

Abstract
Fibroblasts are central to the acute and chronic response of tissues to stress: they are necessary for wound healing, involved in inflammatory responses and critical for long term remodeling of tissue. These diverse roles of fibroblasts arise from the cells’ ability to respond to internal and extracellular cues regarding the physical state of the host tissue. In this article, we review recent evidence for the role of chromatin as a sensor of cellular stress and chromatin-dependent gene regulatory events that may be essential for fibroblast activation in the setting of injury. This emerging evidence highlights chromatin structure and accessibility as features necessary for our understanding of how cell type-specific epigenomes sense and respond to stress.
A genome evolves by rendering itself useful for a multitude of different purposes. A substrate for this usefulness is chromatin and the terms of evolution are the establishment of distinct folding properties1, resulting in transcriptomes as different as neurons, nephrons, fibroblasts and cardiomyocytes. Genomes accomplish cell type specific things with ubiquitous chromatin components and the help of non-ubiquitous transcription factors, giving rise to cell diversity and plasticity.
The mammalian heart is comprised of distinct cell types with vastly different potentiality (Figure 1): cardiomyocytes, for example, are terminally differentiated and highly structured cells that die or expand with stress; fibroblasts, on the other hand, are phenotypically plastic cells that participate in chamber remodeling, wound healing and tissue integrity. This distinct potentiality is underpinned by distinct nuclear topology and chromatin architecture. How these and other resident cardiac cells participate in normal heart function and the response to acute or chronic injury is controlled by the cells’ epigenomes—the collection of molecules responsible for chromatin structure, chromatin accessibility, and gene transcription (Figure 2). One manner to achieve stimulus-specific gene expression in fibroblasts is for direct mechanical sensing of cellular phenotype by chromatin—indeed, these cells’ reason for existing is to sense, respond to and either mitigate or promote tissue stiffening.
Figure 1: Structural considerations for cardiac fibroblast epigenomics.
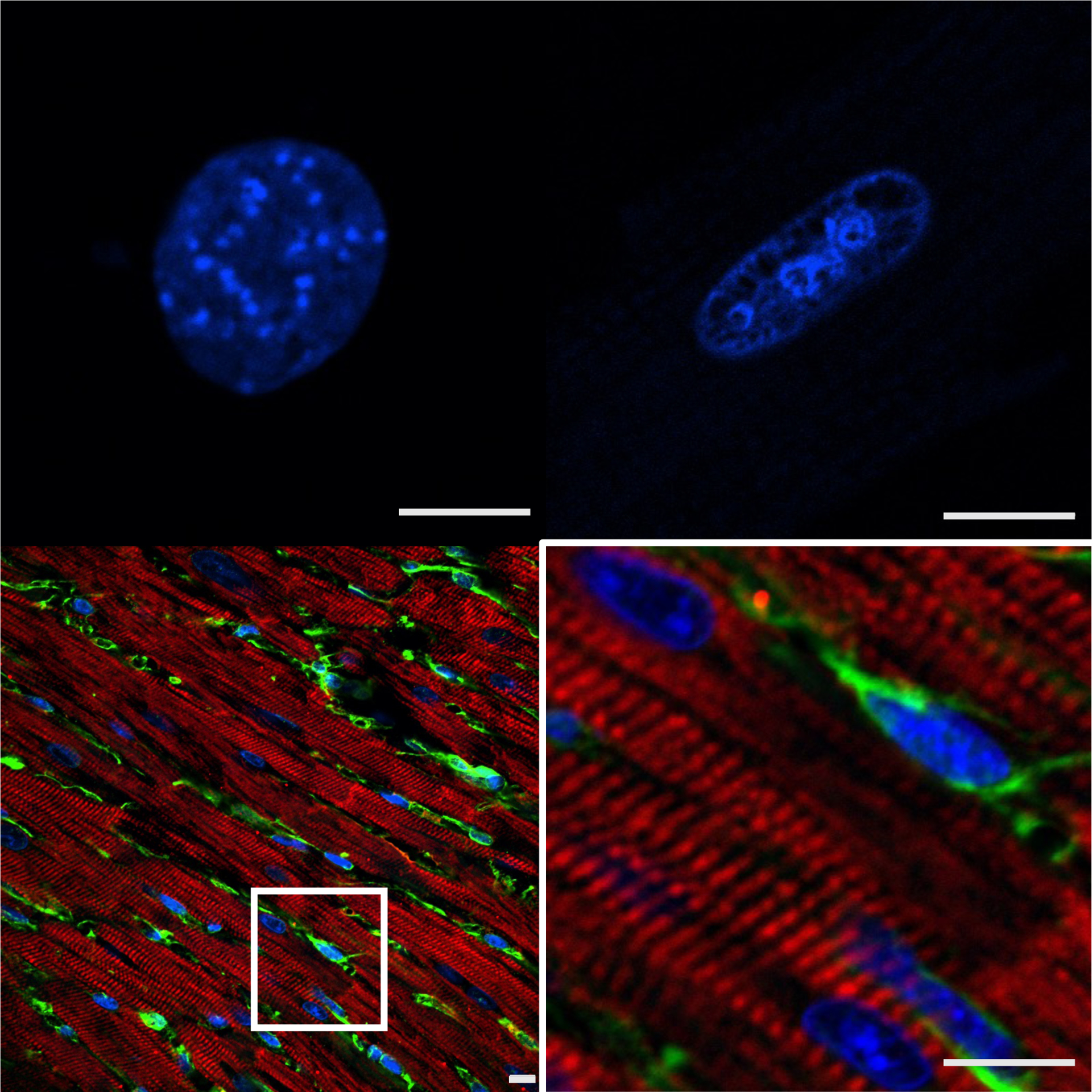
Top left, DAPI stained nucleus of primary cardiac fibroblast in culture. Top right, DAPI stained primary cardiac myocyte nucleus. Bottom left, Cardiac tissue labeled with cardiac troponin T (red, myocytes), vimentin (green, fibroblasts) and DAPI. Bottom right, inset of region of interest in bottom left. All specimens from adult mice; bars=10μm. Fibroblast nuclei have distinct global structural features (compared to myocytes) and the nuclear organization of these cells changes when in culture due to altered extracellular tension and signaling, with implications for the study of gene regulation and chromatin architecture.
