shown to inhibit the interaction between the soluble S-RBD and the ACE2 receptor expressed at the surface of HEK293T-ACE2 cells, confirming their capability to interfere with the formation of the S/ACE2 complex in a cell membrane context. To determine whether the selected drugs could be potential antiviral lead compounds, and to confirm the role of the S/ACE2 interfaces in the entry pathways, we tested them against wild-type viral replication using different cell lines, including naturally susceptible cells. The data indicated that both drugs were able to block, or reduce, the replication of the virus in pulmonary A549-ACE2 and Calu3 cells. While AB-00047476 was found to be less efficient in inhibiting viral replication in naturally susceptible pulmonary Calu3 cells, AB-00011778 maintained an efficient blocking effect, reaching 60 to 40% inhibition at 1 μM. Overall, both drugs showed IC50 values between 0.25 and 5 μM among all the tested cell lines without showing significant cytotoxicity, allowing them to reach selectivity indices of >10. Notably, AB-00011778 showed the best inhibitory effects in all the cell lines, with SI values of >30 to 120. Further analysis of the inhibitory effects of AB-00011778 showed that it could also block the replication of the current major circulating forms (Wuhan, alpha, delta, and omicron) of SARS-CoV-2 in A549-ACE2-TMPRSS2 pulmonary cells. Interestingly, both ACE2 and TMPRSS2 were shown to dictate the viral entry (5). Recent data from the literature showed that the expression of the cellular protease TMPRSS2 may dictate the entry pathway of the virus (5). The similar inhibition properties observed in A549-ACE2 and A549-ACE2-TMPRSS2 cells also indicate that the drug is able to block the different entry routes of the virus, underlining its broad antiviral properties. Taken together, these data identified AB-00011778 as a strong lead compound candidate for further development.
Aside from the recently licensed remdesivir and nirmatrelvir-ritonavir (Paxlovid), no effective drugs have been validated as potential specific anti-COVID treatments. Thus, the compounds reported here constitute the first molecules that act on the SARS-CoV-2 entry step by specifically targeting the S/ACE2 interfaces, making them very promising agents. A comparison of AB-00011778, remdesivir, and ritonavir (one of the two antiproteases included in Paxlovid) showed that while the remdesivir remains the most efficient molecule in our assay, AB-00011778 was more efficient than the ritonavir molecule in inhibiting the SARS-CoV-2 replication in human pulmonary A549-ACE2 cells (see Fig. S9 in the supplemental material). Of note, in contrast to the repurposed remdesivir and ritonavir drugs acting on different replication steps, AB-00011778 is an original molecule that has been specifically selected to block SARS-CoV-2 entry and has not yet been optimized. The low cytotoxicity observed for this molecule in various cell lines also paves the way for future animal model studies and clinical trials to fully evaluate their antiviral properties and therapeutic potency. Our work also provides a strong molecular and structural basis for further structure/function relationship studies to design potential optimized derivatives from the initially selected hits currently developed in our laboratory. Furthermore, these studies will help in the further characterization of the molecular mechanism underlying the inhibition of the S/ACE2 interaction by the selected drugs and will certainly allow optimization of this original antiviral process, leading to new promising agents usable for COVID-19 curative approaches.
MATERIALS AND METHODS
In silico molecular dynamics simulation of the SARS-CoV-2 S/ACE2 complex and virtual screening.
A model was prepared considering the spike protein S1 of SARS-CoV-2 complexed with human ACE2 starting from the X-ray structure 6M0J (resolution, 2.45 Å) (7). This structure contains the receptor binding domain (RBD) of S1 and includes all the amino acid residues of the RBD, with well-defined density between the residues on S1 RDB and ACE2. The model was treated with PropKa (12) to assign protonation states and subjected to molecular dynamics simulations for 400 ns based on the AMBER ff14SB force field as described in detail previously (6). Final trajectories were analyzed in terms of backbone root mean square deviation (RMSD), hydrogen bonds formed, and cluster analysis. From the cluster analysis performed on the ensemble of poses generated from the MD simulations, one representative structure from each of the 8 dominant clusters was selected and analyzed with FPocket software (13). This approached enabled the identification of possible druggable pockets in the interfacial S1-ACE2 region in each of the selected structures and in the initial model prepared from the X-ray structure. These were described in detail previously (6).
