

Abstract
A major contributor to contractile dysfunction in heart failure is remodelling and loss of the cardiomyocyte transverse tubular system (t-system), but underlying mechanisms and signalling pathways remain elusive. It has been shown that dexamethasone promotes t-tubule development in stem cell-derived cardiomyocytes and that cardiomyocyte-specific glucocorticoid receptor (GR) knockout (GRKO) leads to heart failure. Here, we studied if the t-system is altered in GRKO hearts and if GR signalling is required for t-system preservation in adult cardiomyocytes. Confocal and 3D STED microscopy of myocardium from cardiomyocyte-specific GRKO mice revealed decreased t-system density and increased distances between ryanodine receptors (RyR) and L-type Ca2+ channels (LTCC). Because t-system remodelling and heart failure are intertwined, we investigated the underlying mechanisms in vitro. Ventricular cardiomyocytes from failing human and healthy adult rat hearts cultured in the absence of glucocorticoids (CTRL) showed distinctively lower t-system density than cells treated with dexamethasone (EC50 1.1nM) or corticosterone. The GR antagonist mifepristone abrogated the effect of dexamethasone. Dexamethasone improved RyR-LTCC coupling and synchrony of intracellular Ca2+ release, but did not alter expression levels of t-system associated proteins junctophilin-2 (JPH2), bridging integrator-1 (BIN1) or caveolin-3 (CAV3). Rather, dexamethasone upregulated LC3B and increased autophagic flux. The broad-spectrum protein kinase inhibitor staurosporine prevented dexamethasone-induced upregulation of autophagy and t-system preservation, and autophagy inhibitors bafilomycin A and chloroquine accelerated t-system loss. Conversely, induction of autophagy by rapamycin or amino acid starvation preserved the t-system. These findings suggest that GR signalling and autophagy are critically involved in t-system preservation and remodelling in the heart.
1. Introduction
Chronic heart failure is characterized by progressive contractile dysfunction, which results from a vicious circle between cardiac stress and maladaptive myocardial and cellular remodelling. One important characteristic of cardiomyocytes from failing hearts is remodelling of the transverse tubular system (t-system). This remodelling results in reduced t-system density as well as changes in t-tubule shape and orientation [22, 46]. In healthy cardiomyocytes the t-system consists of regularly arranged tubular membrane structures, running from the surface into the myocyte. Its main purpose is to guarantee efficient excitation-contraction coupling by positioning L-type Ca2+ channels (LTCCs), located in the cell membrane, next to ryanodine receptors (RyRs), located in the membrane of the sarcoplasmic reticulum (SR). This allows for fast and synchronous triggering of intracellular Ca2+ release, a prerequisite for efficient contraction. T-system remodelling in heart failure results in reduced LTCC-RyR coupling, asynchronous intracellular Ca2+ release and, consequently, decreased cardiac contractility [14, 27]. The close correlation between t-system remodelling and impaired contractility has been convincingly demonstrated in end-stage failing human hearts, where ventricular regions with preserved contractility still presented a regular t-system whereas regions with severely impaired contractility exhibited a degenerated t-system [9]. Furthermore, it has been reported that the degree of t-system remodelling correlates inversely with functional recovery of human failing hearts after implantation of ventricular assist devices [46]. A preserved t-system may therefore be required for recovery, or, put differently, t-system remodelling might be the result of irreversible myocardial changes. For these and other reasons, the t-system has been suggested as a promising target of prevention, diagnosis and therapy [28, 50].
Mechanisms behind t-system remodelling, however, are poorly understood. Due to the chronic development and complex pathophysiology of heart failure it is particularly difficult to identify causal relationships in animal models or patients [50]. It has been suggested that increased wall stress [12], strain [24] or fibrosis [8, 47] may trigger t-system remodelling and that the remodelling involves downregulation of t-system associated proteins, such as junctophilin 2 (JPH2) [59] or bridging integrator 1 (BIN1) [6]. However, although convincing evidence exists for the involvement of these proteins in t-tubule and junctional organization, it is possible that a loss of t-tubules merely reduces the amounts of proteins preferentially residing in the t-system.
In-vitro studies, which provide more control over experimental parameters, may facilitate a better understanding of t-system remodelling. For instance, primary-isolated cardiomyocytes remodel spontaneously in culture, and this remodelling resembles the structural and functional changes observed in heart failure [2, 27]. Thus, the discovery of interventions which prevent or mitigate these effects of cell culture may provide important clues towards the mechanisms underlying t-system remodelling in vivo. It was recently reported that t-tubules develop in stem cell-derived cardiomyocytes only if thyroxin (T3) and dexamethasone are added to the culture medium [36]. Moreover, cardiomyocyte-specific glucocorticoid receptor (GR) knockout in mice leads to irregular Ca2+ signalling, impaired contractility and heart failure [34]. Based on these studies, we formulated the hypothesis that glucocorticoid signalling is involved in t-system homeostasis and remodelling, possibly by genetic regulation of t-system associated proteins. Furthermore, we hypothesized that impaired or excessive autophagy, as present in situations of cardiomyocyte stress and heart failure [32], may underlie the degradation of t-tubules.
The present study shows that t-tubule density and LTCC-RyR coupling is decreased in hearts from cardiomyocyte-specific GR knockout (GRKO) mice. Effects of GR signalling on the t-system were then investigated in an in-vitro model of t-system loss to circumvent the confounding effects of heart failure. The results indicate that GR agonists prevent t-system loss and preserve excitation-contraction coupling in cultured rat and human cardiomyocytes. Furthermore, this study provides evidence that enhanced autophagy rather than upregulation of JPH2 or BIN1 mediates the effects of GR activation on the t-system and that maintenance of autophagic flux is critical for preservation of the t-system.
2. Methods
Methods are described in detail in the Online Supplementary Material.
2.1. Animals
Female Wistar rats (150–200 g) were obtained from Charles River Germany. Rats were killed by intraperitoneal injection of thiopental-sodium (100 mg/kg), followed by thoracotomy and excision of the heart. All experiments with rats were approved by the Animal Care and Use Committee Mittelfranken, Bavaria, Germany. Female cardiomyocyte-specific glucocorticoid receptor knockout (GRKO) mice were generated and bred as described previously [33, 34]. Mice were killed by cervical dislocation. All experiments with mice were approved by the Animal Care and Use Committee at the National Institute of Environmental Health Sciences (NIEHS), National Institutes of Health (NIH).
2.2. Human cardiac samples
Human cardiac tissue samples were collected from the left-ventricular apical core during implantation of mechanical assist devices or from the free left-ventricular wall of explanted hearts. Collection and use of human cardiac tissue samples was approved by the Institutional Review Boards of the University of Erlangen-Nürnberg and the Ruhr-University Bochum. Studies were conducted according to Declaration of Helsinki principles. Patients gave their written informed consent prior to tissue collection. Patient characteristics are displayed in Online Table 1.
2.3. Cardiomyocyte isolation and culture
Left-ventricular human and rat cardiomyocytes were isolated and investigated immediately (fresh myocytes) or cultured in M199 medium. In some experiments a modified medium, free from amino acids, was used (see Online Supplementary Material). Culture dishes were randomly assigned to treatment groups.
2.4. Confocal imaging of living and fixed cardiomyocytes
Living myocytes were placed into culture medium with 30mmol/L butanedione monoxime (BDM) and incubated with 8μmol/L Di8-ANEPPS at room temperature to stain cell membranes. Myocytes were then imaged with a Zeiss LSM 780 inverted confocal microscope. For immunostaining, cardiomyocytes were fixed with 2% PFA (RyR, LTCC co-staining) or 100% acetone (BIN1, JPH2 co-staining, LC3BII staining), then incubated with the respective primary and secondary antibodies and imaged on the confocal microscope.
2.5. Ca2+ imaging and analysis of Ca2+ signals
Cardiomyocytes were stained with 8μmol/L Di8-ANEPPS and loaded with 10μmol/L Fluo-4 AM, selected randomly by light microscopy, field-stimulated and subjected to a standardized protocol. Confocal line scans were performed at room temperature in the cell centre along the myocyte long axis (1.89ms per line). Line scans were noise-filtered, corrected for spillover from the Di8-ANEPPS channel into the Fluo-4 channel and analysed using a custom-written Matlab script (R2017b, Mathworks). See Online Figure 12 for an example.
2.6. STED microscopy of mouse cardiac tissue
A 3D STED super resolution system (Abberior) with an Olympus U-TB190 inverted microscope with a 100x oil immersion lens was used. Two pulsed laser lines (594nm and 640nm) were used for excitation, together with a 775 nm STED laser (1250mW).
2.7. Statistics
Researchers were blinded against groups when imaging the t-system of living human and rat cardiomyocytes. Technical replicates and repeated measures taken from the same sample, for instance multiple image stacks from one mouse heart, were averaged and then treated as one statistical unit. The two-tailed Welch’s t-test, which does not assume equal variance in the samples, was applied, and resulting p-values subsequently corrected for multiple comparison by the Holm-Bonferroni method. Differences in distributions were tested by the Kolmogorov-Smirnov test. P-values ≤0.05 were considered significant. If not otherwise indicated, data are presented as mean ± standard error.
3. Results
3.1. GR knockout leads to t-system loss and increased RyR-LTCC distance
Because cardiomyocyte-specific GR knockout (GRKO) mice develop heart failure and show alterations in cardiomyocyte calcium handling [33, 34], we investigated whether t-system remodelling as a result of deficient GR signalling may contribute to cardiac dysfunction in these animals. After staining with wheat germ agglutinin (WGA) and antibodies against RyR and LTCC, we acquired 3D confocal images from control (FLOX) and GRKO left-ventricular tissue sections (Figure 1, A–D and F–I). WGA staining of the t-system revealed cell regions with little or absent t-system in GRKO hearts (see Online Figure 1 for example images). However, because WGA staining of the t-system in rodent heart tissue is commonly less reliable than in larger animals and humans, we investigated if GRKO leads to increased RyR-LTCC distances, which would be expected from a loss of t-tubules. After threshold-based segmentation, distances from centres of ryanodine receptor clusters to centres of their closest LTCC cluster were calculated. In control myocardium, most of the RyR clusters were found within 1μm from the closest LTCC cluster (Figure 1E). In contrast, a large proportion of RyR clusters in GRKO myocardium were located farther than 1μm from LTCC clusters (Figure 1J). Three-dimensional representations of RyR-LTCC distances in tissues are provided in Online Figure 2 and as animations (Online Resources 2 and 3). Quantitative image analysis indicated that both mean intracellular sarcolemma distance and mean RyR-sarcolemma distance were significantly increased in GRKO myocardium (Figure 1K–L). The histogram of RyR-LTCC distance was right-shifted in GRKO myocardium, confirming that a greater fraction of RyR clusters exhibited increased distance from their closest LTCC cluster (Figure 1M, Kolmogorov-Smirnov test: p<0.001). Accordingly, mean RyR-LTCC distance was greater in GRKO than in control hearts (0.77±0.04μm vs 0.58±0.04μm, Figure 1N).
Figure 1.
