

Summary
The factors that influence the atherosclerotic disease process in high-risk individuals remain poorly understood. Here, we used a combination of vascular imaging, risk factor assessment, and biomarkers to identify factors associated with 3-year change in carotid disease severity in a cohort of high-risk subjects treated with preventive therapy (n = 865). The results show that changes in intima-media thickness (IMT) are most pronounced in the carotid bulb. Progression of bulb IMT demonstrates independent associations with baseline bulb IMT, the plaque gray scale median (GSM), and the plasma level of platelet-derived growth factor (PDGF) (standardized β-coefficients and 95% confidence interval [CI] −0.14 [−0.06 to −0.02] p = 0.001, 0.15 [0.02–0.07] p = 0.001, and 0.20 [0.03–0.07] p < 0.001, respectively). Plasma PDGF correlates with the plaque GSM (0.23 [0.15–0.29] p < 0.001). These observations provide insight into the atherosclerotic process in high-risk subjects by showing that progression primarily occurs in fibrotic plaques and is associated with increased levels of PDGF.
Keywords: atherosclerosis, biomarkers, carotid ultrasound, intima-media thickness, risk factors, type 2 diabetes
Graphical abstract
Highlights
-
•
High age, male gender, and smoking increases risk of carotid disease progression
-
•
Plaques that progress are more echogenic, indicating an increased degree of fibrosis
-
•
Progression is associated with high plasma levels of pro-fibrotic growth factors
-
•
Regression is most common in large, less fibrotic plaques
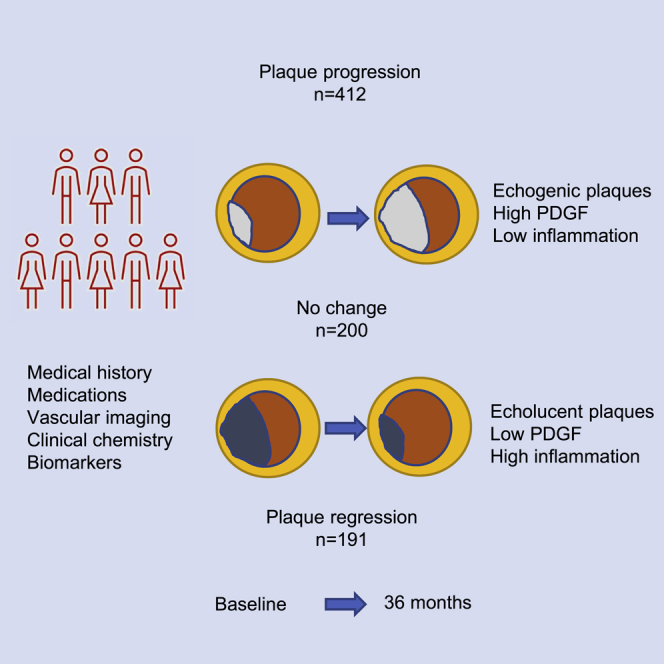
In a cohort with high-risk subjects, Khan et al. show that progression of atherosclerosis is associated with presence of more fibrotic plaques and higher circulating levels of pro-fibrotic growth factors. Regression of atherosclerosis is more common in subjects with large, less fibrotic plaques and is associated with signs of inflammation.
Introduction
Atherosclerotic plaque formation is caused by cardiovascular risk factors, such as hypercholesterolemia, hypertension, smoking, and diabetes, with intimal accumulation and oxidation of low-density lipoprotein (LDL)-derived lipids playing a particularly detrimental role.1, 2, 3 Local activation of inflammation and tissue repair responses with time lead to the formation of atherosclerotic plaques generally consisting of a core of extracellular lipids and cellular debris covered by a fibrous cap of smooth muscle cells. Plaque destabilization through degradation of the fibrous cap may result in plaque rupture, giving rise to acute cardiovascular events, such as myocardial infarction (MI) and stroke.4 Both primary and secondary prevention of cardiovascular disease (CVD) is based on treating atherosclerosis through risk-factor intervention. This approach has been proven successful and explains much of the reduced cardiovascular mortality observed in many developed countries during recent decades.5 However, subjects with established CVD remain at an increased risk of recurrent events despite receiving state-of-the-art preventive treatment. One possible explanation to this could be that alternative factors affect the atherosclerotic disease process in subjects receiving current risk factor intervention therapy.6, 7, 8 In line with this, there is evidence of altered disease characteristics during recent years with a shift from larger ST segment elevation MI (STEMI) towards non-STEMI.9 The latter is frequently caused by plaque erosions occurring on top of an intact fibrous cap rather than plaque rupture.10 Moreover, a study analyzing more than 1,500 atherosclerotic plaques removed at carotid surgery between 2002 and 2011 revealed a trend towards more stable plaques with less lipids and inflammation over time.11
Observational studies in subjects with type 2 diabetes (T2D) also point to the existence of disease mechanisms not targeted by current treatments. Although improved risk factor intervention, including statin treatment, has markedly reduced the incidence of CVD in T2D over the last few decades, the risk still remains 2-fold higher than in the general population.12 Impaired vascular repair has been suggested as one possible explanation to the development of macrovascular complications in T2D.13 Support for a protective role of vascular growth factors in CVD has recently also come from studies demonstrating that subjects with an increased ability to express the endothelial mitogen placental growth factor (PlGF) in response to metabolic stress have a lower risk of MI and stroke.14,15
Here, we used the Surrogate Markers for Micro- and Macro-vascular Hard Endpoints for Innovative Diabetes Tools Vascular Imaging Prediction (SUMMIT VIP) study to identify factors associated with progression of carotid atherosclerosis during a 3-year period in a cohort of subjects receiving CVD preventive therapy. The SUMMIT VIP study was originally designed to identify markers of CVD risk in subjects with T2D and, by comparing with subjects without diabetes, determine to what extent they were specific for T2D.16 The baseline investigation of SUMMIT-VIP included analyses of intima-media thickness (IMT) in the common carotid artery (CCA) and the carotid bulb, gray scale median (GSM) analysis of the largest carotid plaque, arterial stiffness by measuring pulse wave velocity (PWV), endothelial function as assessed by the reactive hyperemia index (RHI) by Endo-PAT, and determination of a number of exploratory biomarkers reflecting the atherosclerotic disease process.