Structure-based virtual screening was performed using the following four molecular libraries: (i) the French National Chemical Library (Chimiothèque Nationale) (70,000 molecules; https://chembiofrance.cn.cnrs.fr/fr/composante/chimiotheque), (ii) the Mu.Ta.Lig. Virtual Chemotheca (ca. 60,000 molecules; http://chemotheca.unicz.it), (iii) the Inhibitors of Protein-Protein Interactions Database (1,956 molecules; https://ippidb.pasteur.fr), and (iv) the ZINC FDA Approved and ZINC In-Trials Database (1,379 and 5,811 molecules for evaluation for drug repurposing; https://zinc.docking.org). These libraries were screened against the SARS-CoV-2 S/ACE2 complex (model 1) (Fig. 1C) and against SARS-CoV-2 S alone (model 2) (Fig. 1D), with two independent docking software programs and scoring functions: AutoDock Vina (14) and GOLD (PLP scoring function) (15). It should be noted that the different docking program results have different values and scales. The score for PLP is dimensionless, and the higher the score, the better the binding affinity. On the other hand, AutoDock Vina uses a metric that resembles binding free energy (in kcal/mol), so a more negative value suggests a stronger affinity.
Compounds were sorted based on their Vina and PLP scores against model 1 and model 2. A selection of 20 compounds based on the most appealing compounds from each library was then made. This selection took into consideration not only the score against both models with Vina and/or PLP but also the chemical diversity among the molecules selected and the physicochemical properties of the molecules (Table S1 and Fig. S2).
Molecular docking of the LEAD compounds.
For the molecular docking of AB-00011778 and AB-00047476, the PLP scoring function from GOLD was used with 500 independent genetic algorithm runs. Molecules were docked into (i) the S-RDB/ACE2 interface, (ii) S-RDB alone, and (iii) ACE2 alone. For (ii) and (iii), the cavity for docking was defined based on the known binding region of the other partner in the complex formed (i.e., of ACE2 for the S-RDB model [ii] and S-RDB for the ACE2 model [iii]) plus a radius of 10 Å. The same procedure was later extended to AB-00011778_2 and AB-00011778_6. Analysis of the interactions was done with the help of the protein-ligand interaction profiler (16).
Chemicals.
Bis-indolyl pyridines and their derivatives have been synthetized as previously reported (17). The tris-benzothiazole triphenylamine derivative AB-00047476 belongs to the Institut Curie/CNRS chemical library (purity of >95% by proton 1H nuclear magnetic resonance [NMR] spectrometry), and its chemical synthesis was previously described (18). All the drugs had been prediluted in 100% dimethyl sulfoxide (DMSO), and the same volume of each serial predilution was added in the assays in order to keep the same DMSO concentrations and to avoid precipitation of the drug.
Proteins and antibodies.
SARS-CoV-2 (438–516) S-RBD(His)6, including the minimal receptor binding motif previously described (7, 19), and biotinylated human ACE2 were purchased from Fisher Scientific (reference no. 16534204 and 16545164, respectively). Monoclonal anti-6×His tag antibody was purchased from Abcam (reference no. ab18184; dilution, 1/200). Anti-α-tubulin antibody was purchased from Sigma (reference no. T6199; dilution, 1/500). Secondary goat anti-mouse antibody coupled to Alexa Fluor 488 was purchased from Fisher Scientific (reference no. Allo2g; dilution, 1/400).
AlphaLISA binding assays.