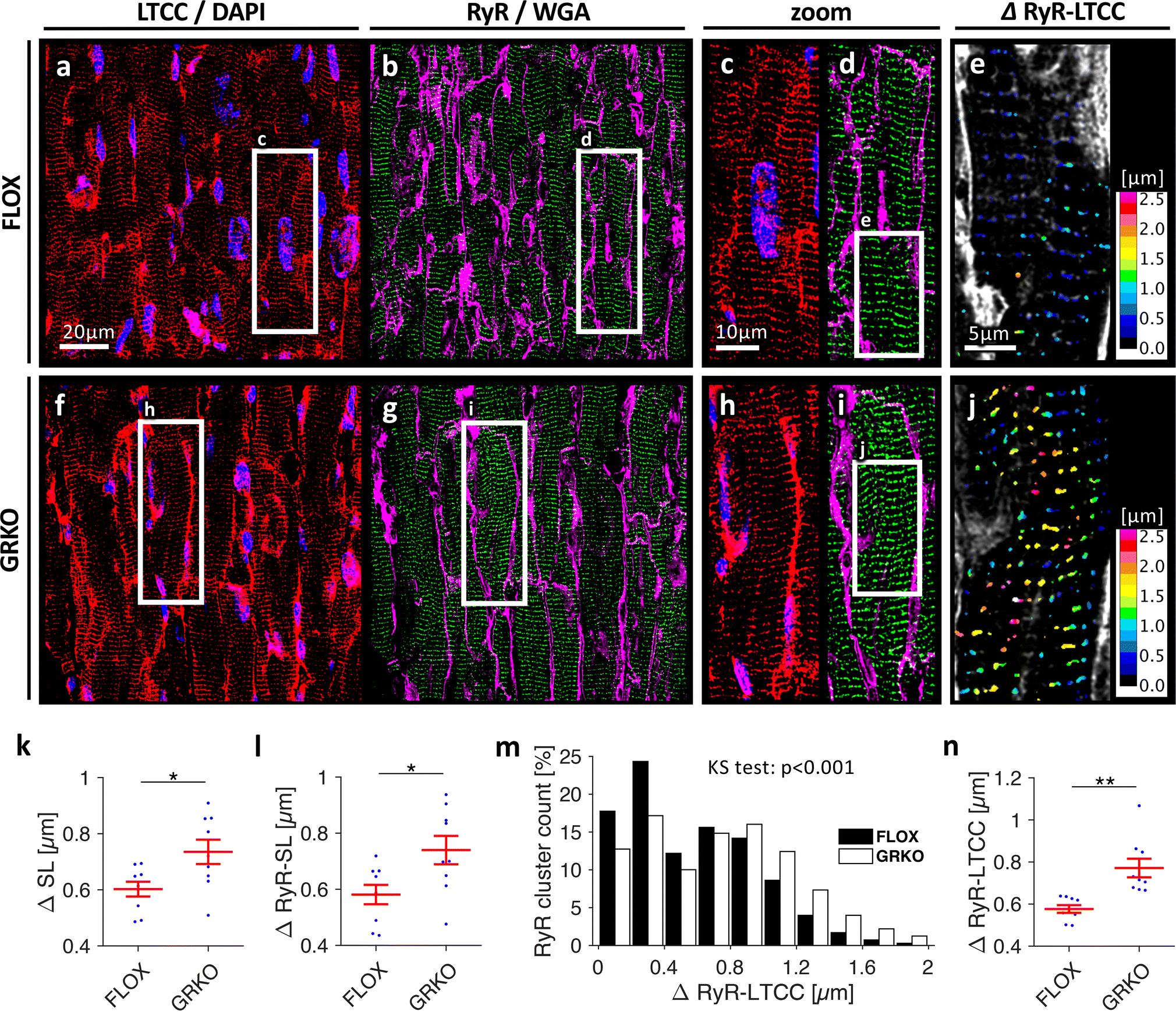
Confocal images of cardiac tissue sections from three-month old mice, A-E, control (FLOX) and, F-J, cardiomyocyte-specific GR knockout (GRKO). Sections were stained for L-type Ca2+ channels (LTCC, red in A, C, F, H), ryanodine receptors (RyR, green in B, D, G, I), nuclei (DAPI, blue in A, C, F, H), and with wheat germ agglutinin (WGA) for the sarcolemma and extracellular matrix (magenta in in B, D, G, I). E and J, Color-coded distances from RyR clusters to their closest LTCC cluster (ΔRyR-LTCC). Scale bar in A also applies to B, F, G. Scale bar in C also applies to D, H, I. Scale bar in E also applies to J. K, Mean intracellular distance to closest sarcolemma (ΔSL) and, L, mean RyR cluster distance to closest sarcolemma (ΔRyR-SL) as measures of t-tubule density. M, Distribution of ΔRyR-LTCC in FLOX and GRKO tissue. The Kolmogorov-Smirnov (KS) test was applied to test for equal distributions. N=478,215/480,486 clusters. N, Mean ΔRyR-LTCC. Data were obtained from n=9/3 samples/animals in each group. *p<0.05 (two-tailed Welch’s t-test)
To better resolve RyR-LTCC distance and verify results from confocal microscopy, we used 3D STED microscopy, an optical super-resolution technique. From each GRKO and control heart, three randomly selected volumes of 6.6×6.6×3μm were scanned and analysed as described for the confocal images (Figure 2). RyR clusters were densely aligned along z-lines in both the GRKO and control myocardium, but LTCC cluster density appeared lower in the GRKO heart (Figure 2B–C vs 2I–J). In particular, we found single RyR clusters in the GRKO heart which were not coupled to LTCC clusters, leading to an increased RyR-LTCC distance (Figure 2, M–N). A 3D representation is provided in the online supplement (Online Figure 3). The histogram of RyR-LTCC distance in STED images, obtained from a total of approximately 400 clusters per group, showed a significant right-shift towards greater values in the GRKO myocardium (Figure 2O), which resulted in an increased mean RyR-LTCC distance (Figure 2P). Together, these findings confirm the results obtained with confocal microscopy that loss of GR signalling in the GRKO heart leads to reduced RyR-LTCC coupling as a result of t-system remodelling.
Figure 2.
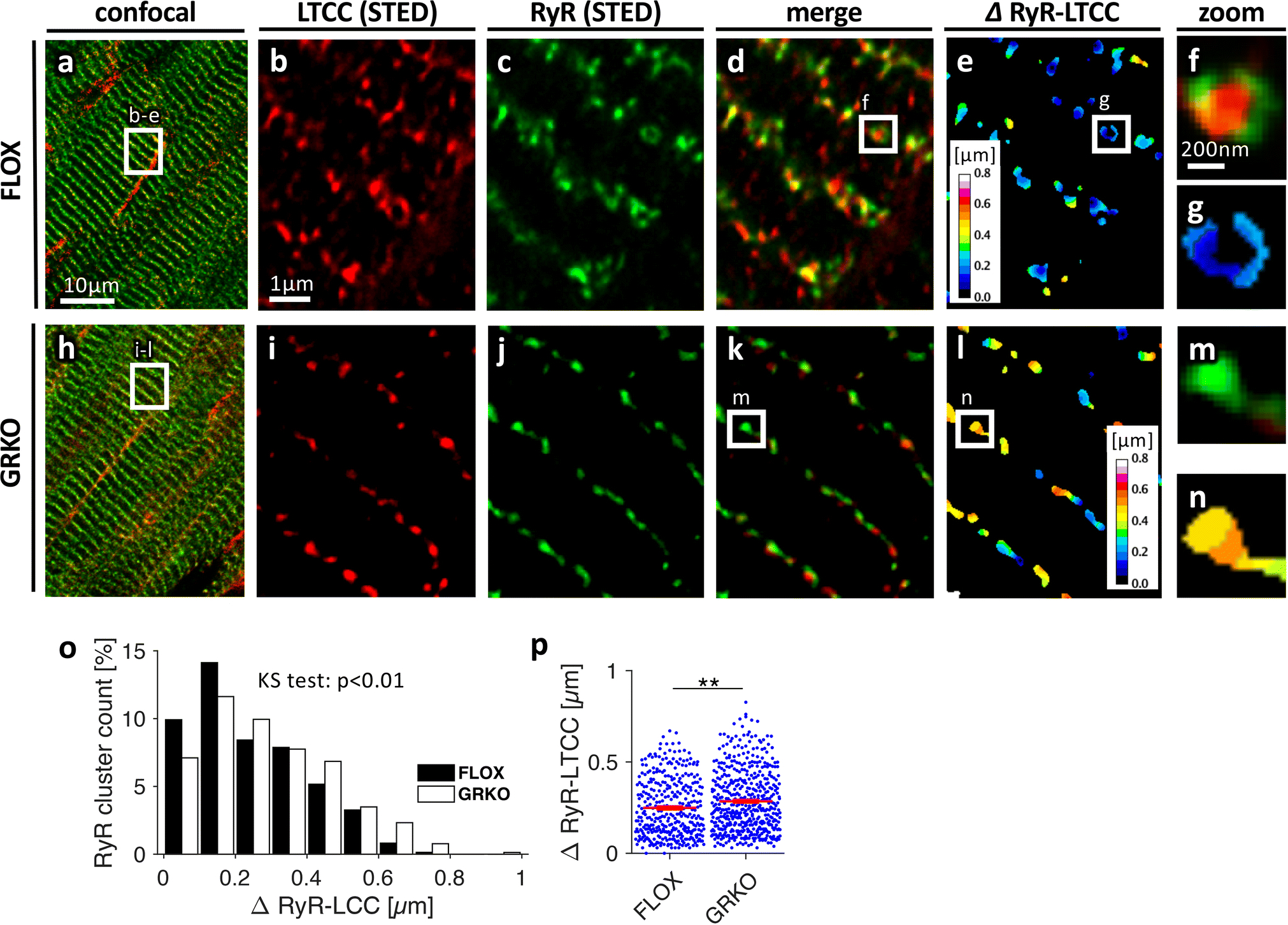
Three-dimensional STED microscopy of cardiac tissue sections from three-month old, A-G, control (FLOX) and, H-N, GRKO mice. Sections were stained for LTCCs (red in A, B, D, F, H, I, K, M) and RyRs (green in A, C, D, F, H, J, K, M). E, G and L, N, Color-coded distances from RyR clusters to their closest LTCC cluster (ΔRyR-LTCC). Scale bar in A also applies to H. Scale bar in B also applies to C-E and I-L. Scale bar in F also applies to G, M, N. O, Distribution of ΔRyR-LTCC in FLOX and GRKO tissue. KS test: Kolmogorov-Smirnov test for equal distribution. P, ΔRyR-LTCC obtained from n=359/494 analysed RyR clusters from 9/9 image stacks (6.6×6.6×3μm3) and 3/3 FLOX or GRKO animals, respectively. **p<0.01 (two-tailed Welchs’s t-test, clusters as statistical units)
3.2. Glucocorticoids preserve the t-system in cultured rat and human cardiomyocytes
A common problem when studying t-system remodelling in vivo is its complex interdependence with heart failure development [50]. Therefore, to avoid confounding effects by heart failure in GRKO mice [34], we used an in-vitro model of t-system remodelling in subsequent experiments to investigate whether t-system loss is caused by a deficiency in cardiomyocyte GR signalling. Primary culture of ventricular cardiomyocytes in serum-free medium has been shown to result in t-system loss [2, 27]. Indeed, three-dimensional confocal images of living ventricular cardiomyocytes isolated from adult rats showed that t-tubules underwent substantial remodelling in serum-free culture, including a decrease in density and an increase in longitudinal components (Online Figure 4). These results are comparable to findings described in other cell culture studies [2] and rodent models of heart failure [12]. We then investigated whether culture-induced t-system remodelling could be prevented by activation of the glucocorticoid receptor (GR). We cultured rat cardiomyocytes for 3 days and applied vehicle as control (CTRL), 1μmol/L dexamethasone (DEX3d), 1μmol/L corticosterone (CORT3d), or 1μmol/L dexamethasone plus 10μmol/L of the GR antagonist mifepristone (DEX+MIFE3d). Confocal images of the t-system showed a preserved t-system in DEX3d and CORT3d cells, but not in CTRL3d or DEX+MIFE3d cells (Figure 3A). In cardiomyocytes isolated from four failing human hearts (see Online Table 1 for patient characteristics), t-system density was low, as expected from previous studies [27, 46]. As observed in rat myocytes, culture of human myocytes caused additional loss of t-tubules (mean t-tubule distance 1.28±0.1μm in fresh myocytes, 2.28±0.13μm after 3d in culture, n=17/17, p<0.01). Importantly, after only 1 day in culture, the t-system appeared denser in human DEX than CTRL cells (Figure 3B). We quantified the t-system by computational analysis of 80–150 rat and 32–34 human cells per group. When comparing DEX3d with CTRL3d rat cells, we found that dexamethasone treatment kept both t-tubule density and mean t-tubule distance at levels of freshly isolated cells, but not the fraction of longitudinal t-tubule components (Figure 3C). Similarly, human cardiomyocytes treated with dexamethasone showed higher t-tubule density and shorter intracellular t-tubule distances than CTRL cells (Figure 3D). We then tested the effects of corticosterone, the main plasma glucocorticoid in the rat, and mifepristone, a GR antagonist, in rat myocytes. Corticosterone reproduced the effects of dexamethasone, whereas mifepristone completely blocked them (Figure 3E). This result suggests that GR is as a major mediator of glucocorticoid effects on the t-system. The mineralocorticoid receptor (MR) antagonist spironolactone only mildly mitigated the effect of dexamethasone (Online Figure 5), indicating negligible contribution of the MR to t-system preservation. Experiments to determine the minimal concentration of dexamethasone required for t-system preservation yielded an EC50 of 1.1 nmol/L, which is consistent with a GR-mediated effect [35]. Additionally, we measured cell size and cell capacitance in CTRL3d and DEX3d rat cells. Cell size was similar in all cultured groups, but cell capacitance, determined by whole-cell patch clamp experiments, was approximately 35% higher in DEX3d than in CTRL3d cells (115.5±4.5pF, n=25, vs 83.3±4.1pF, n=30, p<0.001, Online Figure 6). Because cell capacitance is proportional to cell membrane area including the t-system, this finding confirmed the results obtained from microscopic imaging. In summary, these results indicate that dexamethasone as well as physiological glucocorticoids preserve the t-system via activation of the GR in healthy rat cardiomyocytes and, importantly, also in cardiomyocytes from failing human hearts.
Figure 3.
