

Abstract
A previous bioinformatic analysis predicted that the ysgA open reading frame of Bacillus subtilis encodes an RNA methyltransferase of the SPOUT superfamily. Here we show that YsgA is the 2′-O-methyltransferase that targets position G2553 (Escherichia coli numbering) of the A-loop of 23S rRNA. This was shown by a combination of biochemical and mass spectrometry approaches using both rRNA extracted from B. subtilis wild-type or ΔysgA cells and in vitro synthesized rRNA. When the target G2553 is mutated, YsgA is able to methylate the ribose of adenosine. However, it cannot methylate cytidine nor uridine. The enzyme modifies free 23S rRNA but not the fully assembled ribosome nor the 50S subunit, suggesting that the modification occurs early during ribosome biogenesis. Nevertheless, ribosome subunits assembly is unaffected in a B. subtilis ΔysgA mutant strain. The crystal structure of the recombinant YsgA protein, combined with mutagenesis data, outlined in this article highlights a typical SPOUT fold preceded by an L7Ae/L30 (eL8/eL30 in a new nomenclature) amino-terminal domain.
Keywords: rRNA, modified nucleosides, Bacillus subtilis, methyltransferase
INTRODUCTION
Based on biochemical and structural data, the S-adenosyl-L-methionine (SAM) dependent methyltransferases can be divided into five classes (Schubert et al. 2003) for which members of classes I and IV comprise RNA methyltransferases. The catalytic domain of class I enzymes (called RFM) contains a Rossmann-like fold, whereas class IV enzymes bear a deep trefoil knotted domain in their tertiary structure. These latter enzymes belong to the SPOUT superfamily of methyltransferases (Anantharaman et al. 2002; Tkaczuk et al. 2007). This name originates from its founding members: the tRNA 2′-O-methylguanosine methyltransferase SpoU and the tRNA 1-methylguanosine methyltransferase TrmD (Byström and Björk 1982; Persson et al. 1997; Cavaillé et al. 1999), each being a representative of a subfamily (Koonin and Rudd 1993).
The structural and catalytic features of SpoU (now named TrmH) and TrmD and other SPOUT methyltransferases have recently been the subject of two review articles (Hori 2017; Krishnamohan and Jackman 2019), the first more focusing on tRNA-specific SPOUT methyltransferases. Most SPOUT enzymes are homodimers for which the active site lies at the dimer interface (Swinehart and Jackman 2015; Hori 2017). Helices from both subunits forming this interface are perpendicular or antiparallel for respectively the SpoU (TrmH) and TrmD subfamily (Krishnamohan and Jackman 2019). All identified 2′-O-methyltransferases are homodimers. Based on structural and mutational analysis of Thermus thermophilus TrmH a catalytic mechanism has been proposed for which a highly conserved arginine residue could function as a general base (Nureki et al. 2004).
In contrast to the majority of SPOUT superfamily members, a few function as monomers. The methyltransferases of the Trm10 family, methylating either a specific purine or both purines at position 9 of tRNA, are monomeric (Shao et al. 2014; Van Laer et al. 2016; Krishnamohan and Jackman 2017; Singh et al. 2018) and their activity relies on an alternative catalytic mechanism (Krishnamohan and Jackman 2019). The SPOUT Sfm1 enzyme is also a monomer (Lv et al. 2015), but rather than RNA it methylates a conserved arginine residue of the uS3 protein of the small ribosomal subunit of yeast (Young et al. 2012). This illustrates that some SPOUT methyltransferases can methylate substrates other than RNA. In this context, it is worth mentioning that yeast Trs3 transfers the aminocarboxypropyl moiety of SAM to 18S rRNA. The binding mode of SAM in this enzyme differs from its binding as a methyl donor (Meyer et al. 2016).
The function of various members of this diverse SPOUT enzyme family remains to be determined. As outlined in a multiple alignment of SPOUT methyltransferases, the YsgA protein of Bacillus subtilis bears the conserved motifs typical for the SpoU subfamily of SPOUT enzymes (Anantharaman et al. 2002). Its function and target are however not known. In this article, we demonstrate that YsgA is an rRNA methyltransferase acting on the A-loop of the 23S rRNA. YsgA is a dimer as the other 2′-O-methyltransferases of the SpoU family and its structure shows that the enzyme is composed of a SPOUT domain with an amino-terminal extension.
RESULTS
B. subtilis YsgA methylates 23S rRNA
Based on bioinformatic studies the YsgA protein of B. subtilis was predicted to be an RNA methyltransferase belonging to the SpoU family (Anantharaman et al. 2002). To show that YsgA displays this RNA methyltransferase activity, unfractionated (total) RNA preparations from B. subtilis ΔysgA (in which the methylated nucleoside formed by YsgA should be lacking) and from wild-type strain were tested as substrates for the enzyme. The recombinant YsgA enzyme was produced in E. coli as a His-tagged protein and purified to quasi-homogeneity by immobilized metal ion affinity chromatography followed by gel filtration.
As shown in Figure 1A, recombinant YsgA methylates total RNA from B. subtilis ΔysgA but not B. subtilis wild-type. This observation confirms that YsgA is an RNA methyltransferase.
FIGURE 1.

YsgA is a methyltransferase that forms 2′O-methylguanosine in 23S rRNA in vitro. (A) 50 µg of total RNA extracted from B. subtilis wild-type (▪) or ΔysgA (●) cells or 50 µg of purified rRNA (○) or tRNA (□) from B. subtilis ΔysgA cells were incubated with 0.25 µM [methyl 3H] SAM and 100 ng of purified YsgA. After incubation, reaction mixtures were processed as described in Materials and Methods, and the radioactivity incorporated in the different RNA preparations was measured using a scintillation counter. (B) Total B. subtilis ΔysgA rRNA (20 µg) was incubated with 1 µg of purified YsgA and 1 µCi [methyl 3H] SAM at 37°C. After 30 min incubation, the reaction mixture was loaded in two slots of a 1% agarose gel. After migration, the bands corresponding to 23S and 16S rRNA were cut out of the gel and melted by heating. Scintillation cocktail was added, and radioactivity measured in a scintillation counter. The number of cpm present in 23S and 16S rRNAs is indicated. (C) Autoradiography of 2D chromatograms of P1 hydrolysates of 14C-methylated rRNA. Dotted circles show the migration of pA, pC, pG, and pU nucleotides used as ultraviolet markers. The methylated nucleotides were identified by comparison to reference maps (Grosjean et al. 2007).
To identify which RNA is the YsgA substrate, rRNA and tRNA were isolated from the B. subtilis ΔysgA strain and were tested as substrates. Of note the tRNA preparation also contains the 5S rRNA. The results presented in Figure 1A show that YsgA efficiently methylates rRNA (16S, 23S, or 5S rRNA) but not tRNA. Since 5S rRNA is present in the tRNA preparation, only 16S or 23S rRNA remain as possible substrates.
To determine whether 16S or 23S rRNA is modified by YsgA, total rRNA from B. subtilis ΔysgA was methylated in vitro by the recombinant enzyme in the presence of 3H-SAM. After incubation, RNA was separated on a 1% agarose gel. The gel blocks containing the 16S or 23S rRNA were excised, melted by heating at 100°C and radioactivity was measured by scintillation counting. Figure 1B shows that YsgA methylates the 23S rRNA and not the 16S rRNA.