The AlphaLISA assay development was performed as previously described (5) using the recombinant S minimal binding domain (RBD) fused to a 6×His tag [S-RBD(His)6] and biotinylated human ACE2 protein (ACE2-Biot). The binding conditions were adapted for use in 384-well plates (white OptiPlate; reference no. 6007290; PerkinElmer, Waltham, MA) in a final reaction volume of 20 μL and are fully described in the legend to Fig. S7. Data were analyzed with GraphPad Prism 5.01 software. The AlphaLISA binding data obtained with the compounds were compared to the 1% DMSO control condition. For hit validation, a TruHits counterassay kit (reference no. AL900D; PerkinElmer) was used.
BLI binding assays.
Biolayer interferometry (BLI) experiments were performed on a BLItz instrument (Sartorius, Göttingen, Germany) to measure the binding of S-RBD(His)6 to ACE2-Biot. The BLI assay was set up as previously described (5) with some adaptations for the competition binding analysis with drugs. All sample dilutions and baseline steps were carried out using the same reaction buffer (phosphate-buffered saline, pH 7.4, 0.1% [wt/vol] bovine serum albumin, 1% [vol/vol] DMSO). Streptavidin biosensors (Sartorius; reference no. 18-5019) were first prewetted for 10 min with the reaction buffer, and 1 μM ACE2-Biot was then loaded onto the coated biosensors for 500 s in order to reach a binding value of ~3 nm. For the competition kinetic analyses, protein samples were prepared at a concentration of 50 nM S-RBD(His)6 in accordance with our previous BLI binding studies and were preincubated for 15 min at room temperature with increased concentrations (0 to 25 μM) of AB-00047476 or AB-00011778 drugs. Binding kinetics were divided into three steps. First, the baseline with the reaction buffer was measured for 60 s. For the association step, each loaded biosensor was dipped into S-RBD sample with drugs or DMSO as a control for 300 s with a 2,200-rpm shaking speed. Finally, the dissociation step of the bound S-RBD(His)6 was monitored by dipping the biosensor back into the reaction buffer for 300 s. Systematic baseline drift correction was done by subtracting the shift recorded for the biosensor loaded with ACE2 but incubated with reaction buffer. The association and dissociation experimental curves were locally fitted using a 1:1 binding model with the BLItz Pro 1.1 software, and the kinetic binding parameters (kon, koff) were determined as the means of results of two to three independent experiments. The equilibrium dissociation constant (KD) was calculated as KD = koff/kon. Sensorgram curves were plotted using Prism 5.0 software (GraphPad Software, La Jolla, CA).
Cells and lentiviral vector production.
Lentivirus vector production was done by the service platform Vect'UB, (INSERM US 005–CNRS UMS 3427–TBM-Core, Université de Bordeaux, France). Lentiviral particles were produced by transient transfection of HEK293T (human embryonic kidney) cells according to standard protocols. In brief, subconfluent HEK293T cells were cotransfected with a lentiviral genome (psPAX2) (gift from Didier Trono (Addgene plasmid no. 12260)), with an envelope coding plasmid (pMD2G-VSVG or wild-type SARS-CoV-2 spike protein [HDM_IDTSpike_fixK; kind gift from Bloom’s laboratory] [20]), and with vector constructs (44: pRRLSIN-PPT-hPGK-eGFP-WPRE or pHAGE_EF1alnt_ACE2_WT) by calcium phosphate precipitation. LVs were harvested 48 h posttransfection and concentrated by ultracentrifugation. Viral titers of VSV-g pseudotype pLV lentivectors were determined by transducing HEK293T cells with serial dilutions of viral supernatant, and eGFP expression was quantified 5 days later by flow cytometry analysis. For SARS-CoV-2 spike pseudotype, p24 antigen levels were measured in the concentrated viral supernatants by an enzyme-linked immunosorbent assay (Innotest HIV Ag nAb; Fugibio, France) and viral titers were estimated by comparing p24 antigen levels of each lentiviral supernatant with a similar VSV-g pseudotype lentiviral supernatant produced simultaneously.