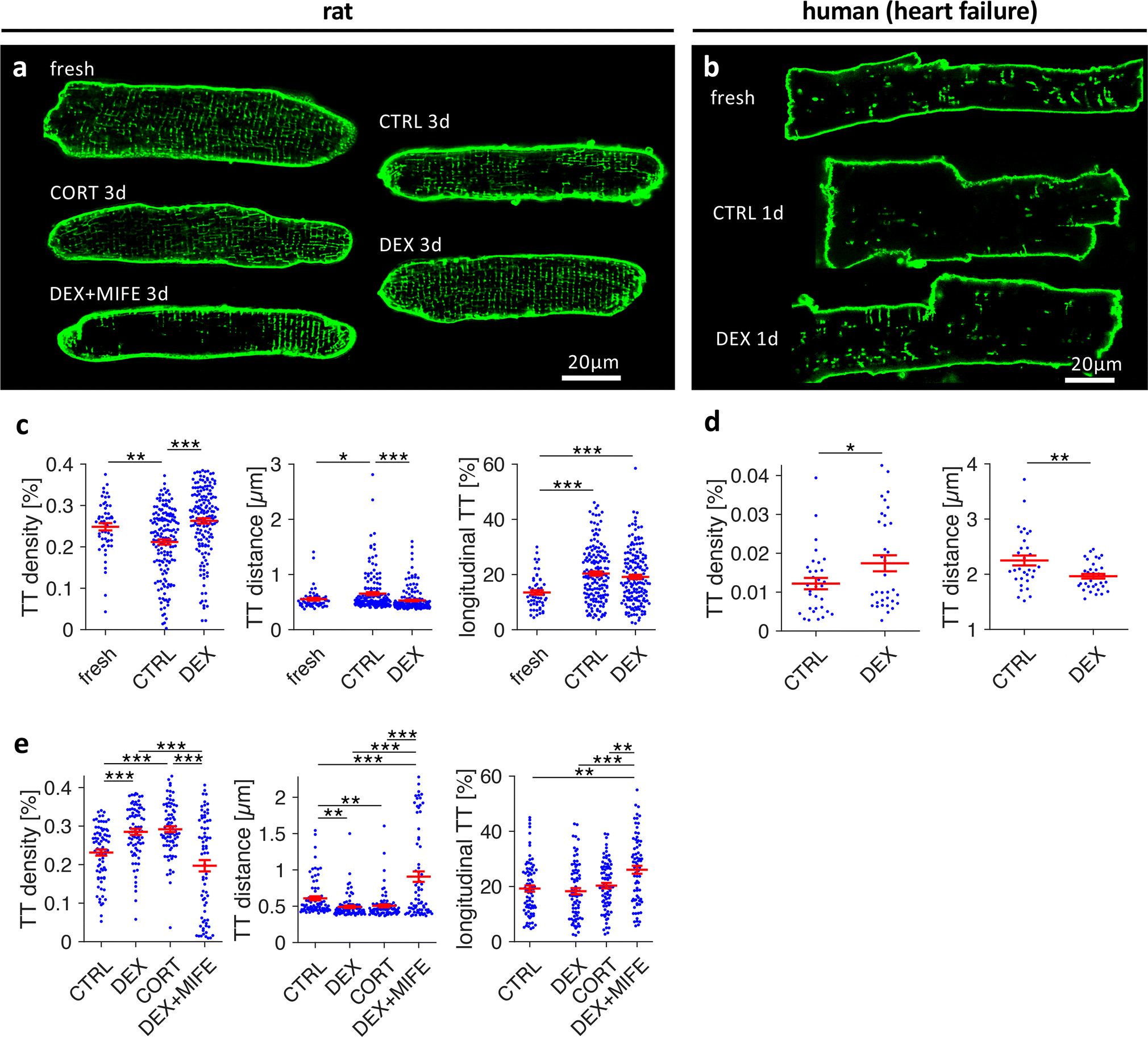
Effect of corticosteroid agonists and antagonists on the t-system in cultured rat and human cardiomyocytes. A, Confocal images of Di8-ANEPPS-stained living rat myocytes of freshly isolated cells (fresh) and cells after 3 days of culture treated with vehicle (CTRL3d), 1μmol/L dexamethasone (DEX3d), 1μmol/L corticosterone (CORT3d), or 1μmol/L dexamethasone + 10μmol/L mifepristone (DEX+MIFE3d). B, Confocal images of freshly isolated and cultured cardiomyocytes from human failing hearts. Cells were cultured for 1 day and treated with vehicle (CTRL1d) or dexamethasone (DEX1d). C, T-tubule density (TT density), mean intracellular distance to nearest t-tubule (TT distance), and fraction of longitudinal t-tubule components (longitudinal TT) in fresh, CTRL3d and DEX3d cells from matched cell isolations. D, TT density and TT distance in CTRL and DEX human myocytes cultured for 1–3 days. E, Same parameters as shown in C in CTRL3d, DEX3d, CORT3d and DEX+MIFE3d cells from matched cell isolations. ***p<0.001, **p<0.01, *p<0.05, n (cells/animals) in C = 53/7, 158/20, 157/20 in fresh, CTRL3d, DEX3d, respectively, n (cells/patients) in D = 32/4, 34/4 in CTRL and DEX, respectively, n (cells/animals) in E = 79/10, 79/10, 80/10, 73/10 in CTRL3d, DEX3d, CORT3d, DEX+MIFE3d, respectively. Two-tailed Welch’s t-test was used for all tests, with multiple comparison correction where required.
3.3. Dexamethasone preserves spatial coupling between RyR and LTCC clusters
We next investigated the spatial coupling of L-type Ca2+ channel (LTCC) clusters and ryanodine receptor (RyR) clusters in fixed rat myocytes co-stained for LTCC and RyR. When compared with fresh cells, LTCC clusters appeared less dense and irregular in CTRL3d cells but nearly unaltered in DEX3d cells (Figures 4A–C). RyR cluster density, however, seemed to be slightly reduced in both CTRL3d and DEX3d cells (Figures 4D–F). As observed in the GRKO myocardium, distances from the centres of ryanodine receptor clusters to the centres of their closest LTCC cluster were increased in cultured control cells (Figures 4G–L). Nearly all RyR clusters in fresh cells were found within 1μm from the closest LTCC cluster (Figure 4J). However, in CTRL3d cells, a large number of non-coupled (non-junctional) RyR clusters with distances >1μm became visible (Figure 4K). This was prevented by dexamethasone treatment (Figure 4L). Statistical analysis of mean cytosolic RyR-LTCC distance (Figure 4M) revealed significantly increased distances in CTRL3d cells (0.71±0.03μm) when compared with fresh cells (0.53±0.02μm), while DEX3d prevented the increase in cluster distance (0.55±0.02μm). To assess RyR-LTCC distances near the surface sarcolemma, we restricted the analysis to regions within 0.5μm from the cell surface. As expected from t-system loss as the primary cause for increased cytosolic RyR-LTCC distances, mean distances near the surface sarcolemma were not significantly altered. RyR cluster density was reduced and a higher percentage of RyRs found near the cell surface in both CTRL3d and DEX3d (Figure 4N), indicating that changes in RyR-LTCC distance did not result from altered RyR expression or distribution. However, when analysing the density and distribution of LTCC (Figure 4O), we found that although LTCC cluster density was reduced in both CTRL3d and DEX3d cells versus fresh cells, DEX3d cells exhibited significantly higher LTCC density than CTRL3d cells, possibly a result of increased LTCC densities within t-tubules [19]. The distribution of LTCC clusters was shifted towards the surface sarcolemma in CTRL3d cells, but not in DEX3d cells, which is in accordance with t-system loss only in CTRL3d cells. Collectively, these findings are consistent with the t-system loss observed in living myocytes (Figure 3) and underscore the importance of the t-system for spatial coupling of LTCC and RyR clusters.
Figure 4.
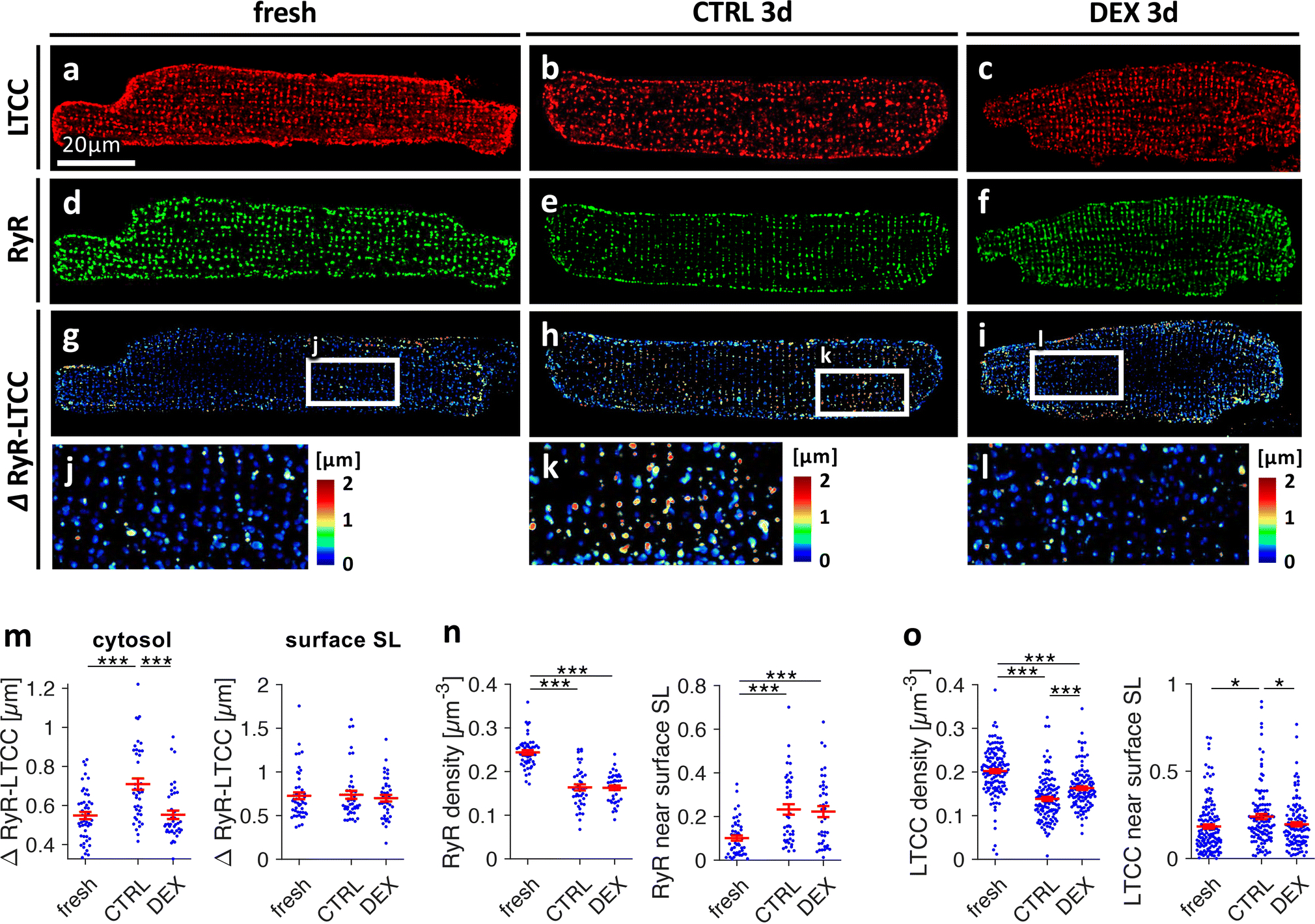
Effect of dexamethasone on distributions and spatial relationship of L-type Ca2+ channels (LTCC) and ryanodine receptors (RyR) in cultured rat cardiomyocytes. Freshly isolated myocytes (fresh) and myocytes cultured for 3d in vehicle (CTRL3d) or 1μmol/L dexamethasone (DEX3d) were fixed, co-immunostained for LTCC and RyR and analysed by 3D confocal microscopy. A-C, LTCC, D-F, RyR, G-L, color-coded distances between RyR clusters and their closest LTCC cluster (ΔRyR-LTCC). M, ΔRyR-LTCC in the cytosol (≥ 0.5μm from the surface sarcolemma, SL) and ΔRyR-LTCC close (< 0.5μm) to the surface SL. N, RyR cluster density (clusters per μm3) and fraction of RyR signal detected close (< 0.5μm) to the surface SL. O, LTCC cluster density and fraction of LTCC signal detected close to the surface SL. Scale bar in A also applies to B-I. ***p<0.001, **p<0.01, *p<0.05, n (cells/animals) = 45/10, 43/7, 40/7 in fresh, CTRL3d, DEX3d, respectively. Two-tailed Welch’s t-test was used for all tests, with multiple comparison correction.