YsgA catalyzes Gm formation at position 2553 of 23S rRNA
In order to identify the nucleoside that is methylated by YsgA, rRNA from B. subtilis ΔysgA was methylated in vitro by recombinant YsgA in the presence of 14C-SAM. After the reaction, RNA was precipitated and hydrolyzed by nuclease P1. The resulting 5′-phosphate-nucleosides were separated by 2D-TLC followed by autoradiography. Figure 1C shows that YsgA forms 2′-O-methylguanosine (Gm).
In contrast to E. coli little is known about the presence and localization of modified nucleosides in rRNA of B. subtilis. Nevertheless, a characterization of part of domain V (comprising the A-loop) of the B. subtilis 23S rRNA was performed showing the presence of Gm at position 2581, which corresponds to position 2553 in E. coli numbering (Hansen et al. 2002). To determine if YsgA can catalyze the formation of Gm2553, different methodologies were used.
A T7 in vitro transcript of the entire 23S rRNA of B. subtilis was modified by YsgA in the presence of 3H-SAM and used in a hybridization-protection study. This was conducted using a DNA oligonucleotide (Hyb-1) that covers a 20 base region centered on G2553 and three oligonucleotides complementary to other regions of the 23S rRNA as controls (Hyb-2, -3 and -4, respectively, centered on U1915, G2252 and U2449, Supplemental Table S1). Oligonucleotide Hyb-3 covers the region where Gm2252 is found in the P-site of E. coli 23S rRNA. The other two control oligonucleotides are the same as those used in Caldas et al. (2000a). The radioactive methylated 23S rRNA transcript was hybridized to each of the different oligonucleotides. The RNA/DNA hybrids were then tested for protection against nuclease T1 hydrolysis. As shown in Figure 2A, the oligonucleotide Hyb-1 (spanning 2543–2562; centered on G2553) confers protection of the methylated region, whereas the other oligonucleotides do not confer any protection. This result shows that the residue modified by YsgA is situated in the 20 base region centered on G2553.
FIGURE 2.

Localization of the position modified by YsgA. (A) Hybridization-protection studies to localize the site of methylation by YsgA. A total of 20 µg of methyl 3H-labeled 23S rRNA (see Materials and Methods) was hybridized with 100 pmol of oligonucleotide and digested by RNase T1. Samples were TCA-precipitated and radioactivity was measured using a scintillation counter; oligo Hyb-1 centered on 2553 (▴), oligo Hyb-2 centered on 1916 (♦), oligo Hyb-3 centered on 2251 (●), and oligo Hyb-4 centered on 2450 (▪). (B) YsgA is able to methylate the ribose of A but not C nor U when G2553 is mutated in 23S rRNA. Autoradiograms of 2D chromatograms of P1 hydrolysates of T7 transcripts of 23S rRNA bearing G2553, A2553, C2553, and U2553 methylated by YsgA in the presence of 14C-SAM. The methylated nucleotides were identified by comparison to reference maps (Grosjean et al. 2007).
To precisely localize the position modified by YsgA, site directed mutagenesis was used to replace G2553 by the three other possible nucleotides. The rationale behind this approach is that the specificity of some 2′-O-methyltransferases is independent of the type of base in the nucleotide. T7 transcripts of wild-type and mutant 23S rRNA were incubated in the presence of YsgA and 14C-SAM. After ethanol precipitation and P1 nuclease hydrolysis the radioactive nucleotides were analyzed by 2D-TLC followed by autoradiography. While Am2553 formation is observed in the transcript in which G2553 is replaced by an A, no 2′-O-ribose methylation is detected when position 2553 is occupied by a pyrimidine (Fig. 2B). This indicates that YsgA modifies G2553 and is purine-specific.
The 23S rRNA of the B. subtilis ΔysgA mutant has lost Gm2553
All the results reported so far indicate that YsgA forms Gm2553 in 23S rRNA. If this would be the case, this modification should be present in the 23S rRNA of the B. subtilis wild-type strain and absent in the ΔysgA mutant. Two different approaches were used to address this question, one using a DNAzyme and the other using RNase digestion coupled to mass spectrometry.
For detection of 2′-O-ribose G2553 methylation by a DNAzyme, rRNA isolated from B. subtilis (wild-type and ΔysgA mutant) was incubated in the presence of a DNA oligonucleotide with DNAzyme activity that targets the phosphodiester bond between nucleotides G2553 and U2554. The DNAzyme (Buchhaupt et al. 2007) needs a free 2′-hydroxyl group to attack the adjacent phosphodiester linkage. This implies that ribose methylation of G2553 would prevent RNA cleavage. Figure 3 shows that 23S rRNA isolated from B. subtilis ΔysgA cells is cleaved by this DNAzyme, contrary to wild-type 23S rRNA. This result is consistent with the presence of Gm2553 in the 23S rRNA from the wild-type strain and its absence in the mutant strain.
FIGURE 3.
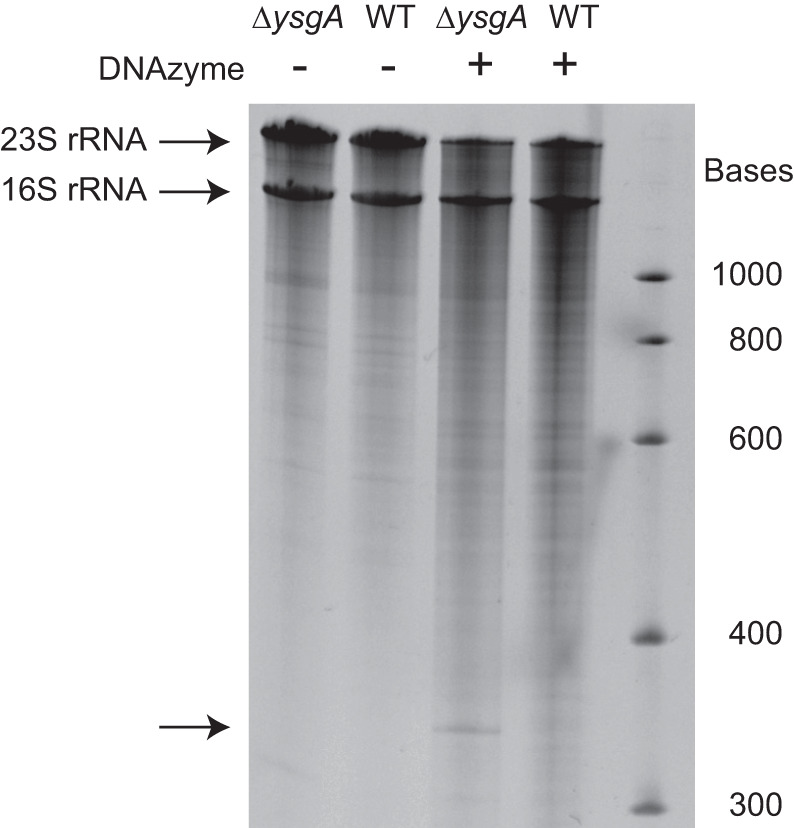
DNAzyme-catalyzed cleavage of 23S rRNA is prevented by the 2′-O-methylation introduced by YsgA. Total RNA from B. subtilis wild-type (WT) or ΔysgA cells was incubated with a 34 base DNAzyme and analyzed by denaturing (8 M urea) 4% polyacrylamide gel electrophoresis, as described by Buchhaupt et al. (2007). The DNAzyme can cleave the 23S rRNA and generate 2579 and 349 nt fragments if G2553 is not 2′-O-methylated. The 2579 nt fragment is not separated from the full length 2928 nt 23S rRNA. The 349 nt fragment is indicated by an arrow.
