

Abstract
T cells play a critical role in the control of cancer. The development of immune checkpoint blockers (ICB) aimed at enhancing antitumor T-cell responses has revolutionized cancer treatment. However, durable clinical benefit is observed in only a subset of patients, prompting research efforts to focus on strategies that target multiple inhibitory signals within the tumor microenvironment (TME) to limit tumor evasion and improve patient outcomes. Adenosine has emerged as a potent immune suppressant within the TME, and CD73 is the major enzyme responsible for its extracellular production. CD73 can be co-opted within the TME to impair T-cell–mediated antitumor immunity and promote tumor growth. To target this pathway and block the formation of adenosine, we designed a novel, selective, and potent class of small-molecule inhibitors of CD73, including AB680 (quemliclustat), which is currently being tested in patients with cancer. AB680 effectively restored T-cell proliferation, cytokine secretion, and cytotoxicity that were dampened by the formation of immunosuppressive adenosine by CD73. Furthermore, in an allogeneic mixed lymphocyte reaction where CD73-derived adenosine had a dominant suppressive effect in the presence of PD-1 blockade, AB680 restored T-cell activation and function. Finally, in a preclinical mouse model of melanoma, AB680 inhibited CD73 in the TME and increased the antitumor activity of PD-1 blockade. Collectively, these data provide a rationale for the inhibition of CD73 with AB680 in combination with ICB, such as anti–PD-1, to improve cancer patient outcomes.
Introduction
Adenosine has a remarkable range of functions in cellular biology and physiology. Importantly, this nucleoside has emerged as a key signaling molecule serving to attenuate excessive immune responses during infection and other inflammatory insults (1, 2). The role of adenosine as a potent immune suppressant within the tumor microenvironment (TME) is now appreciated as a prominent mechanism of immune escape and tumor progression (2–5). CD73, also known as ecto-5′-nucleotidase, converts extracellular AMP into adenosine and organic phosphate and is the major enzyme involved in the generation of adenosine (6). In the absence of disease, CD73 is expressed on lymphocytes and endothelial cells and plays a role in the regulation of endothelial barrier function (7, 8) and cell migration (9, 10). In malignancy, CD73 is overexpressed and has been correlated with poor prognosis (11, 12). While there is a clear role for CD73-expressing cancer cells in promoting tumor growth (13, 14), expression is not restricted to just cancer cells. CD73 is also found on tumor-infiltrating myeloid cells (15, 16), cancer-associated fibroblasts (17), and on T cells, where CD73 enzymatic activity results in local accumulation of adenosine and activation of the adenosine receptor A2AR. The resultant dampening of T cell–mediated antitumor immunity is thus mediated through CD73-derived adenosine and subsequent A2AR signaling in T cells (5, 14, 18–20). Indeed, in vivo reconstitution studies with CD73-deficient regulatory T cells (Treg) in mice demonstrate a critical role for T cell–derived adenosine in creating an immunosuppressive environment and promoting tumor growth (18, 21, 22). CD39, an enzyme that converts extracellular ATP to AMP, is also expressed on many cell types within the TME and cooperates with CD73 to convert extracellular ATP to adenosine. For example, activated Tregs express both CD39 and CD73 (21–23) and have been shown to cooperate with CD73+ CD4+ T cells (19, 24, 25) and other CD73+ suppressive cell types in the TME, such as tumor-associated macrophages, myeloid-derived suppressor cells, cancer cells, and even CD8+ T cells to dampen T cell–mediated immunity (26). Thus, inhibiting CD73 in the TME could be an effective therapeutic approach to promote a robust antitumor T-cell response (27–30).
While the success of immune checkpoint blockade (ICB) targeting T cells initiated a revolutionary shift toward immunotherapeutic approaches to treat cancer, durable clinical benefit is limited to only a subset of patients (31). Along the lines of augmenting tumor-specific immunity, preclinical studies targeting CD73 in addition to other key nodes of the adenosinergic pathway have been shown to increase therapeutic benefits observed PD-1 and CTLA-4 blockade (28, 32, 33), as well as chemotherapies that induce immunogenic cell death (13, 15, 30), lending support for exploring the combination in a clinical setting. Correspondingly, CD73 and adenosine receptor inhibition is being investigated in solid tumor indications in regimens with PD-1 blockade (26, 34). AB680 (quemliclustat) is a potent, reversible, small-molecule competitive inhibitor of human CD73. It exhibits a Ki of 5 pmol/L (35) and is characterized by low plasma clearance and a long half-life in preclinical species, resulting in a pharmacokinetic profile suitable for long-acting parenteral administration (36). In humans, initial data show that AB680 is well tolerated and exhibits a pharmacokinetic profile (long half-life) suitable for intravenous administration (36, 37). AB680 is currently being evaluated in patients with treatment-naïve pancreatic cancer (ref. 37; NCT04104672), metastatic colorectal cancer (NCT04660812), and castrate- and androgen-resistant prostate cancer (NCT04381832). Herein we describe the immunotherapeutic effects of AB680—a novel, selective, and potent inhibitor that blocks adenosine generation by CD73. AB680 reversed adenosine-mediated immunosuppression of human and mouse immune cells in vitro and promoted antitumor immunity as a single agent and in combination with anti-PD-1 in a syngeneic mouse tumor model.
Materials and Methods
Cell lines
B16F10 and EG7.OVA cell lines were obtained from the ATCC and were cultured in DMEM supplemented with 10% FBS, 100 U/mL penicillin/streptomycin, and 1× GlutaMAX or RPMI-1640 supplemented with 10% FBS, 100 U/mL penicillin/streptomycin, 0.4 mg/mL G418, and 1× GlutaMAX, respectively. Cell lines were authenticated using short tandem repeat DNA profiling and tested for pathogen contamination including Mycoplasma spp. (STAT-Myco and IMPACT II testing) via RT-PCR (IDEXX Bioresearch). EG7.OVA cells were fluorescently labeled using the IncuCyte Nuclight Red Lentivirus Reagent (Essen Biosciences), single-cell cloned in the presence of Puromycin (Invivogen), and selected on the basis of red fluorescence intensity by flow cytometry and imaging. K562 cells engineered to express either HLA-A02 alone or HLA-A02 and tumor-specific epitope (Neo-12; ref. 38) were provided by PACT Pharma and were maintained in RPMI-1640 supplemented with 10% FBS, 100 U/mL penicillin/streptomycin, and 1× GlutaMAX. All cell lines were maintained at 37°C, 5% CO2.
Antibodies and reagents
For the human in vitro assays, anti-PD-1 (AB122) was supplied by Arcus Biosciences while purified human IgG4 (anti-β-Gal, Invivogen) isotype control antibody was sourced commercially. Adenosine 5′-monophosphate disodium salt (AMP) and Erythro-9-(2-Hydroxy-3-nonyl) adenine, HCl (EHNA) were purchased from Sigma-Aldrich. For mouse in vivo studies, purified anti-PD-1 (clone RMP1-14) or isotype control (rat IgG2a, clone 2A3) were purchased from BioXCell. CD73 inhibitor AB680 was synthesized by Arcus Biosciences as described previously (39). Flow cytometry was performed on the LSRFortessa (BD Biosciences) and analyzed using FlowJo software (TreeStar). Antibodies used for flow cytometry are summarized in Supplementary Table S1.
Mice
Female 6 to 8 weeks old wild-type (WT), CD73−/− (B6.129S1-Nt5etm1Lft/J), and OT-I [C57BL/6-Tg(TcraTcrb)1100Mjb/J] C57BL/6J mice were purchased from The Jackson Laboratory. Experiments were performed at Arcus Biosciences in accordance with federal, state, and institutional guidelines and were approved by Arcus Biosciences’ Institutional Animal Care and Use Committee.
Human tumor-infiltrating lymphocyte flow cytometry
Dissociated tumor biopsies were obtained from Conversant Bio with informed written consent and according to Institutional Review Board (IRB)-approved guidelines in accordance with the Declaration of Helsinki. Subject information is detailed in Supplementary Table S2. Tumor samples were thawed and washed with PBS, blocked with Human Fc Block (BD Biosciences) before staining with fluorophore-conjugated antibodies for 30 minutes at 4°C. Cells were washed and resuspended in PBS for flow cytometric analysis.
