

ABSTRACT
The bacterium Pseudomonas aeruginosa can colonize the airways of patients with chronic lung disease. Within the lung, P. aeruginosa forms biofilms that can enhance resistance to antibiotics and immune defenses. P. aeruginosa biofilm formation is dependent on the secretion of matrix exopolysaccharides, including Pel and Psl. In this study, recombinant glycoside hydrolases (GHs) that degrade Pel and Psl were evaluated alone and in combination with antibiotics in a mouse model of P. aeruginosa infection. Intratracheal GH administration was well tolerated by mice. Pharmacokinetic analysis revealed that, although GHs have short half-lives, administration of two GHs in combination resulted in increased GH persistence. Combining GH prophylaxis and treatment with the antibiotic ciprofloxacin resulted in greater reduction in pulmonary bacterial burden than that with either agent alone. This study lays the foundation for further exploration of GH therapy in bacterial infections.
KEYWORDS: Pseudomonas aeruginosa, antibiotic, antimicrobial combinations, biofilm, exopolysaccharide, bacteria, Pel, Psl, glycoside hydrolase (GH), acute pulmonary infection
INTRODUCTION
The bacterium Pseudomonas aeruginosa is a Gram-negative organism and a common cause of respiratory infections in adult patients with chronic pulmonary disease, such as chronic obstructive pulmonary disease, cystic fibrosis, bronchiectasis, and asthma (1–4). P. aeruginosa infection accelerates the progressive destruction of the airways and consequent decline in lung function in these chronic pulmonary diseases (2). Despite early therapeutic intervention, P. aeruginosa remains a predominant and persistent airway microbial pathogen that is associated with increased morbidity and mortality rates for patients, which underscores the need for novel treatment strategies (5–7). One approach to improving patient outcomes is to target molecules involved in bacterial pathogenesis.
During infection, P. aeruginosa forms communities of aggregated bacterial cells embedded in an extracellular matrix of self-produced and host-derived components known as a biofilm (8–12). The bacterium-derived biofilm matrix is dependent on the production of macromolecules such as proteins (13, 14), extracellular DNA (15, 16), and exopolysaccharides (11, 17, 18). Of these matrix molecules, exopolysaccharides play a key role in biofilm formation (17, 19). During biofilm formation, the synthesis of three exopolysaccharides, namely, Pel, Psl, and alginate, can be upregulated depending on the P. aeruginosa strain and host-mediated selective pressures (8, 10, 19, 20). Pel is a cationic polymer composed predominantly of N-acetyl-d-galactosamine (GalNAc) and N-acetylated d-glucosamine (GalN) residues, one or both of which are partially de-N-acetylated (18). Psl is a branched neutral polymer composed of pentasaccharide repeats of d-mannose, l-rhamnose, and d-glucose (21). Alginate produced by mucoid Pseudomonas strains is an anionic exopolysaccharide composed of nonrepeating β-1,4-linked partially O-acetylated d-mannuronic acid and l-guluronic acid (22, 23). Pel and/or Psl production is essential for biofilm formation in vitro, as mutants lacking the ability to produce both polymers are impaired in biofilm formation (19, 24), while alginate is dispensable to biofilm architecture (24–29). Psl initiates bacterial surface attachment and maintains biofilm architecture in Psl-dominant strains, whereas Pel mediates cell-cell interactions for multicellular aggregate formation in Pel-dependent strains (30). Redundancy in the structural roles of these polymers has been demonstrated, and Pel and Psl were found to contribute interchangeably to the biofilm architecture (19, 24). The Pel/Psl matrix functions as a barrier to antimicrobial agents and host defenses, thereby enhancing the persistence of P. aeruginosa. Psl provides a barrier to penetration by antibiotics in newly formed biofilms, while protection mediated by Pel has been observed in more established mature biofilms (31). Pel enhances the resistance of Pel-dominant P. aeruginosa biofilms to killing by neutrophil-like leukocytes (32), whereas Psl enhances the resistance of Psl-dominant P. aeruginosa to opsonophagocytosis by neutrophils (33). Production of Psl and/or Pel enhances the fitness of P. aeruginosa in acute models of pulmonary infection (24, 33). The genetic capacity for P. aeruginosa to synthesize biofilm exopolysaccharides with redundant roles may allow bacterial cells to adapt to the selective pressures of chronically inflamed airways.
The dependence of P. aeruginosa on the structural and functional roles of Pel and Psl in bacterial pathogenesis led us to hypothesize that targeting Pel and Psl to disrupt the biofilm matrix encasing P. aeruginosa cells may be an effective therapeutic approach. Within the 7- and 12-gene operons that encode the Pel and Psl biosynthetic machinery, respectively, are genes that are predicted to encode glycoside hydrolases (GHs) (21, 29, 34, 35). The pelA gene within the Pel operon encodes the multidomain enzyme PelA, which contains a GH domain (34). The pslG gene in the Psl operon is predicted to encode the GH PslG (35). Mass spectrometry enzymatic fingerprinting and nuclear magnetic resonance (NMR) studies using the soluble recombinant GH domain from the PelA protein (PelAh) revealed that PelAh is an endo-α-1-4-N-acetyl-d-galactosaminidase from the GH166 family that cleaves GalNAc-GalNAc linkages within acetylated and deacetylated regions of GalN-GalNAc oligomers (36). Although the enzymatic activity of PslG has not been elucidated in detail, structure-function studies with PslGh suggest that the enzyme contains a catalytic groove that resembles endo-acting GHs (35). In biofilm disruption studies performed in vitro, both soluble recombinant PelAh and PslGh hydrolyzed Pel and Psl within Pel- and Psl-dependent biofilms, respectively, and both inhibited biofilm formation and disrupted multicellular aggregates in mature biofilms (32). A combination of PslGh and PelAh (PslGh-PelAh) was more effective than either GH alone in disrupting biofilms formed by a matrix-hyperproducing isolate, demonstrating that these GHs are compatible in combination and effective against strains that are genetically capable of upregulating expression of both exopolysaccharide operons (19, 32, 37).
Early studies have suggested that combining PslGh and PelAh with antimicrobials is a promising approach to treating P. aeruginosa infections. Combining PslGh or PelAh with the antimicrobial peptide colistin reduced the viability of Pel- or Psl-dependent P. aeruginosa (32), and a combination of PslGh and PelAh enhanced the bactericidal activity of the aminoglycosides tobramycin and neomycin and the antimicrobial peptides polymyxin and colistin against Psl-dependent P. aeruginosa biofilms (38). PelAh disruption of mature Pel-dependent biofilms increased the level of killing of P. aeruginosa by leukocyte-like cells, suggesting that PelAh can potentiate the responses of host immune cells (32). In an acute wound model of P. aeruginosa infection, PslGh-tobramycin prophylaxis enhanced bacterial clearance (38). Collectively, these studies demonstrate that GHs can potentiate antimicrobial activity and potentially enhance host cell responses against persistent P. aeruginosa infection (8, 18, 19, 32).
The biofilm-forming mold Aspergillus fumigatus secretes a Pel-like cationic matrix exopolysaccharide galactosaminogalactan (GAG) that is composed of α-1,4-linked d-galactose and partially deacetylated GalNAc residues (39). As with Pel, the GAG biosynthetic pathway includes a GH enzyme, Ega3 (40–43). Ega3 is an endo-α-1-4-d-galactosaminidase with specificity for GalN-GalN linkages, and treatment with the recombinant hydrolase domain of Ega3 (Ega3h) can disrupt both GAG-dependent fungal and Pel-dependent P. aeruginosa biofilms in vitro (42). Interestingly, PelAh also exhibits cross-kingdom antibiofilm activity and can disrupt GAG-dependent A. fumigatus biofilms, suggesting the presence of regions of similar composition in the two polymers (44).
Here, the tolerability and anti-P. aeruginosa activity of recombinant GH prophylaxis with the combination of PslGh and PelAh (PslGh-PelAh) or PslGh and Ega3h (PslGh-Ega3h), alone and with antibiotics, were evaluated in vitro and in vivo. Checkerboard studies were performed to test the activity of PslGh-PelAh or PslGh-Ega3h in combination with the fluoroquinolone ciprofloxacin or the cephalosporin ceftazidime against P. aeruginosa biofilms in vitro. GH combinations potentiated the antibacterial activity of ciprofloxacin and ceftazidime. Intratracheal administration of single-dose combination PslGh-PelAh or PslGh-Ega3h was well tolerated by uninfected mice. In a mouse model of acute pulmonary P. aeruginosa infection, the administration of single-dose PslGh-PelAh at the time of infection potentiated the antibacterial activity of ciprofloxacin but not that of ceftazidime, whereas PslGh-Ega3h failed to potentiate either ciprofloxacin or ceftazidime activity. Administration of GH combinations without antibiotics did not reduce pulmonary bacterial burden and was associated with a trend toward increased hematogenous dissemination of P. aeruginosa. Collectively, these findings suggest that specific GH antibiotic combinations may have potential for the prevention and/or treatment of pulmonary of P. aeruginosa infections.