To further confirm the presence of Gm2553 in wild-type and its absence in B. subtilis ΔysgA we used LC-MS/MS mass spectrometry analysis after RNase T1 digestion. After digestion RNA fragments were separated by liquid chromatography and directly analyzed by MS/MS. MS/MS spectra were manually inspected and sequenced. However, the direct mapping of post-transcriptional modifications by LC-MS/MS in large RNA such as 23S RNA is impossible. Indeed, RNase T1 generates more than 300 fragments, often with different sequences but with the same or close nucleotide composition. In those cases, MS/MS sequencing is difficult. To overcome this problem, we isolated a defined RNA fragment of 57 nt containing G2553 (position 2515 to position 2571). This fragment was obtained by RNase H cleavage after complementary DNA hybridization. The fragment was separated by gel electrophoresis and the corresponding gel band was incubated in the presence of RNase T1. LC-MS/MS analysis of the wild-type revealed the presence of the sequence CU[Gm] UUCGp (mass of 2249.3 Da) and MS/MS fragmentation confirmed methylation at position 2553 (Fig. 4A). This fragment was not detected when using 23S rRNA of the ΔysgA mutant, but the sequence UUCGp (mass of 1279.1 Da) was found instead (Fig. 4B). This sequence is unique in the RNase H fragments and corresponds to the cleavage at G2553. This result confirms the absence of methylation of G2553 in the 23S rRNA of the ΔysgA mutant.
FIGURE 4.

Localization of the YsgA target nucleoside by mass spectrometry. (A) MS/MS sequencing spectra of 2249.3 Da (m/z 1124.13, z = 2−), sequence CU[Gm] UUCGp, obtained from the analysis of B. subtilis wild-type 23S fragment RNase H 2515-2571 after T1 digestion. CID fragmentation shows methylation of G 2553. (B) MS/MS sequencing spectra of 1279.1 Da (m/z 639.07, z = 2−), sequence UUCGp, obtained from the analysis of B. subtilis ΔysgA 23S fragment RNase H 2515–2571 after T1 digestion.
The results obtained with the ΔysgA strain combined with the experiments showing the in vitro enzymatic activity of YsgA demonstrate that YsgA is the B. subtilis methyltransferase forming Gm2553 in 23S rRNA.
YsgA methylates 23S rRNA neither in the fully assembled ribosome nor in the 50S subunit
Fully assembled ribosomes (70S) and the large ribosomal subunit (50S) were isolated to determine whether YsgA could modify guanine 2553 in the 23S rRNA of the assembled particle. Since the methylation efficiency of YsgA for RNA from E. coli and B. subtilis ΔysgA is equivalent (Supplemental Fig. S1), ribosomes (fully assembled 70S and 50S subunit only) were isolated from the E. coli JE28 strain in which the ribosomal L12 protein (bL12 in the new nomenclature [Ban et al. 2014]) bears a hexa-histidine affinity tag at its carboxyl terminus (Ederth et al. 2009). Of note, G2553 of the 23S rRNA of E. coli is naturally not modified. Assays were performed with the fully assembled 70S ribosome, as well as with the isolated 50S ribosomal subunit. For comparison, rRNA extracted from the ribosomal particles by phenol extraction was also tested. The results presented in Figure 5 show that 70S and 50S particles are very poor substrates, whereas deproteinized rRNA is efficiently modified by YsgA.
FIGURE 5.
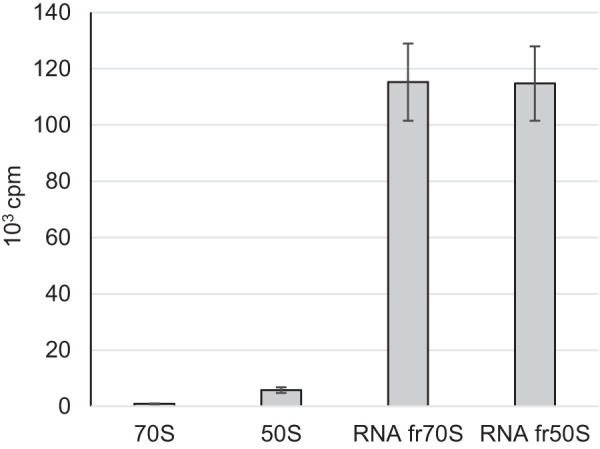
Purified 70S and 50S ribosomal particles are poor substrates of YsgA. A total of 0.5 A260 units of 70S or 50S ribosomal particles were incubated with 1 µg of purified YsgA and 1 µCi [methyl 3H] SAM. Incorporated radioactivity was measured by scintillation counting. RNA fr50S or fr70S are for RNA extracted from 50S or 70S ribosomal particles, respectively.
The loss of YsgA does not affect growth nor lead to ribosome assembly intermediates accumulation
In E. coli, the loss of a particular rRNA modification rarely leads to a phenotypic change. Only the absence of RlmE (also called FtsJ or RrmJ), the enzyme forming Um2552, results in a growth defect and leads to accumulation of a 50S ribosomal subunit assembly intermediate (45S intermediate) (Arai et al. 2015; Pletnev et al. 2020). Since position 2552 is adjacent to the target position of YsgA, growth rates and ribosomal subunits profiles of B. subtilis wild-type and ΔysgA strains were compared. Both strains were grown in rich (Materials and Methods) and in minimal (Spizizen 1958) medium at 37°C. The doubling times for both strains were similar in rich medium (WT, 25 min ± 2 min and ΔysgA, 24 min ± 3 min) as well as in minimal medium (WT, 121 min ± 14 min and ΔysgA, 119 min ± 16 min). For ribosome profiling both strains were grown in rich medium at 37°C and at 22°C. Low temperatures are indeed known to aggravate ribosome assembly defects (Shajani et al. 2011). Cells were lysed and ribosome subunits were separated by sucrose gradient centrifugation in the presence of low (0.5 mM) and high (10 mM) Mg++ concentrations. Figure 6 shows that the ribosomal subunits profile is the same for the two strains and is independent of the growth temperature.
FIGURE 6.

Sucrose density gradient profile of ribosomal subunits from B. subtilis wild-type (WT) and ΔysgA grown at 37°C or 22°C. Free NA: free nucleic acids. The peaks corresponding to 30S and 50S ribosomal subunits are indicated.
Crystal structure of YsgA
Bioinformatic studies predicted that YsgA belongs to the SPOUT superfamily of methyltransferases (Anantharaman et al. 2002). These proteins are characterized by the presence of three sequence motifs (Gustafsson et al. 1996) and a deep trefoil (topological) knot in their catalytic domain (Nureki et al. 2002). To confirm that YsgA is indeed a SPOUT enzyme, its crystal structure was solved. The structure of YsgA reveals an amino-terminal domain (NTD, residues 1–96) connected to a carboxy-terminal domain (CTD, 104–248) by an interdomain linker (residues 97–103). The carboxy-terminal core catalytic domain adopts a typical SPOUT fold and is composed of a β-sheet with seven parallel β-strands sandwiched between five α-helices on one side and two α-helices on the other side (Fig. 7A; Supplemental Fig. S2; Tkaczuk et al. 2007; Hori 2017). As the other members of the SPOUT RNA methyltransferase superfamily, YsgA has a deep trefoil knot which participates in the formation of the SAM-binding pocket and is formed by threading residues 222–248 through a hoop made of residues 180–190, which comprises a portion of β9 strand.
FIGURE 7.