HEK293T-ACE2 cell lines were generated by lentiviral transduction using pHAGE_EF1alnt_ACE2_WT plasmid (kind gift from Bloom’s laboratory [20]). HEK293T cells (200,000 cells) were then transduced with an optimized concentration of ACE2 lentiviral particles in 6-well plates. The efficacy of transduction was assessed using real-time PCR 10 days postinfection. ACE2 provirus DNA from this cell line was quantified by quantitative PCR (qPCR) using the ΔΔCT method in comparison with a 1-copy cell line. DNA from two different cell clones (human 293T and K562 cells), containing a single integrated copy of the provirus, was used as a normalized cell line. HEK293T cells were cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 1% penicillin-streptomycin and 10% fetal bovine serum (FBS).
A549 cells (human alveolar basal epithelial carcinoma), Calu3 (human bronchial epithelial carcinoma), and Vero E6 cells (African green monkey kidney cells) were maintained in DMEM (Invitrogen Life Technologies) supplemented with 10% heat-inactivated FBS, 1% penicillin/streptomycin (P/S), and 1% GlutaMAX (Life Technologies).
The MTT cell viability assay was performed in accordance with the manufacturer’s protocols and is further described in the legend to Fig. S5.
In cellulo imaging of the SARS-CoV-2 S-RBD/ACE2 interaction.
A total of 10,000 HEK293T and HEK293T-ACE2 cells were plated on glass coverslips pretreated with poly-l-lysine solution, 0.01% (Sigma reference no. P4832), for 5 min at room temperature. After 24 h, the RBD recombinant protein (4 to 40 pmol) was added to the cells in 100 μL phosphate-buffered saline (PBS). The cells were then washed and fixed (paraformaldehyde [PFA], 4%) at different time points. Cell imaging was performed as described above.
For quantification of the S-RBD/ACE2 interaction in a cellular context, 45,000 HEK293T and HEK293T-ACE2 cells were incubated for 45 min at 37°C in 100 μL DMEM and increasing concentrations of RBD recombinant protein. One milliliter of PBS was added, and the cells were centrifuged for 5 min at 2,500 rpm. Fifty microliters of anti-His antibody at 1/200 in DMEM was added, and the cells were incubated for a further 45 min at 37°C. After PBS washes, 50 μL of a 1/400 DMEM solution of secondary antibody was added and the cells were incubated for 30 min at 37°C. After PBS washes, the cells were resuspended in 200 μL PBS–2% FBS–2 mM EDTA and the percentage of fluorescein isothiocyanate (FITC)-positive cells was quantified by flow cytometry.
SARS-CoV-2 production and infection assays.
SARS-CoV-2 strain 220_95 (EPI_ISL_469284) was isolated from nasopharyngeal swab specimens collected at Service de Virologie (Hospital Saint Louis, Paris, France) and grown as previously described (21). Briefly, virus was propagated on Vero E6 cells in DMEM-2% (DMEM supplemented with 2% FBS, 1% P/S, 1% GlutaMAX, and 25 mM HEPES). Viruses were purified through a 20% sucrose cushion by ultracentrifugation at 80,000 × g for 2 h at 4°C. Pellets were resuspended in 1× HNE (25 mM HEPES, 100 mM NaCl, 0.5 mM EDTA), aliquoted, and stored at −80°C.
A549-Ace2 and Calu3 cells grown in 12-well plates were challenged with SARS-CoV-2 at a multiplicity of infection (MOI) of 0.05 in the presence of the indicated drug or with DMSO as a control. After 3 h, cells were washed once with PBS and incubated with fresh medium. Compounds were maintained throughout the course of infection. To assess productive viral replication, we quantified the infectious viral particle release during infection for 24 h postinfection by reverse transcription reverse transcriptase (RT)-qPCR. Viruses were first inactivated by incubating the supernatants by volume with 1% Triton X-100 (Sigma) in PBS for 30 min under agitation at room temperature. Yields of viral RNA were quantified by real-time qPCR by using SARS-CoV-2-specific primers targeting the E gene with the Luna universal one-step RT-qPCR kit (New England Biolabs) in a LightCycler 480 thermocycler (Roche) according to the manufacturer’s protocol. The number of viral genomes is expressed in PFU equivalents/mL and was calculated by performing a standard curve with a similarly treated supernatant from a viral stock with a known titer as described by Gordon et al. (22, 23).