Human T-cell activation assays
Total, memory, and naïve human CD4+ or CD8+ T cells were isolated from buffy coats obtained from Stanford Blood Center with informed written consent and according to IRB-approved human sample guidelines in accordance with U.S. Common Rule. To track proliferation, isolated CD4+ T cells were labeled with 5 μmol/L CellTrace Violet (Invitrogen), washed, and plated. 5 × 104 CD4+ T cells and CD8+ T cells were activated with anti-CD2/CD3/CD28 beads (Miltenyi Biotec) at a 1:1 bead-to-cell ratio. AB680 was added at indicated concentrations in a final DMSO concentration of 0.1% and incubated for 1 hour at 37°C. Unless otherwise indicated, 6.25 μmol/L AMP and 2.5 μmol/L EHNA were added and incubated for 3 days at 37°C prior to surface marker staining and for flow cytometric analysis. Cytokine secretion (IFNγ, granzyme B, or IL2) was measured using Cytometric Bead Arrays (CBA, BD Biosciences) per manufacturer's instructions.
Mouse T-cell activation assays
For mouse, CD8+ T cells were isolated from WT or CD73−/− mouse splenocytes and resuspended in complete media containing RPMI, 5% FBS. A total of 5 × 104 cells were activated with anti-CD3/CD28 beads (Miltenyi Biotec) at a 2:1 bead-to-cell ratio in the presence of 50 U/mL recombinant mouse IL2 (Peprotech). CD73 inhibitors were added at indicated concentrations in a final DMSO concentration of 0.1% and incubated for 1 hour at 37°C. A total of 50 μmol/L AMP and 2.5 μmol/L EHNA were added and incubated for 4 days at 37°C. Supernatant was collected for IFNγ quantification by CBA.
Allogeneic mixed lymphocyte reaction
Peripheral blood mononuclear cells were isolated from healthy donor buffy coats obtained from Stanford Blood Center. CD14+ monocytes were isolated using the EasySep Human CD14 Positive Selection Kit (StemCell Technologies). Monocyte-derived dendritic cells (mo-DC) were generated by resuspending CD14+ monocytes in 6-well cell culture plates in RPMI-1640 supplemented with 5% FBS and recombinant human GMCSF (100 ng/mL, R&D Systems) and IL4 (100 ng/mL, Peprotech) for 7 days. mo-DCs were cocultured with CD4+ T cells (isolated as described above) at a ratio of 1:4 (2.5 × 104 mo-DCs, 1 × 105 CD4+ T cells) in X-VIVO-20 media. Where indicated, anti-PD-1 or IgG4 isotype (0.67 or 6.7 nmol/L), AMP (100 μmol/L), and AB680 (100 nmol/L) were added. After 72 hours, cells were harvested for RNA isolation and after 96 hours, supernatant was collected for IFNγ quantification by CBA.
RT-PCR
RNA was isolated using the RNeasy Mini Kit (Qiagen) and converted to cDNA using Superscript IV First Strand Synthesis (Invitrogen). RT-PCR was carried out using Taqman probes (Thermo Fisher Scientific) for CTLA4 (Hs00175480_m1), TBX21 (Hs00894392_m1), PDCD1 (Hs01550088_m1), IFNG (Hs00989291_m1), and HPRT1 (Hs02800695_m1). Relative gene expression was calculated using the formula 2–ΔCt where ΔCt equals the cycle threshold (Ct) for the gene of interest minus the Ct for the reference gene HPRT1.
Immunofluorescence staining
Multiplex fluorescence was performed by Ventana Medical Systems/Roche Tissue Diagnostics using a Ventana Benchmark Ultra System. Formalin-fixed, paraffin-embedded tissue sections of human colorectal cancer were stained with the following antibody clones: CD73 (Cell Signaling Technology, D7F9A), pan-cytokeratin (Roche Tissue Diagnostics, AE1/AE3/PCK26), CD8 (Spring Bioscience, SP239) and imaged on a Zeiss Axio Scan.Z1 slide scanner.
Mouse in vitro CTL assay
Growth Factor Reduced Matrigel (3 mg/mL; Phenol Red-free, LDEV-free, Corning) in RPMI was added at 30 μL/well to coat a flat 96-well cell culture plate. Red fluorescently labeled EG7.OVA cells (1 × 104) were added to the precoated wells and incubated at 37°C overnight. OT-I splenocytes were activated with 1 μg/mL of SIINFEKL peptide (Genscript) 48 hours prior to coculture.. CD8+ T cells were isolated using a mouse CD8α T-cell Negative Isolation Kit (Miltenyi Biotec) and activation status and purity were examined by flow cytometry. Isolated CD8+ T cells were resuspended in RPMI-1640 supplemented with 10% FBS and added to EG7.OVA cells at a 2:1 ratio in the presence of 50 μmol/L AMP and 2.5 μmol/L EHNA with or without 50 nmol/L AB680 in a final DMSO concentration of 0.1%. The coculture was imaged every 4 hours for a total of 48 hours in the IncuCyte ZOOM under a 10× objective. Percent confluence of red fluorescence objects was measured over time and calculated as a percent change relative to day zero.
Human tumor-specific T-cell assay
A tumor-specific T-cell receptor (TCR; Neo12-TCR; ref. 38) was transduced into primary human CD4+ and CD8+ T cells from the same donor using site-specific nucleases in a single-step transfection process to knock out the endogenous TCRβ chain and knock in the Neo12-TCR (40). Efficiency was confirmed by dextramer staining. T cells were added at 1 × 105 cells/well and incubated with 100 nmol/L AB680 for 1 hour at 37°C. AMP (50 μmol/L) and EHNA (2.5 μmol/L) were added along with K562 cells expressing HLA-A2 with the Neo12 epitope at a T cell:K562 cell ratio of 4:1. After 72 hours, activation of T cells was determined by the percent of CD25+ CD69+ cells using flow cytometry. IL2 and IFNγ in the supernatant was quantified by CBA.
AMPase histochemistry
B16F10 tumors were frozen in O.C.T. compound (Tissue-Tek). A modified form of the Wachstein-Meisel phosphatase activity assay (41) was developed and performed. Briefly, sections were fixed with 4% formaldehyde for 5 minutes at 4°C and washed three times in 50 mmol/L tris maleate buffer (pH 7.4). Samples were preincubated for 30 minutes with 50 mmol/L tris maleate buffer, 250 mmol/L sucrose, 2 mmol/L MgCl2, and AB680 (5 μmol/L) at room temperature. For assessment of AMPase activity, sections were incubated in 50 mmol/L tris maleate buffer (pH 7.4) supplemented with AMP (0.3 mmol/L), AB680 (5 μmol/L), MnCl2 (5 mmol/L), and lead acetate (2 mmol/L) for 1 hour at 37°C. Samples were washed with 2% acetic acid and incubated with 1% ammonium sulfide for 3 minutes before being washed with H2O and counterstained with hematoxylin (Vector Laboratories).
CD73 enzymatic assay on T cells
Human or mouse CD8+ T cells were resuspended in assay buffer consisting of 20 mmol/L HEPES, pH 7.4, 137 mmol/L NaCl, 5.4 mmol/L KCl, 1.3 mmol/L CaCl2, 4.2 mmol/L NaHCO3, and 0.1% glucose. Serial dilutions of AB680 were prepared in DMSO, added to assay buffer, and transferred to cells followed by incubation for 30 minutes at 37°C and addition of AMP. Final assay conditions comprised of 2.6 × 104 cells per well in 2% DMSO and 50 μmol/L of AMP substrate. After 120 minutes, supernatant was collected and added to wells preloaded with 20 μL of PiColorLock Gold colorimetric assay reagent (Thermo Fisher Scientific). Inorganic phosphate reaction product was assessed and quantitated using an Envision 2102 Multilabel Reader fitted with a 620 nm filter. CD73 enzymatic activity was evaluated as a correlate of phosphate product levels. Percentage maximum activity in each test well was calculated on the basis of DMSO (maximum activity) and no cell control wells (baseline activity) and the IC50 values were determined from compound dose–response curve fitted using a standard four-parameter fit equation.
CD73 enzymatic 13C5 AMP assay
Implanted B16F10 tumors were excised, snap frozen, and homogenized in protein lysis buffer. Inhibitors including 10 μmol/L EHNA, 625 μmol/L TNAP inhibitor (supplied by Arcus Biosciences), 10 μmol/L 5-iodotubercidin (Sigma-Aldrich), and 4 μmol/L aristeromycin (Sigma-Aldrich) were added to the lysate at 37°C, pH 7.4. AB680 was added exogenously to tumor homogenates and incubated for 1 hour at 37°C before adding 5 μmol/L 13C5 AMP. After 5 minutes, the reaction was quenched with a final concentration of 0.1 mol/L PCA and CD73 activity was calculated from the amount of 13C5 adenosine produced (measured by LC/MS).