3D structure of YsgA. (A) Overall structure of the wild-type YsgA dimer, represented as cartoon. The subunits A and B are colored in blue and orange, respectively, with the amino-terminal domain in a darker color and residues 222–248 of the B subunit in green. The secondary structure elements mentioned in the manuscript are indicated. The cofactor-binding pocket in the B subunit is shown as a van der Waals surface. The single quote symbol refers to the dimerization partner. The side chain of the residues which were mutated to alanine (see panel B) are represented as sticks. (B) Residual methyltransferase activity of several YsgA mutants. The values are the percentage of activity compared to the wild-type enzyme and represent the mean ± SEM of four experiments.
In agreement with the results obtained by size exclusion chromatography (Supplemental Fig. S3), the crystal structure of YsgA consists of a dimer, a usual feature of the SPOUT RNA methyltransferase superfamily. The dimerization interface is formed by the α7 and α13 helices as well as the loop 224–234. The perpendicular organization of the two pairs of helices at the dimer interface constitutes a characteristic of the SpoU (TrmH) family (Fig. 7A).
To further confirm that YsgA is a member of the SPOUT methyltransferase family, residues critical for the activity of other SPOUT methyltransferases were mutated to alanine. These residues are N117 and R123 in motif 1, E206 in motif 2 and N234 in motif 3 (Supplemental Fig. S4; Gustafsson et al. 1996; Hori 2017). This resulted in a dramatic decrease of the methyltransferase activity compared to the wild-type enzyme (Fig. 7B) as shown for other methyltransferases of the same family (Nureki et al. 2004; Mosbacher et al. 2005; Watanabe et al. 2005).
DISCUSSION
The presence of Gm2581 (Gm2553 in E. coli numbering) in the 23S rRNA of B. subtilis was reported (Hansen et al. 2002). However, the enzyme responsible for its formation has remained unidentified. Here we show that YsgA, one of the first predicted SPOUT methyltransferases (Anantharaman et al. 2002), catalyzes this modification. Position 2553 is part of helix 92, a component of the ribosomal A-site. G2553 contacts C75 of the aminoacylated tRNA during protein synthesis (Kim and Green 1999; Polikanov et al. 2015).
The crystal structure of YsgA reported in this work confirms this enzyme to be a member of the SPOUT superfamily. Among the proteins of known structure, the closest homologs in terms of sequence conservation are a SPOUT RNA methyltransferase of Sinorhizobium meliloti (SmMTase) and of Thermus thermophilus HB8 (Ttha0275), with 33% identity over the whole sequence. Despite this low amino acid sequence identity, the tridimensional structure of YsgA superposes well with that of these two homologs and other members of the SPOUT superfamily. A DALI search (Holm 2020) highlighted the RNA methyltransferases Ttha0275, SmMTase, RrmA (Nureki et al. 2002), NHR (Yang et al. 2010), AviRb (Mosbacher et al. 2005), a SPOUT RNA methyltransferase of Porphyromonas gingivalis (PgMTase), Tsr (Dunstan et al. 2009), as well as RlmB (Michel et al. 2002) as closest structural homologs. The NTD and CTD of these enzymes superpose very well, despite important variations in the orientation of the NTD relative to the CTD (Supplemental Fig. S5). Of note, although the NTD of RlmB can be superimposed with that of YsgA, it lacks the 27 first residues compared to YsgA (Michel et al. 2002). The NTD of these methyltransferases show high structural homology with the ribosomal proteins L7Ae/L30 (Mao and Williamson 1999; Kawaguchi et al. 2011; Huang and Lilley 2016) and is supposed to correspond to the RNA binding domain. These ribosomal proteins are called eL8/eL30 in the new nomenclature (Ban et al. 2014).
Although G2553 is a conserved residue, its 2′-O-methylation is not universal. For instance, it is found in B. subtilis, Geobacillus stearothermophilus, Haloarcula marismortui, and various eukaryotes but not in E. coli and Sulfolobus acidocaldarius (Hansen et al. 2002). Moreover, Gm2553 formation is not always catalyzed by homologous enzymes. Contrary to the B. subtilis YsgA and the human mitochondrial MRM3/RNMTL1 enzymes (Lee et al. 2013; Lee and Bogenhagen 2014; Rorbach et al. 2014) which are SPOUT methyltransferases, the functional equivalent yeast 2′-O-methyltransferase Spb1 has an RFM fold (Pintard et al. 2002; Lapeyre and Purushothaman 2004), illustrating a case of convergent evolution. Beside their structures, the two enzymes show many other differences: (i) in the absence of guide snR52, Spb1 can catalyze the formation of Um2552 (as its E. coli homolog RlmE) in addition to Gm2553 (positions 2921 and 2922 in yeast 25S rRNA) (Lapeyre and Purushothaman 2004), whereas YsgA is limited to Gm2553 formation; (ii) Spb1 is an essential protein for yeast, independently of its catalytic activity (Kressler et al. 1999), whereas a ΔysgA B. subtilis mutant is viable; (iii) S. cerevisiae Spb1 acts at late stages of ribosome formation (as its E. coli homolog RlmE), whereas B. subtilis YsgA modifies free 23S rRNA. This is consistent with YsgA as an early actor in ribosome assembly. Indeed, as mentioned previously (Siibak and Remme 2010; Sergeeva et al. 2015), most bacterial 23S modifications were shown to occur at early steps of ribosome biogenesis.
G2553 is adjacent to U2552 which can be 2′-O-methylated in various organisms but not in B. subtilis. In E. coli, Um2552 is formed by RlmE, a methyltransferase belonging to the RFM superfamily (Caldas et al. 2000a). A mutant deficient in RlmE shows alterations in ribosomal 50S subunit assembly (Arai et al. 2015; Pletnev et al. 2020) leading to translational defects (Caldas et al. 2000b). This contrasts with the B. subtilis ΔysgA mutant, which does not show any apparent defect in 50S subunit assembly. Therefore, the role played by Gm2553 remains to be elucidated.
Although G2553 is a universally conserved residue in 23S rRNA, we have shown using synthetic RNA transcripts that YsgA can also 2′-O-methylate an adenosine at this position. However, pyrimidine nucleosides are not methylated by the enzyme. Such broadened specificity is sometimes found among RNA 2′-O-methyltransferases of the SpoU (TrmH) family. The tRNA methyltransferase TrmL of E. coli can methylate the ribose of U34 and C34 (Liu et al. 2013) and the tRNA methyltransferase TrmJ of E. coli can methylate the four canonical nucleosides while its homolog in Sulfolobus acidocaldarius can only methylate a cytidine (Somme et al. 2014).
In conclusion, YsgA is the B. subtilis methyltransferase forming Gm2553 in 23S rRNA. Therefore we propose to rename YsgA as RlmP, according to the bacterial nomenclature of rRNA methyltransferases. Future work will consist in elucidating the role played by Gm2553 and in studying the recognition of 23S rRNA by RlmP.
MATERIALS AND METHODS
General procedures
Ampicillin was used at a concentration of 50 µg/mL and kanamycin at 30 µg/mL. Restriction endonucleases and T4 DNA ligase were purchased from Thermo Fisher Scientific. [methyl-14C] SAM and [methyl-3H] SAM were from PerkinElmer. Oligonucleotides were synthesized by Merck (Supplemental Table S1). Nuclease P1 and RNase T1 were from Sigma. Site-directed mutagenesis was performed using the QuikChange Kit (Agilent).
Bacterial strains and culture media
Bacillus subtilis 168 was used as the wild-type strain. B. subtilis was grown in an in house rich medium, medium 853, composed of 10 g/L bacto tryptone, 5 g/L yeast extract, 5 g/L NaCl, 1g/L glucose, 0.7 g/L K2HPO4, and 0.3 g/L KH2PO4 and in minimal medium (Spizizen 1958).