Results
Change in carotid IMT in subjects with and without T2D
Out of the original 1,500 participants in the SUMMIT-VIP study, 865 were included in the present follow-up imaging study. Reasons for not participating included death during follow-up (n = 46), lost to follow-up (n = 45), and declined to participate (n = 539; Figure S1). Five subjects that completed the follow-up examination were excluded from the present study because of carotid surgery during the study period. The baseline clinical characteristics of the study participants and non-participants are shown in Table 1. Almost two-thirds of the participants had T2D, and close to 40% had prevalent CVD. Non-participants were younger, more often males and smokers, and had higher BMI and blood pressure and lower high-density lipoprotein (HDL). When comparing the 3-year follow-up period in terms of mm IMT change, the variation appeared more pronounced in the carotid bulb than in the CCA (Figures 1A and 1B). However, this difference was less pronounced when comparing the relative change from baseline (Figure 1C). There was no significant difference in CCA or bulb IMT change between subjects with and without T2D (0.016 ± 0.121 versus 0.018 ± 0.111 mm and 0.056 ± 0.278 versus 0.051 ± 0.255 mm, respectively). Moreover, there was also no significant difference in CCA or bulb IMT change between subjects with and without prevalent CVD at baseline (0.019 ± 0.119 versus 0.015 ± 0.116 mm and 0.061 ± 0.317 versus 0.050 ± 0.237 mm, respectively). There was only a weak correlation between IMT changes in the CCA and in the bulb (r = 0.17; p < 0.001). The latter observation suggests that different factors drive IMT change in the CCA and the carotid bulb.
Table 1.
Baseline clinical characteristics of participants and non-participants in the follow-up imaging study
| Participants (n = 865) | Non-participants (n = 630) | p | |
|---|---|---|---|
| Age (years) | 68.1 ± 8.4 | 66.5 ± 9.7 | 0.001 |
| Gender (% males) | 63.2 | 74.4 | 0.005 |
| Ethnicity (n) white, Indian-Asian, Afro-Caribbean | 860 3 2 | 623 2 2 | |
| Current smokers (%) | 8.0 | 13.8 | 0.001 |
| T2D (%) | 65.4 | 66.5 | 0.54 |
| Prevalent CVD (%) | 40.0 | 58.0 | <0.001 |
| BMI (kg/m−2) | 29.1 ± 4.9 | 29.8 ± 5.2 | 0.006 |
| Medications | |||
| Insulin (%) | 16.6 | 14.5 | 0.32 |
| Statin (%) | 65.8 | 67.9 | 0.30 |
| ACE inhibitors (%) | 36.9 | 40.5 | 0.13 |
| Metformin (%) | 45.9 | 44.9 | 0.78 |
| Betablockers (%) | 31.6 | 38.1 | 0.009 |
| Metabolic factors | |||
| HbA1c (mmol/mmol) | 50.8 ± 14.0 | 52.1 ± 14.9 | 0.08 |
| LDL (mmol/L) | 2.43 ± 0.93 | 2.42 ± 0.90 | 0.76 |
| HDL (mmol/L) | 1.36 ± 0.42 | 1.28 ± 0.37 | <0.001 |
| Triglycerides (mmol/L) | 1.30 (0.94–1.87) | 1.35 (0.85–1.87) | 0.06 |
| Blood pressure | |||
| Systolic (mmHg) | 135 ± 17 | 137 ± 19 | 0.006 |
| Diastolic (mmHg) | 76 ± 9 | 78 ± 10 | <0.001 |
| Renal function | |||
| eGFR (mL/min−1 per 1.73 m2) | 80.1 ± 19.2 | 82.0 ± 22.1 | 0.12 |
Variables with normal distribution are shown as mean ± SD and skewed variables as median and interquartile range. Differences between means of normally distributed continuous variables were assessed with independent sample t tests and between skewed variables with the Mann-Whitney U-test. χ2 test was used for categorical variables. eGFR, estimated glomerular filtration rate.
Figure 1.

Changes in carotid artery IMT
(A) Ultrasound image of a carotid artery. Bar indicates the border between the CCA and the bulb.
(B and C) Distribution of IMT changes in the carotid bulb and the CCA expressed in (B) mm change and (C) percent change during the study period. Values represent the means of the IMT in the left and right carotid arteries.