RESULTS
GHs enhance antibiotic activity against P. aeruginosa biofilms in vitro.
Previous studies of PslGh and PelAh produced in Escherichia coli or Ega3h produced in Pichia pastoris (Ega3h) demonstrated that these soluble recombinant GH domains could disrupt Psl-dependent or Pel-dependent P. aeruginosa biofilms by degrading Pel or Psl, respectively (32, 35, 42). To extend these findings, dose-response matrix analyses were performed to evaluate the antimicrobial activity of GH-antibiotic combinations against P. aeruginosa biofilms. Given the genetic capacity of the P. aeruginosa PAO1 strain to produce Psl and Pel, combinations of Psl-degrading PslGh and either the Pel-degrading PelAh or Ega3h were tested for their ability to augment the antimicrobial activity of ciprofloxacin and ceftazidime. In comparison to ciprofloxacin treatment alone, greater biofilm biomass reduction was observed with increasing doses of PslGh-PelAh beginning at 0.625 μM combined with ciprofloxacin at ≥250 μg/mL (Fig. 1). In contrast, additive biomass reduction was observed with concentrations of PslGh-Ega3h beginning at 0.625 μM combined with ciprofloxacin at concentrations of ≥15.65 μg/mL (Fig. 1). These results suggest that, while ciprofloxacin activity against biofilm biomass was potentiated by either PslGh-PelAh or PslGh-Ega3h, PslGh-Ega3h exhibited greater potency than PslGh-PelAh. In the case of ceftazidime, PslGh-PelAh or PslGh-Ega3h combinations beginning at 0.625 μM enhanced the activity of ceftazidime at concentrations of ≥3.9 μg/mL (Fig. 1). Together, these observations suggest that PslGh-PelAh and PslGh-Ega3h had greater potentiation effects when used in combination with ceftazidime, compared with ciprofloxacin.
FIG 1.

GHs potentiate antibiotic activity against P. aeruginosa biofilms in vitro. Dose-response matrix analyses of P. aeruginosa biofilms cotreated with 2-fold serial dilutions of either PslGh-PelAh or PslGh-Ega3h and 4-fold serial dilutions of ceftazidime or ciprofloxacin in a MBEC high-throughput assay as indicated are shown. Biofilm biomass was quantified with crystal violet staining. Colored squares represent the ratio of treated to untreated biofilms of ≥2 independent experiments. Lighter yellow boxes represent high biofilm biomass and darker green boxes represent low biofilm biomass, compared with untreated control wells.
Intratracheal GH treatment is well tolerated by mice.
Previously, we found that the intratracheal administration of a single dose of up to 500 μg PelAh or Ega3h alone was well tolerated by mice (45). To extend this work, the tolerability of intratracheal PelAh or Ega3h in combination with PslGh was tested. BALB/c mice were administered a single intratracheal dose of PslGh-PelAh or PslGh-Ega3h (up to 250/250 μg of each GH), monitored for changes in weight and temperature, and then euthanized 6 days later for measurement of pulmonary injury and inflammation. Treatment with a single dose of up to 250/250 μg of PslGh-PelAh or PslGh-Ega3h was well tolerated by mice, without signs of respiratory distress or death. GH-treated mice exhibited no difference in body weight and temperature, compared with mice treated with buffer alone (see Fig. S1 and S2 in the supplemental material).
To test whether intratracheal GH therapy induced pulmonary injury, pulmonary damage was assessed by measuring lactate dehydrogenase activity in bronchoalveolar lavage (BAL) fluid from mouse lungs. No significant increase in lactate dehydrogenase activity was detected in the BAL fluid from mice treated with a 250/250-μg dose of PslGh-PelAh or PslGh-Ega3h, compared with mice treated with buffer alone (Fig. 2A), suggesting that single-dose GH treatment does not induce pulmonary injury in mice. Consistent with these findings, histological examination of pulmonary tissues did not reveal any differences between GH-treated and buffer-treated mice (see Fig. S3).
FIG 2.

Intratracheal GH therapy does not induce pulmonary damage. Immunocompetent BALB/c mice were intratracheally administered a single dose of 250/250 μg PslGh-PelAh or PslGh-Ega3h. (A) Lactate dehydrogenase activity was quantified in BAL fluid. (B) Pulmonary leukocyte populations, including lymphocytes, macrophages, eosinophils, and neutrophils, were quantified by flow cytometry 7 days after GH treatment. Bars represent the means ± standard errors of 4 independent experiments with ≥13 mice per group. Asterisks indicate significant differences (P < 0.0020), relative to the buffer-treated group and where there are no asterisks no significant differences were observed as compared with the buffer-treated group (P > 0.7794), as determined by two-way ANOVA with Dunnett’s multiple-comparison test.
To further probe the host response to intratracheal GH treatment, pulmonary leukocytes from GH-treated mice were quantified by flow cytometry (Fig. 2B). PslGh-PelAh administration to mice at a 250/250-μg dose had no effect on macrophage, eosinophil, and neutrophil numbers. A significant increase in pulmonary lymphocyte numbers was observed following treatment with 250/250 μg of PslGh-PelAh. Treatment with PslGh-Ega3h was associated with a significant increase in pulmonary eosinophils and a small but significant increase in neutrophils in mice at the 250/250-μg dose. Treatment with PslGh-Ega3h at the 250/250-μg dose resulted in effects on macrophage and lymphocyte populations similar to those observed with PslGh-PelAh treatment. Taken together, these data suggest that, while 250/250-μg doses of PslGh-PelAh and PslGh-Ega3h are likely nearing the maximal tolerated intratracheal dose, the PslGh-Ega3h combination induces a greater inflammatory response than PslGh-PelAh and may be less well tolerated by mice.
Combined GHs have longer pulmonary half-lives than individual GHs alone.
To inform the design of efficacy studies, the pulmonary pharmacokinetics of GHs alone and in combination were determined. Mice were given a single intratracheal dose of 500 μg PelAh, PslGh, or Ega3h, and at select time points their lungs were harvested and homogenized in a cocktail of protease inhibitors to prevent degradation of GHs. Lung homogenates were assayed by Western blotting using specific rabbit anti-GH antibodies and quantified by densitometry. PslGh displayed the longest half-life, i.e., 18 h, compared to PelAh and Ega3h with half-lives of 3 h and 9 h, respectively (Fig. 3A). To evaluate the stability of each GH in GH combinations, mice were given a single 250/250-μg intratracheal dose of PslGh-PelAh or PslGh-Ega3h. The half-life of PslGh in combination therapy was lower than that when administered alone (16 h with PelAh and 12 h with Ega3h) (Fig. 3B and C). In contrast, compared with GH monotherapy, the half-lives of PelAh and Ega3h in combination with PslGh increased to 5 h from 3 h and to 12 h from 9 h, respectively (Fig. 3B and C). These data suggest that, although the persistence of PslGh is marginally reduced in combination with either PelAh or Ega3h, PelAh and Ega3h are more stable in combination with PslGh than when administered alone.
FIG 3.