3.4. Synchrony of cytosolic Ca2+ rise is improved by dexamethasone
Subsequently, we studied the effects of GR activation on excitation-contraction coupling by confocal imaging of cytosolic [Ca2+] (Figure 5). Living rat cardiomyocytes were stained with Di8-ANEPPS and loaded with Fluo-4 as a Ca2+ indicator. Line scanning was performed at the centre of each cell along the myocyte long axis (dotted lines in Figures 5A–C). The time of Ca2+ rise was defined as the time of maximum upstroke of Fluo-4 intensity, detected by pixel-wise curve fitting. In some experiments, we additionally stained myocyte nuclei, but exclusion of intranuclear pixels from analysis did not have significant effects (Online Figure 14). Possessing a dense t-system, fresh cells exhibited a synchronous initiation of Ca2+ rise along the scanned line (Figure 5A), indicating simultaneous opening of RyR clusters. In contrast, the Ca2+ rise in CTRL3d cells appeared asynchronous, due to regions of significant delay. Upon close inspection of Figure 5B, it becomes clear that delays in Ca2+ rise were especially pronounced in regions devoid of t-tubules, which is consistent with delayed or absent Ca2+ release from the SR as a result of altered coupling between RyRs and LTCCs. In DEX3d myocytes, however, the Ca2+ rise appeared nearly as synchronous as in fresh cells (Figure 5C), as expected from the preserved t-tubule density in these cells. Analysis of the distribution of Ca2+ rise times (Figure 5D) confirmed these observations. While the mode (maximum value) of the histogram ranged from 20–30ms in all shown example cells, both the standard deviation and maximum of Ca2+ rise time were considerably increased in CTRL3d cells but not in DEX3d cells. Statistical analysis revealed significantly increased standard deviations of Ca2+ rise time from 8.5±0.7ms in fresh to 15.9±1.7ms in CTRL3d cells but not in DEX3d cells (10.6±1.2ms, Figure 5E). Similarly, maximum cellular delay of Ca2+ rise after stimulation was larger in CTRL3d cells (83±7ms) than in fresh (56±3ms) or DEX3d (67±5ms) cells (Figure 5F). Analysis of whole-line Ca2+ transient kinetics and cell shortening (Online Figure 7) yielded no significant difference in maximum relative Fluo4 signal (F/F0), but did reveal faster upstroke velocity (d(F/F0)/dt) and decay rate in dexamethasone-treated myocytes, which is consistent with higher t-system density. Maximum cell shortening was reduced in CTRL3d cells, but not in DEX3d cells, when compared with fresh myocytes. In summary, although Ca2+ entering via LTCC may have contributed to the observed effects, the results indicate that asynchronous and delayed Ca2+ rise in CTRL3d cells followed mainly from the loss of t-tubules and increased RyR-LTCC distances. Thus, by preserving the t-system, GR activation also preserved excitation-contraction coupling.
Figure 5.
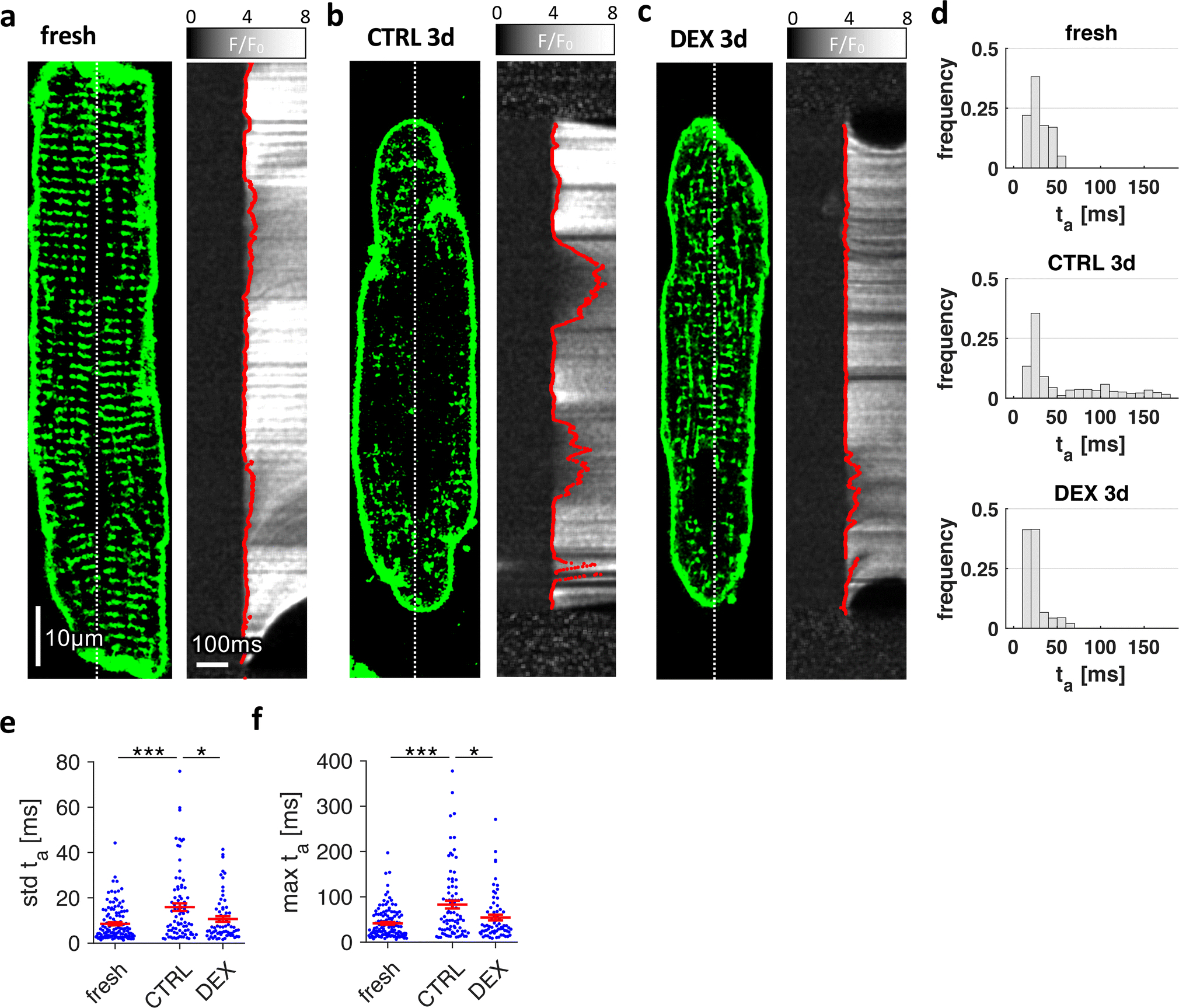
Effect of dexamethasone on stimulated cytosolic Ca2+ rise in cultured rat cardiomyocytes. Freshly isolated myocytes (fresh) and myocytes cultured for 3 days in vehicle (CTRL3d) or 1μmol/L dexamethasone (DEX3d) were stained with Di-8-ANEPPS, loaded with Fluo-4-AM as Ca2+ indicator and then field-stimulated. A-C, Examples of 2D confocal images with corresponding line scans recorded along the dotted lines with 1.89ms/line (530Hz). F/F0 shows the relative fluorescence intensity with respect to baseline intensity before stimulation. Red lines show the points of maximum increase in fluorescence intensity (dF/dtmax), determined by curve fitting algorithms. Local activation time of Ca2+ rise, ta, was defined as the time of dF/dtmax. D, Histograms of ta of the cells shown in A-C. E, Standard deviation (std) of ta, and, F, maximum local ta, obtained from fresh and cultured cells. Scale bar in A also applies to B and C. ***p<0.001, **p<0.01, *p<0.05, n (cells/animals) = 100/9, 82/8, 67/8 in fresh, CTRL3d, DEX3d, respectively. Two-tailed Welch’s t-test was used for all tests, with multiple comparison correction.
3.5. BIN1, JPH2 or CAV3 are not upregulated by dexamethasone
To identify mechanisms responsible for t-system loss in culture and its preservation by dexamethasone, we studied cellular distribution and expression levels of t-system associated proteins (Figure 6). First, we co-immunostained BIN1 and JPH2 and found a dense, regular arrangement of both proteins along the z-line in fresh cells (Figure 6A and 6D), but irregular arrangement in cultured cells (Figures 6B and 6E). BIN1 was no longer restricted to z-lines, and JPH2 appeared shifted to the surface sarcolemma. Dexamethasone treatment had little effect on JPH2 and BIN1 remodelling, although both appeared somewhat denser (Figures 6C and 6F). Quantification of BIN1 immunofluorescence (Figure 6G) revealed a greater BIN1 volume density in both CTRL3d and DEX3d cells versus fresh cells, but no significant increase in BIN1 staining near the surface sarcolemma. However, BIN1 regularity, measured by analysis of the Fourier spectral density corresponding to z-line distances (BIN1 spectral density) was strikingly reduced in both CTRL3d and DEX3d cells. When analysing JPH2, we found a pronounced reduction in JPH2 volume density (Figure 6H), which was in contrast to the increase seen in BIN1 volume density. This coincided with a redistribution of JPH2 towards the surface sarcolemma, suggesting that JPH2 was decreased mainly in the cell interior. Reduced spectral density in both CTRL3d and DEX3d cells versus fresh cells confirmed the observed loss of a regular z-line pattern. Again, no significant effects of dexamethasone were detected. In addition, we investigated BIN1 and JPH2 protein and mRNA expression levels. BIN1 immunoblotting yielded an approximately 50% increase in BIN1 protein levels in both CTRL3d and DEX3d cells versus fresh cells (Figure 6I), which was consistent with increased BIN1 density obtained from immunostaining. To test if this increase resulted from upregulation of BIN1 mRNA in cell culture, we applied qPCR to fresh, CTRL1d and DEX1d cells. Indeed, BIN1 mRNA was significantly increased in CTRL1d cells when compared with fresh cells, but, surprisingly, not in DEX1d cells. In contrast to BIN1, JPH2 protein and mRNA expression were downregulated to less than 30% of levels in fresh cells in both CTRL3d and DEX3d cells (Figure 6J), suggesting that low abundance of JPH2 protein resulted from decreased gene expression. We also analysed protein expression of caveolin-3 (CAV3), but could not detect any significant upregulation by dexamethasone (Online Figure 8). Based on these data, we conclude that t-system preservation by dexamethasone was not mediated by upregulation of BIN1, JPH2 or CAV3. To test for differences in junctional proteins in vivo, we analysed protein expressions of JPH2, BIN1 and CAV3 in 8 control and 7 GRKO hearts from 3-month old mice (Online Figure 9). JPH2 abundance was slightly decreased by approximately 25%, but CAV3 and BIN1 were unaltered. Taking into account the results from a recent RNA sequencing study carried out by Cruz-Tropete et al. [10], where JPH2 mRNA was not altered in 3 month cardiomyocyte-specific GRKO mice, our findings suggest little to no regulation of BIN1, CAV3 or JPH2 by glucocorticoids.
Figure 6.