ACKNOWLEGDMENTS
This work was developed by a consortium of scientists who signed up for the Crowdfight COVID-19 initiative (https://crowdfightcovid19.org) to help in the global effort against the COVID-19 pandemic.
We thank the Ligue Contre le Cancer du Grand Ouest (Comités des Deux Sèvres, du Finistère, de l’Ile et Villaine, du Loir et Cher, de Loire Atlantique, du Loiret, de la Vienne) for their financial support. This work was also supported by national funds from Fundação para a Ciência e a Tecnologia (grant numbers UIDP/04378/2020, UIDB/04378/2020, and 2020.01423.CEECIND/CP1596/CT0003). Some of the calculations were produced with the support of the INCD funded by the FCT and FEDER under project 01/SAICT/2016 number 022153 and project numbers CPCA/A00/7140/2020 and CPCA/A00/7145/2020.
The manuscript was edited for English by the Nature language editing service.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Walls AC, Park Y-J, Tortorici MA, Wall A, McGuire AT, Veesler D. 2020. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181:281–292.e6. 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M. 2003. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426:450–454. 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu J, Lazartigues E. 2020. Expression of ACE2 in human neurons supports the neuro-invasive potential of COVID-19 virus. Cell Mol Neurobiol 42:305–309. 10.1007/s10571-020-00915-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamming I, Timens W, Bulthuis M, Lely A, Navis G, van Goor H. 2004. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 203:631–637. 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koch J, Uckeley ZM, Doldan P, Stanifer M, Boulant S, Lozach P. 2021. TMPRSS2 expression dictates the entry route used by SARS‐CoV‐2 to infect host cells. EMBO J 40:e107821. 10.15252/embj.2021107821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lapaillerie D, Charlier C, Fernandes HS, Sousa SF, Lesbats P, Weigel P, Favereaux A, Guyonnet-Duperat V, Parissi V. 2021. In silico, in vitro and in cellulo models for monitoring SARS-CoV-2 spike/human ACE2 complex, viral entry and cell fusion. Viruses 13:365. 10.3390/v13030365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q, Shi X, Wang Q, Zhang L, Wang X. 2020. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581:215–220. 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 8.Shapovalov MV, Dunbrack RL. 2011. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 19:844–858. 10.1016/j.str.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Serrano C, Teixeira CSS, Cooper DN, Carneiro J, Lopes-Marques M, Stenson PD, Amorim A, Prata MJ, Sousa SF, Azevedo L. 2021. Compensatory epistasis explored by molecular dynamics simulations. Hum Genet 140:1329–1342. 10.1007/s00439-021-02307-x. [DOI] [PubMed] [Google Scholar]
- 10.Ferreira P, Sousa SF, Fernandes PA, Ramos MJ. 2017. Improving the catalytic power of the DszD enzyme for the biodesulfurization of crude oil and derivatives. Chemistry 23:17231–17241. 10.1002/chem.201704057. [DOI] [PubMed] [Google Scholar]
- 11.Lupala CS, Ye Y, Chen H, Su X-D, Liu H. 2022. Mutations on RBD of SARS-CoV-2 Omicron variant result in stronger binding to human ACE2 receptor. Biochem Biophys Res Commun 590:34–41. 10.1016/j.bbrc.2021.12.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olsson MHM, Søndergaard CR, Rostkowski M, Jensen JH. 2011. PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J Chem Theory Comput 7:525–537. 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- 13.Le Guilloux V, Schmidtke P, Tuffery P. 2009. Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics 10:168. 10.1186/1471-2105-10-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trott O, Olson AJ. 2010. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korb O, Stützle T, Exner TE. 2009. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J Chem Inf Model 49:84–96. 10.1021/ci800298z. [DOI] [PubMed] [Google Scholar]