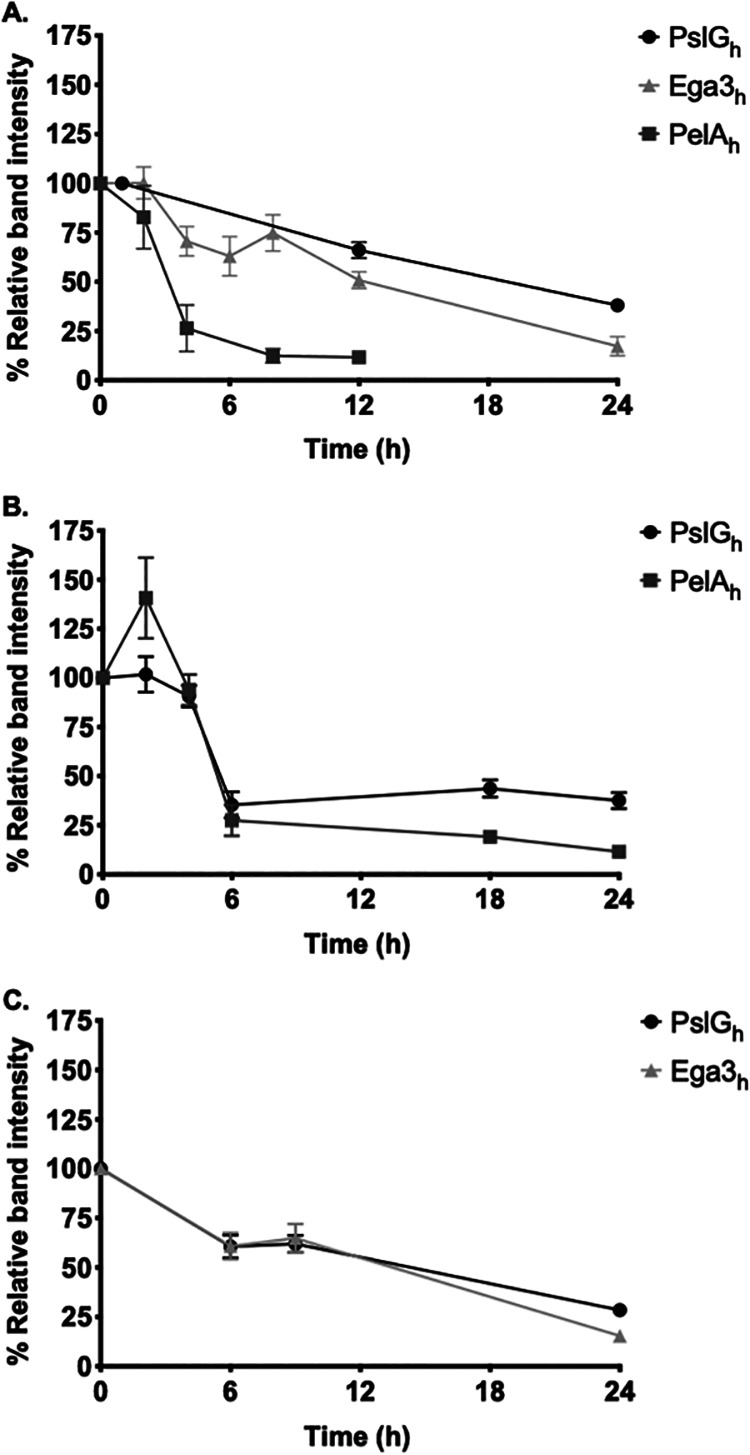
Pulmonary pharmacokinetics of intratracheally administered GHs. Mice were treated intratracheally with a single dose of 500 μg PelAh, Ega3h, or PslGh (A) or 250/250 μg each of a combination of PslGh and PelAh (B) or PslGh and Ega3h (C) and then sacrificed at the indicated time points. Lung homogenates were assessed by Western blotting. Dots represent the means ± standard errors of band intensities normalized to total band intensity at 0 h from 2 independent experiments with ≥5 mice per time point.
To determine whether the short half-lives of the GHs might be a consequence of proteolytic degradation, 10 μg of PelAh and/or PslGh was treated with 100 μg/mL neutrophil elastase in vitro. Degradation of PelAh and PslGh by neutrophil elastase was detectable as early as 1 h (see Fig. S4). Consistent with the shorter half-life of PelAh in vivo, PelAh exhibited greater sensitivity to elastase than PslGh and was no longer detectable after 24 h of coincubation.
Attenuation of bacterial virulence by prophylaxis with GHs in combination with antibiotic treatment is drug specific in an acute model of pulmonary P. aeruginosa infection.
PslGh-Ega3h induced a greater inflammatory response than PslGh-PelAh. In previous studies of Ega3 for therapy of invasive aspergillosis, we observed that administration of a soluble recombinant form of Ega3 produced in a human embryonic kidney (HEK) cell line (Ega3h-HEK) was less inflammatory than Ega3h produced in yeast (45). Therefore, for subsequent in vivo efficacy studies, we used Ega3h-HEK in combination with PslGh to reduce inflammatory responses that might have been generated against fungal glycosylation patterns or trace amounts of fungal β-glucan in Pichia-derived Ega3h preparations (46, 47).
The antibacterial effects of intratracheal GHs alone and in combination with antibiotics were assessed in a mouse model of acute P. aeruginosa infection. Mice were intratracheally infected with P. aeruginosa and administered a single dose of 250/250 μg PslGh-PelAh or PslGh-Ega3h-HEK alone or in combination with a subtherapeutic dose of ciprofloxacin (10 mg/kg every 8 h). Twenty-four hours following infection, pulmonary and blood CFU were quantified as a measure of bacterial burden. Prophylaxis with PslGh-PelAh or PslGh-Ega3h-HEK alone did not reduce pulmonary bacterial burden, compared with buffer-treated mice (Fig. 4A and B). Blood cultures of infected mice revealed a higher rate of bacteremia in animals that received PslGh-PelAh, in comparison to those that received PslGh-Ega3h-HEK or buffer (30% versus 10% versus 12.5%, respectively) (Tables 1 and 2). These observations are consistent with histological examinations of pulmonary tissues, which revealed increased inflammation in PslGh-PelAh-treated animals, compared with animals that received PslGh-Ega3h-HEK (see Fig. S5). These data suggest that GH therapy alone may increase bacterial dispersion and actually worsen outcomes for pulmonary P. aeruginosa infections. In contrast, a significant reduction in pulmonary bacterial burden was observed in infected mice that received PslGh-PelAh-ciprofloxacin in combination, compared to ciprofloxacin or PslGh-PelAh alone (Fig. 4A). This observation suggests that GH pretreatment potentiated the antimicrobial activity of ciprofloxacin in vivo and that combination therapy might be more effective than ciprofloxacin therapy alone (Fig. 4A). Bacteremia was not observed in mice treated with the PslGh-PelAh-ciprofloxacin combination (Table 1), suggesting that coadministration of antibiotics can also protect against GH-mediated bacterial dissemination. The pulmonary bacterial burden of mice treated with PslGh-Ega3h-HEK-ciprofloxacin was not significantly different from that of mice treated with ciprofloxacin alone (Fig. 4B), suggesting that GH potentiation of ciprofloxacin activity is dependent on the combination of GHs.
FIG 4.

Specific GH-antibiotic combinations reduce bacterial burden in an acute mouse model of pulmonary P. aeruginosa infection. Mice were intratracheally infected with 3 × 107 P. aeruginosa CFU coadministered with or without PslGh-PelAh (A) or PslGh-Ega3h-HEK (B) and then treated as indicated with 10 mg/kg ciprofloxacin or 25 mg/kg ceftazidime every 8 h for 1 day. Pulmonary bacterial burden was determined by CFU quantification. Bars represent the medians with interquartile ranges of at least 2 independent experiments with ≥16 mice per group. *, significant difference (P = 0.0347) between combinations of GHs and ciprofloxacin and ciprofloxacin alone; ns, no significant difference (P > 0.9999), as determined by the Kruskal-Wallis test with Dunn’s multiple-comparison test. Cipro, ciprofloxacin; Cefta, ceftazidime.
TABLE 1.
Single-dose combination PslGh-PelAh prophylaxis disseminates bacteria in an acute mouse model of pulmonary P. aeruginosa infection
| Treatment | No. of mice with bacteremia | Total no. of mice | % of mice with bacteremia | Odds ratio (95% confidence interval) | Corrected Pa |
|---|---|---|---|---|---|
| Buffer | 5 | 41 | 12.2 | 3.09 (1.03–8.61) | 0.18 |
| PslGh-PelAh | 12 | 40 | 30 | ||
| Ciprofloxacin | 0 | 24 | 0 | NAb | NA |
| Ciprofloxacin + PslGh-PelAh | 0 | 24 | 0 | ||
| Ceftazidime | 1 | 16 | 6.3 | 2.143 (0.22–32.86) | >0.99 |
| Ceftazidime + PslGh-PelAh | 2 | 16 | 12.5 |
P values were corrected by the Bonferroni method for three comparisons.
NA, not applicable.
TABLE 2.
Single-dose combination PslGh-Ega3h-HEK prophylaxis disseminates bacteria in an acute mouse model of pulmonary P. aeruginosa infection
| Treatment | No. of mice with bacteremia | Total no. of mice | % of mice with bacteremia | Odds ratio (95% confidence interval) | Corrected Pa |
|---|---|---|---|---|---|
| Buffer | 5 | 40 | 12.5 | 0.78 (0.22–2.94) | >0.99 |
| PslGh-Ega3h-HEK | 4 | 40 | 10 | ||
| Ciprofloxacin | 0 | 24 | 0 | NAb | NA |
| Ciprofloxacin + PslGh-Ega3h-HEK | 0 | 24 | 0 | ||
| Ceftazidime | 2 | 16 | 12.5 | 0.000 (0.000–2.12) | >0.99 |
| Ceftazidime + PslGh-Ega3h-HEK | 0 | 16 | 0 |
P values were corrected by the Bonferroni method for three comparisons.