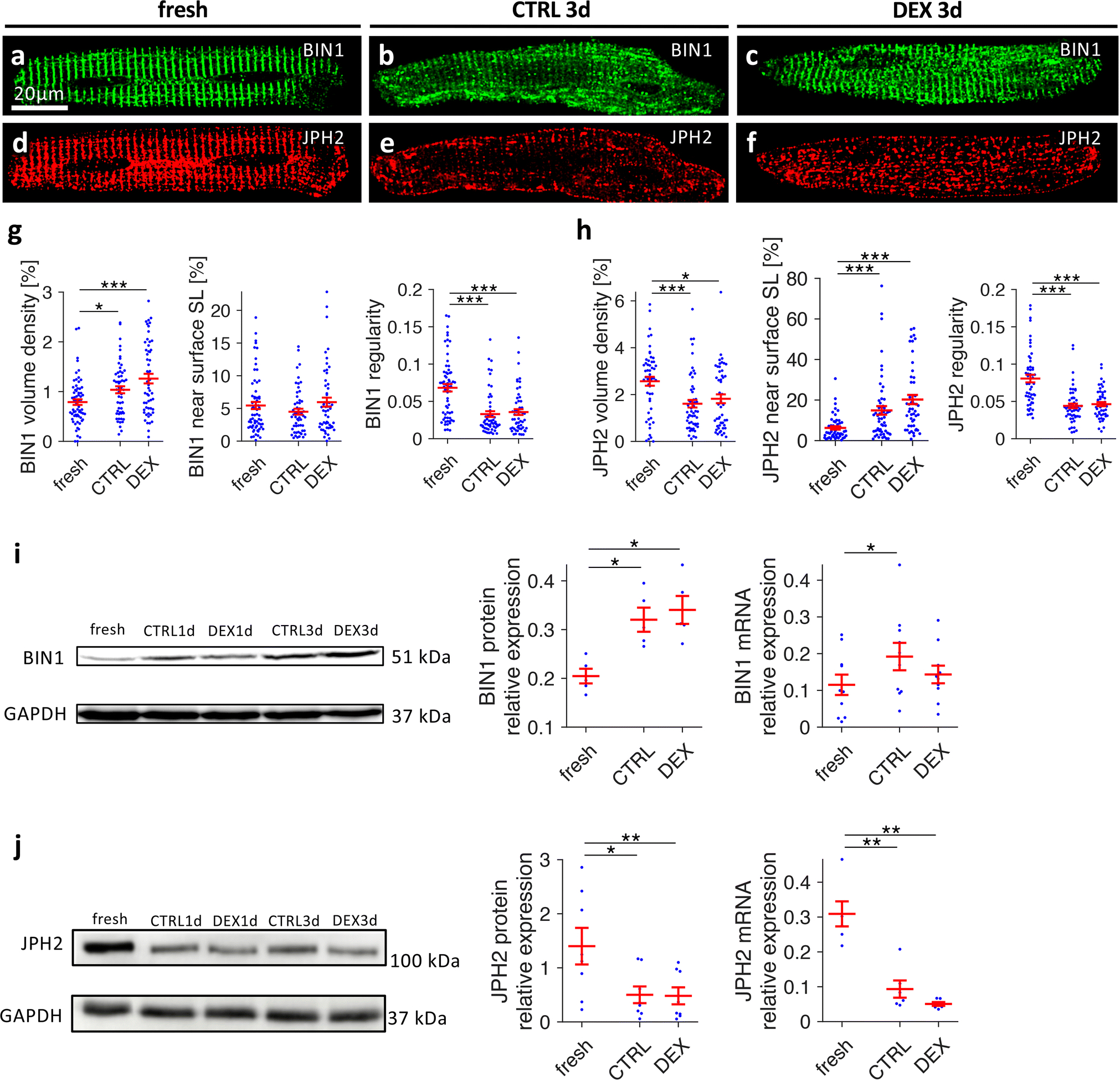
Effects of dexamethasone on distribution and expression of BIN1 and JPH2 in cultured rat cardiomyocytes. Freshly isolated myocytes (fresh) and myocytes cultured for 3 days in vehicle (CTRL3d) or 1μmol/L dexamethasone (DEX3d) were fixed, co-immunostained for BIN1 (green) and JPH2 (red), and analysed by three-dimensional confocal microscopy. A-C, BIN1, D-F, JPH2. Scale bar in A also applies to B-F. G, Quantitative analysis of BIN1 volume density, percentage of BIN1 near (< 0.5μm) the surface sarcolemma (SL), and BIN1 spectral density as a measure of spatial regularity. n (cells/animals) = 65/8, 56/8, 55/8 in fresh, CTRL3d, DEX3d, respectively, from matched cell isolations. H, Quantitative analyses of JPH2 immunostaining, as shown for BIN1 in G. n (cells/animals) = 53/7, 56/7, 50/7 in fresh, CTRL3d, DEX3d, respectively, from matched cell isolations (two-tailed Welch’s t-test with multiple comparison correction). I, BIN1 protein and mRNA expression levels. BIN1 protein expression was assessed by Western blot quantification (normalization by Ponceau staining) in fresh, CTRL3d and DEX3d cells, n=5 matched cell isolations. BIN1 mRNA expression assessed by quantitative RT-PCR in fresh, CTRL1d and DEX1d cells (normalized to reference genes, see Methods), n=10 matched cell isolations (paired t-test with multiple comparison correction). J, JPH2 protein and mRNA expression levels as described for BIN1 in I. Western blot: n=8 matched cell isolations, qPCR: n=6 matched cell isolations (paired t-test with multiple comparison correction). ***p<0.001, **p<0.01, *p<0.05
3.6. Dexamethasone upregulates autophagic flux
Because dexamethasone has been suggested to upregulate autophagic flux in skeletal muscle [54], we asked if dexamethasone might prevent t-system loss by influencing autophagy in cardiac cells. To answer this question, we immunostained microtubule-associated proteins 1A/1B light chain 3B (LC3B), a protein located predominantly in the membrane of autophagosomes (Figure 7A), and found reduced amounts of LC3B in CTRL1d cells when compared with fresh cardiomyocytes. DEX1d cells, however, exhibited increased LC3B staining and larger LC3B-positive vesicular structures. Quantitative analysis (Figure 7B) revealed significantly lower LC3B volume density in CTRL1d cells than in fresh cells (0.25±0.03% vs 0.4±0.04%), whereas dexamethasone preserved LC3B density (0.49±0.05%). This could be attributed mainly to a marked decrease in LC3B vesicle size in CTRL1d cells. Dexamethasone prevented the reduction in vesicle size, while vesicle number was unaffected. Thus, autophagosomes were larger in cells treated with dexamethasone. LC3B vesicle size or number is generally not sufficient to assess autophagic flux because LC3B accumulates also if autophagosome degradation is blocked [58]. Therefore, we carried out autophagic flux assays based on the short-term application of bafilomycin A1, a potent blocker of autophagosome degradation. Because LC3BII (the membrane-bound form of LC3B) and SQSTM-1 (sequestosome-1, ubiquitin-binding protein p62) are degraded together with autophagosomes, the differences in LC3BII and SQSTM-1 levels before and after application of bafilomycin provide an estimate of how much autophagic cargo is degraded over time, which translates into autophagosome turnover rate or flux [58]. We applied bafilomycin for 2.5h to fresh, CTRL1d, DEX1d or rapamycin-treated cells from matched cell isolations and then determined LC3BII or SQSTM-1 protein levels by Western blotting. Figure 7C shows that basal LC3BII levels were significantly higher in DEX1d than in fresh or CTRL1d cells, and that, as expected, LC3BII increased in response to bafilomycin in all groups. Importantly, the amount of accumulated LC3BII was nearly three times higher in DEX1d cells than in CTRL1d cells. SQSTM-1 showed lower basal levels in cells treated with dexamethasone or 1μmol/L rapamycin, an inductor of autophagy (Figure 7D), suggesting that increased autophagic flux leads to faster degradation of SQSTM-1. As found for LC3BII, the amount of SQSTM-1 accumulated after application of bafilomycin was considerably higher in dexamethasone- and rapamycin-treated cells than in control cells. We also observed that basal SQSTM-1 protein levels were 3–4fold higher in cultured CTRL than in fresh cells (Online Figure 10), whereas cultured DEX cells exhibited much lower basal SQSTM-1 levels (Figure 7D, mid panel). This suggests that dexamethasone maintained SQSTM-1 flux on a level comparable to fresh cells. Altogether, these results indicate that dexamethasone increases autophagic flux in ventricular cardiomyocytes.
Figure 7.
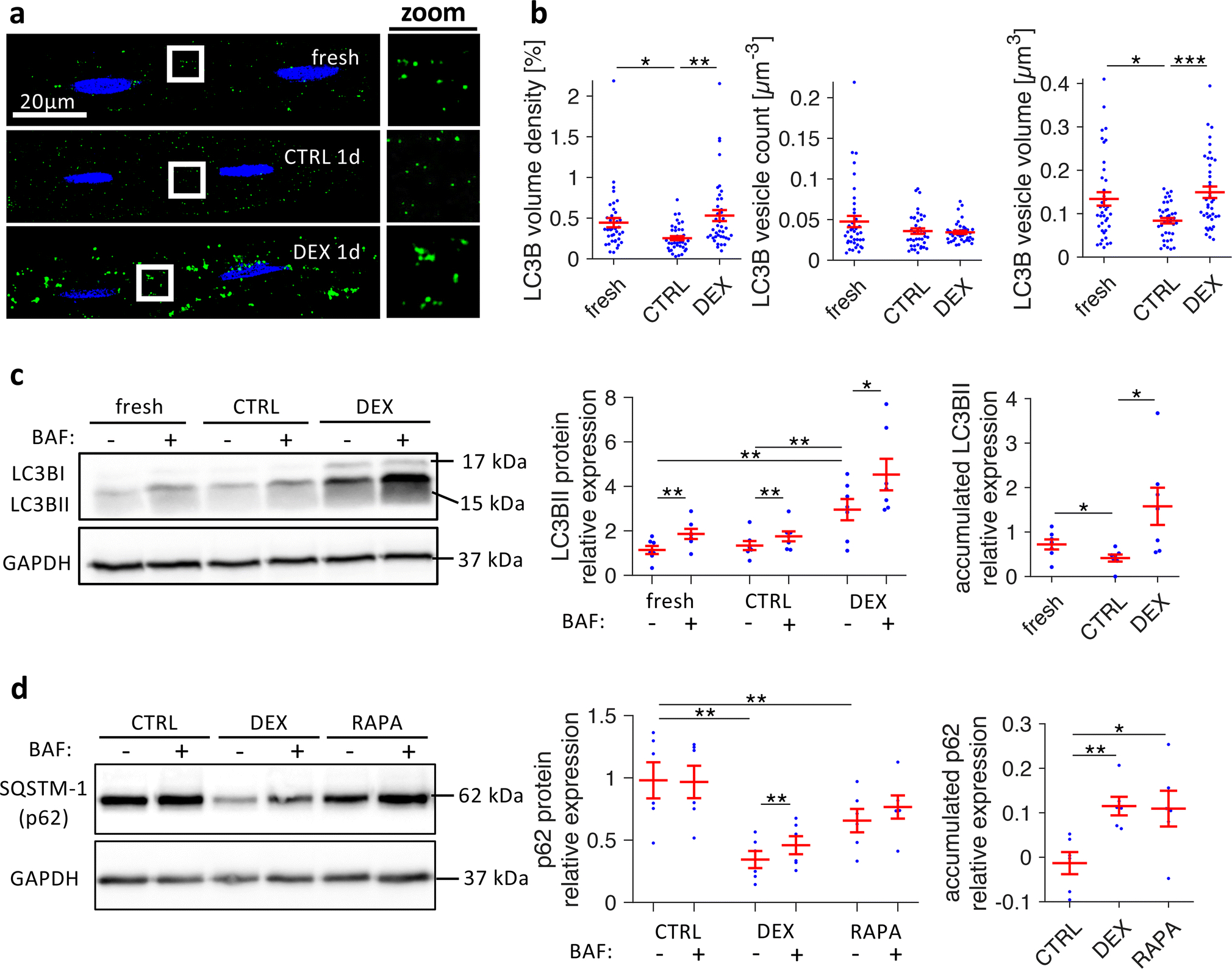
Effects of dexamethasone on autophagic flux in cultured rat cardiomyocytes. A, Freshly isolated myocytes (fresh) and myocytes cultured for 1 day in vehicle (CTRL1d) or 1μmol/L dexamethasone (DEX1d) were fixed, co-stained for LC3BII (green) and nuclei (blue), and imaged with confocal microscopy. Boxed regions are shown as magnified views next to each image (zoom). B, Quantitative analysis of LC3BII confocal image stacks, showing LC3BII volume density, LC3BII vesicle count (number of vesicles per μm3), and LC3BII vesicle size (volume of vesicles in μm3). n (cells/animals) = 39/5, 40/5, 40/5 in fresh, CTRL1d, DEX1d, respectively, from matched cell isolations. ***p<0.001, **p<0.01, *p<0.05 (two-tailed Welch’s t-test with multiple comparison correction). C, Example and quantitative analysis of LC3BII Western blots from fresh myocytes and myocytes cultured for 1d (CTRL, DEX) that were treated with either 100nmol/L bafilomycin A1 (BAF+) or with vehicle (BAF-) for 2.5 h before freezing, to assess LC3BII accumulation as a measure of autophagic flux. Accumulated LC3BII due to BAF treatment was calculated by pairwise subtraction of BAF− from BAF+ LC3BII protein levels from n = 7 matched cell isolations. D, Example and quantitative analysis of SQSTM-1 (p62) Western blots from myocytes cultured for 1d (CTRL, DEX, 1μmol/L rapamycin (RAPA)), treated with or without bafilomycin A1 for 2.5 h. Accumulated SQSTM-1 due to BAF treatment was calculated by pairwise subtraction of BAF− from BAF+ SQSTM-1 protein levels from n = 6 matched cell isolations. Protein expressions of LC3BII and SQSTM-1 were quantified by normalization to Ponceau staining and a reference sample. **p<0.01, *p<0.05 (paired t-tests)
3.7. Autophagy is critical for t-system preservation
Finally, to investigate whether increased autophagic flux may underlie t-system preservation in dexamethasone-treated cells, we tested if enhancing autophagic flux reproduced and blocking autophagic flux inhibited the effects of dexamethasone on the t-system (Figure 8). We found that enhancing autophagy by rapamycin, an mTOR inhibitor or amino acid starvation [30] significantly increased t-tubule density in comparison to cultured CTRL cells (Figures 8A). Conversely, blocking autophagic flux by chloroquine or bafilomycin A resulted in a distinct reduction of t-tubule density, also in the presence of dexamethasone (Figures 8B). Furthermore, staurosporine, a broad-spectrum protein kinase inhibitor, not only inhibited t-system preservation by dexamethasone (Figures 8B), but also prevented dexamethasone-induced upregulation of autophagic flux, while upregulation of basal LC3B levels was not affected (LC3BII autophagic flux assay, Figure 8C). In summary, these findings reveal a critical role of autophagy in t-system maintenance and suggest that upregulation of autophagic flux is a mechanism by which glucocorticoid receptor activation preserves the t-system.