Figure 2: Interrelationship between chromatin structure, chromatin accessibility, gene expression and phenotype.
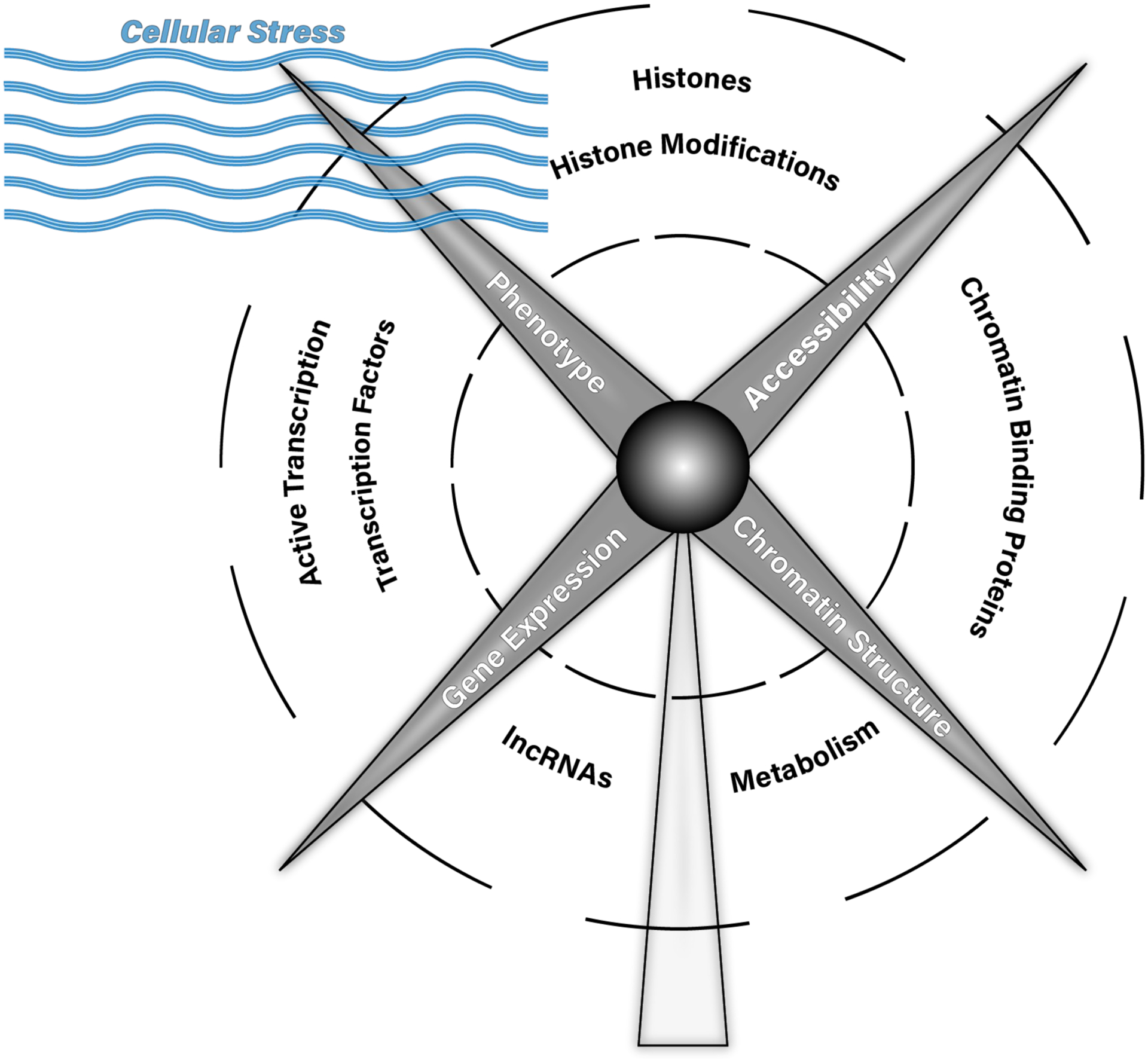
In this cartoon, chromatin structure, chromatin accessibility, gene expression and phenotype are blades of a windmill, representing inter-related components that influence each other and that themselves are directly acted upon by cellular stress (represented here as the wind). These cellular stresses (e.g. extracellular environment, physical forces, intercellular signaling) act through the features labeled along the blades’ trajectory (the act of transcription itself, transcription factors, chromatin binding proteins, histones, histone modifications, DNA modifications, metabolism and lncRNAs) to influence chromatin structure, chromatin accessibility, gene expression and phenotype, and vice versa.