NA, not applicable.
To determine whether GHs could potentiate another class of P. aeruginosa antibiotics in vivo, these experiments were repeated with ceftazidime (25 mg/kg) in place of ciprofloxacin. While ceftazidime treatment alone reduced the pulmonary bacterial burden, compared with that of buffer-treated mice, neither PslGh-PelAh nor PslGh-Ega3h-HEK in combination with ceftazidime exhibited increased antimicrobial activity, compared with ceftazidime alone (Fig. 4A and B). Collectively, these results suggest that the observed GH potentiation of antibiotic activity is dependent on the combination of GHs with antibiotic. Bacteremia was not observed in mice treated with PslGh-Ega3h-HEK-ceftazidime (Table 2), but a similar number of mice that received PslGh-PelAh-ceftazidime developed bacteremia, compared with buffer-treated mice (12.5% versus 12.2%) (Table 1).
DISCUSSION
In this study, GHs PslGh-PelAh and PslGh-Ega3h were found to potentiate the antimicrobial activity of the commonly used antibiotics ciprofloxacin and ceftazidime against P. aeruginosa biofilms in vitro. Single-dose pulmonary administration of PslGh-PelAh or PslGh-Ega3h was demonstrated to be well tolerated and induced minimal immune responses in uninfected mice. While PelAh and Ega3h alone have relatively short half-lives in vivo, the stability of these GHs increased when coadministered with PslGh. In a pulmonary model of acute P. aeruginosa infection, the GH combination PslGh-PelAh potentiated the antibacterial activity of ciprofloxacin. In vivo augmentation of antibiotics was demonstrated to be dependent on GH-antibiotic combinations, as neither PslGh-PelAh-ceftazidime nor PslGh-Ega3h-HEK-ciprofloxacin or -ceftazidime combinations reduced pulmonary bacterial burden more than antibiotic therapy alone. GH prophylaxis alone did not lead to reduced pulmonary bacterial burden and was associated with increased rates of P. aeruginosa hematogenous dissemination.
Single-dose GH therapy was well tolerated and resulted in minimal changes in the pulmonary inflammatory response in the absence of infection. Although the PelAh and Ega3h GHs exhibited short half-lives when administered as monotherapy, combining these enzymes with PslGh prolonged their pulmonary half-lives. It is unlikely that the stability of the GHs is a reflection of quantitative effects, as equimolar amounts of the GHs were administered in these experiments. Previously, we demonstrated that, in a leukopenic mouse model, the half-life of Ega3h was similar to that in the current study; however, the half-life of PelAh was longer (45) than in immunocompetent mice. Together, these findings suggest that, while some GHs may be less stable in mice with an intact immune system, combining GHs increases GH stability, possibly through the formation of a complex in which the GHs are less susceptible to proteolytic degradation. While repeated topical PslGh dosing was previously reported to be well tolerated in a chronic wound model of P. aeruginosa infection in immunocompetent mice (38, 48), the effects of repeated pulmonary GH administrations remain to be evaluated, and more detailed immunotoxicity studies of multiple combined GH doses and anti-GH antibody responses are required to advance these agents toward use in clinical trials.
Different combinations of GH enzymes with antibiotics exhibited differences in efficacy. In this study, PslGh-PelAh prophylaxis potentiated the activity of ciprofloxacin but not that of ceftazidime, and neither PslGh-Ega3h-HEK prophylaxis in combination with ciprofloxacin nor that with ceftazidime resulted in reduction of the pulmonary bacterial burden. These observations contrast with the in vitro checkerboard potentiation experiments, in which combinations of either PslGh-PelAh or PslGh-Ega3h-HEK with either ciprofloxacin or ceftazidime resulted in biofilm biomass reduction. These observations suggest that in vivo bacterial susceptibility to the therapy and biofilm disruption by GHs may be influenced by host determinants (48, 49). While these observations suggest that specific GH-antibiotic combinations exhibit unique efficacy, the observation that PslGh-PelAh monotherapy was associated with the highest rates of bacterial dissemination suggests that the specific ability of PslGh-PelAh to enhance ciprofloxacin activity may simply reflect greater antibiofilm activity. In support of this hypothesis, pulmonary histopathology demonstrated that PslGh-PelAh monotherapy was uniquely associated with an increased inflammatory response, consistent with augmented bacterial dispersion following exopolysaccharide degradation.
The mechanism by which PslGh-PelAh potentiates the antibacterial activity of ciprofloxacin remains to be determined. Pharmacokinetic analyses revealed that PslGh and PelAh have relatively short half-lives in vivo. Previously, we demonstrated that single-dose administration of the recombinant form of the fungal GH Sph3, Sph3h, potentiated the antifungal posaconazole in reducing fungal burden in a model of A. fumigatus pulmonary infection, despite the fact that this enzyme exhibited a short pulmonary half-life relative to predicted timing of GAG exopolysaccharide expression in vivo (45, 50). Furthermore, a catalytically inactive variant of Sph3h, D166AAC, exhibited activity levels similar to those of enzymatically active Sph3h, suggesting that cleavage of GAG was dispensable for the activity of this enzyme in vivo. Possible explanations for these observations include direct activation of pulmonary immune cells by GH enzymes prior to significant amounts of GAG production (45) or direct lectin-like interactions of catalytically inactive Sph3h with GAG that interfere with the virulence properties of this polymer (32). In contrast to the fungal system, Psl and Pel secretion by P. aeruginosa occurs well within the period during which PslGh and PelAh enzymes are present (11, 30). The higher rates of bacterial dissemination observed in mice that received PslGh-PelAh alone suggest that at least one of the Pel or Psl polymers is present during GH exposure and is cleaved by these enzymes. However, the inability of GH therapy alone to decrease the pulmonary bacterial burden suggests that exopolysaccharide cleavage alone is insufficient to attenuate bacterial virulence during pulmonary infection. These findings are consistent with observations in previous studies demonstrating that treatment with PslGh or other GHs, such as α-amylase and β-cellulase, alone was insufficient to reduce the bacterial burden in a wound model of acute P. aeruginosa infection (38, 48).
Disruption of the extracellular matrix by biofilm-targeting therapies has the potential to lead to bacterial dissemination and worsening of clinical outcomes. Approximately one-third of mice that received PslGh-PelAh exhibited bacteremia, although the addition of ciprofloxacin was effective in preventing dissemination. Bacterial dissemination has been reported in vivo with manipulation of P. aeruginosa biofilm regulatory pathways (51), as well as enzymatic treatment of P. aeruginosa biofilms with the GHs α-amylase and β-cellulase (48, 52). Interestingly, GH-mediated dissemination was not observed during treatment of experimental pulmonary A. fumigatus infection (45). These differences in outcomes between bacterial and mold infections may stem from their unique morphologies and capacity for motility. Molds grow as multicellular filamentous hyphae that intertwine and thus are unlikely to passively disperse and disseminate like unicellular bacteria (53). Furthermore, unlike P. aeruginosa and many other bacteria with motility appendages, including type IV pili or flagella, A. fumigatus hyphae are not motile and therefore are unable to actively disseminate from the site of infection (54–58). The results of a study using a nonmotile bacterial species, Staphylococcus aureus, support this hypothesis, as bacterial dissemination was not observed following α-amylase- and β-cellulase-mediated disruption of bacterial biofilms in a chronic wound model (52). In the study here, degradation of exopolysaccharides might have facilitated P. aeruginosa dissemination mediated by flagellar swarming (59) or possibly through intraleukocyte transport (the so-called Trojan horse mechanism) (60–62). Collectively, these data suggest that the result of biofilm-directed therapies may partly depend on the morphology and capacity for motility of an organism.
The results of this study demonstrate that intratracheal administration of PslGh-PelAh in combination with ciprofloxacin can improve the outcomes of P. aeruginosa infection in a mouse model of acute bacterial infection. While this proof-of-concept preclinical study provides evidence for the efficacy of combination GH-antibiotic prophylaxis in acute P. aeruginosa pulmonary infection, further studies are required to understand the therapeutic utility of GH therapy. The results of these studies lay the foundation for future work to elucidate the molecular mechanisms by which combinations of antibiotics with these enzymes and other forms of recombinant microbial GHs limit bacterial growth in vivo. Developing easily administrable aerosolizable formulations of GH enzymes will be critical to advancing their clinical development, including expanded multidose immunotoxicity, GH efficacy in established infection models, and pharmacokinetic studies.