(D) Correlations between IMT change in the CCA and the bulb. Correlation was calculated using the Spearman rank test.
(E) Representative ultrasound images of an echolucent and an echogenic plaque. The arrow indicates the luminal surface of the echolucent plaque. Scale bars in (A) and (E) are 5 mm.
Relationships between conventional risk factors and medications with change in carotid IMT
Using the change in IMT as a continuous variable, age, male gender, smoking, and LDL were all positively associated with change in bulb IMT in subjects with T2D (Table 2). Since both progression and regression of IMT had occurred during the study period (Figures 1B and 1C), we next categorized subjects into those with regression, no change, or progression, using a cutoff of ±0.05 mm to define no change. Using these categorized variables, age and male gender were associated with progression of bulb IMT (Table 2). The associations between conventional risk factors and IMT change were weaker in the CCA than in the bulb, with significant correlations observed only for BMI and HDL (Table 2).
Table 2.
Relationships between baseline clinical characteristics with change in bulb IMT
| Mean bulb IMT |
|||||
|---|---|---|---|---|---|
| R | p | Regression n = 191 | No change n = 200 | Progression n = 412 | |
| Age (years) | 0.11 | 0.001 | 67.9 ± 8.3 | 66.6 ± 8.8 | 69.0 ± 8.3a |
| Male gender (%) | 0.14 | 0.0001 | 56.5 | 58.0 | 70.1a |
| Current smokers (%) | 0.11 | 0.02 | 6.3 | 9.0 | 8.6 |
| BMI (kg/m−2) | 0.01 | 0.84 | 29.5 ± 5.0b | 28.2 ± 4.7 | 28.9 ± 4.6 |
| Metabolic factors | |||||
| HbA1c (mmol/mmol) | 0.01 | 0.85 | 48.0 (41.0–60.3) | 47.0 (39.0–57.0) | 48.0 (40.0–60.0) |
| LDL (mmol/L) | 0.10 | 0.007 | 2.28 ± 0.96 | 2.48 ± 0.94 | 2.52 ± 0.90 |
| HDL (mmol/L) | −0.01 | 0.83 | 1.35 ± 0.41 | 1.41 ± 0.45 | 1.37 ± 0.41 |
| TG (mmol/L) | −0.04 | 0.26 | 1.37 (0.97–1.95) | 1.28 (0.88–1.93) | 1.28 (0.96–1.69) |
| Blood pressure | |||||
| Systolic (mmHg) | 0.04 | 0.32 | 137 ± 17a | 131 ± 17 | 136 ± 17a |
| Diastolic (mmHg) | −0.04 | 0.28 | 77 ± 9 | 75 ± 9 | 76 ± 10 |
| Renal function | |||||
| eGFR | 0.03 | 0.36 | 79.5 ± 9.5 | 82.3 ± 16.5 | 79.3 ± 19.8 |
| Mean CCA IMT |
|||||
|---|---|---|---|---|---|
| R | p | Regression n = 181 | No change n = 379 | Progression n = 299 | |
| Age (years) | −0.02 | 0.48 | 68.2 ± 8.3 | 68.6 ± 8.3 | 67.7 ± 8.6 |
| Male gender (%) | 0.02 | 0.63 | 66.9 | 65.4 | 61.8 |
| Current smokers (%) | 0.02 | 0.21 | 7.2 | 7.8 | 8.8 |
| BMI (kg/m−2) | −0.08 | 0.02 | 29.8 ± 5.2 | 29.1 ± 5.0 | 28.7 ± 4.6 |
| Metabolic factors | |||||
| HbA1c (mmol/mmol) | −0.01 | 0.86 | 49.0 (40.0–59.0) | 48.6 (40.0–60.0) | 47. 0 (40.0–56.0) |
| LDL (mmol/L) | 0.06 | 0.10 | 2.38 ± 0.99 | 2.38 ± 0.89 | 2.50 ± 0.93 |
| HDL (mmol/L) | 0.09 | 0.008 | 1.27 ± 0.39a | 1.38 ± 0.44 | 1.40 ± 0.41 |
| TG (mmol/L) | −0.06 | 0.07 | 1.49 (1.10–2.10) | 1.28 (0.95–1.83) | 1.30 (0.90–1.80) |
| Blood pressure | |||||
| Systolic (mmHg) | −0.04 | 0.21 | 137 ± 18 | 134 ± 17 | 134 ± 17 |
| Diastolic (mmHg) | −0.05 | 0.18 | 77 ± 9b | 75 ± 9 | 76 ± 10 |
| Renal function | |||||
| eGFR | 0.07 | 0.05 | 76.7 ± 20.4 | 79.9 ± 18.9 | 82.0 ± 17.9 |
Variables with normal distribution are shown as mean ± standard deviation and skewed variables as median and interquartile range. eGFR: mL/min−1 per 1.73 m2. Correlations are shown as Spearman rank correlation coefficients. Weighted linear trends across IMT change categories (regression, no change, and progression) were calculated with one-way ANOVA and significance of differences versus no change with Scheffe’s post hoc test. Linear trends for categorical variables were calculated using chi-square test. TG, triglycerides.
p < 0.01.
p < 0.05.