The ΔysgA strain, derivating from strain 168, was obtained from the Bacillus Genetic Stock Center (ref BKK28650). E. coli JE28 was a kind gift of Suparna Sanyal (Uppsala University).
Cloning of the B. subtilis ysgA open reading frame
A synthetic B. subtilis ysgA open reading frame (ORF), respecting the codon usage of E. coli, was synthetically generated (GeneArt, Thermo Fisher Scientific). The sequence of ysgA was flanked by the restriction sites NdeI and XhoI to facilitate cloning into the pET28 expression vector. This plasmid allows T7 expression in E. coli of the B. subtilis YsgA protein bearing an amino-terminal His-tag.
Expression and purification of YsgA
The E. coli BL21(DE3) strain was transformed with the expression vector and grown in 1 L of Luria broth at 37°C until an optical density of 0.6 was reached. Then 0.2 mM isopropyl-β-d-thiogalactopyranoside was added to the culture which was maintained at 37°C for 3 h. The cells were harvested by centrifugation and resuspended in a total volume of 40 mL of buffer A (50 mM Tris, 1 M NaCl, pH 8.0), and lyzed by sonication for 20 min at 4°C using a Branson 250 sonicator (duty cycle 80%, output control 4). After a centrifugation (20,000g, 30 min, 4°C) to clear the lysate, the supernatant was submitted to further purification. The sample was loaded on a Chelating Sepharose Fast Flow column (1 × 30 cm, GE Healthcare) charged with Ni2+ previously equilibrated with buffer A. After washing the column with buffer A supplemented with imidazole 10 mM, an imidazole gradient was applied to elute the recombinant YsgA protein (10 mM to 500 mM, 10 column volumes). Pooled fractions containing YsgA were concentrated by ultrafiltration (Amicon, 10 kDa cut-off, Millipore Merck) and further purified by gel filtration chromatography, using a Superose 12 10/300 GL column (GE Healthcare) in buffer B (20 mM Tris, 100 mM NaCl, 2 mM DTT, pH 8.0). The fractions of interest were concentrated by ultrafiltration to reach a concentration of 19 mg/mL. The purity of the protein was assessed by SDS-PAGE with Coomassie blue staining and shown to be above 95%. Glycerol 10% (v/v) was added to the sample, which was frozen in liquid nitrogen and stored at −80°C until use. The different variants of YsgA were purified using the same procedure.
Preparation of B. subtilis total RNA
Bacillus subtilis cells were grown to late exponential phase at 37°C in 1 L of Luria broth. Cells were harvested by centrifugation and the pellet was resuspended in 20 mL buffer C (50 mM Tris-HCl, 10 mM MgCl2, pH 8.0). The cells were disrupted by sonication at 4°C for 15 min using a Branson 250 Sonicator (same conditions as described above). The lysate was cleared by centrifugation (12,000g for 20 min at 4°C). After two extractions with nonbuffered phenol, total RNA was precipitated with 1/10 volume of AcONa 3 M pH 6.5 and an equal volume of isopropanol. The pellet was dissolved in H2O.
Preparation of B. subtilis rRNA
Bacillus subtilis cells were grown to late exponential phase at 37°C in 1 L of Luria broth. Cells were harvested by centrifugation and the pellet was resuspended in 20 mL buffer C. The cells were disrupted by sonication at 4°C using a Branson 250 Sonicator (same conditions as described above). The lysate was cleared by centrifugation (12,000g for 20 min). PEG6000 was then added to a concentration of 10% to precipitate the ribosomes. After centrifugation (12,000g for 20 min at 4°C), the rRNAs were extracted by nonbuffered phenol, and the rRNAs were precipitated with 1/10 volume of AcONa 3 M pH 6.5 and an equal volume of isopropanol. The pellet was dissolved in H2O.
Preparation of B. subtilis tRNA
Bacillus subtilis cells were grown to late exponential phase at 37°C in 2 L of Luria broth. Cells were harvested and the pellet was resuspended in H2O. Lysozyme was added to a concentration of 10 mg/mL and the sample was incubated for 20 min at 37°C. Then an equal volume of buffer D (100 mM NaOAc, 20 mM MgCl2, 300 mM NaCl, pH 4.5) was added. After two extractions with nonbuffered phenol, nucleic acids were precipitated with 1/10 volume of 20% KOAc pH 4.5 and an equal volume of isopropanol. The pellet was dissolved in buffer E (30 mM NaOAc, 10 mM MgCl2, pH 5.5) and applied to a DEAE Sepharose Fast Flow column (1 × 5 cm, GE Healthcare) equilibrated with buffer E. The column was washed with the same buffer supplemented with 400 mM NaCl, and tRNAs were eluted with a linear gradient of NaCl (400 to 800 mM, 10 column volumes). The fractions containing the tRNAs were pooled. These fractions also contain the 5S rRNA.
Cloning and T7 in vitro transcription of the B. subtilis 23S rRNA gene
The B. subtilis rrnO gene encoding 23S rRNA was amplified by PCR using Pfu DNA polymerase and oligonucleotides rrnO-F and rrnO-R (Supplemental Table S1). The obtained fragment was cloned in the pJET2.1 vector. The different rrnO mutants were generated by site-directed mutagenesis using the oligonucleotides listed in Supplemental Table S1. T7 in vitro transcription was performed according to the instructions of the RiboMAX Kit (Ambion). The 23S rRNA transcripts were purified using illustra MicroSpin G-25 columns (GE Healthcare).
Hybridization-protection studies
Hybridization-protection studies were performed as described previously (Caldas et al. 2000a) using 100 pmol of oligonucleotide Hyb-1, Hyb-2, Hyb-3, or Hyb-4 (Supplemental Table S1) and 20 µg 3H-methyl-labeled B. subtilis 23S rRNA produced by YsgA methylation of a T7 in vitro transcript.
Purification of His-tagged ribosomes from E. coli JE28
Ribosome purification was performed as described previously (Ederth et al. 2009). Briefly, E. coli JE28 cells were grown to late log phase in 1 L Luria broth. Pelleted cells were resuspended in 40 mL of buffer F (20 mM Tris-HCl, 10 mM MgCl2, 150 mM KCl, 30 mM NH4Cl, pH7.6) prior to cell disruption by sonication at 4°C using a Branson 250 Sonicator (same conditions as above). The subsequent steps were as for YsgA purification except that the column was equilibrated in buffer F and that ribosomes were eluted with a linear gradient (from 0 to 0.5 M, 10 column volumes) of imidazole in buffer F. The fractions containing 70S ribosomes were dialyzed against buffer F. Purification of the 50S ribosomal subunit was as for the complete ribosomal particle except that a low Mg2+ concentration (1 mM) was used.
Analysis of ribosomal subunits by sucrose density gradient
The analysis of ribosomal subunits was performed by sucrose density gradient (Arai et al. 2015; Pletnev et al. 2020). B. subtilis cells were cultivated in 1 L of LB medium at 22°C and 37°C until A660 reached 0.5, chilled on ice and harvested by centrifugation. The cells were suspended in 20 mL of buffer G [20 mM Hepes-KOH, 0.5 mM Mg(OAc)2, 200 mM NH4Cl, 6 mM β-mercaptoethanol, pH 7.6]. Cells were sonicated at 4°C using a Branson 250 Sonicator. The lysate was cleared by centrifugation (16,000g for 30 min at 4°C), and 15 A260 units were layered on top of a sucrose gradient [10%–40% (w/v)] in buffer G and separated by ultracentrifugation in a Beckman SW-41Ti Rotor at 35,000 rpm for 5 h at 4°C. Ribosomal subunits were fractionated on a Piston Gradient Fractionator, and the A260 was measured using a UV monitor.