MATERIALS AND METHODS
Strain and growth conditions.
The P. aeruginosa PAO1 strain was grown on Luria-Bertani (LB) agar plates overnight at 37°C, from −80°C stocks. For experiments, P. aeruginosa was cultured overnight and subcultured in LB broth (BD Difco Miller LB) to mid-log phase at 37°C. Bacteria were resuspended to 3 × 108 CFU/mL in phosphate-buffered saline (PBS) for intratracheal infections.
Recombinant GH expression and purification.
His-tagged PelAh and PslGh were expressed in ClearColi cells grown in Terrific Broth (BioShop) or autoinduction medium with 50 μg/mL kanamycin (Bio Basic) as described previously (32, 35, 41). Bacterial cultures in Terrific Broth were induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) (Bio Basic) when the cells reached an optical density at 600 nm (OD600) of 1.2 to 1.4. The cells were incubated postinduction overnight at 18°C with shaking at 200 rpm before being harvested by centrifugation at 5,000 × g for 30 min at 4°C. Both proteins were purified by fast protein liquid chromatography using Ni-nitrilotriacetic acid columns (GE Healthcare) followed by buffer exchange, and the purity of the protein was confirmed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) as described previously (35).
Expression of Ega3h in the PichiaPink system was optimized as described previously (42). To generate a glycosylated form of Ega3 that would mimic mammalian glycosylation patterns (63), Ega3h-HEK was expressed in a cell line of HEK cells (HEK293) as described previously (45). The culture supernatants containing the secreted proteins were harvested at 6 days (45). His-tagged protein was purified from the supernatant by affinity chromatography followed by gel filtration using a HiLoad 16/600 Superdex 200 preparation-grade column (GE Healthcare).
Checkerboard studies.
Bacterial biofilm susceptibility to killing by antibiotics and GH potentiation of antibiotics were quantified in a minimum biofilm eradication concentration (MBEC) high-throughput assay, modified from that described previously (64). Bacterial biofilms were grown in MBEC/Calgary Biofilm Device plates (Innovotech) for 20 h at 37°C. The biofilm-coated peg lids were rinsed with water, placed in new base plates containing serial 2-fold and 4-fold dilutions of 4,000-μg/mL concentrations of the antibiotics ciprofloxacin or ceftazidime and 10 μM concentrations of the GH combinations PslGh-PelAh or PslGh-Ega3h-Pp, respectively, in a two-dimensional checkerboard, and incubated overnight at 37°C. To quantify biofilm biomass, peg lids were separated from base plates and stained with 150 μL 0.1% crystal violet (BioShop) for 10 min. Crystal violet was solubilized with 150 μL ethanol (BioShop) for 10 min, and the absorbance of 150 μL of the solution was measured at 595 nm (SpectraMax M2).
Mice.
Six- to 8-week-old BALB/c female mice (Charles River Laboratories Inc, Senneville, QC, Canada and Kingston, NY, USA) were used for animal studies. Mice were anaesthetized with 4% isoflurane prior to intratracheal infection and prophylaxis/treatment with GHs/drugs. Body weight was measured using a top-loading balance, and surface body temperature was measured on the abdomen using a digital infrared thermometer. Moribund animals were euthanized by isoflurane and CO2 overdose.
Tolerability studies.
Immunocompetent mice were treated intratracheally with a single dose of PslGh-PelAh or PslGh-Ega3h at 10/10 μg or 250/250 μg in 50 μL PBS or with PBS alone. Mice were monitored daily for 6 days for signs of illness, and body weights and temperatures were recorded. For histopathology studies, lungs from immunocompetent mice were inflated with 10% buffered formalin (Thermo Fisher Scientific) and fixed in formalin as described previously (65). Lungs were then embedded in paraffin, and 4-μm-thick sections were stained with hematoxylin and eosin. Scanned sections (Aperio scanner; Leica Biosystems) were analyzed with QuPath v0.1.2 software (66).
Pulmonary leukocyte quantification.
Immunocompetent mice were treated intratracheally with a single dose of the GHs PslGh-PelAh or PslGh-Ega3h at 10/10 μg or 250/250 μg in 50 μL PBS or with PBS alone. Seven days after treatment, mice were euthanized, and their lungs were washed in PBS, minced in RPMI 1640 medium (Wisent) containing 5% (vol/vol) fetal bovine serum (FBS) (Wisent), and then digested with 150 U/mL collagenase (Sigma) (65, 67). The resulting suspension was passed through a 70-μm cell strainer and treated with ammonium-chloride-potassium (ACK) buffer (Gibco). Approximately 1 × 106 leukocytes were resuspended in a fixable viability dye (eBioscience) and washed, and their Fc receptors were blocked by unlabeled anti-CD16/32 antibodies (FcBlock; BD Pharmingen) as described previously (45, 65). Cell surface components were then stained with fluorescently labeled antibodies (BD Biosciences) as described previously (45, 65). Leukocytes were washed, fixed with paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA), and then resuspended in PBS as described previously (45, 65). Data were acquired on an LSR Fortessa flow cytometer with FACSDiva software (BD Biosciences) and analyzed with FlowJo software v10 (FlowJo, LLC). Immune cell subsets were defined as described previously (45, 65). Total cell populations were calculated using CountBright absolute counting beads (Invitrogen).
Densitometry and antibody production.
SDS-PAGE and Western blotting techniques were used to assess pulmonary GH pharmacokinetics. Rabbit polyclonal antibodies specific for each of the GHs were produced by Cedarlane (Burlington, Canada) as described previously (35). Mice were treated intratracheally with a single dose of 500 μg of each GH or 250/250 μg of each GH for a combination of PslGh-PelAh or PslGh-Ega3h, and then they were euthanized and their lungs were harvested at the indicated time points. Lungs were homogenized in a cocktail of protease inhibitors (Roche), and pulmonary GH concentrations were quantified by Western blotting with rabbit anti-GH antibodies. Goat anti-rabbit horseradish peroxidase-conjugated secondary antibody (Bio-Rad) binding was detected with a chemiluminescent substrate (Thermo Fisher Scientific). The half-lives of the GHs were determined by densitometric analysis using ImageJ software. Band intensity at each time point was normalized to the intensity at the zero-hour time point. The half-life was determined as the time to 50% relative intensity of the bands, compared to the zero-hour time point.
SDS-PAGE and Western blotting techniques were used to confirm the sensitivity of GHs to proteolytic degradation in vitro. PelAh or PslGh (10 μg) was treated with 100 μg/mL neutrophil elastase. Proteolysis was stopped with SDS loading buffer, and GHs were detected by Western blotting using rabbit anti-GH antibodies as described above.
Pulmonary damage.
Mice were treated intratracheally with a single dose of PslGh-PelAh or PslGh-Ega3h at 10/10 μg or 250/250 μg in 50 μL PBS or with PBS alone. Seven days after treatment, mice were euthanized and their lungs were lavaged twice with 1 mL PBS as described previously (45, 65). Lactate dehydrogenase activity was measured in the pooled BAL fluid by commercial assay (CytoTox 96 nonradioactive cytotoxicity assay; Promega), according to the manufacturer’s instructions.
Mouse model of acute pulmonary P. aeruginosa infection.
Mice were intratracheally infected with a 50-μL suspension of 1.5 × 107 P. aeruginosa CFU in PBS and treated with either a single dose of PslGh-PelAh or PslGh-Ega3h-HEK at 250/250 μg or PBS alone. For antibiotic combination studies, mice were treated by intraperitoneal injection with 10 mg/kg ciprofloxacin or subcutaneous injection with 25 mg/kg ceftazidime or 0.1 N NaOH in PBS reconstitution solution every 8 h beginning 4 h after infection (see Fig. S6 in the supplemental material). One day after infection, mice were euthanized and their blood and lungs were harvested for bacterial burden assessment, modified from the method described previously (68).
(i) Pulmonary bacterial burden.
Lungs were harvested and homogenized in 5 mL PBS with a Polytron tissue homogenizer. Homogenates were serially diluted at a 1:5 ratio and spot plated on Pseudomonas isolation agar plates (Sigma-Aldrich). Pulmonary bacterial burden was determined by colony quantification.
(ii) Blood bacterial burden.
Blood was diluted in PBS at a 1:10 ratio, serially diluted at a 1:5 ratio, and spot plated on Pseudomonas isolation agar (Sigma-Aldrich). Blood bacterial burden was determined by colony quantification.