As the association between conventional risk factors and change in carotid IMT may be affected by medical treatment, we analyzed the association between medication and progression of carotid IMT. However, we found no significant associations with medication (Table S2). As previously reported, only minor changes in medication occurred during the study period.17
Taken together, these observations show that atherosclerotic plaque changes in this high-risk cohort tended to be more pronounced in the carotid bulb than in the CCA and that conventional risk factors, such as age, male gender, smoking, and LDL cholesterol, predict progression of carotid bulb atherosclerosis. In the CCA, there was an unexpected association between low HDL and regression of IMT.
Biomarkers predicting change in carotid bulb IMT
The biological processes that determine the balance between regression and progression of atherosclerotic plaques include inflammation, cell death, degradation of extracellular matrix, and activation of endothelial and connective tissue repair responses.1,3,18 To explore the pathophysiological processes involved in plaque regression and progression, we analyzed relationships between baseline levels of biomarkers included in the OLINK CVD I panel reflecting inflammation (high-sensitivity C-reactive protein [hsCRP], interleukin-6 [IL-6], monocyte chemoattractant protein-1 [MCP-1], and macrophage inflammatory protein-1α [MIP-1α]), degradation of extracellular matrix (matrix metalloproteinase [MMP]-3, -7, and -12), cell death by apoptosis (soluble tumor necrosis factor receptor-1 [TNFR-1], soluble TNF-related apoptosis-inducing ligand receptor-2 [TRAILR-2], and soluble Fas), connective tissue cell growth (platelet-derived growth factor [PDGF], epidermal growth factor [EGF], and heparin-binding EGF [HBEGF]), and endothelial growth factors (hepatocyte growth factor [HGF], PlGF, and vascular endothelial growth factor [VEGF]) with change in carotid IMT. It has previously been shown that the plasma levels of TNFRR-1, TRAILR-2, and Fas reflect ongoing apoptosis and that high levels of these circulating death receptors are associated with an increased risk of MI, stroke, and cardiovascular death.19 Moreover, increased release of endothelial growth factors has recently been shown to be part of the endothelial cell response to stress and to predict the risk of cardiovascular events.14
We found significant associations between high plasma levels of connective tissue cell growth factors and progression of bulb IMT, and these associations remained significant when adjusting for age, gender, smoking, and baseline LDL cholesterol and bulb IMT in linear regression models (Table 3). Circulating levels of both PDGF and EGF were twice as high in those with bulb IMT progression compared with those with regression. In contrast, we found inverse associations between progression of bulb IMT and the plasma levels of MCP-1, MIP-1α, MMP-3, TNFR-1, TRAILR-2, HGF, PlGF, and VEGF. Also, these associations remained significant when adjusting for age, gender, smoking, and baseline LDL cholesterol and bulb IMT in linear regression models (Table 3).
Table 3.
Relationships between biomarkers and change in carotid bulb IMT
| Correlation | p | p adjusted | Regression (n = 191) | No change (n = 200) | Progression (n = 412) | |
|---|---|---|---|---|---|---|
| Inflammatory | ||||||
| hsCRP (mg/L) | 0.00 | 0.98 | 0.28 | 1.40 (0.67–2.90) | 1.16 (0.60–2.54) | 1.30 (0.67–2.69) |
| IL-6 | −0.04 | 0.29 | 0.19 | 47.5 (26.9–64.0)a | 31.7 (22.0–50.4) | 33.8 (22.9–52.3) |
| MCP-1 | −0.09 | 0.02 | 0.014 | 13.3 (10.1–17.0)b | 11.5 (8.6–15.4) | 11.5 (8.9–14.7) |
| MIP-1α | −0.17 | 4.0 × 10−6 | 6.1 × 10−6 | 4.92 (4.08–6.19) | 4.71 (3.97–5.39) | 4.59 (3.86–5.66) |
| Matrix proteases | ||||||
| MMP-3 | −0.08 | 0.03 | 2.8 × 10−4 | 2.51 (1.91–3.40) | 2.38 (1.90–2.97) | 2.38 (1.91–3.03) |
| MMP-7 | 0.12 | 0.005 | 0.73 | 468 (282–676) | 387 (283–559) | 446 (311–690)a |
| MMP-12 | −0.07 | 0.06 | 0.01 | 154 (99–232)b | 122 (91–169) | 136 (98–199)a |
| Apoptosis | ||||||
| TNFR-1 | −0.07 | 0.04 | 0.02 | 6,608 (5,185–8,192)a | 6,039 (5,008–7,473) | 6,295 (5,185–7,643) |
| TRAILR-2 | −0.13 | 3.6 × 10−4 | 0.002 | 3.68 (2.51–4.63)a | 3.24 (2.35–4.21) | 3.18 (2.31–4.33) |
| Fas | −0.12 | 0.01 | 0.001 | 226 (177–280)a | 197 (168–242) | 198 (167–244) |
| SMC growth factors | ||||||
| PDGF | 0.28 | 1.8 × 10−15 | 5.3 × 10−11 | 70.0 (29.2–166.8)c | 132.5 (61.2–246.0) | 174.8 (90.4–263.2)a |
| EGF | 0.26 | 3.1 × 10−13 | 8.7 × 10−9 | 35.6 (11.0–73.5)c | 51.1 (21.1–116.2) | 67.6 (32.4–121.1)a |
| HBEGF | 0.12 | 0.001 | 0.08 | 22.8 (18.9–29.0)b | 25.9 (21.0–34.8) | 26.7 (21.6–34.5) |
| EC growth factors | ||||||
| HGF | −0.12 | 0.001 | 0.001 | 121 (93–150)b | 101 (83–130) | 103 (87–127) |
| PlGF | −0.15 | 3.3 × 10−6 | 3.0 × 10−6 | 191 (144–246)b | 163 (139–202) | 168 (139–207) |
| VEGF | −0.10 | 0.005 | 0.008 | 1,510 (1,193–1,965) | 1,370 (1,097–1,783) | 1,370 (1,097–1,739) |
All biomarkers except hsCRP are expressed as arbitrary units, and values are shown as median and IQR. Correlations are shown as Spearman correlation coefficients. Linear regression models were used to adjust correlations for age, gender, smoking, LDL cholesterol, and baseline carotid bulb IMT (p adjusted). Differences between IMT change categories (regression and progression versus no change) were calculated with one-way ANOVA and Scheffe’s post hoc test. If correcting for multiple comparisons in the present table, the threshold for significance is p < 0.004. EC, endothelial cell; SMC, smooth muscle cell.