Fibrosis and its resolution require plasticity, impermanence. A necessary response to a wound or infarct, fibrosis can also play a detrimental role in the long-term healing process. Several recent investigations reviewed in this article have examined the interplay of flexibility and stability at the level of chromatin, to investigate the relationship between myofibroblast transcriptome activation in the setting of cardiac stress and the chromatin remodeling events that enable it.
Chromatin Regulation During Fibroblast Activation: Current Understanding
The wall of the ventricle is a complex environment beset by constant physical stress due to contraction, necessitating bidirectional communication between the cytoskeleton and the nucleus. To investigate how tissue-like aggregation of cells in 3D alters the phenotype of cardiac fibroblasts, these cells were cultured in either 2D or 3D conditions [1]. In parallel experiments, cells were started in 3D conditions, transitioned to 2D and then back to 3D conditions, in all situations measuring changes in chromatin accessibility (ATAC-seq) and transcription [1]. Unsurprisingly, culturing in 2D versus 3D conditions produced substantial differences in gene expression (~1000 up-regulated genes in 3D)—remarkably, the gene expression signature was almost completely reversed when the cells were transitioned back to the 3D culture situation in the 3D to 2D to 3D experiment, evincing tight coupling between extracellular mechanical and chemical cues and the fibroblast transcriptome. Interesting, while some (18–23%) of the genes with altered expression also exhibited altered accessibility with changing culture conditions, most (77–82%) did not [1]. When cardiac fibroblast transcription and chromatin accessibility were likewise assessed after myocardial infarction, ATAC-detectable accessibility at the majority (69%) of promoters did not correlate with gene expression changes [2], demonstrating that changes in transcription, particularly in non-differentiating conditions, are often not associated with changes in accessibility (this phenomenon has also been reported in myocytes [3])2. ATAC-detectable accessibility changes thus may reflect structural/accessibility changes on nearby (in 3D) regions of the genome undergoing transcription separate from the genes whose accessibility is being measured, i.e. a structural change unrelated to transcription at the given locus.
Chromatin states require cellular machinery to decode and enact distinct transcriptomes, properties often attributed to reader proteins. One such reader, bromodomain containing protein 4 (BRD4) binds acetylated lysines, including those on histones, which are often found at active genes and their enhancers. BRD4 in turn acts by recruiting the Mediator complex and positive transcriptional elongation factor (pTEFb) to release transcriptional pausing. In cardiac fibroblasts, BRD4 has—amongst the other BRD isoforms expressed in the heart—a sanctioned role in binding enhancers and super-enhancers (as defined by histone H3 K27 acetylation ChIP-seq) and controlling expression of the genes they regulate. Genetic or pharmacologic manipulation of BRD4 binding modulates transcriptional activation in myocytes [4,5] and fibroblasts: in the latter, its binding is necessary for TGF-β induced fibroblast activation and pathologic gene expression [6]. In each case, the activity of the chromatin reader is tuned to enact the desired transcriptome of host cell type (e.g. in myocytes, BRD4 localizes to myocyte genes such as Nppa/Nppb [5]; in fibroblasts, it localizes to fibroblast genes such as Acta2 [6]). The activities of BRD4 in distinct populations of cardiac cells was recently further elaborated using single cell ATAC-seq and single cell RNA-seq [7]: across myocytes, fibroblasts and other cardiac cells, the binding of BRD4 (as assessed by the presence or absence of the BRD-inhibitor, JQ-1; n.b., JQ-1 inhibits other BET/Bromodomain proteins with varying Kds and there are thus isoform-specific roles amongst this family of proteins that may not be fully elucidated by its use) was reversibly associated with transcription, suggesting dynamic modulation of transcriptional activity solely due to the presence of the reader. Some of the regulatory regions (identified by precision nuclear run on sequencing, PRO-seq, which maps active RNA-polymerases) activated by TGF-β in cardiac fibroblasts also exhibited altered ATAC-detectable accessibility (some of which was similarly sensitive to BRD4 binding), enabling identification of Meox1 as a positive regulator of cardiac fibroblast activation (notably, Meox1 enhancer accessibility was altered by JQ1 in fibroblasts, but not myeloid or endothelial cells, demonstrating cell type specific tuning of chromatin remodeling in vivo) [7]. An open question resulting from the many single cell studies of the heart that have appeared in the last few years is what are the relative stabilities of the distinct cell populations captured by single cell technologies? One model addressing this through “state space” theory [8] may indicate that epigenetic regulation, similar to the original Waddington thesis, controls not only differentiation but also instantaneous potentiality in every somatic cell, along with the cell’s response to stress.