RNA methyltransferase assays
A semiquantitative method to follow RNA methylation in vitro consisted in measuring the amount of 3H or 14C transferred to B. subtilis or E. coli RNA, or to rRNA transcripts, using [methyl-3H or 14C] SAM as the methyl donor. The reaction mixture (200 µL) consisted of 50 mM Tris-HCl, 5 mM MgCl2, 1 µCi [methyl-3H] SAM (18 Ci/mmol), or 20 nCi [methyl-14C] SAM (50 mCi/mmol), pH 8.0, and the amount of RNA and enzyme is specified in the legend of the figures. The mixture was incubated for 30 min at 37°C. The reaction was stopped by phenol extraction and the nucleic acids were TCA-precipitated. Radioactive methylated RNA was captured on a Whatman GF/C filter and washed three times with ethanol prior to the measurement of radioactivity in a scintillation counter.
For modified nucleotide identification by two-dimensional thin-layer chromatography (2D TLC), the RNA was ethanol precipitated after phenol extraction and thereafter hydrolyzed by nuclease P1. Modified nucleotides were analyzed by 2D-TLC on cellulose plates (Merck). The first dimension was with solvent A (isobutyric acid/concentrated NH4OH/water; 66/1/33; v/v/v); the second dimension was with solvent B (0.1 M sodium phosphate at pH 6.8/solid (NH4)2SO4/n-propanol; 100/60/2; v/w/v). The migration pattern was visualized by autoradiography. The nucleotides were identified using a reference map (Grosjean et al. 2007).
DNAzyme reactions and analysis
DNAzyme reactions and analysis were performed as outlined previously (Buchhaupt et al. 2007) using 4 µg rRNA from B. subtilis wild-type or ΔysgA strains and 200 pmol of DNAzyme (Supplemental Table S1).
Mass spectrometry
Isolation of a specific RNA fragment
For LC-MS/MS analysis, a 56 nt fragment of B. subtilis 23S rRNA containing G2553 (position 2515 to position 2571) was obtained by RNase H (Thermo Fisher Scientific) cleavage of RNA regions complementary to the DNA oligonucleotides MS-1 and MS-2 (Supplemental Table S1). The 23S rRNA digestion was performed in RNase H buffer (20 mM Tris-HCl, 40 mM KCl, 8mM MgCl2, 1 mM DTT, pH 7.8) for 2 min at 80°C, followed by slow cooling to 50°C and incubation with 0.5 U of RNase H for 20 min at 50°C. The RNase H fragment was isolated by denaturing (8M urea) 10% polyacrylamide gel electrophoresis. The corresponding band was excised under UV light for LC-MS/MS analysis.
T1 digestion and mass spectrometry analysis
LC-MS/MS analysis was done as previously described (Antoine and Wolff 2020). Briefly, gel pieces containing the RNase H fragment were digested by 20 µL of 0.1 U/µL RNase T1 (Thermo Fisher Scientific) during 4 h at 50°C. Samples were desalted using ZipTip C18 (Millipore) by several washes with 200 mM ammonium acetate and elution with 50% acetonitrile in milli-Q water and finally dried under vacuum. The pellet containing RNase digestion products was resuspended in 3 µL of milli-Q water. The products were separated on an Acquity Peptide BEH C18 Column (130 Å, 1.7 µm, 75 µm × 200 mm) using a nanoAcquity System (Waters). The column was equilibrated in a buffer containing 7.5 mM triethylammonium acetate, 7.0 mM triethylamine, and 200 mM hexafluoroisopropanol at a flow rate of 300 nL/min. The column was washed using a gradient from 15% to 35% methanol for 2 min, and the oligonucleotides were eluted with an increase of methanol up to 50% in 20 min. MS and MS/MS analysis was performed using a SYNAPT G2-S instrument (Waters). All experiments were performed in negative mode with a capillary voltage set at 2.6 kV and a sample cone voltage set at 30 V. The source was heated to 130°C. Samples were analyzed over an m/z range from 500 to 1500 for the full scan, followed by a fast data direct acquisition scan (Fast DDA). Collision induced dissociation (CID) spectra were deconvoluted using MassLynx Software (Waters) and manually sequenced by following the y and/or c series.
Crystallization, data collection, and structural characterization of YsgA
Crystallogenesis was performed at 20°C by the under-oil crystallization procedure, using 18 µL paraffin oil to cover the crystallization drops. Crystals of YsgA were obtained by mixing 1 µL of pure YsgA (19 mg/mL) with 1 µL of crystallization solution (2% PEG3350, 200 mM MES, pH 5.5). Streak seeding was used to improve crystallization, using crystals generated with higher precipitant concentrations. Crystals typically grew within a week and were analyzed at the SOLEIL Proxima 1 and 2 beamlines. Crystals were soaked for 60 sec in a cryoprotectant solution (200 mM MES, 10% PEG3350, 20% glycerol, pH 6.0). The XDS (Kabsch 2010) and Phenix (Liebschner et al. 2019) packages were used for data processing and model building and refinement, respectively. The structure was solved by molecular replacement, with SmMTase (pdb code: 5kzk) as search model. All the residues of YsgA could be modeled, except for the side chain atoms of E84 (chain A) and D62, E84, K99, and Q185 (chain B), due to the poor electron density. Data collection and refinement statistics are presented in Supplemental Table S2. PyMOL was used as a molecular graphic tool to prepare the figures (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC). The PDBSum software was used to create a 2D topology diagram (Laskowski et al. 2018). The coordinates of the YsgA tridimensional structure were deposited in the Protein Data Bank (pdb entry: 7QIU). The pdb entries of the structural homologs discussed in this work are the following: Ttha0275 (4 × 3m and 4 × 3l [H Demirci, R Bernadinelli, and G Jogl, unpubl.]), RlmB (1gz0 [Michel et al. 2002]), SmMTase (5kzk and 5l0z [D Dey, RP Hedge, SC Almo, et al., unpubl.]), PgMTase (2i6d [ME Cuff, KE Mussar, H Li, et al., unpubl.]), AviRb (1 × 7o [Mosbacher et al. 2005]), RrmA (1ipa [Nureki et al. 2002]), NHR (3nk7 [Yang et al. 2010]), Tsr (3gyq [Dunstan et al. 2009]), Archaeoglobus fulgidus eL8 (5g4v [Huang and Lilley 2016]), human eL30 (3vi6 [Kawaguchi et al. 2011]), and yeast eL30 (1ck2 [Mao and Williamson 1999]).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Suparna Sanyal for providing the JE28 strain. C.G., V.K., and L.D. were supported by a grant from the Fonds J. Brachet. P.W. was supported by the French National Program Investissement d'Avenir (Labex NetRNA) administered by the Agence Nationale de la Recherche (ANR-10-LABX-0036_ NETRNA). Access to Proxima 2 beamline (SOLEIL synchrotron) was within Block Allocation Groups 20191372. The authors are grateful to Proxima 2 staff for assistance during the experiments.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079131.122.