p < 0.05.
p < 0.01.
p < 0.001.
Relationships between biomarkers and change in CCA IMT
There were fewer significant associations between biomarkers and change in CCA IMT than observed for the carotid bulb (Table 4). However, in line with what was seen in the bulb regression of CCA IMT (as compared with the no change group) associated with higher plasma levels of TRAILR-2 and lower levels PDGF, the inverse association between CCA IMT progression and TRAILR-2 levels remained significant when adjusting for age, gender, smoking, and LDL cholesterol and bulb IMT in linear regression models (Table 4).
Table 4.
Relationships between biomarkers and change in common carotid IMT
| Correlation | p | p adjusted | Regression (n = 181) | No change (n = 379) | Progression (n = 299) | |
|---|---|---|---|---|---|---|
| Inflammatory | ||||||
| hsCRP (mg/L) | 0.00 | 0.98 | 0.95 | 1.20 (0.66–2.40) | 1.30 (0.62–2.90) | 1.37 (0.70–2.59) |
| IL-6 | 0.01 | 0.84 | 0.74 | 38.6 (24.3–58.7) | 34.3 (23.0–53.0) | 35.8 (24.3–55.4) |
| MCP-1 | 0.00 | 0.92 | 0.85 | 12.7 (9.4–16.3) | 11.5 (8.9–15.1) | 12.0 (9.0–15.5) |
| MIP-1α | 0.04 | 0.25 | 0.34 | 4.86 (3.86–5.80) | 4.76 (4.00–5.74) | 4.69 (3.95–5.82) |
| Matrix proteases | ||||||
| MMP-3 | −0.03 | 0.41 | 0.28 | 2.59 (2.01–3.16) | 2.33 (1.87–3.04) | 2.46 (1.93–3.23) |
| MMP-7 | 0.08 | 0.03 | 0.07 | 413 (256–676) | 434 (305–650) | 448 (309–657) |
| MMP-12 | −0.02 | 0.60 | 0.74 | 133 (98–202) | 140 (101–201) | 137 (94–205) |
| Apoptosis | ||||||
| TNFR-1 | −0.06 | 0.07 | 0.07 | 6,562 (5,349–8,079) | 6,383 (5,113–7,899) | 6,165 (5,113–7,630) |
| TRAILR-2 | −0.12 | 0.001 | 0.004 | 3.76 (2.86–4.87)a | 3.27 (2.22–4.35) | 3.07 (2.45–4.14) |
| Fas | −0.05 | 0.16 | 0.09 | 209 (172–260) | 204 (169–244) | 206 (169–260) |
| SMC growth factors | ||||||
| PDGF | 0.10 | 0.005 | 0.25 | 94.4 (42.6–204.4)a | 145.0 (65.3–249.0) | 146.5 (65.8–240.5) |
| EGF | 0.04 | 0.28 | 0.70 | 47.2 (16.7–108.8) | 54.0 (23.3–111.4) | 53.4 (20.4–104.7) |
| HBEGF | 0.07 | 0.05 | 0.41 | 24.1 (19.0–32.8) | 25.4 (20.9–32.7) | 25.8 (21.0–33.6) |
| EC growth factors | ||||||
| HGF | 0.00 | 0.99 | 0.88 | 111 (89–139) | 108 (87–133) | 107 (87–136) |
| PlGF | −0.06 | 0.07 | 0.06 | 184 (146–232) | 171 (142–211) | 169 (139–219) |
| VEGF | −0.02 | 0.54 | 0.48 | 1,468 (1,193–1,820) | 1,448 (1,123–1,820) | 1,389 (1,105–1,758) |
All biomarkers except hsCRP are expressed as arbitrary units, and values are shown as median and IQR. Correlations are shown as Spearman correlation coefficients. Linear regression models were used to adjust correlations for age, gender, smoking, LDL cholesterol, and baseline CCA IMT (p adjusted). Differences between IMT change categories (regression and progression versus no change) were calculated with one-way ANOVA and Scheffe’s post hoc test. If correcting for multiple comparisons in the present table, the threshold for significance is p < 0.004.
p < 0.01.