Statistical analysis.
Data are presented and statistical significance was calculated as indicated. All graphs were generated and statistical analyses were performed with GraphPad Prism v9.0.0 software. Significant differences between values were compared by two-way analysis of variance (ANOVA) with Dunnett’s multiple-comparison test, the Kruskal-Wallis test with Dunn’s multiple-comparison test, and Fischer’s exact test with Bonferroni correction for multiple comparisons. Odds ratios were calculated by the Baptista-Pike method.
Ethics statement.
All procedures involving mice were approved by the animal care committees of the Institutional Animal Care and Use Committee (IACUC) of the McGill University Health Centre (protocol number 2016-7808) and the Animal Care and Use Review Office (ACURO) of the U.S. Army Medical Research and Materiel Command (USAMRMC).
ACKNOWLEDGMENTS
D.R., I.L., S.G., P.S., N.C.B., and P.B. designed plasmid constructs and synthesized and purified recombinant GHs. D.R. performed and analyzed checkerboard studies. M.L. performed in vivo assays for tolerability, processed samples, and acquired and analyzed monitoring and immunophenotyping data. R.C. performed in vivo assays for GH efficacy studies. R.C. processed samples and acquired bacterial burden data. D.R., I.L., S.G., and P.S. performed and acquired and analyzed data for pharmacokinetic studies. H.O., D.R., F.N.G., P.L.H., and D.C.S. interpreted the tolerability, pharmacokinetic, and efficacy data. H.O. assembled figures and performed statistical analyses on tolerability, pharmacokinetic, and GH efficacy data. H.O. wrote the original draft of the manuscript. H.O., D.R., P.L.H., and D.C.S. reviewed and edited the final draft of the manuscript. P.L.H. and D.C.S. conceptualized the study and acquired funding for the project.
This work was performed with the support of the Research Institute of the McGill University Health Centre histopathology and immunophenotyping platforms. Research described in this paper was supported by grants from the USAMRMC (grant W81XWH-15-PRMRP-IIRA to D.C.S. and P.L.H.), Cystic Fibrosis Canada (CFC) (grant 558692 to D.C.S. and P.L.H.), and the Canadian Glycomics Network (GlycoNet) (grant AM-15 to D.C.S. and P.L.H.). P.L.H. was the recipient of a Tier I Canada Research Chair (2006 to 2020). D.C.S. is supported by a Chercheur-Boursier Award from the Fonds de Recherche Quebec Santé (FRSQ). The funders have no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Supplemental material is available online only.
Contributor Information
P. Lynne Howell, Email: howell@sickkids.ca.
Donald C. Sheppard, Email: donald.sheppard@mcgill.ca.
REFERENCES
- 1.Purcell P, Jary H, Perry A, Perry JD, Stewart CJ, Nelson A, Lanyon C, Smith DL, Cummings SP, De Soyza A. 2014. Polymicrobial airway bacterial communities in adult bronchiectasis patients. BMC Microbiol 14:130. 10.1186/1471-2180-14-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cuthbertson L, Walker AW, Oliver AE, Rogers GB, Rivett DW, Hampton TH, Ashare A, Elborn JS, De Soyza A, Carroll MP, Hoffman LR, Lanyon C, Moskowitz SM, O'Toole GA, Parkhill J, Planet PJ, Teneback CC, Tunney MM, Zuckerman JB, Bruce KD, van der Gast CJ. 2020. Lung function and microbiota diversity in cystic fibrosis. Microbiome 8:45. 10.1186/s40168-020-00810-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Monsó E, Garcia-Aymerich J, Soler N, Farrero E, Felez MA, Antó JM, Torres A, EFRAM Investigators . 2003. Bacterial infection in exacerbated COPD with changes in sputum characteristics. Epidemiol Infect 131:799–804. 10.1017/s0950268803008872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Q, Illing R, Hui CK, Downey K, Carr D, Stearn M, Alshafi K, Menzies-Gow A, Zhong N, Fan Chung K. 2012. Bacteria in sputum of stable severe asthma and increased airway wall thickness. Respir Res 13:35. 10.1186/1465-9921-13-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eklöf J, Sørensen R, Ingebrigtsen TS, Sivapalan P, Achir I, Boel JB, Bangsborg J, Ostergaard C, Dessau RB, Jensen US, Browatzki A, Lapperre TS, Janner J, Weinreich UM, Armbruster K, Wilcke T, Seersholm N, Jensen JUS. 2020. Pseudomonas aeruginosa and risk of death and exacerbations in patients with chronic obstructive pulmonary disease: an observational cohort study of 22 053 patients. Clin Microbiol Infect 26:227–234. 10.1016/j.cmi.2019.06.011. [DOI] [PubMed] [Google Scholar]
- 6.Chalmers JD, Aliberti S, Filonenko A, Shteinberg M, Goeminne PC, Hill AT, Fardon TC, Obradovic D, Gerlinger C, Sotgiu G, Operschall E, Rutherford RM, Dimakou K, Polverino E, De Soyza A, McDonnell MJ. 2018. Characterization of the “frequent exacerbator phenotype” in bronchiectasis. Am J Respir Crit Care Med 197:1410–1420. 10.1164/rccm.201711-2202OC. [DOI] [PubMed] [Google Scholar]
- 7.Emerson J, Rosenfeld M, McNamara S, Ramsey B, Gibson RL. 2002. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol 34:91–100. 10.1002/ppul.10127. [DOI] [PubMed] [Google Scholar]
- 8.Starkey M, Hickman JH, Ma L, Zhang N, De Long S, Hinz A, Palacios S, Manoil C, Kirisits MJ, Starner TD, Wozniak DJ, Harwood CS, Parsek MR. 2009. Pseudomonas aeruginosa rugose small-colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J Bacteriol 191:3492–3503. 10.1128/JB.00119-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pedersen SS, Høiby N, Espersen F, Koch C. 1992. Role of alginate in infection with mucoid Pseudomonas aeruginosa in cystic fibrosis. Thorax 47:6–13. 10.1136/thx.47.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mathee K, Ciofu O, Sternberg C, Lindum PW, Campbell JIA, Jensen P, Johnsen AH, Givskov M, Ohman DE, Søren M, Høiby N, Kharazmi A. 1999. Mucoid conversion of Pseudomonas aeruginosa by hydrogen peroxide: a mechanism for virulence activation in the cystic fibrosis lung. Microbiology (Reading) 145:1349–1357. 10.1099/13500872-145-6-1349. [DOI] [PubMed] [Google Scholar]
- 11.Jennings LK, Dreifus JE, Reichhardt C, Storek KM, Secor PR, Wozniak DJ, Hisert KB, Parsek MR. 2021. Pseudomonas aeruginosa aggregates in cystic fibrosis sputum produce exopolysaccharides that likely impede current therapies. Cell Rep 34:108782. 10.1016/j.celrep.2021.108782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walker TS, Tomlin KL, Worthen GS, Poch KR, Lieber JG, Saavedra MT, Fessler MB, Malcolm KC, Vasil ML, Nick JA. 2005. Enhanced Pseudomonas aeruginosa biofilm development mediated by human neutrophils. Infect Immun 73:3693–3701. 10.1128/IAI.73.6.3693-3701.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reichhardt C, Jacobs HM, Matwichuk M, Wong C, Wozniak DJ, Parsek MR. 2020. The versatile Pseudomonas aeruginosa biofilm matrix protein CdrA promotes aggregation through different extracellular EPS interactions. J Bacteriol 202:e00216-20. 10.1128/JB.00216-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Borlee BR, Goldman AD, Murakami K, Samudrala R, Wozniak DJ, Parsek MR. 2010. Pseudomonas aeruginosa uses a cyclic-di-GMP-regulated adhesin to reinforce the biofilm extracellular matrix. Mol Microbiol 75:827–842. 10.1111/j.1365-2958.2009.06991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whitchurch CB, Tolker-Nielsen T, Ragas PC, Mattick JS. 2002. Extracellular DNA required for bacterial biofilm formation. Science 295:1487. 10.1126/science.295.5559.1487. [DOI] [PubMed] [Google Scholar]
- 16.Allesen-Holm M, Barken KB, Yang L, Klausen M, Webb JS, Kjelleberg S, Molin S, Givskov M, Tolker-Nielsen T. 2006. A characterization of DNA release in Pseudomonas aeruginosa cultures and biofilms. Mol Microbiol 59:1114–1128. 10.1111/j.1365-2958.2005.05008.x. [DOI] [PubMed] [Google Scholar]