In vivo treatments
C57BL/6 female mice were injected subcutaneously on the right flank with B16F10 cells in 100 μL PBS (3 × 105). For single-agent efficacy, mice were treated with vehicle (1% HPMC) or AB680 (10 mg/kg), subcutaneous (between the shoulder blades) daily starting on the day of tumor injection and continued throughout the duration of the study. For combination efficacy, once tumors were established (50 mm3), mice were randomized into groups based on tumor size and treated with anti-PD-1 [2.5 mg/kg, intraperitoneal] every 3 days for a total of four administrations with or without AB680 (10 mg/kg, s.c.) daily. Tumor volume was monitored with digital calipers and volume was calculated using the formula: (Length × Width2)/2. For survival, a predetermined tumor volume of 1,000 mm3 was used as the endpoint.
Mouse tumor-infiltrating lymphocyte phenotyping
B16F10 tumors were minced with scissors and dissociated with tumor digestion buffer [RPMI-1640 supplemented with 20% FBS, 0.25 mg/mL Collagenase D (Roche), and 100 KUnits/mL Type IV DNase I (Sigma-Aldrich)] using the GentleMACS Octo Dissociator (Miltenyi Biotec). Single-cell tumor suspensions were passed through a 70 μm cell strainer and washed. A total of 1 × 106 cells per well were incubated for 15 minutes at room temperature with Live-Dead Aqua and anti-mouse CD16/CD32 Fc block followed by surface staining for 30 minutes at 4°C. Cells were washed, fixed/permeabilized using FoxP3 Transcription Factor Staining Buffer Set (eBioscience), and stained intracellularly with an anti-mouse FoxP3 for 30 minutes at 4°C. Cells were washed, resuspended in HBSS, and analyzed by flow cytometry.
Statistical analysis
Statistical analyses were completed using Prism (GraphPad). Differences between the control and treated groups were analyzed by ANOVA and either Dunnett test, Tukey test, or t test as stated in figure legends. P values less than or equal to 0.05 were considered significant. For survival experiments, Kaplan–Meier survival plots were generated and three comparisons between groups were conducted using a family-wise significance level of 5%. As such, P values less than or equal to 0.016 were considered significant.
Data availability statement
The data generated by the authors in this study are available within the article and its Supplementary Data files.
Results
CD73 is expressed by T cells in the TME
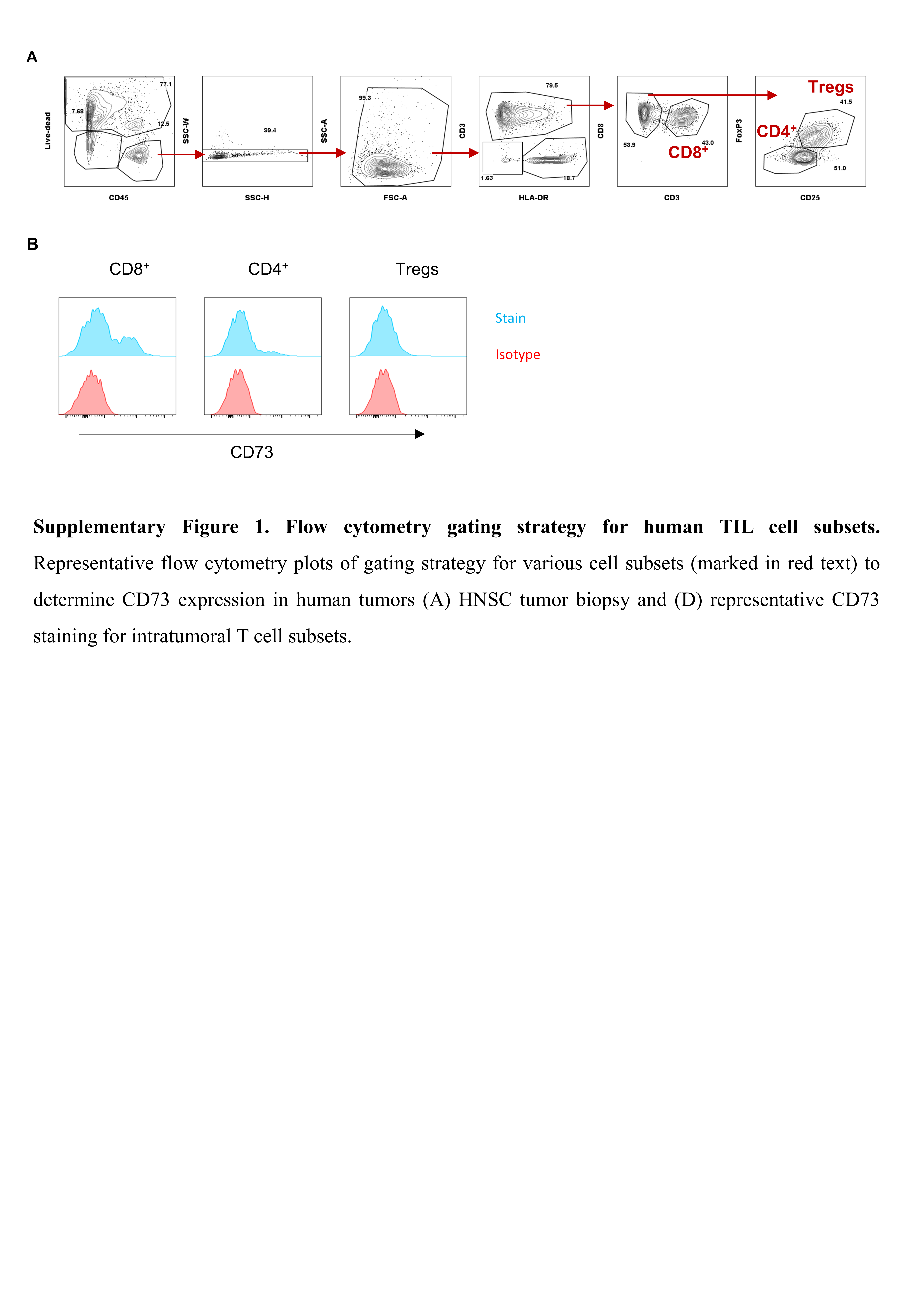
CD73 is expressed on both hematopoietic and non-hematopoietic cell types (18). At the mRNA level, CD73 expression varied quite dramatically across different indications in The Cancer Genome Atlas (TCGA; Fig. 1A). Adenocarcinomas of the colon/rectum (COAD, READ), lung (LUAD), and pancreas (PAAD) were among the solid tumors with the highest overall CD73 expression, while cancers such as prostate and squamous non–small cell lung carcinoma (NSCLC, LUSC) were among the lowest. Despite distinctions based on the median expression level, it is important to note that there was a wide distribution of CD73 expression even within the same indication. To determine expression of CD73 protein on intratumoral cell subsets, human tumor tissue samples representing the spectrum of CD73 expression observed in TCGA dataset—specifically, melanoma (SKCM), colorectal cancer (CRC), head and neck (HNSC), renal cell carcinomas (KICH; chromaphobe, KIRC; clear cell), and NSCLC (LUSC)—were dissociated and interrogated by flow cytometry (Fig. 1B; Supplementary Figs. S1A and S1B). Intratumoral CD8+ T cells exhibited a greater percent of CD73 positivity compared with CD4+ CD25+ FoxP3+ Tregs within each indication (Fig. 1B). In addition, CD73 protein expression in the CD45− subset, encompassing both stromal and cancer cells, was reflective of TCGA transcriptional data as levels varied both across and within the five cancer types examined (Fig. 1A and B). These features were confirmed and visualized by IHC for CD73 (Fig. 1C and D). In a primary rectal tumor biopsy, panCK+ colorectal cancers cells exhibited intense staining for CD73 (Fig. 1C). In contrast, a metastatic colorectal cancer tumor biopsy exhibited little to no staining of CD73 on cancer cells, yet epithelial cells and peripheral T cells in the normal adjacent tissue were positive for CD73 (Fig. 1D, yellow arrow and white arrows). Factors that may contribute to the wide range of CD73 expression between individuals and indications include the proportion of immune infiltrate as well as variations at the cellular level in stromal and/or cancer cell compartments. Collectively, bioinformatic, flow cytometry, and IHC approaches demonstrated that a number of tumor types express an appreciable amount of CD73 transcript and protein.