Vascular characteristics associated with change in carotid IMT
Using the change in IMT as a continuous variable, there were significant positive associations between the GSM of carotid plaques at baseline and progression of IMT in the bulb as well as in the CCA, and these associations remained significant when controlling for the same factors as above as well as for baseline IMT for the respective site in linear regression models (Table 5). Analysis of GSM is a standardized way of measuring the ability of tissue to reflect ultrasound waves. Plaques that are rich in fibrous tissue have a high ability to reflect ultrasound waves and become echogenic20,21 and are associated with a lower risk of CVD events.22,23 Our observations thus suggest that lesion growth primarily occurs in arteries with more echogenic (high GSM value) plaques. Representative ultrasound images of echolucent and echogenic plaques are shown in Figure 1E. There were associations between the RHI and progression of IMT that remained significant when controlling for confounders (Table 5). The association between plaque GSM and progression of carotid was primarily due to lower GSM values in the groups with IMT regression (Table 5). This group also had the largest plaque at baseline as assessed by the bulb IMT. These findings show that regression of IMT in the bulb primarily occurred in subjects with larger and more echolucent plaques. Importantly, subjects with regression of IMT in the bulb still had more echolucent plaques at the follow-up investigation, suggesting that they still had a low content of fibrous tissue (Table 5).
Table 5.
Relationships between baseline vascular measurements and change in carotid bulb IMT
| Carotid bulb |
||||||
|---|---|---|---|---|---|---|
| Correlation | p | p adjusted | Regression | No change | Progression | |
| CCA | n = 191 | n = 200 | n = 412 | |||
| Mean IMT (mm) | 0.03 | 0.41 | 0.02 | 0.88 (0.80–1.02) | 0.85 (0.76–0.98) | 0.89 (0.7–1.02) |
| Bulb | ||||||
| Mean IMT (mm) | −0.08 | 0.03 | 0.001 | 1.22 (1.01–1.56)a | 1.03 (0.88–1.20) | 1.05 (0.89–1.34) |
| GSM baseline | 0.26 | 1.9 × 10−11 | 3.7 × 10−9 | 44 (31–70)a | 67 (49–89) | 68 (50–87) |
| GSM follow-up | 0.26 | 4.5 × 10−8 | 0.003 | 53 (38–71)b | 67 (54–90) | 72 (50–96) |
| Arterial stiffness | ||||||
| PWV (m/s) | −0.02 | 0.68 | 0.17 | 10.8 (9.2–13.1) | 9.8 (8.5–11.5) | 10.5 (8.8–12.4) |
| Endothelial function | ||||||
| RHI | 0.02 | 0.58 | 0.76 | 1.97 (1.68–2.45) | 2.11 (1.66–2.58) | 2.04 (1.65–2.57) |
| Common carotid artery | ||||||
| CCA | n = 181 | n = 379 | n = 299 | |||
| Mean IMT (mm) | −0.24 | 9.9 × 10−13 | 7.2 × 10−15 | 0.97 (0.85–1.13)a | 0.87 (0.77–0.99) | 0.85 (0.74–0.97) |
| Bulb | ||||||
| Mean IMT (mm) | −0.01 | 0.86 | 0.36 | 1.10 (0.93–1.40) | 1.06 (0.91–1.33) | 1.09 (0.92–1.34) |
| GSM baseline | 0.10 | 0.01 | 0.04 | 53 (37–78)c | 65 (45–85) | 65 (44–84) |
| GSM follow-up | 0.10 | 0.04 | 0.23 | 54 (44–75)c | 70 (46–91) | 62 (47–85) |
| Arterial stiffness | ||||||
| PWV (m/s) | −0.10 | 0.007 | 0.37 | 11.1 (9.6–13.3)c | 10.1 (8.7–12.4) | 10.3 (8.7–12.1) |
| Endothelial function | ||||||
| RHI | 0.04 | 0.21 | 0.43 | 2.04 (1.76–2.53) | 2.13 (1.76–2.62) | 2.16 (1.82–2.67) |
Gray scale median (GSM) value is the median of all gray levels of the pixels in the plaque. Correlations are shown as Spearman rank correlation coefficients. Linear regression models were used to adjust correlations for age, gender, smoking, LDL cholesterol, and baseline CCA or bulb IMT as appropriate (p adjusted). Differences between IMT change categories (regression and progression versus no change) were calculated with one-way ANOVA and Scheffe’s post hoc test. Values are shown as median and IQR. If correcting for multiple comparisons in the present table, the threshold for significance is p < 0.004. PWV, pulse wave velocity; RHI, reactive hyperemia index.
p < 0.001.
p < 0.01.
p < 0.05.
Associations between biomarkers and carotid plaque GSM
We next investigated whether there were associations between circulating biomarkers and plaque GSM. We identified highly significant correlations between the plasma level of PDGF and the plaque GSM (r = 0.37; p < 0.001; Figure 2A) as well as an inverse association between the plasma level of HGF and plaque GSM (r = 0.20; p < 0.001; Figure 2B). Also, the plasma concentrations of MIP1-α and TRAILR-2 were inversely, although more weakly, correlated with plaque GSM (r = −0.09, p = 0.004 and r = 0.11, p < 0.001, respectively; Figures 2C and 2D).