REFERENCES
- Anantharaman V, Koonin EV, Aravind L. 2002. SPOUT: a class of methyltransferases that includes spoU and trmD RNA methylase superfamilies, and novel superfamilies of predicted prokaryotic RNA methylases. J Mol Microbiol Biotechnol 4: 71–75. [PubMed] [Google Scholar]
- Antoine L, Wolff P. 2020. Mapping of posttranscriptional tRNA modifications by two-dimensional gel electrophoresis mass spectrometry. Methods Mol Biol 2113: 101–110. 10.1007/978-1-0716-0278-2_8 [DOI] [PubMed] [Google Scholar]
- Arai T, Ishiguro K, Kimura S, Sakaguchi Y, Suzuki T, Suzuki T. 2015. Single methylation of 23S rRNA triggers late steps of 50S ribosomal subunit assembly. Proc Natl Acad Sci 112: E4707–E4716. 10.1073/pnas.1415046112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban N, Beckmann R, Cate JH, Dinman JD, Dragon F, Ellis SR, Lafontaine DL, Lindahl L, Liljas A, Lipton JM, et al. 2014. A new system for naming ribosomal proteins. Curr Opin Struct Biol 24: 165–169. 10.1016/j.sbi.2014.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchhaupt M, Peifer C, Entian KD. 2007. Analysis of 2′-O-methylated nucleosides and pseudouridines in ribosomal RNAs using DNAzymes. Anal Biochem 361: 102–108. 10.1016/j.ab.2006.11.001 [DOI] [PubMed] [Google Scholar]
- Byström AS, Björk GR. 1982. The structural gene (trmD) for the tRNA(m1G)methyltransferase is part of a four polypeptide operon in Escherichia coli K-12. Mol Gen Genet 188: 447–454. 10.1007/BF00330047 [DOI] [PubMed] [Google Scholar]
- Caldas T, Binet E, Bouloc P, Costa A, Desgres J, Richarme G. 2000a. The FtsJ/RrmJ heat shock protein of Escherichia coli is a 23 S ribosomal RNA methyltransferase. J Biol Chem 275: 16414–16419. 10.1074/jbc.M001854200 [DOI] [PubMed] [Google Scholar]
- Caldas T, Binet E, Bouloc P, Richarme G. 2000b. Translational defects of Escherichia coli mutants deficient in the Um2552 23S ribosomal RNA methyltransferase RrmJ/FTSJ. Biochem Biophys Res Commun 271: 714–718. 10.1006/bbrc.2000.2702 [DOI] [PubMed] [Google Scholar]
- Cavaillé J, Chetouani F, Bachellerie JP. 1999. The yeast Saccharomyces cerevisiae YDL112w ORF encodes the putative 2'-O-ribose methyltransferase catalyzing the formation of Gm18 in tRNAs. RNA 5: 66–81. 10.1017/S1355838299981475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunstan MS, Hang PC, Zelinskaya NV, Honek JF, Conn GL. 2009. Structure of the thiostrepton resistance methyltransferase • S-adenosyl-L-methionine complex and its interaction with ribosomal RNA. J Biol Chem 284: 17013–17020. 10.1074/jbc.M901618200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ederth J, Mandava CS, Dasgupta S, Sanyal S. 2009. A single-step method for purification of active His-tagged ribosomes from a genetically engineered Escherichia coli. Nucleic Acids Res 37: e15. 10.1093/nar/gkn992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosjean H, Droogmans L, Roovers M, Keith G. 2007. Detection of enzymatic activity of transfer RNA modification enzymes using radiolabeled tRNA substrates. Methods Enzymol 425: 55–101. 10.1016/S0076-6879(07)25003-7 [DOI] [PubMed] [Google Scholar]
- Gustafsson C, Reid R, Greene PJ, Santi DV. 1996. Identification of new RNA modifying enzymes by iterative genome search using known modifying enzymes as probes. Nucleic Acids Res 24: 3756–3762. 10.1093/nar/24.19.3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen MA, Kirpekar F, Ritterbusch W, Vester B. 2002. Posttranscriptional modifications in the A-loop of 23S rRNAs from selected archaea and eubacteria. RNA 8: 202–213. 10.1017/S1355838202013365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L. 2020. DALI and the persistence of protein shape. Protein Sci 29: 128–140. 10.1002/pro.3749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori H. 2017. Transfer RNA methyltransferases with a SpoU-TrmD (SPOUT) fold and their modified nucleosides in tRNA. Biomolecules 7: 23. 10.3390/biom7010023 [DOI] [Google Scholar]
- Huang L, Lilley DM. 2016. A quasi-cyclic RNA nano-scale molecular object constructed using kink turns. Nanoscale 8: 15189–15195. 10.1039/C6NR05186C [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. 2010. XDS. Acta Crystallogr D Biol Crystallogr 66: 125–132. 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi A, Ose T, Yao M, Tanaka I. 2011. Crystallization and preliminary X-ray structure analysis of human ribosomal protein L30e. Acta Crystallogr Sect F Struct Biol Cryst Commun 67: 1516–1518. 10.1107/S1744309111045131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DF, Green R. 1999. Base-pairing between 23S rRNA and tRNA in the ribosomal A site. Mol Cell 4: 859–864. 10.1016/S1097-2765(00)80395-0 [DOI] [PubMed] [Google Scholar]
- Koonin EV, Rudd KE. 1993. SpoU protein of Escherichia coli belongs to a new family of putative rRNA methylases. Nucleic Acids Res 21: 5519. 10.1093/nar/21.23.5519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kressler D, Rojo M, Linder P, Cruz J. 1999. Spb1p is a putative methyltransferase required for 60S ribosomal subunit biogenesis in Saccharomyces cerevisiae. Nucleic Acids Res 27: 4598–4608. 10.1093/nar/27.23.4598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamohan A, Jackman JE. 2017. Mechanistic features of the atypical tRNA m1G9 SPOUT methyltransferase, Trm10. Nucleic Acids Res 45: 9019–9029. 10.1093/nar/gkx620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamohan A, Jackman JE. 2019. A family divided: distinct structural and mechanistic features of the SpoU-TrmD (SPOUT) methyltransferase superfamily. Biochemistry 58: 336–345. 10.1021/acs.biochem.8b01047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapeyre B, Purushothaman SK. 2004. Spb1p-directed formation of Gm2922 in the ribosome catalytic center occurs at a late processing stage. Mol Cell 16: 663–669. 10.1016/j.molcel.2004.10.022 [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Jabłońska J, Pravda L, Vařeková RS, Thornton JM. 2018. PDBsum: structural summaries of PDB entries. Protein Sci 27: 129–134. 10.1002/pro.3289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Bogenhagen DF. 2014. Assignment of 2′-O-methyltransferases to modification sites on the mammalian mitochondrial large subunit 16 S ribosomal RNA (rRNA). J Biol Chem 289: 24936–24942. 10.1074/jbc.C114.581868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Okot-Kotber C, LaComb JF, Bogenhagen DF. 2013. Mitochondrial ribosomal RNA (rRNA) methyltransferase family members are positioned to modify nascent rRNA in foci near the mitochondrial DNA nucleoid. J Biol Chem 288: 31386–31399. 10.1074/jbc.M113.515692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkóczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ, et al. 2019. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol 75: 861–877. 10.1107/S2059798319011471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu RJ, Zhou M, Fang ZP, Wang M, Zhou XL, Wang ED. 2013. The tRNA recognition mechanism of the minimalist SPOUT methyltransferase, TrmL. Nucleic Acids Res 41: 7828–7842. 10.1093/nar/gkt568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv F, Zhang T, Zhou Z, Gao S, Wong CC, Zhou JQ, Ding J. 2015. Structural basis for Sfm1 functioning as a protein arginine methyltransferase. Cell Discov 1: 15037. 10.1038/celldisc.2015.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H, Williamson JR. 1999. Local folding coupled to RNA binding in the yeast ribosomal protein L30. J Mol Biol 292: 345–359. 10.1006/jmbi.1999.3044 [DOI] [PubMed] [Google Scholar]