Long noncoding RNAs
Long noncoding RNAs (lncRNAs) have been touted as one solution to the problem of specifying cell type transcriptomes: some are indeed noncoding and thus have actions presumably at the RNA level, many localize to the nucleus and interact with chromatin3, and many are cell type specific—qualifying as conditions necessary for targeting of protein machinery for gene regulation and chromatin opening. Maternally expressed gene 3 (Meg3), a lncRNA originally identified as TGF-β-responsive in breast cancer where it was shown to bind chromatin, overlap with EZH2 (a member of the polycomb repressive complex, which deposits the histone H3 K27 silencing mark) on some regions of the genome, and form triple helices with DNA [10], is one such example of an epigenetic lncRNA capable of determining protein complex localization and/or formation (inhibition of this lncRNA attenuated pressure overload induced cardiac remodeling in vivo and TGF-β-induced fibroblast activation in vitro [11]). Loss of the cardiac enriched lncRNA H19 ameliorated remodeling after myocardial infarction and was found to bind to Y box binding protein (YB-1) and to in turn negatively regulate YB-1’s association with the Col1a1 promoter in the nucleus of cardiac fibroblasts [12]. In a global analysis of fibroblast enriched lncRNAs, Wisper was identified as a lncRNA expressed from a super-enhancer (classified by extensive histone H3 K27ac deposition) specific to cardiac, and to a lesser extent, lung fibroblasts [13]. Wisper was upregulated after myocardial infarction, its inhibition prevented wound closure in vitro and its inhibition in vivo attenuated infarction-induced fibrosis [13]. Although not shown to interact with chromatin or regulate transcription by direct physical interaction, Wisper’s localization to the nucleus (~40% of total RNA in fibroblasts) and transcription from super-enhancer decorated regions provides an interesting candidate of lncRNA-mediated chromatin organization by virtue (solely or mostly) of the act of its transcription, rather than by the lncRNA functioning by precise molecular interactions once transcribed. Safe is a nuclear-localized lncRNA that binds to other mRNAs and whose inhibition blunts cardiac fibrosis in vitro and in vivo [14]. Pro-cardiac fibrotic lncRNA (PCFL) has been shown to promote cardiac fibrosis [15], likely through interactions with miRNAs and not via chromatin. Similarly, the lncRNAs Cfast [16] and Myhrt [17] have been shown to regulate cardiac fibrosis in vivo through actions that involve binding miRNAs and non-nuclear proteins.
Histone-modifying enzymes
The histone demethylase Jumonji domain-containing protein 3 (JMJD3), which targets histone H3 K27me3, was found to be upregulated after infarction in mouse hearts and isolated cardiac fibroblasts following angiotensin II. Following Ang II treatment, H3K27me3 abundance was decreased in the promoter of β-catenin, a necessary component of the fibrotic response, and siRNA knockdown of JMJD3 attenuated β-catenin expression [18], although global targets of JMJD3 and direct effects on transcription (as well as mechanisms of genomic targeting), remain unknown. Global deletion of the histone lysine demethylase KDM3A prevents pressure overload induced fibrosis (in addition to ameliorating hypertrophy and chamber dilation), in the process blocking activation of a host of profibrotic genes [19]. Pharmacologic inhibition of KDM3A with JIB-04 recapitulates these phenotypic effects [19], increasing global H3K9me2 and H3K9me3 in mouse hearts, although direct effects on cardiac fibroblasts are unclear. In vivo knockdown of the histone methyltransferase disruptor of telomeric silencing 1-like (DOT1L) reduced infarct size and fibrosis in mice, while also inhibiting fibroblast activation in cell culture [20] whereas the p300 acetyltransferase-associated factor PCAF was activated by isoproterenol (although its protein expression decreases) in vivo and its inhibition attenuated TGF-β-induced Acta2 and Col1a1 expression in isolated fibroblasts [21]. Silencing of EZH2 (enhancer of zeste 2) in human atrial fibroblasts prevented AngII-induced activation and decreased overall histone H3K27 trimethylation, regardless of AngII stimulation [22].
Among histone mark erasers, histone deacetylases (HDACs) are the best studied class of proteins with regard to their ability to regulate cardiac physiology. Beyond the beneficial effects to the myocyte, HDAC inhibition blocks recruitment of the pause-release protein BRD4 to super-enhancers upstream of the pro-fibrotic gene Sertad4, perhaps indicative of the broader means by which HDAC inhibition simultaneously reversing the organ, cell, gene expression and proteomic level phenotypic changes associated with diastolic heart failure in the unilateral nephrectomy plus deoxycorticosterone acetate–salt hypertensive mouse model [23]. HDAC activity was also shown to be directed by the binding of the E-box binding family of zinc finger transcription repressor Twist1 (specifically on the gene Clca2, necessary for conversion to myofibroblasts), in the setting of TGF-β-induced cardiac fibroblast activation [24]. Insights into direct mechanical coupling between the extracellular matrix, the nucleus and chromatin were revealed from studies in which cardiac fibroblasts were subjected to culture conditions of increasing stiffness. Stiff matrices induced fibroblast activation and the longer the cell was kept in a stiff environment, the more committed to activation it became: if switched to soft media after 1–3 days, the cells deactivated, but if switched from stiff to soft after 5 days or more, the myofibroblast phenotype was maintained notwithstanding the soft environment extracellular environment. These cellular phenotypic changes were mirrored by chromatin condensation changes (measured by whole nuclear DAPI intensity and quantified using a chromatin condensation parameter) in which as cells transitioned from quiescence to transient activation, chromatin decondensed, but if they further transitioned to persistent activation, chromatin again condensed [25]. This behavior was sensitive to HDAC inhibition with trichostatin A, which alone decreased chromatin condensation and in combination with transition to soft matrix, was effective to deactivate the cells even after 7 days on stiff matrix. Interestingly, the progression from transient to persistent activation was associated with decreased ATAC-detectable chromatin accessibility [25]. These findings are direct evidence that fibroblast chromatin is sensitive to extracellular physical stress which is sensed in part by global changes in nuclear architecture and chromatin accessibility. Hypoxia has been shown to regulate DNA methylation enzymes in cardiac fibroblasts the setting of myocardial ischemia and fibrosis, including increased DNMT1 and DNMT3 (DNA methyltransferases) expression and hypermethylation [26]. Although treatment with 5-aza-2’-deoxycytidine, the DNMT inhibitor, attenuated Acta2 (in agreement with other studies showing regulation of this gene by DNA methylation [27]) and Col1a1 expression, the totality of targeted loci and mechanisms at chromatin remain unknown. Indeed, a role for DNA methylation as active regulator of global gene expression and chromatin structure in somatic cells is questionable.