Figure 1.

CD73 expression is variable at the tissue and cellular level across and within cancer indications. A, Total CD73 mRNA expression in various cancer indications from TCGA. B, Quantification of frequency of CD73+ T cells and CD45− cells in dissociated tumor biopsy samples from various indications. Each data point is an individual donor. Flow cytometry data pooled from at least two replicate experiments. Data represented as mean ± range. HNSC, head and neck squamous cell carcinoma; SKCM, skin cutaneous melanoma; CRC, colorectal carcinoma; NSCLC, non–small cell lung cancer; KIRC, kidney renal clear cell carcinoma. C and D, Fluorescent IHC staining for panCK (red), CD8 (magenta), and CD73 (teal) on two independent human colorectal cancer tumors. Pockets of panCK-positive cancer cells are outlined in yellow and marked “Tumor.” Yellow arrows denote CD73-positive epithelial cells in the normal adjacent tissue, and white arrows show CD73-positive CD8 T cells. CRC, colorectal carcinoma; HNSC, head and neck squamous cell carcinoma; KIRC, kidney renal clear cell carcinoma; NSCLC, non–small cell lung cancer; SKCM, skin cutaneous melanoma.
AB680 inhibits the enzymatic activity of CD73 and prevents CD73-mediated suppression of T-cell functionality
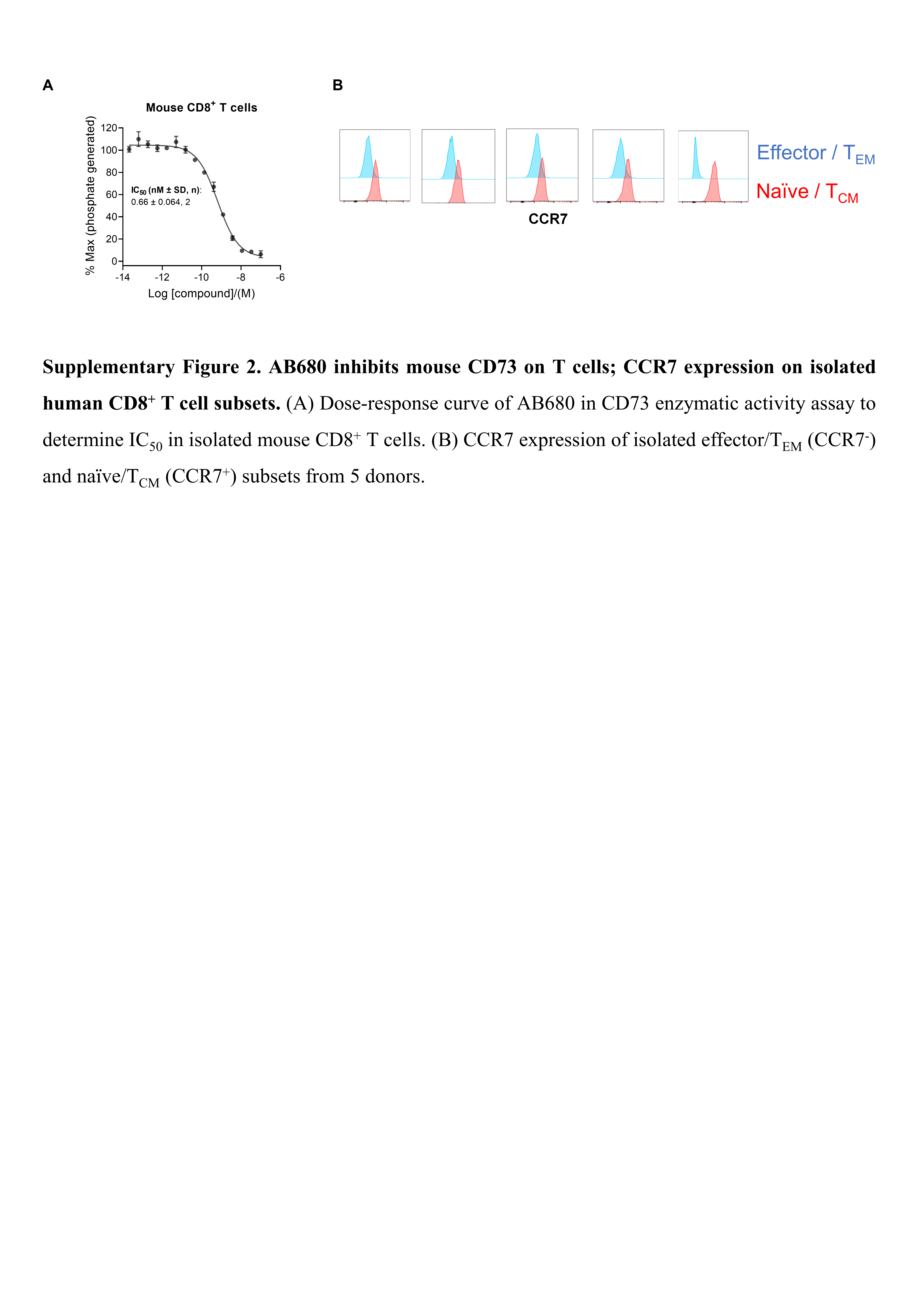
AB680 is a novel small-molecule CD73 inhibitor (39) that potently inhibited the enzymatic activity of CD73 on human and mouse CD8+ T cells in a dose-dependent manner (Fig. 2A; Supplementary Fig. S2A). In humans, both central memory (TCM)/naive (CCR7+) and effector memory (TEM)/effector (CCR7−) CD8+ T-cell subsets express CD73. When the TCR was stimulated in the presence of varying concentrations of AMP (and EHNA to prevent adenosine degradation), both subsets exhibited decreased IFNγ secretion in all donors tested (Fig. 2B; Supplementary Fig. S2B). Importantly, AB680 prevented AMP-mediated suppression of human CD4+ (Fig. 2C and D) and CD8+ (Fig. 2E) T-cell proliferation and effector cytokine secretion in all donors tested. To genetically validate that CD73 was responsible for T-cell inhibitory effects in the presence of AMP, AMP hydrolysis (a surrogate measure for the formation of adenosine) was quantified in WT and CD73−/− mouse splenic T-cell cultures. Mouse CD73−/− CD8+ T cells had a significantly impaired ability to hydrolyze AMP compared with WT CD8+ T cells (Fig. 2F, left). In addition, AMP suppressed activation (as measured by IFNγ) of WT, but not CD73−/− T cells—an effect that was reversed by AB680 (Fig. 2F, right). Taken together, these results confirm that the formation of adenosine via T cell–derived CD73 can suppress activated T cells and demonstrates the capacity of CD73 inhibition by AB680 to restore T-cell functionality in the presence of AMP.
Figure 2.

The CD73 inhibitor AB680 rescues the inhibitory effects of adenosine generation on T-cell activation and function. A, Dose–response curve of AB680 in CD73 enzymatic activity assay to determine the IC50 in isolated human CD8+ T cells. B, Left: Frequency of CD73+ on human naïve/central memory (TCM; CD8+ CCR7+) and effector/effector memory (TEM; CD8+ CCR7−) CD8+ T cells determined by flow cytometry. Right three panels: Total, naïve, or memory CD8+ T cells were activated with anti-CD2/CD3/CD28 beads in the presence of indicated concentrations of AMP (+10 μmol/L EHNA). Secreted IFNγ was measured after 48 hours. *, P < 0.05; **, P < 0.01; ***, P < 0.005; ****, P < 0.0001, Dunnett multiple comparisons test versus activation alone. Human CD4+ (C and D) and CD8+ (E) T cells were activated with anti-CD2/CD3/CD28 beads in the presence of AMP and EHNA ± AB680 (200 nmol/L). C, Proliferation was measured by cell trace violet after 96 hours and shown as representative histograms (left) and quantified for 4 donors (right). D and E, Secreted IFNγ (CD4+ and CD8+ T cells), IL2 (CD4+ T cells), and granzyme B (CD8+ T cells) was measured after 72 hours. For B–E, bar graphs with data points are individual donors pooled from multiple independent experiments. Paired t test; *, P < 0.05; **, P < 0.01; ***, P < 0.005; ****, P < 0.0001. F, Left: mouse CD8+ T cells were isolated from splenocytes from either WT or CD73−/− mice as indicated, and AMP hydrolysis was measured with AMP-Glo. ****, P < 0.0001, Sidak multiple comparisons test versus WT. Right: WT or CD73−/− CD8+ T cells were activated with anti-CD3/CD28 beads + IL2 in the presence of 50 μmol/L of AMP + 2.5 μmol/L EHNA ± AB680 (200 nmol/L). Secretion of IFNγ was measured after 96 hours to determine T-cell activation. Results were repeated in an independent experiment with another CD73 inhibitor. *, P < 0.05; **, P < 0.01, Dunnett multiple comparisons test versus AMP alone.