Figure 8.
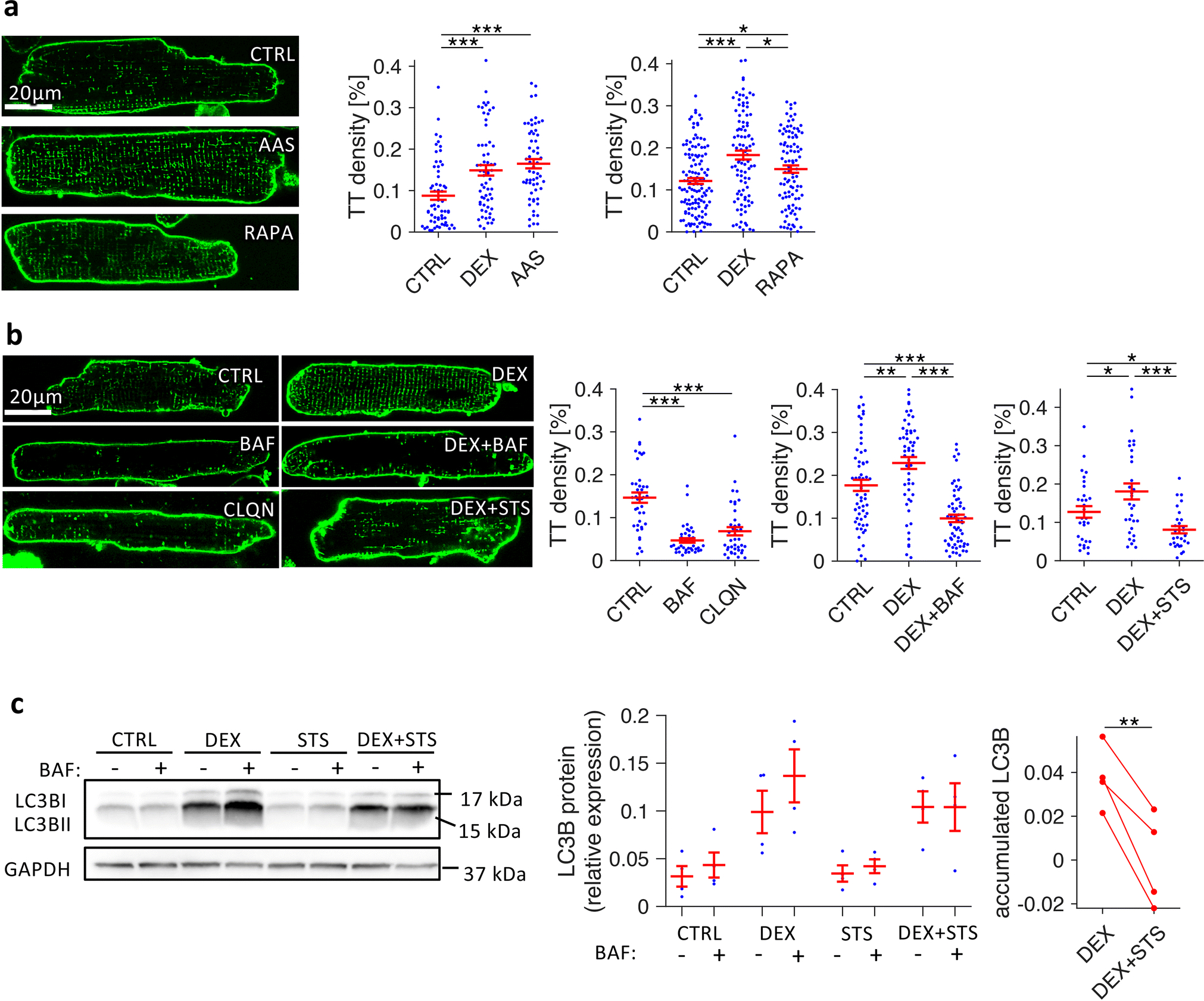
Effects of autophagy enhancers and blockers on t-system density in isolated rat cardiomyocytes. A, Confocal images and quantification of t-tubule (TT) skeleton density of Di8-ANEPPS-stained myocytes after 3 days in culture, treated with vehicle (CTRL), amino-acid free medium (amino acid starvation, AAS), or 1μmol/L rapamycin (RAPA). n (cells/animals) = 38/5, 40/5, 38/5 in CTRL, DEX, AAS, respectively, and 126/14, 97/14, 97/14 in CTRL, DEX, RAPA, respectively. B, Confocal images and quantification of TT density of myocytes after 2 days in culture, treated with vehicle (CTRL), 100nmol/L bafilomycin A1 (BAF), 5μmol/L chloroquine (CLQN), 1μmol/L dexamethasone (DEX), DEX + 100nmol/L bafilomycin A1 (DEX+BAF) or DEX + 10nmol/L staurosporine (DEX+STS). n (cells/animals) = 40/5, 40/5, 40/5 in CTRL, BAF, CLQN, respectively, and 60/8, 61/8, 61/8 in CTRL, DEX, DEX+BAF, respectively, and 30/4, 31/4, 29/4 in CTRL, DEX, DEX+STS, respectively. Example images were chosen to be representative of mean TT densities. ***p<0.001, **p<0.05 (Welch’s t-test with multiple comparison correction) C, Example and quantitative analysis of LC3BII Western blots from myocytes cultured for 1d (CTRL, DEX, 10 nmol/L staurosporine (STS), DEX+STS) that were treated with either 100nmol/L bafilomycin A1 (BAF+) or with vehicle (BAF−) for 2.5 h before freezing, to assess LC3BII accumulation as a measure of autophagic flux. Accumulated LC3BII due to BAF treatment was calculated by pairwise subtraction of BAF− from BAF+ LC3BII protein levels from n = 4 matched cell isolations. **p<0.01 (paired t-test)
4. Discussion
Because of its pathophysiological importance, t-system remodelling is considered a promising future target in the prevention and therapy of heart failure. However, the level of mechanistic understanding needed to design preventive or therapeutic strategies has not yet been reached. As heart failure development and t-system remodelling intertwine closely, it is particularly challenging to study underlying mechanisms in vivo. Here, we investigated if GR signalling is important for t-system preservation by first studying the t-system, RyR and LTCC clusters in tissues from GRKO hearts and then investigating GR effects and mechanisms of t-system remodelling in vitro. T-system density and RyR-LTCC coupling were decreased in hearts from GRKO mice. In cultured rat and human cardiomyocytes, GR activation prevented the loss of t-tubules and preserved excitation-contraction coupling. We discovered that GR signalling leads to distinct upregulation of autophagic flux in ventricular cardiomyocytes. Furthermore, we showed that induction of autophagy preserved the t-system independently of glucocorticoids, whereas inhibition of autophagy accelerated t-system loss and blocked the effects of dexamethasone. These results provide evidence that t-system preservation via glucocorticoids is mediated by increased autophagy and that autophagy is critically involved in t-system remodelling.
4.1. GR signalling and t-system
GR signalling is required for normal cardiac development [42] and for proper function of the adult heart [33, 34, 40]. Cardiomyocyte-specific GRKO mice start developing signs of contractile dysfunction by 2–3 months of age and die due to heart failure by 6–12 months. This is associated with decreased RyR expression and defects in Ca2+ handling [34]. Conversely, overexpression of the GR increases Ca2+ currents and myocyte contractility [44]. However, neither of these studies investigated the t-system.
In the present study, we studied the t-system and associated proteins in hearts from cardiomyocyte-specific GRKO mice [34]. We found lower t-tubule density and reduced coupling between LTCCs and RyRs, which suggests that GR activation is required for t-system maintenance. It also provides a possible explanation for impaired cardiac function in GRKO animals. However, because these animals develop heart failure [34], and because the absence of cardiac GR signalling may lead to secondary effects, such as an imbalance between GR and mineralocorticoid signalling [33], it is difficult to distinguish between specific and non-specific effects of GR deficiency in vivo. Thus, we decided to culture adult cardiomyocytes in serum-free media, which can be considered as a model of t-system loss [2, 27]. Consistently, we found decreased t-tubule density and asynchronous Ca2+ transients in myocytes cultured without glucocorticoids, whereas dexamethasone as well as physiological concentrations of corticosterone prevented these changes. However, dexamethasone did not prevent an increase in longitudinal tubules, suggesting that other factors may lead to changes in t-tubule orientation, for example remodelling of the cytoskeleton [53]. GR agonists did not alter cell size (Online Figure 5), which rules out the possibility that higher t-tubule density may have resulted from cell shrinkage or atrophy in response to dexamethasone. Because mifepristone blocked the effects of dexamethasone, whereas spironolactone had only marginal effects, it is most likely that GR activation was responsible for t-system preservation. In addition, larger transmembrane L-type Ca2+ currents induced by dexamethasone [44] may activate Ca2+-dependent signalling cascades, but neither verapamil nor nifedipine blunted dexamethasone effects on the t-system (Online Figure 11).
4.2. T-system associated proteins
Although other proteins have been proposed to stabilize the t-system in cardiac and skeletal muscle [1] we focused on JPH2, CAV3 and BIN1 because they have been investigated most extensively [3, 5, 6, 21]. Consistent with previous reports [59], we found downregulation of JPH2 protein and mRNA, and redistribution of JPH2 to the surface sarcolemma in cell culture. However, while redistribution of JPH2 was suggested to be responsible for t-system loss in cell culture and heart failure [59], our finding that dexamethasone preserved the t-system without altering JPH2 abundance, mRNA expression or distribution does not support this idea. Although JPH2 protein levels were slightly reduced in GRKO hearts (Online Figure 9), a recent study using the same animals found no effects of GRKO on JPH2 mRNA levels [10], which provides additional evidence that JPH2 is not regulated by glucocorticoids. Instead, JPH2 behaviour paralleled that of RyR, where we observed a similar reduction in cluster density and redistribution to the surface sarcolemma, which also remained unaffected by dexamethasone (compare Figures 4N and 6H). Given the close relationship between JPH2 and RyR clusters seen here and reported by others [17, 18], we suggest that JPH2 alterations are not directly associated with t-system loss, but rather with changes in RyR distribution and expression.
We did not find evidence for changes in CAV3 protein expression in cell culture or in response to glucocorticoids or in GRKO hearts, although recent studies suggest that CAV3 is important for t-system regulation in vivo [5, 21]. It is possible that alterations in cellular CAV3 distribution or turnover rates may have contributed to t-system remodelling, which was, however, not assessed here.
When studying effects of cell culture and glucocorticoids on BIN1, we unexpectedly found that the expression of BIN1 was increased at both the protein and mRNA level in CTRL myocytes. This was surprising, because BIN1 downregulation has been proposed to cause t-system loss [6, 23]. However, other studies indicate that BIN1 is particularly critical for t-tubule development [11, 15], and a recent study did not find significant changes in BIN1 protein abundance in a rat model of heart failure, although t-system remodelling was present [25]. Moreover, upregulation of BIN1 has been associated with t-tubule remodelling in porcine myocardial infarction [37]. Increased amounts of BIN1 protein found here may therefore have distinct causes: In cultured CTRL cardiomyocytes, BIN1 may be upregulated as a result of increased gene expression, possibly reflecting a compensating mechanism for t-tubule loss. In dexamethasone-treated cells, however, increased BIN1 abundance may result from altered turnover or recycling of BIN1, because mRNA levels did not differ from fresh cells. In GRKO hearts, we were not able to detect differences in BIN1 protein levels when compared with control hearts, which also contradicts the hypothesis that BIN1 is involved in t-system loss or regulated by glucocorticoids. However, because we investigated GRKO hearts in 3-month old mice, which is before they develop severe heart failure [34], we cannot exclude differences in BIN1 protein expression in older animals.