Systematic analyses of rat cardiac myocyte and fibroblast chromatin by ChIP-seq and ATAC-seq was used to define cis-regulatory regions in these cells and to explore differential logic for transcription factor binding at individual genes between different cell types in the same organ [28]. Cell type-specific cis regulatory elements (defined using HOMER-based differential ATAC-seq peak analysis) were identified around transcription start sites, away from proximal promoters and tended to reside in cell type-specific genes. Likewise putative enhancers, decorated with histone H3K27acetylation, were also enriched around cell type specific genes in cardiac fibroblasts and tended to colocalize with ATAC-defined cis regulatory elements[28]. Transcriptome remodeling in fibroblasts in the mouse heart following myocardial infarction preferentially involves activation of a subset of cardiac transcription factors in addition to garden variety immune and inflammatory genes such as Zpf57, Wt1, Jun, and Npnt [29]. Other studies have shown that cardiac fibroblast ATAC-detectable chromatin accessibility is dynamic over time after myocardial infarction [30], although the role of this accessibility change in gene expression remains opaque: expression of some genes can be explained by accessibility changes whereas others cannot.
The high mobility group (HMG) family of proteins have been shown to exhibit isoform specific roles in chromatin conformation and gene expression in a variety of cells and organs, including the heart [31]. There is evidence that directly, or through competition with other chromatin structural proteins such as histones or CTCF (which has a well-established role in myocyte chromatin architecture [32,33]), HMGs increase local chromatin compaction in neonatal myocytes [34]. In fibroblasts, HMGA1 was induced by isoproterenol in vivo or TGF-β in vitro, and its periostin-dependent overexpression in vivo promoted cardiac dysfunction, whereas knockdown inhibited fibroblast activation in isolated fibroblasts [35]. The Hippo pathway is a powerful regulator of organ size and cellular proliferation, including in the heart, where its inhibition promotes myocyte proliferation in vivo, including in pigs [36]. In cardiac fibroblasts, Hippo signaling through its downstream effector molecules Lats1/2 modulates basal fibrotic response [37]: deletion of Lats1/2 induces fibrosis and prevent proper chromatin localization of the Lats1/2 target, Yap. Furthermore, Yap has been found (using ATAC and CUT&RUN [a higher resolution ChIP-seq approach]) to control a subset of histone H3K27ac decorated enhancers associated with immune response genes, potentially facilitating their interaction with nearby promoters. Indeed, most Yap peaks (90%) associated with enhancers and were demonstrated, by chromatin conformation measurement, to interact primarily with other enhancers (60% total enhancer-associated Yap peaks) or promoters (32%) [37]. In the realm of therapeutic reprogramming of cardiac fibroblasts to myocytes, chromatin regulation has been a long-sought target to augment the actions of reprogramming transcription factors Gata4, Mef2c, and Tbx5 (this cocktail has also been supplemented with other transcription factors including Hand2 and Myocardin, as well as with signaling molecules, like Akt) [38,39]. The chromatin reader Phf7, which acts via interaction with histone H3K4me2/3 marks to facilitate transcription, was found to be enriched in Mesp1+ myocyte progenitors [39]. Overexpression of PHF7 enhances in vitro reprogramming of fibroblasts to myocytes through actions that involved association with SWI/SNF chromatin remodeling complexes, localization to histone H3K27ac enhancers in cardiac fibroblasts and increased ATAC-detectable accessibility around genes with gene ontology terms enriched in cardiac muscle processes [39]. Despite these insights, the roles of many chromatin structural proteins in cardiac fibroblast phenotype have not been explored with in vivo gain/loss of function approaches.
Perspective
Our natural predilection to extrapolate behavior based on well-defined examples means that we do not know whether all genes involved in myofibroblast activation are regulated by epigenetic machinery in the same manner as the archetypical Acta2, Col1a1 and others that are directly tested in many studies. The phenotypic effects of epigenetic machinery (chromatin writers, erasers and readers in particular) demonstrated by genetic gain/loss of function approaches in the absence of unbiased measurement of genome-wide histone modification and accessibility changes, leave open the question of to what extent the actions of these proteins operate via colloquial histone code dogma. Non-compliance of ATAC-seq findings to said dogma provide a cautionary tale. The means to establish cell type specific gene expression thus include: (a) altered transcription factor binding; (b) altered localization of chromatin remodeling complexes, chromatin readers and/or Mediator and RNA polymerase complexes; (c) altered accessibility resulting from (a) or (b), preceding (a) or (b) or independent of (a) and (b); and (d) altered chromatin structure, defined as changes in chromatin contacts detectable by HiC (or comparable technique), changes in global chromatin compaction, or changes in orientation of relevant regions of DNA within the nucleus with respect to other loci or nuclear features (e.g. lamina, transcription factories or central heterochromatin centers). Empirical evidence for a positive correlation between ATAC-detectable accessibility and gene expression is modest, demonstrating that ATAC is measuring something other than active transcription—which itself is more accurately captured by techniques such as GRO-seq. There are no rules yet discovered for how local accessibility, as measured by ATAC, nuclease-dependent (e.g. MNase) techniques, or microscopy, relate to higher order structure and thus it is impossible at present to infer, based on local accessibility, how a region of chromatin positions within the nucleus4. Likewise with the opposite: the structural basis for how distinct chromatin neighborhoods use nucleosome positioning and higher order structure to segregate transcriptional active from silent regions follow a logic yet to be revealed. There is abundant evidence for a correlation between histone post-translational modifications (and the actions of the writers and erasers that control them) and expression of some genes. In limited cases, this correlation has been shown to extend to some regions of accessibility and in still more limited cases, to select features of higher order structure. However, we do not know the rules of structural catenation defining an invariant relationship between local compaction and global structure.