Pharmacologic inhibition of CD73 enables maximal T-cell activation, cytokine secretion, and cytolytic capabilities associated with antigen-specific TCR engagement
We next asked whether CD73 inhibitors could block AMP-mediated T-cell suppression in the context of antigen-specific TCR activation. Human CD4+ and CD8+ T cells engineered to express a tumor antigen-specific TCR (Fig. 3A) were cocultured with peptide-presenting K562 cells in the presence or absence of AMP and AB680. AMP significantly decreased the percent of CD25+ CD69+ activated T cells and cytokine secretion in the cocultures, and addition of AB680 in the presence of AMP restored T-cell activation and functionality (Fig. 3B). We also assessed whether CD73 inhibition could restore any AMP-mediated impairment in cancer cell killing by cytotoxic CD8+ T cells. To do this, we utilized the ovalbumin mouse model antigen system by coculturing OT-I cytotoxic T cells with a fluorescently labeled mouse lymphoma line expressing ovalbumin (EG7.OVA) and killing was measured by fluorescent image analysis. While the EG7.OVA cells did not express CD73, approximately 34% of preactivated OT-I T cells were CD73+ (Fig. 3C). OT-I T cells were able to efficiently kill EG7.OVA cells; however, T-cell cytolytic capacity was significantly diminished in the presence of AMP (Fig. 3D). Like the human system, AB680 facilitated antigen-specific T-cell functionality in the presence of AMP. These results demonstrate that the cytotoxic capacity of T cells can be suppressed through the local accumulation of adenosine via T cell–derived CD73 and that pharmacologic inhibition of CD73 may enhance the capacity of antigen-specific CD8+ T cells to kill cancer cells in the context of an adenosine-rich TME.
Figure 3.

Pharmacologic inhibition of CD73 enhances activation and cytolytic capabilities of tumor-specific T cells. A, Human CD4+ and CD8+ T cells from a healthy donor were transduced with a Neo-12 TCR and examined for CD73 expression and antigen specificity (dextramer staining) by flow cytometry. Greater than 95% of transduced cells expressed CD73, and 48.8% of CD4+ and 51.3% of CD8+ T cells expressed the transduced TCR as measured by dextramer staining. B, T cells from A were cocultured with K562 cells expressing HLA-A2 alone or HLA-A2 with the Neo12 epitope in the presence of AMP + EHNA ± AB680 for 72 hours. Frequency of CD25+ CD69+ T cells and secretion of IL2 (CD4+ T cells) and IFNγ (CD8+ T cells) were quantified. ***, P < 0.001; ****, P < 0.0001, Dunnett multiple comparisons test versus AMP alone. C, Representative flow plots displaying activated phenotype (CD44+, CD25+, CD62Lhi) of mouse OT-I CD8+ T cells and frequency of CD73+ cells on both activated OT-I CD8+ T cells and EG7.OVA tumor cells. Stained samples (red) overlaid on the isotype controls (blue). D, CTL killing assay showing time course (top) and 48 hours quantification (bottom) of tumor cell growth measured by the IncuCyte of red fluorescently labeled EG7.OVA cell confluence when cocultured with preactivated OT-I CD8+ T cells in the presence of 50 μmol/L AMP + 2.5 μmol/L EHNA ± 50 nmol/L AB680. Data presented as mean ± SEM and representative of two independent experiments. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001, Dunnett multiple comparisons test versus AMP alone.
Adenosine signaling results in a dominant suppression of T-cell activation in the presence of PD-1 blockade that can be restored by inhibiting CD73 with AB680
Using an allogeneic mixed lymphocyte reaction (MLR), we evaluated whether the suppressive effects of CD73-derived adenosine were dominant in the presence of PD-1 checkpoint blockade in primary human cells. While anti-PD-1 did facilitate transcriptional upregulation of CTLA-4 and PD-1, IFNγ, and T-bet, this effect was superseded by the addition of AMP, indicating that the suppressive effect of adenosine signaling is not reversed by PD-1 blockade. However, T-cell activation was restored by addition of AB680 (Fig. 4A and B), and IFNγ was similarly regulated at the protein level (Fig. 4C). To determine whether the suppressive effect of CD73-derived adenosine on IFNγ production could affect an ongoing T-cell response, a similar MLR was conducted with the addition of AMP and AB680 48 hours after coculture was initiated. Even after the MLR had been established, CD73-derived adenosine was able to significantly suppress IFNγ secretion, and inhibition of CD73 reversed this effect (Fig. 4D). These results indicate that the suppressive effect of AMP upon CD73+ primary human T cells not only operates in the presence of PD-1 blockade, but can override T-cell activation mediated though blockade of the PD-1–PD-L1 axis. This suggests that, in an adenosine-rich TME, single agent anti-PD-1 treatment may not be sufficient to potentiate tumor-specific T-cell responses to the fullest capability and provides a rationale for combining PD-1 blockade with AB680.
Figure 4.

Adenosine signaling results in a dominant suppression of T-cell activation in the presence of PD-1 blockade that can be restored by blocking CD73 with AB680. A–D, Human moDCs were cultured with CD4+ T cells in an allogeneic MLR for 96 hours in the presence of anti–PD-1, AMP and AB680 as indicated. A and B, Gene expression measured by quantitative RT-PCR from an MLR treated with IgG4 isotype (0.67 nmol/L), anti–PD-1 (0.67 nmol/L), AMP (100 μmol/L), and AB680 (100 nmol/L) as indicated for 72 hours. Data shown from one representative donor pair. Similar results obtained in another donor pair. C, Secretion of IFNγ was measured to determine T-cell functionality after 96 hours treatment with IgG4 isotype, anti–PD-1 (0.67 or 6.7 nmol/L), AMP (100 μmol/L) ± AB680 (100 nmol/L) as indicated. Data shown of IFNγ in one donor pair (left) and of normalized IFNγ of 16 donor pairs from three independent experiments (right). Each symbol represents a unique donor pairing. D, Secretion of IFNγ was measured after 96 hours treatment with IgG4 isotype or anti–PD-1 (0.67 or 6.7 nmol/L) ± the addition of AMP (100 μmol/L) and AB680 (100 nmol/L) at the beginning of the coculture or after 48 hours. Data shown of IFNγ in one donor pair (left) and normalized IFNγ of nine donor pairs from two independent experiments (right). Data represented as mean ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001, (A–C) Dunnett multiple comparisons test versus AMP + anti–PD-1. D, Sidak multiple comparisons test.
Anti–PD-1 treatment in combination with pharmacologic inhibition of CD73 with AB680 enhances antitumoral immunity in syngeneic B16F10 tumor model
To investigate the efficacy of AB680 alone or in combination with ICB in vivo, we utilized the B16F10 syngeneic mouse melanoma model. B16F10 cells are reported to be CD73 negative (29), assigning any effect of AB680 to cancer cell–extrinsic (immune, stromal, and soluble) CD73 inhibition and thereby supporting therapeutic applicability of AB680 to patients and indications irrespective of CD73 expression by cancer cells. We first sought to characterize cellular CD73 expression in this model and to evaluate the ability of AB680 to inhibit mouse CD73 enzymatic activity in tumors. Using a modified form of the Wachstein–Meisel enzyme histochemistry method, staining representative of AMP hydrolysis by CD73 was detected along the vasculature and surrounding the blood vessels (Fig. 5A, red arrows), corroborating findings that CD73 is highly expressed on endothelial cells (18). We were not able to assess the relative contribution of CD73-mediated AMP hydrolysis by lymphocytes, as these cells were not well detected histologically in poorly infiltrated B16F10 tumors (42). However, similarly to the human setting (Fig. 1B), CD73 was expressed on mouse intratumoral T-cell subsets as assessed by flow cytometry, albeit the relative expression levels differed as mouse melanoma Tregs had the highest median percent expression (Fig. 5B; Supplementary Fig. S3A). Both methods produced results consistent with the fact that B16F10 cells do not express CD73 (Fig. 5A and B). Finally, using a carbon radioisotope-labeled AMP substrate, CD73 enzymatic activity was measured within the total tumor homogenate to determine the capability of AB680 to block CD73 present in the TME (Fig. 5C). AB680 robustly decreased AMP hydrolysis with an IC50 of 0.704 nmol/L.