- 17.Flemming HC, Wingender J. 2010. The biofilm matrix. Nat Rev Microbiol 8:623–633. 10.1038/nrmicro2415. [DOI] [PubMed] [Google Scholar]
- 18.Jennings LK, Storek KM, Ledvina HE, Coulon C, Marmont LS, Sadovskaya I, Secor PR, Tseng BS, Scian M, Filloux A, Wozniak DJ, Howell PL, Parsek MR. 2015. Pel is a cationic exopolysaccharide that cross-links extracellular DNA in the Pseudomonas aeruginosa biofilm matrix. Proc Natl Acad Sci USA 112:11353–11358. 10.1073/pnas.1503058112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Colvin KM, Irie Y, Tart CS, Urbano R, Whitney JC, Ryder C, Howell PL, Wozniak DJ, Parsek MR. 2012. The Pel and Psl polysaccharides provide Pseudomonas aeruginosa structural redundancy within the biofilm matrix. Environ Microbiol 14:1913–1928. 10.1111/j.1462-2920.2011.02657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramsey DM, Wozniak DJ. 2005. Understanding the control of Pseudomonas aeruginosa alginate synthesis and the prospects for management of chronic infections in cystic fibrosis. Mol Microbiol 56:309–322. 10.1111/j.1365-2958.2005.04552.x. [DOI] [PubMed] [Google Scholar]
- 21.Byrd MS, Sadovskaya I, Vinogradov E, Lu H, Sprinkle AB, Richardson SH, Ma L, Ralston B, Parsek MR, Anderson EM, Lam JS, Wozniak DJ. 2009. Genetic and biochemical analyses of the Pseudomonas aeruginosa Psl exopolysaccharide reveal overlapping roles for polysaccharide synthesis enzymes in Psl and LPS production. Mol Microbiol 73:622–638. 10.1111/j.1365-2958.2009.06795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evans LR, Linker A. 1973. Production and characterization of the slime polysaccharide of Pseudomonas aeruginosa. J Bacteriol 116:915–924. 10.1128/jb.116.2.915-924.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hentzer M, Teitzel GM, Balzer GJ, Heydorn A, Molin S, Givskov M, Parsek MR. 2001. Alginate overproduction affects Pseudomonas aeruginosa biofilm structure and function. J Bacteriol 183:5395–5401. 10.1128/JB.183.18.5395-5401.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang L, Hengzhuang W, Wu H, Damkiaer S, Jochumsen N, Song Z, Givskov M, Høiby N, Molin S. 2012. Polysaccharides serve as scaffold of biofilms formed by mucoid Pseudomonas aeruginosa. FEMS Immunol Med Microbiol 65:366–376. 10.1111/j.1574-695X.2012.00936.x. [DOI] [PubMed] [Google Scholar]
- 25.Nivens DE, Ohman DE, Williams J, Franklin MJ. 2001. Role of alginate and its O acetylation in formation of Pseudomonas aeruginosa microcolonies and biofilms. J Bacteriol 183:1047–1057. 10.1128/JB.183.3.1047-1057.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stapper AP, Narasimhan G, Ohman DE, Barakat J, Hentzer M, Molin S, Kharazmi A, Høiby N, Mathee K. 2004. Alginate production affects Pseudomonas aeruginosa biofilm development and architecture, but is not essential for biofilm formation. J Med Microbiol 53:679–690. 10.1099/jmm.0.45539-0. [DOI] [PubMed] [Google Scholar]
- 27.Wozniak DJ, Wyckoff TJ, Starkey M, Keyser R, Azadi P, O'Toole GA, Parsek MR. 2003. Alginate is not a significant component of the extracellular polysaccharide matrix of PA14 and PAO1 Pseudomonas aeruginosa biofilms. Proc Natl Acad Sci USA 100:7907–7912. 10.1073/pnas.1231792100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ryder C, Byrd M, Wozniak DJ. 2007. Role of polysaccharides in Pseudomonas aeruginosa biofilm development. Curr Opin Microbiol 10:644–648. 10.1016/j.mib.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friedman L, Kolter R. 2004. Genes involved in matrix formation in Pseudomonas aeruginosa PA14 biofilms. Mol Microbiol 51:675–690. 10.1046/j.1365-2958.2003.03877.x. [DOI] [PubMed] [Google Scholar]
- 30.Colvin KM, Gordon VD, Murakami K, Borlee BR, Wozniak DJ, Wong GC, Parsek MR. 2011. The Pel polysaccharide can serve a structural and protective role in the biofilm matrix of Pseudomonas aeruginosa. PLoS Pathog 7:e1001264. 10.1371/journal.ppat.1001264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Billings N, Millan M, Caldara M, Rusconi R, Tarasova Y, Stocker R, Ribbeck K. 2013. The extracellular matrix component Psl provides fast-acting antibiotic defense in Pseudomonas aeruginosa biofilms. PLoS Pathog 9:e1003526. 10.1371/journal.ppat.1003526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baker P, Hill PJ, Snarr BD, Alnabelseya N, Pestrak MJ, Lee MJ, Jennings LK, Tam J, Melnyk RA, Parsek MR, Sheppard DC, Wozniak DJ, Howell PL. 2016. Exopolysaccharide biosynthetic glycoside hydrolases can be utilized to disrupt and prevent Pseudomonas aeruginosa biofilms. Sci Adv 2:e1501632. 10.1126/sciadv.1501632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mishra M, Byrd MS, Sergeant S, Azad AK, Parsek MR, McPhail L, Schlesinger LS, Wozniak DJ. 2012. Pseudomonas aeruginosa Psl polysaccharide reduces neutrophil phagocytosis and the oxidative response by limiting complement-mediated opsonization. Cell Microbiol 14:95–106. 10.1111/j.1462-5822.2011.01704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Colvin KM, Alnabelseya N, Baker P, Whitney JC, Howell PL, Parsek MR. 2013. PelA deacetylase activity is required for Pel polysaccharide synthesis in Pseudomonas aeruginosa. J Bacteriol 195:2329–2339. 10.1128/JB.02150-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baker P, Whitfield GB, Hill PJ, Little DJ, Pestrak MJ, Robinson H, Wozniak DJ, Howell PL. 2015. Characterization of the Pseudomonas aeruginosa glycoside hydrolase PslG reveals that its levels are critical for Psl polysaccharide biosynthesis and biofilm formation. J Biol Chem 290:28374–28387. 10.1074/jbc.M115.674929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le Mauff F, Bamford NC, Alnabelseya N, Zhang Y, Baker P, Robinson H, Codee JDC, Howell PL, Sheppard DC. 2019. Molecular mechanism of Aspergillus fumigatus biofilm disruption by fungal and bacterial glycoside hydrolases. J Biol Chem 294:10760–10772. 10.1074/jbc.RA119.008511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wolfgang MC, Kulasekara BR, Liang X, Boyd D, Wu K, Yang Q, Miyada CG, Lory S. 2003. Conservation of genome content and virulence determinants among clinical and environmental isolates of Pseudomonas aeruginosa. Proc Natl Acad Sci USA 100:8484–8489. 10.1073/pnas.0832438100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pestrak MJ, Baker P, Dellos-Nolan S, Hill PJ, Passos da Silva D, Silver H, Lacdao I, Raju D, Parsek MR, Wozniak DJ, Howell PL. 2019. Treatment with the Pseudomonas aeruginosa glycoside hydrolase PslG combats wound infection by improving antibiotic efficacy and host innate immune activity. Antimicrob Agents Chemother 63:e00234-19. 10.1128/AAC.00234-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fontaine T, Delangle A, Simenel C, Coddeville B, van Vliet SJ, van Kooyk Y, Bozza S, Moretti S, Schwarz F, Trichot C, Aebi M, Delepierre M, Elbim C, Romani L, Latge JP. 2011. Galactosaminogalactan, a new immunosuppressive polysaccharide of Aspergillus fumigatus. PLoS Pathog 7:e1002372. 10.1371/journal.ppat.1002372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee MJ, Geller AM, Bamford NC, Liu H, Gravelat FN, Snarr BD, Le Mauff F, Chabot J, Ralph B, Ostapska H, Lehoux M, Cerone RP, Baptista SD, Vinogradov E, Stajich JE, Filler SG, Howell PL, Sheppard DC. 2016. Deacetylation of fungal exopolysaccharide mediates adhesion and biofilm formation. mBio 7:e00252-16. 10.1128/mBio.00252-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bamford NC, Snarr BD, Gravelat FN, Little DJ, Lee MJ, Zacharias CA, Chabot JC, Geller AM, Baptista SD, Baker P, Robinson H, Howell PL, Sheppard DC. 2015. Sph3 is a glycoside hydrolase required for the biosynthesis of galactosaminogalactan in Aspergillus fumigatus. J Biol Chem 290:27438–27450. 