Communication between phenotype and chromatin—unlike the central dogma—need not be hierarchical (Figure 2). Changes in transcription are sufficient to alter chromatin structure: one way evolution may use lncRNAs is to position them throughout the genome in a manner that facilitates differential phase separation based on the accretion of protein and RNA molecules near active transcription factories [40] [41]. Physical distortion of the cell can alter chromatin structure (and thereby transcription) and the rigidity of the nucleus itself can contribute to the phenotype of the cell independent of transcription. In such a model, the cell uses the pre-existing guides from the physical positioning of gene and noncoding regions in the genome, along with the RNA and proteins species unique to cell type, to fold the genome appropriately for the given phenotype. Extranuclear stimuli influence the nature of this folding, which in turn endows the cell with the physical and chemical phenotypes that define it.
Acknowledgements:
We regret that the abbreviated format of this article precluded citation of many studies that have influenced our thinking on these matters. We thank Todd Kimball (UCLA) for helpful discussions and with design and creation of figures. Research in the Vondriska laboratory is supported by the National Institutes of Health (HL 105699; HL 150225), the David Geffen School of Medicine and the Department of Anesthesiology & Perioperative Medicine at UCLA.
Footnotes
Declaration of Interest: None.
By analogy, origami, the art of folding paper into visually appealing shapes, transforms a flat sheet of paper in two ways: first, the artist creates an interesting shape like an animal or perhaps a flower; second, the 3D space of an ellipsoid occupying the folded paper is significantly smaller than that of the unfolded paper.
These regions are probably best understood as ATAC-accessible regions that are either transcriptionally on or off due to other chromatin features that do not alter availability to the transposase.
Whether lncRNAs act via chromatin is rarely directly tested and much of the rubric for chromatin-based lncRNA actions come from the prototypical lncRNA, Xist, which silences the X chromosome through extensive binding to it and recruitment of the polycomb repressive complex (polycomb itself interacts with perhaps ~1000 other lncRNAs 9. Lee JT: Epigenetic regulation by long noncoding RNAs. Science 2012, 338:1435–1439.).
The fractal properties of chromatin, elsewhere described by the principles of phase separation, are not invariant but neither are they random. That is to say, we reason chromatin structure is governed by rules that can be understood and tested, for example with genetic and pharmacologic approaches to manipulate stoichiometry of chromatin proteins, so long as the right chromatin level endpoints (e.g. global structure and accessibility) are measured.
References
- 1.Yu J, Seldin MM, Fu K, Li S, Lam L, Wang P, Wang Y, Huang D, Nguyen TL, Wei B, et al. : Topological Arrangement of Cardiac Fibroblasts Regulates Cellular Plasticity. Circ Res 2018, 123:73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li C, Sun J, Liu Q, Dodlapati S, Ming H, Wang L, Li Y, Li R, Jiang Z, Francis J, et al. : The landscape of accessible chromatin in quiescent cardiac fibroblasts and cardiac fibroblasts activated after myocardial infarction. Epigenetics 2021:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chapski DJ, Cabaj M, Morselli M, Mason RJ, Soehalim E, Ren S, Pellegrini M, Wang Y, Vondriska TM, Rosa-Garrido M: Early adaptive chromatin remodeling events precede pathologic phenotypes and are reinforced in the failing heart. J Mol Cell Cardiol 2021, 160:73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anand P, Brown JD, Lin CY, Qi J, Zhang R, Artero PC, Alaiti MA, Bullard J, Alazem K, Margulies KB, et al. : BET bromodomains mediate transcriptional pause release in heart failure. Cell 2013, 154:569–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stratton MS, Lin CY, Anand P, Tatman PD, Ferguson BS, Wickers ST, Ambardekar AV, Sucharov CC, Bradner JE, Haldar SM, et al. : Signal-Dependent Recruitment of BRD4 to Cardiomyocyte Super-Enhancers Is Suppressed by a MicroRNA. Cell Rep 2016, 16:1366–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stratton MS, Bagchi RA, Felisbino MB, Hirsch RA, Smith HE, Riching AS, Enyart BY, Koch KA, Cavasin MA, Alexanian M, et al. : Dynamic Chromatin Targeting of BRD4 Stimulates Cardiac Fibroblast Activation. Circ Res 2019, 125:662–677. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study is the first to link actions of histone reader proteins (BRD4) to fibroblast activation through mechanisms involving chromatin remodeling.