Figure 5.

Inhibition of CD73 enzymatic activity in a syngeneic B16F10 tumor model enhances antitumoral immunity. A, AMPase activity was assessed on B16F10 tumor sections using a modified form of the Wachstein–Meisel method of enzyme histochemistry. Red arrows indicate regions with active AMP hydrolysis. Data repeated in independent experiment with related CD73 inhibitor AB1421. B, Tumors from B16F10 tumor-bearing mice examined by flow cytometry for CD73 expression on B16F10 cancer cells (CD45−) as well as T-cell subsets. n = 11, mean ± SEM. C, Dose-dependent inhibition of CD73 enzymatic activity by AB680 was quantified in B16F10 tumor homogenates using 13C5-AMP hydrolysis method. Data presented as mean ± SD. D, Top: tumor growth curve showing C57Bl6/J mice implanted subcutaneously with B16F10 cells and subsequently treated with vehicle or AB680 at 10 mg/kg every day. Data representative of three independent experiments, *, P < 0.05, mixed effects analysis, Sidak multiple comparisons test for each timepoint. Bottom: percent of CD8+ T cells and the CD8+ T cell to Treg ratio within tumors from each treatment group were determined by flow cytometry and plotted. n = 9–15, mean ± SEM, *, P < 0.05, Student t test. E, Left: tumor growth and survival curve showing B16F10 tumor-bearing mice treated with anti–PD-1 (2.5 mg/kg) and AB680 (10 mg/kg) as indicated. Data representative of three independent experiments, ns = not significant, ***, P < 0.001. Tumor growth curve: mixed effects analysis, Tukey multiple comparisons test. Kaplan–Meier survival plot: multiple comparisons conducted using family-wise significance level of 5%. Right: percent of CD8+ T cells and the CD8+ T cell to Treg ratio within tumors from each treatment group were determined by flow cytometry and plotted. n = 9–15 mice, mean ± SEM. Tumor-infiltrating lyphocyte data repeated in independent experiment, *, P < 0.05; **, P < 0.01; ***, P < 0.001, Sidak multiple comparisons test, vehicle versus anti–PD-1, anti–PD-1 versus anti–PD-1/AB680, vehicle versus anti–PD-1/AB680.
Next, mice injected in the flank with B16F10 cells were given 10 mg/kg AB680 once daily to assess the effect of blocking CD73 upon tumor growth and composition. AB680-treated mice exhibited a statistically significant delay in tumor growth that was accompanied by an increase in tumor-infiltrating CD8+ T cells and CD8+ T cell to immunosuppressive Treg ratio (CD8+:Treg; Fig. 5D). Similar B16F10 tumor growth inhibition was observed in RAG−/− mice reconstituted with CD73−/− T cells (Supplementary Figs. S3B–S3F), suggesting that the efficacy observed with AB680 was a result of inhibiting the suppressive effects of T cell–derived adenosine and are also consistent with findings from CD73-deficient bone marrow reconstitution studies (18, 21, 22).
Finally, we assessed whether treating mice with established B16F10 tumors with AB680 and anti-PD-1 resulted in greater antitumor efficacy than anti-PD-1 alone. Whereas anti-PD-1 exhibited minimal single-agent efficacy, mice treated with a combination of anti-PD-1 and AB680 had significantly decreased tumor burden and increased survival compared with mice treated with vehicle alone (Fig. 5E). Similar to treatment with AB680 alone (Fig. 5D), the decrease in tumor burden observed with the combination treatment was accompanied by a significant increase in the frequency of tumor-infiltrating CD8+ T cells, and the addition of AB680 increased the CD8+:Treg ratio significantly more than anti-PD-1 alone (Fig. 5E). Collectively, these in situ, ex vivo, and in vivo data demonstrate that AB680 blocks CD73 enzymatic activity in the TME, and this activity correlates with decreased tumor burden and increased CD8+ T-cell infiltration in both single agent and anti-PD-1 combination settings in a mouse melanoma model.
Discussion
In this article, we tested the effect of inhibiting CD73 activity with AB680 in a range of human and mouse in vitro T-cell assays as well as in vivo in a mouse syngeneic tumor model. With a subnanomolar IC50 in both mouse and human T cells, AB680 potently blocks CD73 enzymatic activity, and the data presented here complement related preclinical studies where both AB680 (43) and a significantly less potent CD73 small-molecule inhibitor (44), adenosine 5′-(α,β-methylene)diphosphate were shown to promote antitumor activity (18, 33). Furthermore, we demonstrated the restorative effect of CD73 inhibition by AB680 on human and mouse T-cell function, which is consistent with other preclinical studies where an anti-CD73 mAb and/or genetic mutants were utilized to limit adenosine-mediated inhibitory effects on T-cell activity, thus validating CD73-mediated biology (27, 30). Extending these findings to the activation and cytotoxic potential of tumor-specific T cells, we also demonstrated the importance of inhibiting CD73 enzymatic activity locally in the TME to enhance tumoricidal T-cell responses. Finally, potentiation of T-cell responses was corroborated in vivo, where treatment of B16F10 tumor-bearing mice with AB680 drove a compositional change to a pro-immunogenic TME, chiefly characterized by decreased tumor burden and increases in infiltrating CD8+ T cells. Thus, AB680 has the potential to be a first-in-class small-molecule CD73 inhibitor therapeutic for cancer (35).
Our data also highlight potential implications for assessing indications in which patients would benefit from CD73 inhibition. Many preclinical studies have demonstrated the efficacy of CD73 inhibition in mouse models where the cancer cells overexpress CD73 (13, 20, 27). In this setting, it is difficult to distinguish the cancer cell–intrinsic versus cancer cell–extrinsic effect of CD73 inhibition. Like others that have established the role of CD73 in immunosuppression (27, 28, 32), we took the opportunity to exploit the fact that many commonly used murine cancer cell lines, exemplified by B16F10 (29, 45), exhibit low or no CD73. While the cancer cells lack CD73, surrounding vasculature and T cells express CD73 and are capable of synthesizing adenosine. Thus, we were able to isolate efficacy observed with AB680 to soluble, immune, and/or stromal CD73 enzymatic activity. Addressing this question experimentally was important, given the number of human tumors with CD73 expression restricted to stromal and/or immune cells. Data leveraging mouse models presented here and elsewhere (26) highlight the importance of cancer cell–extrinsic CD73 inhibition in antitumor immunity. Given that the direct effect of AB680 upon T cells is consistent between mouse and human, our data support broadening the therapeutic applicability of AB680 from patients with high CD73-expressing tumors to patients and indications with peripheral and intratumoral T-cell infiltrate, irrespective of cancer cell–intrinsic CD73 expression.
Another important facet to the clinical application of new immunotherapeutic agents is how these agents can serve to complement or enhance existing immunogenic or immuno-oncology drugs, namely ICB. To further characterize the biological effects of adenosine in the context of ICB, we used in vitro systems to evaluate the activation and effector function of T cells in an adenosine-rich environment. We found that, in the presence of anti-PD-1, or even after TCR signaling had been initiated, human T-cell function was appreciably reduced in a CD73-dependent manner. This result suggests that the potential therapeutic benefit of PD-1 blockade may be restricted in the context of the levels of adenosine that have been reported within the TME (26) and provides one explanation for resistance to ICB treatment. Consistent with previous publications reporting enhanced antitumor efficacy when ICB were combined with CD73-targeting agents (27, 28, 33, 46), combining PD-1 blockade with AB680 overcame suppressive effects of adenosine upon human T cells in vitro and enhanced antitumor efficacy in vivo.
Additional facets to contemplate when considering the therapeutic applicability of CD73 inhibition are combinations with therapeutics that can either alter the expression of CD73 or induce immunogenic cell death associated with the release of ATP (the penultimate substrate for CD73). It has been demonstrated that CD39 and CD73 expression is significantly upregulated in human breast cancer, melanoma, and leukemia cell lines upon treatment with doxorubicin, oxaliplatin, and cisplatin chemotherapies (13, 47). In vivo, combination of doxorubicin with a CD73-neutralizing antibody resulted in a T cell–dependent reduction in tumor growth (13), suggesting that CD73 may play a role in resistance to chemotherapies known to induce immunogenic cell death. Radiotherapy has also been shown to upregulate expression of CD73 and promote immunogenicity via ATP release, and likewise has also been shown to improve antitumor efficacy when used in combination with anti-CD73 (48). Furthermore, maintaining an immunostimulatory ATP pool by blocking CD39 may also enhance antitumor efficacy; indeed, a preclinical study has demonstrated that combination of anti-CD73 with anti-CD39 promotes T-cell activation (30).