Figure 2.

Associations between carotid plaque GSM and biomarkers
Scatterplots demonstrating associations between baseline bulb carotid plaque GSM and plasma levels of (A) PDGF, (B) HGF, (C) MIP-1α, and (D) soluble TRAIL receptor-2. Correlations were calculated using the Pearson correlation method. Lnn2 transformed biomarker values were used in the graphs to improve the visibility of associations.
Treatment with statins has previously been shown to enhance the echogenicity (i.e., increase GSM) of carotid plaques,24 but we found no significant difference in carotid plaque GSM between subjects with or without statins (median and interquartile range [IQR] 62 gray scale units [40–82] versus 67 gray scale units [46–85]).
Multifactorial analyses of associations with change in carotid IMT
We next entered age, gender, smoking, LDL cholesterol, mean baseline CCA IMT (for CCA only), mean baseline bulb IMT (for carotid bulb only), baseline GSM, MIP-1α, TRAILR-2, PDGF, PlGF, and HGF into linear regression models to study independent associations with IMT change. For continuous variables, standardized Z scores were used to calculate standardized β-coefficients and 95% CI. In these analyses, baseline bulb IMT (−0.14 [−0.06 to −0.02] p = 0.001), baseline plaque GSM (0.15 [0.02–0.07] p = 0.001), and PDGF (0.20 [0.03–0.09] p < 0.001) were independently associated with change of IMT in the bulb. Baseline CCA IMT (−0.30 [−0.45 to −0.26] p < 0.001), baseline plaque GSM (0.09 [0.00–0.02] p = 0.04), smoking (0.08 [0.00–0.03] p = 0.04), MIP-1α (0.11 [0.00–0.03] p = 0.03), and HGF (0.12 [0.00–0.03] p = 0.03) were independently associated with change of IMT in the CCA.
In view of the identified association between circulating levels of PDGF and change in bulb IMT, we next analyzed how plasma PDGF related to conventional risk factors. In a linear regression model, using standardized Z scores for continuous variables, age, and LDL demonstrated independent associations with plasma PDGF (standardized β-coefficients and 95% CI were 0.17 [0.10–0.25] and 0.23 [0.15–0.29], respectively, p < 0.001 for both; the other variables in the model were gender, current smoking, HbA1c, HDL, triglycerides, and systolic blood pressure). Release from aggregating platelets represents one possible source of plasma growth factors. Comparing subjects treated with (n = 381) and without (n = 456) anti-platelet medication, we found that subjects on anti-platelet medication had lower plasma levels of EGF (median and IQR 47.8 [19.4–96.7] versus 57.3 [23.3–118] arbitrary units [a.u.]; p = 0.012 and HBEGF 24.4 [20.3–30.5] versus 26.4 [20.7–34.2] a.u.; p = 0.008), while there was no significant difference in the plasma level of PDGF (134 [52–237] versus 141 [61–242] a.u. as assessed by the Mann-Whitney U test). The association of PDGF with age and LDL could imply that PDGF is released from platelets interacting with the endothelium of arteries more affected by atherosclerosis. Although we found no correlation between PDGF levels and baseline IMT in the CCA or the bulb (r = −0.04, p = 0.20 and r = 0.05, p = 0.16, respectively), there was a weak association with impaired endothelial function as assessed by the RHI (r = 0.09; p = 0.01).
As there is no established cutoff for defining regression and progression of carotid IMT, we also performed sensitivity analyses using a cutoff of ±0.1 mm to define no change. Using this cutoff, there were only minor effects on the associations described above (Tables S3–S6). The associations also remained significant when all subjects without prevalent CVD were excluded in sensitivity analyses. The pattern of associations of biomarkers and vascular measurements with IMT change were similar in subjects with and without T2D (Tables S7–S12). We also compared biomarkers and vascular measurements between T2D subjects with and without insulin treatment (Tables S13 and S14). Those treated with insulin had higher levels of hsCRP, IL-6, MMP-7, MMP-12, TNFR-1, and TRAILR-2, indicating increased inflammation, extracellular matrix remodeling, and cell death by apoptosis. They also had increased bulb IMT and PWV at baseline.
Carotid IMT and incident cardiovascular events
Sixty-seven of the study subjects suffered from at least one of the pre-defined incident cardiovascular events (non-fatal acute MI, hospitalized unstable angina, resuscitated cardiac arrest, any coronary revascularization procedure, non-fatal stroke, transient ischemic attack confirmed by a specialist, lower extremity artery disease defined as ankle brachial pressure index [ABPI] < 0.9 with intermittent claudication, angioplasty, or above ankle amputation) during the 3-year study period. Those that suffered a cardiovascular event were older (70.9 ± 6.9 versus 67.9 ± 8.5 years; p = 0.001) and more often had prevalent CVD (62.3% versus 37.3%; p < 0.001). They also had increased baseline IMT both in the CCA (1.00 ± 0.25 versus 0.90 ± 0.21 mm; p = 0.001) and in the bulb (1.37 ± 0.30 versus 1.18 ± 0.43 mm; p = 0.003). There were no differences in IMT change in the CCA (0.00 ± 0.11 versus 0.02 ± 0.12 mm; p = 0.24) or the bulb (−0.01 ± 0.36 versus 0.06 ± 0.26 mm; p = 0.18) between those with and without an incident cardiovascular event. There was also no difference in gender, smoking, T2D, plasma PDGF, and LDL and HDL cholesterol between those with and without an incident cardiovascular event.