- Meyer B, Wurm JP, Sharma S, Immer C, Pogoryelov D, Kötter P, Lafontaine DL, Wöhnert J, Entian KD. 2016. Ribosome biogenesis factor Tsr3 is the aminocarboxypropyl transferase responsible for 18S rRNA hypermodification in yeast and humans. Nucleic Acids Res 44: 4304–4316. 10.1093/nar/gkw244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel G, Sauvé V, Larocque R, Li Y, Matte A, Cygler M. 2002. The structure of the RlmB 23S rRNA methyltransferase reveals a new methyltransferase fold with a unique knot. Structure 10: 1303–1315. 10.1016/S0969-2126(02)00852-3 [DOI] [PubMed] [Google Scholar]
- Mosbacher TG, Bechthold A, Schulz GE. 2005. Structure and function of the antibiotic resistance-mediating methyltransferase AviRb from Streptomyces viridochromogenes. J Mol Biol 345: 535–545. 10.1016/j.jmb.2004.10.051 [DOI] [PubMed] [Google Scholar]
- Nureki O, Shirouzu M, Hashimoto K, Ishitani R, Terada T, Tamakoshi M, Oshima T, Chijimatsu M, Takio K, Vassylyev DG, et al. 2002. An enzyme with a deep trefoil knot for the active-site architecture. Acta Crystallogr D Biol Crystallogr 58: 1129–1137. 10.1107/S0907444902006601 [DOI] [PubMed] [Google Scholar]
- Nureki O, Watanabe K, Fukai S, Ishii R, Endo Y, Hori H, Yokoyama S. 2004. Deep knot structure for construction of active site and cofactor binding site of tRNA modification enzyme. Structure 12: 593–602. 10.1016/j.str.2004.03.003 [DOI] [PubMed] [Google Scholar]
- Persson BC, Jäger G, Gustafsson C. 1997. The spoU gene of Escherichia coli, the fourth gene of the spoT operon, is essential for tRNA (Gm18) 2′-O-methyltransferase activity. Nucleic Acids Res 25: 4093–4097. 10.1093/nar/25.20.4093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintard L, Lecointe F, Bujnicki JM, Bonnerot C, Grosjean H, Lapeyre B. 2002. Trm7p catalyses the formation of two 2′-O-methylriboses in yeast tRNA anticodon loop. EMBO J 21: 1811–1820. 10.1093/emboj/21.7.1811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletnev P, Guseva E, Zanina A, Evfratov S, Dzama M, Treshin V, Pogorel'skaya A, Osterman I, Golovina A, Rubtsova M, et al. 2020. Comprehensive functional analysis of Escherichia coli ribosomal RNA methyltransferases. Front Genet 11: 97. 10.3389/fgene.2020.00097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polikanov YS, Melnikov SV, Söll D, Steitz TA. 2015. Structural insights into the role of rRNA modifications in protein synthesis and ribosome assembly. Nat Struct Mol Biol 22: 342–344. 10.1038/nsmb.2992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorbach J, Boesch P, Gammage PA, Nicholls TJ, Pearce SF, Patel D, Hauser A, Perocchi F, Minczuk M. 2014. MRM2 and MRM3 are involved in biogenesis of the large subunit of the mitochondrial ribosome. Mol Biol Cell 25: 2542–2555. 10.1091/mbc.e14-01-0014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert HL, Blumenthal RM, Cheng X. 2003. Many paths to methyltransfer: a chronicle of convergence. Trends Biochem Sci 28: 329–335. 10.1016/S0968-0004(03)00090-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeeva OV, Bogdanov AA, Sergiev PV. 2015. What do we know about ribosomal RNA methylation in Escherichia coli? Biochimie 117: 110–118. 10.1016/j.biochi.2014.11.019 [DOI] [PubMed] [Google Scholar]
- Shajani Z, Sykes MT, Williamson JR. 2011. Assembly of bacterial ribosomes. Annu Rev Biochem 80: 501–526. 10.1146/annurev-biochem-062608-160432 [DOI] [PubMed] [Google Scholar]
- Shao Z, Yan W, Peng J, Zuo X, Zou Y, Li F, Gong D, Ma R, Wu J, Shi Y, et al. 2014. Crystal structure of tRNA m1G9 methyltransferase Trm10: insight into the catalytic mechanism and recognition of tRNA substrate. Nucleic Acids Res 42: 509–525. 10.1093/nar/gkt869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siibak T, Remme J. 2010. Subribosomal particle analysis reveals the stages of bacterial ribosome assembly at which rRNA nucleotides are modified. RNA 16: 2023–2032. 10.1261/rna.2160010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RK, Feller A, Roovers M, Van Elder D, Wauters L, Droogmans L, Versées W. 2018. Structural and biochemical analysis of the dual-specificity Trm10 enzyme from Thermococcus kodakaraensis prompts reconsideration of its catalytic mechanism. RNA 24: 1080–1092. 10.1261/rna.064345.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somme J, Van Laer B, Roovers M, Steyaert J, Versées W, Droogmans L. 2014. Characterization of two homologous 2′-O-methyltransferases showing different specificities for their tRNA substrates. RNA 20: 1257–1271. 10.1261/rna.044503.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spizizen J. 1958. Transformation of biochemically deficient strains of Bacillus subtilis by deoxyribonucleate. Proc Natl Acad Sci 44: 1072–1078. 10.1073/pnas.44.10.1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinehart WE, Jackman JE. 2015. Diversity in mechanism and function of tRNA methyltransferases. RNA Biol 12: 398–411. 10.1080/15476286.2015.1008358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkaczuk KL, Dunin-Horkawicz S, Purta E, Bujnicki JM. 2007. Structural and evolutionary bioinformatics of the SPOUT superfamily of methyltransferases. BMC Bioinformatics 8: 73. 10.1186/1471-2105-8-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Laer B, Roovers M, Wauters L, Kasprzak JM, Dyzma M, Deyaert E, Kumar Singh R, Feller A, Bujnicki JM, Droogmans L, et al. 2016. Structural and functional insights into tRNA binding and adenosine N1-methylation by an archaeal Trm10 homologue. Nucleic Acids Res 44: 940–953. 10.1093/nar/gkv1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Nureki O, Fukai S, Ishii R, Okamoto H, Yokoyama S, Endo Y, Hori H. 2005. Roles of conserved amino acid sequence motifs in the SpoU (TrmH) RNA methyltransferase family. J Biol Chem 280: 10368–10377. 10.1074/jbc.M411209200 [DOI] [PubMed] [Google Scholar]
- Yang H, Wang Z, Shen Y, Wang P, Jia X, Zhao L, Zhou P, Gong R, Li Z, Yang Y, et al. 2010. Crystal structure of the nosiheptide-resistance methyltransferase of Streptomyces actuosus. Biochemistry 49: 6440–6450. 10.1021/bi1005915 [DOI] [PubMed] [Google Scholar]
- Young BD, Weiss DI, Zurita-Lopez CI, Webb KJ, Clarke SG, McBride AE. 2012. Identification of methylated proteins in the yeast small ribosomal subunit: a role for SPOUT methyltransferases in protein arginine methylation. Biochemistry 51: 5091–5104. 10.1021/bi300186g [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.