4.3. Autophagy and t-system
Autophagy is a regulated cellular process which is essential for the degradation and recycling of cellular components, thereby playing an important role in cellular homeostasis. Upregulation of autophagy is an adaptive response to cardiac stress [31], and both impaired and excessive autophagy have been associated with heart failure [32]. Modulating autophagy could therefore represent a therapeutic approach [41]. In this study, we demonstrate that glucocorticoids induce autophagy in ventricular cardiomyocytes. Furthermore, we show a link between autophagy and cardiac t-system remodelling. In a study using skeletal muscle myotubes it was reported that dexamethasone upregulates autophagy-related genes, such as LC3B or beclin-1, and increases autophagic flux [54]. It has also been reported that autophagy is critically involved in the assembly and disassembly of t-tubules in drosophila muscle cells [11]. This agrees with the effects of glucocorticoids on cardiomyocytes presented here. We found LC3B upregulation, increased autophagosome size, higher autophagic flux and t-system preservation. Intriguingly, stimulating autophagy by rapamycin or amino acid starvation preserved the t-system, while inhibiting autophagy by chloroquine or bafilomycin A abrogated t-system preservation by dexamethasone. This suggests that high autophagic flux is crucial for t-system homeostasis in the cardiac muscle rather than a mechanism underlying the loss of t-tubular membrane. We observed that t-system preservation by rapamycin was less pronounced than by glucocorticoids or amino acid starvation, which may result from distinct activation pathways. Rapamycin induces autophagy primarily by inhibiting the mammalian target of rapamycin complex 1 (mTORC1) and only partial inhibits mTORC2 [45], whereas amino acid starvation may induce autophagy additionally by effects independent of mTORC1 [13, 20, 52, 55]. Glucocorticoids, on the other hand, not only inhibit mTOR, for example by upregulating REDD1, but also increase the expression of key proteins and regulators of autophagy, as found in the skeletal muscle [49]. Consistently, basal LC3B protein levels were significantly upregulated by dexamethasone, even over the level of fresh cardiomyocytes (Figure 7C).
Another important finding was that broad-spectrum protein kinase inhibition by staurosporine blocked t-system preservation by dexamethasone and the dexamethasone-induced increase in autophagic flux, but did not blunt the increase in basal LC3B levels. This suggests that protein kinases upregulated or activated by GR signalling are involved in the increase of autophagic flux, but not in the increase of key proteins of autophagy, such as LC3B. The identity of the responsible kinases and signalling cascades will need to be addressed in future studies. Here, however, we can conclude that blockage of autophagic flux in the presence of glucocorticoids inhibits t-system preservation, which is strong evidence for the hypothesis that glucocorticoids preserve the t-tubular system in ventricular cardiomyocytes by upregulation of autophagic flux.
4.4. Translational perspective
Although the results of this study will first need to be validated in animal models of cardiac disease, the positive effects of glucocorticoids on cardiomyocyte structure and function demonstrated here raise the question whether dexamethasone and related drugs might be used clinically to improve outcomes in patients suffering from heart failure. We found an EC50 of dexamethasone on the t-system of 1.1 nmol/L, with the maximum effect size seen at approximately 10 nmol/L (Online Figure 5F). This would be close to plasma levels reached in patients [43]. In animal models of myocardial infarction and cardiac ischemia, it has been reported that dexamethasone treatment reduces infarct size, alleviates cardiomyocyte damage and improves lysosomal function [51, 57]. Similar observations were made in children undergoing cardiopulmonary bypass, where dexamethasone significantly reduced postoperative cardiac troponin I levels, suggesting a cardioprotective effect of glucocorticoids also in humans [7]. However, while these studies investigated short-term application of dexamethasone, long-term application would likely be necessary for the treatment of chronic heart failure. Glucocorticoids exert systemic side effects, including elevated blood pressure and insulin resistance, and may also activate mineralocorticoid receptors, for instance in endothelial cells, which has been suggested to have negative cardiac effects [26, 33]. Therefore, identifying and directly targeting effects downstream of the glucocorticoid receptor might be a more promising approach. We suggest that enhancers of autophagy should be investigated with respect to effects on cardiomyocytes and the transverse tubular system in animal models and the goal to improve cardiomyocyte excitation-contraction coupling in heart failure.
4.5. Limitations
In this study we cultivated isolated cardiomyocytes as an in-vitro model of t-system loss in order to separate t-system remodelling from secondary effects of heart failure and to gain better control over hormonal and cellular signalling. Major implications, that is, t-system preservation by glucocorticoids and upregulation of autophagy, will need to be verified in vivo. It is possible that the mechanisms underlying t-system loss in isolated myocytes differ from the mechanisms that drive t-system remodelling in cardiac disease. For example, the absence of extracellular matrix may contribute to the rapid loss of t-tubules in culture, as well as missing direct or cytokine-mediated interactions between myocytes and fibroblasts [8, 16, 39, 47]. Furthermore, culture media used in this study did not contain serum or growth factors, and we cannot exclude that their presence in vivo might alter the effects of glucocorticoids. We also acknowledge that isolation and culture may induce stress responses in cardiomyocytes, which could render autophagy particularly important for maintaining cellular homeostasis. Nevertheless, although cardiomyocyte culture differs in several aspects from in-vivo models of heart failure, it was shown that there are high similarities regarding gene expression, structure and function [25, 27, 38]. Moreover, stress responses occur in cardiac disease, where autophagy was shown to be essential [31]. Drugs enhancing autophagy, such as rapamycin, have been shown to exert positive effects in models of heart failure and pressure overload by antagonizing excessive hypertrophy [4, 29], and the severity of cardiac hypertrophy closely correlates with the degree of t-tubule remodelling [48, 56]. We suggest that future studies investigate the signalling pathways involved in GR-mediated t-system preservation, and if altered autophagy correlates with pathological t-system remodelling and defective excitation-contraction coupling in vivo. Clarifying how autophagy and related processes are involved in t-system homeostasis and remodelling may advance our pathophysiological understanding of heart failure and eventually lead to new treatment options.
Supplementary Material
6. Acknowledgements
We would like to thank Celine Grüninger, Lorenz Reeh, Jessica Rinke and Ralf Rinke for excellent technical support, and Philipp Tripal from the Optical Imaging Centre Erlangen (OICE) for excellent assistance with STED microscopy.
5. Funding
This work was supported by grants from the Interdisciplinary Centre for Clinical Research (IZKF) at the University Hospital of the University of Erlangen-Nuremberg.
Footnotes
Conflict of Interest
None declared.
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
8 References
- 1.Al-Qusairi L, Laporte J (2011) T-tubule biogenesis and triad formation in skeletal muscle and implication in human diseases. Skelet Muscle 1:26 doi: 10.1186/2044-5040-1-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banyasz T, Lozinskiy I, Payne CE, Edelmann S, Norton B, Chen B, Chen-Izu Y, Izu LT, Balke CW (2008) Transformation of adult rat cardiac myocytes in primary culture. Exp Physiol 93:370–382 doi: 10.1113/expphysiol.2007.040659 [DOI] [PubMed] [Google Scholar]
- 3.Beavers DL, Landstrom AP, Chiang DY, Wehrens XH (2014) Emerging roles of junctophilin-2 in the heart and implications for cardiac diseases. Cardiovasc Res 103:198–205 doi: 10.1093/cvr/cvu151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bishu K, Ogut O, Kushwaha S, Mohammed SF, Ohtani T, Xu X, Brozovich FV, Redfield MM (2013) Anti-remodeling effects of rapamycin in experimental heart failure: dose response and interaction with angiotensin receptor blockade. PLoS One 8:e81325 doi: 10.1371/journal.pone.0081325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bryant SM, Kong CHT, Watson JJ, Gadeberg HC, Roth DM, Patel HH, Cannell MB, James AF, Orchard CH (2018) Caveolin-3 KO disrupts t-tubule structure and decreases t-tubular ICa density in mouse ventricular myocytes. Am J Physiol Heart Circ Physiol 315:H1101–H1111 doi: 10.1152/ajpheart.00209.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caldwell JL, Smith CE, Taylor RF, Kitmitto A, Eisner DA, Dibb KM, Trafford AW (2014) Dependence of cardiac transverse tubules on the BAR domain protein amphiphysin II (BIN-1). Circ Res 115:986–996 doi: 10.1161/CIRCRESAHA.116.303448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Checchia PA, Backer CL, Bronicki RA, Baden HP, Crawford SE, Green TP, Mavroudis C (2003) Dexamethasone reduces postoperative troponin levels in children undergoing cardiopulmonary bypass. Crit Care Med 31:1742–1745 doi: 10.1097/01.CCM.0000063443.32874.60 [DOI] [PubMed] [Google Scholar]
- 8.Crossman DJ, Shen X, Jullig M, Munro M, Hou Y, Middleditch M, Shrestha D, Li A, Lal S, Dos Remedios CG, Baddeley D, Ruygrok PN, Soeller C (2017) Increased collagen within the transverse tubules in human heart failure. Cardiovasc Res 113:879–891 doi: 10.1093/cvr/cvx055 [DOI] [PubMed] [Google Scholar]
- 9.Crossman DJ, Young AA, Ruygrok PN, Nason GP, Baddelely D, Soeller C, Cannell MB (2015) T-tubule disease: Relationship between t-tubule organization and regional contractile performance in human dilated cardiomyopathy. J Mol Cell Cardiol 84:170–178 doi: 10.1016/j.yjmcc.2015.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cruz-Topete D, Oakley RH, Carroll NG, He B, Myers PH, Xu X, Watts MN, Trosclair K, Glasscock E, Dominic P, Cidlowski JA (2019) Deletion of the Cardiomyocyte Glucocorticoid Receptor Leads to Sexually Dimorphic Changes in Cardiac Gene Expression and Progression to Heart Failure. J Am Heart Assoc 8:e011012 doi: 10.1161/JAHA.118.011012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De La Mata A, Tajada S, O’Dwyer S, Matsumoto C, Dixon RE, Hariharan N, Moreno CM, Santana LF (2019) BIN1 Induces the Formation of T-Tubules and Adult-Like Ca(2+) Release Units in Developing Cardiomyocytes. Stem Cells 37:54–64 doi: 10.1002/stem.2927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frisk M, Ruud M, Espe EK, Aronsen JM, Roe AT, Zhang L, Norseng PA, Sejersted OM, Christensen GA, Sjaastad I, Louch WE (2016) Elevated ventricular wall stress disrupts cardiomyocyte t-tubule structure and calcium homeostasis. Cardiovasc Res 112:443–451 doi: 10.1093/cvr/cvw111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo X, Huang C, Lian K, Wang S, Zhao H, Yan F, Zhang X, Zhang J, Xie H, An R, Tao L (2016) BCKA down-regulates mTORC2-Akt signal and enhances apoptosis susceptibility in cardiomyocytes. Biochem Biophys Res Commun 480:106–113 doi: 10.1016/j.bbrc.2016.09.162 [DOI] [PubMed] [Google Scholar]
- 14.Heinzel FR, Bito V, Biesmans L, Wu M, Detre E, von Wegner F, Claus P, Dymarkowski S, Maes F, Bogaert J, Rademakers F, D’Hooge J, Sipido K (2008) Remodeling of T-tubules and reduced synchrony of Ca2+ release in myocytes from chronically ischemic myocardium. Circ Res 102:338–346 doi: 10.1161/CIRCRESAHA.107.160085 [DOI] [PubMed] [Google Scholar]