Collectively, these data support the hypothesis that inhibition of CD73 with AB680 may benefit patients with cancer in several clinical settings and provide rationale for exploring combination of adenosine path inhibitors with standard-of-care ICB, chemotherapy, and radiotherapy regimens.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The CD73 transcriptional data across tumor types published here are based upon data generated by TCGA Research Network: https://www.cancer.gov/tcga. We would also like to acknowledge and thank Bhamini Purandare, Michael Bethune, and colleagues at PACT Pharma for generously providing Neo12-TCR-transduced human T cells and K562 cells engineered to express both HLA-A2 and the Neo12-epitope for the human tumor-specific T-cell assay.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/).
Authors' Disclosures
D. Piovesan reports personal fees from Arcus Biosciences and other support from Arcus Biosciences during the conduct of the study; personal fees from Arcus Biosciences and other support from Arcus Biosciences outside the submitted work. J.B.L. Tan reports personal fees from Arcus Biosciences and other support from Arcus Biosciences outside the submitted work; in addition, J.B.L. Tan has a patent for WO2020185859A1 pending. A. Becker reports personal fees from Arcus Biosciences and other support from Arcus Biosciences during the conduct of the study; personal fees from Arcus Biosciences and other support from Arcus Biosciences outside the submitted work. J. Banuelos reports personal fees from Arcus Biosciences and other support from Arcus Biosciences during the conduct of the study; personal fees from Arcus Biosciences and other support from Arcus Biosciences outside the submitted work. N. Narasappa reports personal fees from Arcus Biosciences and other support from Arcus Biosciences during the conduct of the study; personal fees from Arcus Biosciences and other support from Arcus Biosciences outside the submitted work. D. DiRenzo reports personal fees from Arcus Biosciences and other support from Arcus Biosciences during the conduct of the study; in addition, D. DiRenzo has a patent for WO 2020/205527 pending. K. Zhang reports personal fees from Arcus Biosciences and other support from Arcus Biosciences during the conduct of the study; personal fees from Arcus Biosciences and other support from Arcus Biosciences outside the submitted work. A. Chen reports personal fees from Arcus Biosciences and other support from Arcus Biosciences during the conduct of the study; personal fees from Arcus Biosciences and other support from Arcus Biosciences outside the submitted work. E. Ginn reports personal fees from Arcus Biosciences and other support from Arcus Biosciences during the conduct of the study; personal fees from Arcus Biosciences and other support from Arcus Biosciences outside the submitted work. A.R. Udyavar reports personal fees from Arcus Biosciences and other support from Arcus Biosciences during the conduct of the study; personal fees from Arcus Biosciences and other support from Arcus Biosciences outside the submitted work; in addition, A.R. Udyavar has a patent for WO 2020/205527 A1 pending. F. Yin reports personal fees from Arcus Biosciences and other support from Arcus Biosciences during the conduct of the study; personal fees from Arcus Biosciences and other support from Arcus Biosciences outside the submitted work. S.L. Paprcka reports personal fees from Arcus Biosciences and other support from Arcus Biosciences during the conduct of the study; personal fees from Arcus Biosciences and other support from Arcus Biosciences outside the submitted work. T.W. Park reports personal fees from Arcus Biosciences and other support from Arcus Biosciences during the conduct of the study; personal fees from Arcus Biosciences and other support from Arcus Biosciences outside the submitted work. N. Kimura reports personal fees from Arcus Biosciences and other support from Arcus Biosciences during the conduct of the study; personal fees from Arcus Biosciences and other support from Arcus Biosciences outside the submitted work. J. Kalisiak reports personal fees from Arcus Biosciences and other support from Arcus Biosciences during the conduct of the study; personal fees from Arcus Biosciences and other support from Arcus Biosciences outside the submitted work; in addition, J. Kalisiak has a patent for US10981944 issued, a patent for US11058704 issued, a patent for US20200062797 pending, and a patent for US20200405629 pending. S.W. Young reports personal fees from Arcus Biosciences and other support from Arcus Biosciences during the conduct of the study; personal fees from Arcus Biosciences and other support from Arcus Biosciences outside the submitted work; in addition, S.W. Young has a patent for WO 2020/205527 A1 pending and a patent for WO 2020/185859 A1 pending. J.P. Powers reports other support from Arcus Biosciences and other support from Arcus Bioscienes during the conduct of the study; other support from Arcus Biosciences and Arcus Biosciences outside the submitted work; in addition, J.P. Powers has a patent 11267845 issued to Arcus Biosciences, a patent 11001603 issued to Arcus Biosciences, and a patent 10981944 issued to Arcus Biosciences. U. Schindler reports other support from Arcus Biosciences during the conduct of the study; other support from Arcus Biosciences outside the submitted work. K.E. Sivick reports personal fees from Arcus Biosciences and other support from Arcus Biosciences during the conduct of the study; personal fees from Arcus Biosciences and other support from Arcus Biosciences outside the submitted work. M.J. Walters reports personal fees from Arcus Biosciences and other support from Arcus Biosciences during the conduct of the study; personal fees from Arcus Biosciences and other support from Arcus Biosciences outside the submitted work; in addition, M.J. Walters has a patent for PCT/US2020/025242 pending. No disclosures were reported by the other authors.
Authors' Contributions
D. Piovesan: Conceptualization, data curation, formal analysis, visualization, writing–original draft, writing–review and editing. J.B.L. Tan: Conceptualization, formal analysis, supervision, investigation, visualization, project administration. A. Becker: Formal analysis, investigation. J. Banuelos: Conceptualization, investigation. N. Narasappa: Formal analysis, investigation. D. DiRenzo: Investigation. K. Zhang: Investigation, visualization. A. Chen: Investigation. E. Ginn: Investigation. A.R. Udyavar: Conceptualization, software, formal analysis. F. Yin: Formal analysis, investigation. S.L. Paprcka: Investigation. B. Purandare: Methodology, providing reagents. T.W. Park: Investigation. N. Kimura: Investigation. J. Kalisiak: Investigation. S.W. Young: Resources, supervision, project administration, writing–review and editing. J.P. Powers: Project administration, writing–review and editing. U. Schindler: Supervision, project administration, writing–review and editing. K.E. Sivick: Conceptualization, resources, data curation, supervision, visualization, writing–original draft, writing–review and editing. M.J. Walters: Conceptualization, resources, data curation, supervision, project administration, writing–review and editing.