- 7.Alexanian M, Przytycki PF, Micheletti R, Padmanabhan A, Ye L, Travers JG, Gonzalez-Teran B, Silva AC, Duan Q, Ranade SS, et al. : A transcriptional switch governs fibroblast activation in heart disease. Nature 2021, 595:438–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reichardt IM, Robeson KZ, Regnier M, Davis J: Controlling cardiac fibrosis through fibroblast state space modulation. Cell Signal 2021, 79:109888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee JT: Epigenetic regulation by long noncoding RNAs. Science 2012, 338:1435–1439. [DOI] [PubMed] [Google Scholar]
- 10.Mondal T, Subhash S, Vaid R, Enroth S, Uday S, Reinius B, Mitra S, Mohammed A, James AR, Hoberg E, et al. : MEG3 long noncoding RNA regulates the TGF-beta pathway genes through formation of RNA-DNA triplex structures. Nat Commun 2015, 6:7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piccoli MT, Gupta SK, Viereck J, Foinquinos A, Samolovac S, Kramer FL, Garg A, Remke J, Zimmer K, Batkai S, et al. : Inhibition of the Cardiac Fibroblast-Enriched lncRNA Meg3 Prevents Cardiac Fibrosis and Diastolic Dysfunction. Circ Res 2017, 121:575–583. [DOI] [PubMed] [Google Scholar]
- 12.Choong OK, Chen CY, Zhang J, Lin JH, Lin PJ, Ruan SC, Kamp TJ, Hsieh PCH: Hypoxia-induced H19/YB-1 cascade modulates cardiac remodeling after infarction. Theranostics 2019, 9:6550–6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Micheletti R, Plaisance I, Abraham BJ, Sarre A, Ting CC, Alexanian M, Maric D, Maison D, Nemir M, Young RA, et al. : The long noncoding RNA Wisper controls cardiac fibrosis and remodeling. Sci Transl Med 2017, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hao K, Lei W, Wu H, Wu J, Yang Z, Yan S, Lu XA, Li J, Xia X, Han X, et al. : LncRNA-Safe contributes to cardiac fibrosis through Safe-Sfrp2-HuR complex in mouse myocardial infarction. Theranostics 2019, 9:7282–7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun F, Zhuang Y, Zhu H, Wu H, Li D, Zhan L, Yang W, Yuan Y, Xie Y, Yang S, et al. : LncRNA PCFL promotes cardiac fibrosis via miR-378/GRB2 pathway following myocardial infarction. J Mol Cell Cardiol 2019, 133:188–198. [DOI] [PubMed] [Google Scholar]
- 16.Zhang F, Fu X, Kataoka M, Liu N, Wang Y, Gao F, Liang T, Dong X, Pei J, Hu X, et al. : Long noncoding RNA Cfast regulates cardiac fibrosis. Mol Ther Nucleic Acids 2021, 23:377–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lang M, Ou D, Liu Z, Li Y, Zhang X, Zhang F: LncRNA MHRT Promotes Cardiac Fibrosis via miR-3185 Pathway Following Myocardial Infarction. Int Heart J 2021, 62:891–899. [DOI] [PubMed] [Google Scholar]
- 18.Long F, Wang Q, Yang D, Zhu M, Wang J, Zhu Y, Liu X: Targeting JMJD3 histone demethylase mediates cardiac fibrosis and cardiac function following myocardial infarction. Biochem Biophys Res Commun 2020, 528:671–677. [DOI] [PubMed] [Google Scholar]
- 19.Zhang QJ, Tran TAT, Wang M, Ranek MJ, Kokkonen-Simon KM, Gao J, Luo X, Tan W, Kyrychenko V, Liao L, et al. : Histone lysine dimethyl-demethylase KDM3A controls pathological cardiac hypertrophy and fibrosis. Nat Commun 2018, 9:5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li F, Li L, Zhang J, Yang X, Liu Y: Histone methyltransferase DOT1L mediates the TGF-beta1/Smad3 signaling pathway through epigenetic modification of SYK in myocardial infarction. Hum Cell 2022, 35:98–110. [DOI] [PubMed] [Google Scholar]
- 21.Lim Y, Jeong A, Kwon DH, Lee YU, Kim YK, Ahn Y, Kook T, Park WJ, Kook H: P300/CBP-Associated Factor Activates Cardiac Fibroblasts by SMAD2 Acetylation. Int J Mol Sci 2021, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song S, Zhang R, Mo B, Chen L, Liu L, Yu Y, Cao W, Fang G, Wan Y, Gu Y, et al. : EZH2 as a novel therapeutic target for atrial fibrosis and atrial fibrillation. J Mol Cell Cardiol 2019, 135:119–133. [DOI] [PubMed] [Google Scholar]
- 23.Travers JG, Wennersten SA, Pena B, Bagchi RA, Smith HE, Hirsch RA, Vanderlinden LA, Lin YH, Dobrinskikh E, Demos-Davies KM, et al. : HDAC Inhibition Reverses Preexisting Diastolic Dysfunction and Blocks Covert Extracellular Matrix Remodeling. Circulation 2021, 143:1874–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This study identifies a previously unknown form of fibrotic protein activation in the heart and demonstrates that HDAC inhibition is sufficient to block it.
- 24.Shao T, Xue Y, Fang M: Epigenetic Repression of Chloride Channel Accessory 2 Transcription in Cardiac Fibroblast: Implication in Cardiac Fibrosis. Front Cell Dev Biol 2021, 9:771466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walker CJ, Crocini C, Ramirez D, Killaars AR, Grim JC, Aguado BA, Clark K, Allen MA, Dowell RD, Leinwand LA, et al. : Nuclear mechanosensing drives chromatin remodelling in persistently activated fibroblasts. Nat Biomed Eng 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This paper uses a host of unconventional approaches for studying chromatin to demonstrate that chromatin itself can sense the extracellular environment of fibroblasts and that extracellular stiffness controls gene expression by changes in chromatin compaction and accessibility.