- 16.Adasme MF, Linnemann KL, Bolz SN, Kaiser F, Salentin S, Haupt VJ, Schroeder M. 2021. PLIP 2021: expanding the scope of the protein-ligand interaction profiler to DNA and RNA. Nucleic Acids Res 49:W530–W534. 10.1093/nar/gkab294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacquemard U, Dias N, Lansiaux A, Bailly C, Logé C, Robert J-M, Lozach O, Meijer L, Mérour J-Y, Routier S. 2008. Synthesis of 3,5-bis(2-indolyl)pyridine and 3-[(2-indolyl)-5-phenyl]pyridine derivatives as CDK inhibitors and cytotoxic agents. Bioorg Med Chem 16:4932–4953. 10.1016/j.bmc.2008.03.034. [DOI] [PubMed] [Google Scholar]
- 18.Bordeau G, Lartia R, Teulade-Fichou M-P. 2010. meta-Substituted triphenylamines as new dyes displaying exceptionally large Stokes shifts. Tetrahedron Lett 51:4429–4432. 10.1016/j.tetlet.2010.06.082. [DOI] [Google Scholar]
- 19.Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, Abiona O, Graham BS, McLellan JS. 2020. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367:1260–1263. 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crawford KHD, Eguia R, Dingens AS, Loes AN, Malone KD, Wolf CR, Chu HY, Tortorici MA, Veesler D, Murphy M, Pettie D, King NP, Balazs AB, Bloom JD. 2020. Protocol and reagents for pseudotyping lentiviral particles with SARS-CoV-2 spike protein for neutralization assays. Viruses 12:513. 10.3390/v12050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Onodi F, Bonnet-Madin L, Meertens L, Karpf L, Poirot J, Zhang S-Y, Picard C, Puel A, Jouanguy E, Zhang Q, Le Goff J, Molina JM, Delaugerre C, Casanova JL, Amara A, Soumelis V. 2021. SARS-CoV-2 induces human plasmacytoid predendritic cell diversification via UNC93B and IRAK4. J Exp Med 218:e20201387. 10.1084/jem.20201387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gordon DE, Hiatt J, Bouhaddou M, Rezelj VV, Ulferts S, Braberg H, Jureka AS, Obernier K, Guo JZ, Batra J, Kaake RM, Weckstein AR, Owens TW, Gupta M, Pourmal S, Titus EW, Cakir M, Soucheray M, McGregor M, Cakir Z, Jang G, O’Meara MJ, Tummino TA, Zhang Z, Foussard H, Rojc A, Zhou Y, Kuchenov D, Hüttenhain R, Xu J, Eckhardt M, Swaney DL, Fabius JM, Ummadi M, Tutuncuoglu B, Rathore U, Modak M, Haas P, Haas KM, Naing ZZC, Pulido EH, Shi Y, Barrio-Hernandez I, Memon D, Petsalaki E, Dunham A, Marrero MC, Burke D, Koh C, Vallet T, QCRG Structural Biology Consortium, et al. 2020. Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science 370. 10.1126/science.abe9403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, O'Meara MJ, Guo JZ, Swaney DL, Tummino TA, Hüttenhain R, Kaake RM, Richards AL, Tutuncuoglu B, Foussard H, Batra J, Haas K, Modak M, Kim M, Haas P, Polacco BJ, Braberg H, Fabius JM, Eckhardt M, Soucheray M, Bennett MJ, Cakir M, McGregor MJ, Li Q, Naing ZZC, Zhou Y, Peng S, Kirby IT, Melnyk JE, Chorba JS, Lou K, Dai SA, Shen W, Shi Y, Zhang Z, Barrio-Hernandez I, Memon D, Hernandez-Armenta C, Mathy CJP, Perica T, Pilla KB, Ganesan SJ, Saltzberg DJ, Ramachandran R, Liu X, Rosenthal SB, et al. 2020. A SARS-CoV-2-human protein-protein interaction map reveals drug targets and potential drug-repurposing. bioRxiv. 10.1101/2020.03.22.002386. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S9. Download aac.00083-22-s0001.pdf, PDF file, 1.3 MB (1.3MB, pdf)