References
- 1. Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 2001;414:916–20. [DOI] [PubMed] [Google Scholar]
- 2. Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med 2012;367:2322–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MKK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U S A 2006;103:13132–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vigano S, Alatzoglou D, Irving M, Ménétrier-Caux C, Caux C, Romero P, et al. Targeting adenosine in cancer immunotherapy to enhance T-cell function. Front Immunol 2019;10:925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang S, Apasov S, Koshiba M, Sitkovsky M. Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood 1997;90:1600–10. [PubMed] [Google Scholar]
- 6. Yegutkin GG. Nucleotide- and nucleoside-converting ectoenzymes: important modulators of purinergic signalling cascade. Biochim Biophys Acta 2008;1783:673–94. [DOI] [PubMed] [Google Scholar]
- 7. Lennon PF, Taylor CT, Stahl GL, Colgan SP. Neutrophil-derived 5’-adenosine monophosphate promotes endothelial barrier function via CD73-mediated conversion to adenosine and endothelial A(2B) receptor activation. J Exp Med 1998;188:1433–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thompson LF, Eltzschig HK, Ibla JC, Van De Wiele CJ, Resta R, Morote-Garcia JC, et al. Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med 2004;200:1395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Adzic M, Nedeljkovic N. Unveiling the role of Ecto-5’-nucleotidase/CD73 in astrocyte migration by using pharmacological tools. Front Pharmacol 2018;9:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Takedachi M, Qu D, Ebisuno Y, Oohara H, Joachims ML, McGee ST, et al. CD73-generated adenosine restricts lymphocyte migration into draining lymph nodes. J Immunol 2008;180:6288–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhao J, Soto LMS, Wang H, Katz MH, Prakash LR, Kim M, et al. Overexpression of CD73 in pancreatic ductal adenocarcinoma is associated with immunosuppressive tumor microenvironment and poor survival. Pancreatology 2021;21:942–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Messaoudi N, Cousineau I, Arslanian E, Henault D, Stephen D, Vandenbroucke-Menu F, et al. Prognostic value of CD73 expression in resected colorectal cancer liver metastasis. 2020;9:1746138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Loi S, Pommey S, Haibe-Kains B, Beavis PA, Darcy PK, Smyth MJ, et al. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc Natl Acad Sci U S A 2013;110:11091–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jin D, Fan J, Wang L, Thompson LF, Liu A, Daniel BJ, et al. CD73 on tumor cells impairs anti-tumor T cell responses: a novel mechanism of tumor-induced immune suppression. Cancer Res 2010;70:2245–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li J, Wang L, Chen X, Li L, Li Y, Ping Y, et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-β-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology 2017;6:e1320011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goswami S, Walle T, Cornish AE, Basu S, Anandhan S, Fernandez I, et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat Med 2020;26:39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yu M, Guo G, Huang L, Deng L, Chang CS, Achyut BR, et al. CD73 on cancer-associated fibroblasts enhanced by the A2B-mediated feedforward circuit enforces an immune checkpoint. Nat Comm 2020;11:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang L, Fan J, Thompson LF, Zhang Y, Shin T, Curiel TJ, et al. CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J Clin Invest 2011;121:2371–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chalmin F, Mignot G, Bruchard M, Chevriaux A, Végran F, Hichami A, et al. Stat3 and Gfi-1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity 2012;36:362–73. [DOI] [PubMed] [Google Scholar]
- 20. Armstrong JM, Chen JF, Schwarzschild MA, Apasov S, Smith PT, Caldwell C, et al. Gene dose effect reveals no Gs-coupled A2A adenosine receptor reserve in murine T-lymphocytes: studies of cells from A2A-receptor-gene-deficient mice. Biochem J 2001;354:123–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 2007;204:1257–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol 2017;18:1332–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jarvis LB, Rainbow DB, Coppard V, Howlett SK, Georgieva Z, Davies JL, et al. Therapeutically expanded human regulatory T-cells are super-suppressive due to HIF1A induced expression of CD73. Commun Biol 2021;4:1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mandapathil M, Hilldorfer B, Szczepanski MJ, Czystowska M, Szajnik M, Ren J, et al. Generation and accumulation of immunosuppressive adenosine by human CD4+CD25highFOXP3+ regulatory T cells. J Biol Chem 2010;285:7176–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gourdin N, Bossennec M, Rodriguez C, Vigano S, Machon C, Jandus C, et al. Autocrine adenosine regulates tumor polyfunctional CD73+CD4+ effector T cells devoid of immune checkpoints. Cancer Res 2018;78:3604–18. [DOI] [PubMed] [Google Scholar]
- 26. Allard B, Allard D, Buisseret L, Stagg J. The adenosine pathway in immuno-oncology. Nat Rev Clin Oncol 2020;17:611–29. [DOI] [PubMed] [Google Scholar]
- 27. Allard B, Pommey S, Smyth MJ, Stagg J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin Cancer Res 2013;19:5626–35. [DOI] [PubMed] [Google Scholar]
- 28. Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, McGlinchey KA, et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology 2016;5:e1208875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yegutkin GG, Marttila-Ichihara F, Karikoski M, Niemelä J, Laurila JP, Elima K, et al. Altered purinergic signaling in CD73-deficient mice inhibits tumor progression. Eur J Immunol 2011;41:1231–41. [DOI] [PubMed] [Google Scholar]
- 30. Perrot I, Michaud H-A, Giraudon-Paoli M, Vivier E, Paturel C, Bonnefoy Correspondence N. Blocking antibodies targeting the CD39/CD73 immunosuppressive pathway unleash immune responses in combination cancer therapies. Cell Rep 2019;27:2411–25. [DOI] [PubMed] [Google Scholar]
- 31. Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol 2018;8:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Beavis PA, Milenkovski N, Henderson MA, John LB, Allard B, Loi S, et al. Adenosine receptor 2A blockade increases the efficacy of anti-PD-1 through enhanced antitumor T-cell responses. Cancer Immunol Res 2015;3:506–17. [DOI] [PubMed] [Google Scholar]
- 33. Iannone R, Miele L, Maiolino P, Pinto A, Morello S. Adenosine limits the therapeutic effectiveness of anti-CTLA4 mAb in a mouse melanoma model. Am J Cancer Res 2014;4:172–81. [PMC free article] [PubMed] [Google Scholar]
- 34. Fong L, Hotson A, Powderly JD, Sznol M, Heist RS, Choueiri TK, et al. Adenosine 2A receptor blockade as an immunotherapy for treatment-refractory renal cell cancer. Cancer Discov 2020;10:40–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bowman CE, Da Silva RG, Pham A, Young SW. An exceptionally potent inhibitor of human CD73. Biochemistry 2019;58:3331–4. [DOI] [PubMed] [Google Scholar]
- 36. Lawson KV, Kalisiak J, Lindsey EA, Newcomb E, Leleti MR, Debien L, et al. Discovery of AB680 – a potent and selective inhibitor of CD73. J Med Chem 2020;63:1148–68. [DOI] [PubMed] [Google Scholar]
- 37. Manji GA, Wainberg ZA, Krishnan K, Giafis N, Udyavar A, Quah CS, et al. ARC-8: Phase I/Ib study to evaluate safety and tolerability of AB680 + chemotherapy + zimberelimab (AB122) in patients with treatment-naive metastatic pancreatic adenocarcinoma (mPDAC). J Clin Oncol 2020;39(3 suppl):404.
- 38. Li G, Bethune MT, Wong S, Joglekar AV, Leonard MT, Wang JK, et al. T cell antigen discovery via trogocytosis. Nat Methods 2019;16:183–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Beatty JW, Lindsey EA, Thomas-Tran R, Debien L, Mandal D, Jeffrey JL, et al. Discovery of potent and selective non-nucleotide small molecule inhibitors of CD73. J Med Chem 2020;63:3935–55. [DOI] [PubMed] [Google Scholar]
- 40. Jacoby K, Lu W, Nguyen D, Sennino B, Conroy A, Purandare B, et al. Non-viral genome engineering method allows highly efficient, single-step removal and precise insertion of multiple large genes [abstract]. In: Proceedings of the Annual Meeting of the American Association for Cancer Research 2020; 2020 Apr 27–28 and Jun 22–24. Philadelphia (PA): AACR; Cancer Res 2020;80(16 Suppl):Abstract nr2192. [Google Scholar]
- 41. Wachstein M, Meisel E. Histochemistry of hepatic phosphatases of a physiologic pH; with special reference to the demonstration of bile canaliculi. Am J Clin Pathol 1957;27:13–23. [DOI] [PubMed] [Google Scholar]
- 42. Lechner MG, Karimi SS, Barry-Holson K, Angell TE, Murphy KA, Church CH, et al. Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J Immunother 2013;36:477–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim M, Min YK, Jang J, Park H, Lee S, Lee CH. Single-cell RNA sequencing reveals distinct cellular factors for response to immunotherapy targeting CD73 and PD-1 in colorectal cancer. J Immunother Cancer 2021;9:e002503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sowa NA, Voss MK, Zylka MJ. Recombinant ecto-5’-nucleotidase (CD73) has long lasting antinociceptive effects that are dependent on adenosine A1receptor activation. Mol Pain 2010;6:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Burghoff S, Gong X, Viethen C, Jacoby C, Flögel U, Bongardt S, et al. Growth and metastasis of B16-F10 melanoma cells is not critically dependent on host CD73 expression in mice. BMC Cancer 2014;14:898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen S, Fan J, Zhang M, Qin L, Dominguez D, Long A, et al. CD73 expression on effector T cells sustained by TGF-β facilitates tumor resistance to anti-4-1BB/CD137 therapy. Nat Commun 2019;10:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Samanta D, Park Y, Ni X, Li H, Zahnow CA, Gabrielson E, et al. Chemotherapy induces enrichment of CD47+/CD73+/PDL1+ immune evasive triple-negative breast cancer cells. Proc Natl Acad Sci U S A 2018;115:E1239–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. De Leve S, Wirsdörfer F, Jendrossek V. Targeting the immunomodulatory CD73/adenosine system to improve the therapeutic gain of radiotherapy. Front Immunol 2019;10:698. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated by the authors in this study are available within the article and its Supplementary Data files.