- 26.Watson CJ, Collier P, Tea I, Neary R, Watson JA, Robinson C, Phelan D, Ledwidge MT, McDonald KM, McCann A, et al. : Hypoxia-induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast-like phenotype. Hum Mol Genet 2014, 23:2176–2188. [DOI] [PubMed] [Google Scholar]
- 27.He Y, Ling S, Sun Y, Sheng Z, Chen Z, Pan X, Ma G: DNA methylation regulates alpha-smooth muscle actin expression during cardiac fibroblast differentiation. J Cell Physiol 2019, 234:7174–7185. [DOI] [PubMed] [Google Scholar]
- 28.Golan-Lagziel T, Lewis YE, Shkedi O, Douvdevany G, Caspi LH, Kehat I: Analysis of rat cardiac myocytes and fibroblasts identifies combinatorial enhancer organization and transcription factor families. J Mol Cell Cardiol 2018, 116:91–105. [DOI] [PubMed] [Google Scholar]
- 29.Quaife-Ryan GA, Sim CB, Ziemann M, Kaspi A, Rafehi H, Ramialison M, El-Osta A, Hudson JE, Porrello ER: Multicellular Transcriptional Analysis of Mammalian Heart Regeneration. Circulation 2017, 136:1123–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruiz-Villalba A, Romero JP, Hernandez SC, Vilas-Zornoza A, Fortelny N, Castro-Labrador L, San Martin-Uriz P, Lorenzo-Vivas E, Garcia-Olloqui P, Palacio M, et al. : Single-Cell RNA Sequencing Analysis Reveals a Crucial Role for CTHRC1 (Collagen Triple Helix Repeat Containing 1) Cardiac Fibroblasts After Myocardial Infarction. Circulation 2020, 142:1831–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fedele M, Fidanza V, Battista S, Pentimalli F, Klein-Szanto AJ, Visone R, De Martino I, Curcio A, Morisco C, Del Vecchio L, et al. : Haploinsufficiency of the Hmga1 gene causes cardiac hypertrophy and myelo-lymphoproliferative disorders in mice. Cancer Res 2006, 66:2536–2543. [DOI] [PubMed] [Google Scholar]
- 32.Rosa-Garrido M, Chapski DJ, Schmitt AD, Kimball TH, Karbassi E, Monte E, Balderas E, Pellegrini M, Shih TT, Soehalim E, et al. : High-Resolution Mapping of Chromatin Conformation in Cardiac Myocytes Reveals Structural Remodeling of the Epigenome in Heart Failure. Circulation 2017, 136:1613–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee DP, Tan WLW, Anene-Nzelu CG, Lee CJM, Li PY, Luu TDA, Chan CX, Tiang Z, Ng SL, Huang X, et al. : Robust CTCF-Based Chromatin Architecture Underpins Epigenetic Changes in the Heart Failure Stress-Gene Response. Circulation 2019, 139:1937–1956. [DOI] [PubMed] [Google Scholar]
- 34.Monte E, Rosa-Garrido M, Karbassi E, Chen H, Lopez R, Rau CD, Wang J, Nelson SF, Wu Y, Stefani E, et al. : Reciprocal Regulation of the Cardiac Epigenome by Chromatin Structural Proteins Hmgb and Ctcf: IMPLICATIONS FOR TRANSCRIPTIONAL REGULATION. J Biol Chem 2016, 291:15428–15446. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study demonstrates the locus-specific role of distinct chromatin structural proteins in controlling local accessibility, gene expression and global nuclear organization.
- 35.Xie Q, Yao Q, Hu T, Cai Z, Zhao J, Yuan Y, Wu QQ, Tang QZ: High-Mobility Group A1 Promotes Cardiac Fibrosis by Upregulating FOXO1 in Fibroblasts. Front Cell Dev Biol 2021, 9:666422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu S, Li K, Wagner Florencio L, Tang L, Heallen TR, Leach JP, Wang Y, Grisanti F, Willerson JT, Perin EC, et al. : Gene therapy knockdown of Hippo signaling induces cardiomyocyte renewal in pigs after myocardial infarction. Sci Transl Med 2021, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiao Y, Hill MC, Li L, Deshmukh V, Martin TJ, Wang J, Martin JF: Hippo pathway deletion in adult resting cardiac fibroblasts initiates a cell state transition with spontaneous and self-sustaining fibrosis. Genes Dev 2019, 33:1491–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang H, Yang Y, Qian Y, Liu J, Qian L: Delineating chromatin accessibility re-patterning at single cell level during early stage of direct cardiac reprogramming. J Mol Cell Cardiol 2021, 162:62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garry GA, Bezprozvannaya S, Chen K, Zhou H, Hashimoto H, Morales MG, Liu N, Bassel-Duby R, Olson EN: The histone reader PHF7 cooperates with the SWI/SNF complex at cardiac super enhancers to promote direct reprogramming. Nat Cell Biol 2021, 23:467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adriaens C, Serebryannyy LA, Feric M, Schibler A, Meaburn KJ, Kubben N, Trzaskoma P, Shachar S, Vidak S, Finn EH, et al. : Blank spots on the map: some current questions on nuclear organization and genome architecture. Histochem Cell Biol 2018, 150:579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This thoughtful review article highlights many of the challenges facing the field of chromatin-dependent gene regulation, advancing novel hypotheses with comprehensive review of the literature supporting structure-function relationships between chromatin and transcription.
- 41.Mele M, Rinn JL: “Cat’s Cradling” the 3D Genome by the Act of LncRNA Transcription. Mol Cell 2016, 62:657–664. [DOI] [PubMed] [Google Scholar]