10.1074/jbc.M115.679050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bamford NC, Le Mauff F, Subramanian AS, Yip P, Millán C, Zhang Y, Zacharias C, Forman A, Nitz M, Codée JDC, Usón I, Sheppard DC, Howell PL. 2019. Ega3 from the fungal pathogen Aspergillus fumigatus is an endo-α-1,4-galactosaminidase that disrupts microbial biofilms. J Biol Chem 294:13833–13849. 10.1074/jbc.RA119.009910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bamford NC, Le Mauff F, Van Loon JC, Ostapska H, Snarr BD, Zhang Y, Kitova EN, Klassen JS, Codée JDC, Sheppard DC, Howell PL. 2020. Structural and biochemical characterization of the exopolysaccharide deacetylase Agd3 required for Aspergillus fumigatus biofilm formation. Nat Commun 11:2450. 10.1038/s41467-020-16144-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee MJ, Liu H, Barker BM, Snarr BD, Gravelat FN, Al Abdallah Q, Gavino C, Baistrocchi SR, Ostapska H, Xiao T, Ralph B, Solis NV, Lehoux M, Baptista SD, Thammahong A, Cerone RP, Kaminskyj SG, Guiot MC, Latge JP, Fontaine T, Vinh DC, Filler SG, Sheppard DC. 2015. The fungal exopolysaccharide galactosaminogalactan mediates virulence by enhancing resistance to neutrophil extracellular traps. PLoS Pathog 11:e1005187. 10.1371/journal.ppat.1005187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ostapska H, Raju D, Lehoux M, Lacdao I, Gilbert S, Sivarajah P, Bamford NC, Baker P, Nguyen TTM, Zacharias CA, Gravelat FN, Howell PL, Sheppard DC. 2021. Preclinical evaluation of recombinant microbial glycoside hydrolases in the prevention of experimental invasive Aspergillosis. mBio 12:e02446-21. 10.1128/mBio.02446-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dell A, Galadari A, Sastre F, Hitchen P. 2010. Similarities and differences in the glycosylation mechanisms in prokaryotes and eukaryotes. Int J Microbiol 2010:148178. 10.1155/2010/148178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steele C, Rapaka RR, Metz A, Pop SM, Williams DL, Gordon S, Kolls JK, Brown GD. 2005. The beta-glucan receptor dectin-1 recognizes specific morphologies of Aspergillus fumigatus. PLoS Pathog 1:e42. 10.1371/journal.ppat.0010042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Redman WK, Welch GS, Williams AC, Damron AJ, Northcut WO, Rumbaugh KP. 2021. Efficacy and safety of biofilm dispersal by glycoside hydrolases in wounds. Biofilm 3:100061. 10.1016/j.bioflm.2021.100061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nguyen D, Joshi-Datar A, Lepine F, Bauerle E, Olakanmi O, Beer K, McKay G, Siehnel R, Schafhauser J, Wang Y, Britigan BE, Singh PK. 2011. Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science 334:982–986. 10.1126/science.1211037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mowat E, Butcher J, Lang S, Williams C, Ramage G. 2007. Development of a simple model for studying the effects of antifungal agents on multicellular communities of Aspergillus fumigatus. J Med Microbiol 56:1205–1212. 10.1099/jmm.0.47247-0. [DOI] [PubMed] [Google Scholar]
- 51.Christensen LD, van Gennip M, Rybtke MT, Wu H, Chiang WC, Alhede M, Høiby N, Nielsen TE, Givskov M, Tolker-Nielsen T. 2013. Clearance of Pseudomonas aeruginosa foreign-body biofilm infections through reduction of the cyclic di-GMP level in the bacteria. Infect Immun 81:2705–2713. 10.1128/IAI.00332-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fleming D, Rumbaugh K. 2018. The consequences of biofilm dispersal on the host. Sci Rep 8:10738. 10.1038/s41598-018-29121-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Powers-Fletcher MV, Kendall BA, Griffin AT, Hanson KE. 2016. Filamentous fungi. Microbiol Spectr 4:4.3.23. 10.1128/microbiolspec.DMIH2-0002-2015. [DOI] [PubMed] [Google Scholar]
- 54.Merz AJ, So M, Sheetz MP. 2000. Pilus retraction powers bacterial twitching motility. Nature 407:98–102. 10.1038/35024105. [DOI] [PubMed] [Google Scholar]
- 55.Kearns DB. 2010. A field guide to bacterial swarming motility. Nat Rev Microbiol 8:634–644. 10.1038/nrmicro2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Toutain CM, Zegans ME, O'Toole GA. 2005. Evidence for two flagellar stators and their role in the motility of Pseudomonas aeruginosa. J Bacteriol 187:771–777. 10.1128/JB.187.2.771-777.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ingham CJ, Kalisman O, Finkelshtein A, Ben-Jacob E. 2011. Mutually facilitated dispersal between the nonmotile fungus Aspergillus fumigatus and the swarming bacterium Paenibacillus vortex. Proc Natl Acad Sci USA 108:19731–19736. 10.1073/pnas.1102097108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Drake D, Montie TC. 1988. Flagella, motility and invasive virulence of Pseudomonas aeruginosa. J Gen Microbiol 134:43–52. 10.1099/00221287-134-1-43. [DOI] [PubMed] [Google Scholar]
- 59.Zhang J, He J, Zhai C, Ma LZ, Gu L, Zhao K. 2018. Effects of PslG on the surface movement of Pseudomonas aeruginosa. Appl Environ Microbiol 84:e00219-18. 10.1128/AEM.00219-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gutiérrez-Jiménez C, Mora-Cartín R, Altamirano-Silva P, Chacón-Díaz C, Chaves-Olarte E, Moreno E, Barquero-Calvo E. 2019. Neutrophils as Trojan horse vehicles for Brucella abortus macrophage infection. Front Immunol 10:1012. 10.3389/fimmu.2019.01012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.John B, Hunter CA. 2008. Neutrophil soldiers or Trojan horses? Science 321:917–918. 10.1126/science.1162914. [DOI] [PubMed] [Google Scholar]
- 62.van Zandbergen G, Klinger M, Mueller A, Dannenberg S, Gebert A, Solbach W, Laskay T. 2004. Cutting edge: neutrophil granulocyte serves as a vector for Leishmania entry into macrophages. J Immunol 173:6521–6525. 10.4049/jimmunol.173.11.6521. [DOI] [PubMed] [Google Scholar]
- 63.Eckart MR, Bussineau CM. 1996. Quality and authenticity of heterologous proteins synthesized in yeast. Curr Opin Biotechnol 7:525–530. 10.1016/s0958-1669(96)80056-5. [DOI] [PubMed] [Google Scholar]
- 64.Habash MB, Goodyear MC, Park AJ, Surette MD, Vis EC, Harris RJ, Khursigara CM. 2017. Potentiation of tobramycin by silver nanoparticles against Pseudomonas aeruginosa biofilms. Antimicrob Agents Chemother 61:e00415-17. 10.1128/AAC.00415-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Snarr BD, Baker P, Bamford NC, Sato Y, Liu H, Lehoux M, Gravelat FN, Ostapska H, Baistrocchi SR, Cerone RP, Filler EE, Parsek MR, Filler SG, Howell PL, Sheppard DC. 2017. Microbial glycoside hydrolases as antibiofilm agents with cross-kingdom activity. Proc Natl Acad Sci USA 114:7124–7129. 10.1073/pnas.1702798114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, James JA, Salto-Tellez M, Hamilton PW. 2017. QuPath: open source software for digital pathology image analysis. Sci Rep 7:16878. 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Urb M, Snarr BD, Wojewodka G, Lehoux M, Lee MJ, Ralph B, Divangahi M, King IL, McGovern TK, Martin JG, Fraser R, Radzioch D, Sheppard DC. 2015. Evolution of the immune response to chronic airway colonization with Aspergillus fumigatus hyphae. Infect Immun 83:3590–3600. 10.1128/IAI.00359-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Crandon JL, Schuck VJ, Banevicius MA, Beaudoin ME, Nichols WW, Tanudra MA, Nicolau DP. 2012. Comparative in vitro and in vivo efficacies of human simulated doses of ceftazidime and ceftazidime-avibactam against Pseudomonas aeruginosa. Antimicrob Agents Chemother 56:6137–6146. 10.1128/AAC.00851-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S6. Download aac.00052-22-s0001.pdf, PDF file, 2.4 MB (2.4MB, pdf)