- 15.Hong T, Yang H, Zhang SS, Cho HC, Kalashnikova M, Sun B, Zhang H, Bhargava A, Grabe M, Olgin J, Gorelik J, Marban E, Jan LY, Shaw RM (2014) Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia. Nat Med 20:624–632 doi: 10.1038/nm.3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Humeres C, Frangogiannis NG (2019) Fibroblasts in the Infarcted, Remodeling, and Failing Heart. JACC Basic Transl Sci 4:449–467 doi: 10.1016/j.jacbts.2019.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jayasinghe ID, Baddeley D, Kong CH, Wehrens XH, Cannell MB, Soeller C (2012) Nanoscale organization of junctophilin-2 and ryanodine receptors within peripheral couplings of rat ventricular cardiomyocytes. Biophys J 102:L19–21 doi: 10.1016/j.bpj.2012.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones PP, MacQuaide N, Louch WE (2018) Dyadic Plasticity in Cardiomyocytes. Front Physiol 9:1773 doi: 10.3389/fphys.2018.01773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawai M, Hussain M, Orchard CH (1999) Excitation-contraction coupling in rat ventricular myocytes after formamide-induced detubulation. American Journal of Physiology-Heart and Circulatory Physiology 277:H603–H609 doi: 10.1152/ajpheart.1999.277.2.H603 [DOI] [PubMed] [Google Scholar]
- 20.Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13:132–141 doi: 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kong CHT, Bryant SM, Watson JJ, Roth DM, Patel HH, Cannell MB, James AF, Orchard CH (2019) CARDIAC-SPECIFIC OVEREXPRESSION OF CAVEOLIN-3 PRESERVES T-TUBULAR ICa DURING HEART FAILURE IN MICE. Exp Physiol doi: 10.1113/EP087304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kostin S, Scholz D, Shimada T, Maeno Y, Mollnau H, Hein S, Schaper J (1998) The internal and external protein scaffold of the T-tubular system in cardiomyocytes. Cell and tissue research 294:449–460 [DOI] [PubMed] [Google Scholar]
- 23.Lawless M, Caldwell JL, Radcliffe EJ, Smith CER, Madders GWP, Hutchings DC, Woods LS, Church SJ, Unwin RD, Kirkwood GJ, Becker LK, Pearman CM, Taylor RF, Eisner DA, Dibb KM, Trafford AW (2019) Phosphodiesterase 5 inhibition improves contractile function and restores transverse tubule loss and catecholamine responsiveness in heart failure. Sci Rep 9:6801 doi: 10.1038/s41598-019-42592-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Lichter JG, Seidel T, Tomaselli GF, Bridge JH, Sachse FB (2015) Cardiac Resynchronization Therapy Reduces Subcellular Heterogeneity of Ryanodine Receptors, T-Tubules, and Ca2+ Sparks Produced by Dyssynchronous Heart Failure. Circ Heart Fail 8:1105–1114 doi: 10.1161/CIRCHEARTFAILURE.115.002352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lipsett DB, Frisk M, Aronsen JM, Norden ES, Buonarati OR, Cataliotti A, Hell JW, Sjaastad I, Christensen G, Louch WE (2019) Cardiomyocyte substructure reverts to an immature phenotype during heart failure. J Physiol doi: 10.1113/JP277273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lother A, Deng L, Huck M, Furst D, Kowalski J, Esser JS, Moser M, Bode C, Hein L (2018) Endothelial cell mineralocorticoid receptors oppose VEGF-induced gene expression and angiogenesis. J Endocrinol doi: 10.1530/JOE-18-0494 [DOI] [PubMed] [Google Scholar]
- 27.Louch WE, Bito V, Heinzel FR, Macianskiene R, Vanhaecke J, Flameng W, Mubagwa K, Sipido KR (2004) Reduced synchrony of Ca2+ release with loss of T-tubules-a comparison to Ca2+ release in human failing cardiomyocytes. Cardiovasc Res 62:63–73 doi: 10.1016/j.cardiores.2003.12.031 [DOI] [PubMed] [Google Scholar]
- 28.Manfra O, Frisk M, Louch WE (2017) Regulation of Cardiomyocyte T-Tubular Structure: Opportunities for Therapy. Curr Heart Fail Rep 14:167–178 doi: 10.1007/s11897-017-0329-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T, Izumo S (2004) Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation 109:3050–3055 doi: 10.1161/01.CIR.0000130641.08705.45 [DOI] [PubMed] [Google Scholar]
- 30.Mizushima N, Yoshimori T, Levine B (2010) Methods in Mammalian Autophagy Research. Cell 140:313–326 doi: 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K (2007) The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 13:619–624 doi: 10.1038/nm1574 [DOI] [PubMed] [Google Scholar]
- 32.Nishida K, Otsu K (2016) Autophagy during cardiac remodeling. J Mol Cell Cardiol 95:11–18 doi: 10.1016/j.yjmcc.2015.12.003 [DOI] [PubMed] [Google Scholar]
- 33.Oakley RH, Cruz-Topete D, He B, Foley JF, Myers PH, Xu X, Gomez-Sanchez CE, Chambon P, Willis MS, Cidlowski JA (2019) Cardiomyocyte glucocorticoid and mineralocorticoid receptors directly and antagonistically regulate heart disease in mice. Sci Signal 12 doi: 10.1126/scisignal.aau9685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oakley RH, Ren R, Cruz-Topete D, Bird GS, Myers PH, Boyle MC, Schneider MD, Willis MS, Cidlowski JA (2013) Essential role of stress hormone signaling in cardiomyocytes for the prevention of heart disease. Proc Natl Acad Sci U S A 110:17035–17040 doi: 10.1073/pnas.1302546110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oakley RH, Revollo J, Cidlowski JA (2012) Glucocorticoids regulate arrestin gene expression and redirect the signaling profile of G protein-coupled receptors. Proc Natl Acad Sci U S A 109:17591–17596 doi: 10.1073/pnas.1209411109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parikh SS, Blackwell DJ, Gomez-Hurtado N, Frisk M, Wang L, Kim K, Dahl CP, Fiane A, Tonnessen T, Kryshtal DO, Louch WE, Knollmann BC (2017) Thyroid and Glucocorticoid Hormones Promote Functional T-Tubule Development in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Circ Res 121:1323–1330 doi: 10.1161/CIRCRESAHA.117.311920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pinali C, Malik N, Davenport JB, Allan LJ, Murfitt L, Iqbal MM, Boyett MR, Wright EJ, Walker R, Zhang Y, Dobryznski H, Holt CM, Kitmitto A (2017) Post-Myocardial Infarction T-tubules Form Enlarged Branched Structures With Dysregulation of Junctophilin-2 and Bridging Integrator 1 (BIN-1). J Am Heart Assoc 6 doi: 10.1161/JAHA.116.004834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poindexter BJ, Smith JR, Buja LM, Bick RJ (2001) Calcium signaling mechanisms in dedifferentiated cardiac myocytes: comparison with neonatal and adult cardiomyocytes. Cell Calcium 30:373–382 doi: 10.1054/ceca.2001.0249 [DOI] [PubMed] [Google Scholar]
- 39.Quinn TA, Camelliti P, Rog-Zielinska EA, Siedlecka U, Poggioli T, O’Toole ET, Knopfel T, Kohl P (2016) Electrotonic coupling of excitable and nonexcitable cells in the heart revealed by optogenetics. Proc Natl Acad Sci U S A 113:14852–14857 doi: 10.1073/pnas.1611184114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richardson RV, Batchen EJ, Thomson AJ, Darroch R, Pan X, Rog-Zielinska EA, Wyrzykowska W, Scullion K, Al-Dujaili EA, Diaz ME, Moran CM, Kenyon CJ, Gray GA, Chapman KE (2017) Glucocorticoid receptor alters isovolumetric contraction and restrains cardiac fibrosis. J Endocrinol 232:437–450 doi: 10.1530/JOE-16-0458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riquelme JA, Chavez MN, Mondaca-Ruff D, Bustamante M, Vicencio JM, Quest AF, Lavandero S (2016) Therapeutic targeting of autophagy in myocardial infarction and heart failure. Expert Rev Cardiovasc Ther 14:1007–1019 doi: 10.1080/14779072.2016.1202760 [DOI] [PubMed] [Google Scholar]
- 42.Rog-Zielinska EA, Thomson A, Kenyon CJ, Brownstein DG, Moran CM, Szumska D, Michailidou Z, Richardson J, Owen E, Watt A, Morrison H, Forrester LM, Bhattacharya S, Holmes MC, Chapman KE (2013) Glucocorticoid receptor is required for foetal heart maturation. Hum Mol Genet 22:3269–3282 doi: 10.1093/hmg/ddt182 [DOI] [PubMed] [Google Scholar]
- 43.Rohdewald P, Mollmann H, Barth J, Rehder J, Derendorf H (1987) Pharmacokinetics of dexamethasone and its phosphate ester. Biopharm Drug Dispos 8:205–212 [DOI] [PubMed] [Google Scholar]
- 44.Sainte-Marie Y, Nguyen Dinh Cat A, Perrier R, Mangin L, Soukaseum C, Peuchmaur M, Tronche F, Farman N, Escoubet B, Benitah JP, Jaisser F (2007) Conditional glucocorticoid receptor expression in the heart induces atrio-ventricular block. FASEB J 21:3133–3141 doi: 10.1096/fj.07-8357com [DOI] [PubMed] [Google Scholar]
- 45.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM (2006) Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22:159–168 doi: 10.1016/j.molcel.2006.03.029 [DOI] [PubMed] [Google Scholar]
- 46.Seidel T, Navankasattusas S, Ahmad A, Diakos NA, Xu WD, Tristani-Firouzi M, Bonios MJ, Taleb I, Li DY, Selzman CH, Drakos SG, Sachse FB (2017) Sheet-Like Remodeling of the Transverse Tubular System in Human Heart Failure Impairs Excitation-Contraction Coupling and Functional Recovery by Mechanical Unloading. Circulation 135:1632–1645 doi: 10.1161/CIRCULATIONAHA.116.024470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seidel T, Sankarankutty AC, Sachse FB (2017) Remodeling of the transverse tubular system after myocardial infarction in rabbit correlates with local fibrosis: A potential role of biomechanics. Prog Biophys Mol Biol 130:302–314 doi: 10.1016/j.pbiomolbio.2017.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shah SJ, Aistrup GL, Gupta DK, O’Toole MJ, Nahhas AF, Schuster D, Chirayil N, Bassi N, Ramakrishna S, Beussink L, Misener S, Kane B, Wang D, Randolph B, Ito A, Wu M, Akintilo L, Mongkolrattanothai T, Reddy M, Kumar M, Arora R, Ng J, Wasserstrom JA (2014) Ultrastructural and cellular basis for the development of abnormal myocardial mechanics during the transition from hypertension to heart failure. Am J Physiol Heart Circ Physiol 306:H88–100 doi: 10.1152/ajpheart.00642.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shimizu N, Yoshikawa N, Ito N, Maruyama T, Suzuki Y, Takeda S, Nakae J, Tagata Y, Nishitani S, Takehana K, Sano M, Fukuda K, Suematsu M, Morimoto C, Tanaka H (2011) Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab 13:170–182 doi: 10.1016/j.cmet.2011.01.001 [DOI] [PubMed] [Google Scholar]
- 50.Smith CER, Trafford AW, Caldwell JL, Dibb KM (2018) Physiology and pathophysiology of the cardiac transverse tubular system. Current Opinion in Physiology 1:153–160 doi: 10.1016/j.cophys.2017.11.002 [DOI] [Google Scholar]
- 51.Spath JA, Lefer AM (1975) Effects of dexamethasone on myocardial cells in the early phase of acute myocardial infarction. Am Heart J 90:50–55 doi: 10.1016/0002-8703(75)90256-2 [DOI] [PubMed] [Google Scholar]
- 52.Tato I, Bartrons R, Ventura F, Rosa JL (2011) Amino acids activate mammalian target of rapamycin complex 2 (mTORC2) via PI3K/Akt signaling. J Biol Chem 286:6128–6142 doi: 10.1074/jbc.M110.166991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tian Q, Pahlavan S, Oleinikow K, Jung J, Ruppenthal S, Scholz A, Schumann C, Kraegeloh A, Oberhofer M, Lipp P, Kaestner L (2012) Functional and morphological preservation of adult ventricular myocytes in culture by sub-micromolar cytochalasin D supplement. J Mol Cell Cardiol 52:113–124 doi: 10.1016/j.yjmcc.2011.09.001 [DOI] [PubMed] [Google Scholar]
- 54.Troncoso R, Paredes F, Parra V, Gatica D, Vasquez-Trincado C, Quiroga C, Bravo-Sagua R, Lopez-Crisosto C, Rodriguez AE, Oyarzun AP, Kroemer G, Lavandero S (2014) Dexamethasone-induced autophagy mediates muscle atrophy through mitochondrial clearance. Cell Cycle 13:2281–2295 doi: 10.4161/cc.29272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vlahakis A, Graef M, Nunnari J, Powers T (2014) TOR complex 2-Ypk1 signaling is an essential positive regulator of the general amino acid control response and autophagy. Proc Natl Acad Sci U S A 111:10586–10591 doi: 10.1073/pnas.1406305111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wei S, Guo A, Chen B, Kutschke W, Xie YP, Zimmerman K, Weiss RM, Anderson ME, Cheng H, Song LS (2010) T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res 107:520–531 doi: 10.1161/CIRCRESAHA.109.212324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu B, Strom J, Chen QM (2011) Dexamethasone induces transcriptional activation of Bcl-xL gene and inhibits cardiac injury by myocardial ischemia. Eur J Pharmacol 668:194–200 doi: 10.1016/j.ejphar.2011.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yoshii SR, Mizushima N (2017) Monitoring and Measuring Autophagy. Int J Mol Sci 18 doi: 10.3390/ijms18091865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang C, Chen B, Guo A, Zhu Y, Miller JD, Gao S, Yuan C, Kutschke W, Zimmerman K, Weiss RM, Wehrens XH, Hong J, Johnson FL, Santana LF, Anderson ME, Song LS (2014) Microtubule-mediated defects in junctophilin-2 trafficking contribute to myocyte transverse-tubule remodeling and Ca2+ handling dysfunction in heart failure. Circulation 129:1742–1750 doi: 10.1161/CIRCULATIONAHA.113.008452 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.