Discussion
The aim of this study was to identify biomarkers and plaque characteristics predicting change in carotid atherosclerosis in subjects with high CVD risk. We also analyzed whether subjects with or without T2D differed in this respect. We focused our studies on IMT changes in the carotid bulb, as this is the most common location of clinically significant plaques in the carotids. Our findings reveal unexpected associations between biomarkers reflecting the atherosclerotic disease process and the progression as well as the regression of carotid plaques. Subjects with progression of carotid plaques were older, more often males and current smokers, and had higher LDL cholesterol levels, but the statistically strongest associations were with the plasma concentration of the connective tissue cell growth factors PDGF and EGF. Moreover, the associations between the conventional risk factors and plaque progression were no longer statistically significant when adjusting for plasma PDGF levels. It is well known that intimal smooth muscle cells and fibrous proteins synthesized by these cells constitute an important part of atherosclerotic plaques. Accordingly, it is likely that factors that stimulate smooth-muscle-cell growth and extracellular matrix synthesis may contribute to plaque growth. However, the association between smooth-muscle-cell growth factors in plasma and plaque growth is still unexpected because the intimal activation of smooth muscle cells is considered to be driven by growth factors released by immune cells in the plaque.1,25 Although it cannot be completely excluded that such processes result in a diffusion of growth factors into the circulation, it appears unlikely that this would be large enough to affect the circulating level of these factors. There were also no signs of increased inflammatory activation in subjects with progression of carotid bulb IMT, and the plaques had relatively high GSM values, suggesting that they are fibrotic rather than lipid rich and inflammatory. An alternative explanation to the association between the plasma concentration of PDGF and progression of carotid bulb IMT is that an increased systemic expression of PDGF results in diffusion of PDGF into the arterial wall, stimulating the proliferative and synthetic activity of plaque smooth muscle cells. In line with this, we have previously shown that increased circulating PDGF, EGF, and HBEGF are associated with higher carotid plaque contents of both collagen and elastin.26 The causes of a systemic activation of PDGF expression remains to be explored, but the present study implicates a possible role of increasing age and hypercholesterolemia. Release from aggregating platelets represents one possible source of plasma PDGF, EGF, and HBEGF. Accordingly, we tested the hypothesis that anti-platelet medication would be associated with lower plasma levels of these growth factor. In line with this notion, we found that subjects on anti-platelet medication had lower plasma levels of EGF and HBEGF. However, since there was no significant difference in plasma PDGF between subjects with and without anti-platelet medication, it appears less likely that release from platelets is a major source of PDGF in plasma. There was an association between PDGF and impaired endothelial function still indicating a possibility of release of PDGF from platelets interacting with a dysfunctional endothelium. Another possibility that should be further investigated is that the level of circulating PDGF is regulated by genetic factors. The possibility that plasma PDGF is an artefact due to a release from platelets during blood sampling also needs to be considered.
Almost one out of four study subjects demonstrated regression of carotid bulb IMT during the study period. Regression was not associated with conventional risk factors or medication. However, regression of plaques in the bulb was associated with plaque characteristics and biomarkers that markedly differed from those associated with plaque progression. Subjects in which plaque regression occurred had lower circulating amounts of growth factors for connective tissue cells as well as increased amounts of biomarkers reflecting inflammation, cell death by apoptosis, connective tissue degradation, and endothelial cell activation. Their plaques were also larger and had a lower GSM score (more echolucent) at baseline. Plaques that are rich in lipids and have a large necrotic core have a low ability to reflect ultrasound and become echolucent.20,21 Presence of echolucent plaques is associated with CVD risk factors and increased risk for CVD events.22,23 Our findings suggest that regression of atherosclerosis in the carotid bulb mainly occurs in larger and more vulnerable lesions. Importantly, these lesions remained more echolucent at the follow-up investigation, indicating the plaque regression was not accompanied by plaque stabilization. The notion that regression primarily takes place in more vulnerable lesions is not entirely unexpected in view of previous studies using analyses of 14C levels in human carotid plaques, demonstrating that the tissue turnover time in fibrous parts of the lesion is more than 10 years.27
Based on the well-documented association between carotid IMT and CVD risk, it has been logical to assume that the same is true also for change in carotid IMT and that this can be used as a surrogate marker in clinical trials. However, the results of studies relating mean change in carotid IMT to risk for development of acute cardiovascular events have been inconsistent, and a meta-analysis of 16 population studies involving 36,985 subjects failed to identify any evidence for an association.28 Similarly, a recent meta-analysis of 31 cohorts, including 89,070 subjects with high CVD risk, found no association between change in carotid IMT and the incidence of cardiovascular events.29 The findings of the present study indicating that regression is more common in large plaques with vulnerable phenotype as assessed by its echogenicity may provide an explanation to this paradox. Even though one might consider that the regression of an echolucent plaque could be beneficial and indicate stabilization of atherosclerotic disease, the fact that these plaques remain more echolucent when they have regressed suggests, to some extent, that regression is a marker of vulnerable plaques and not an indicator of an improvement in cardiovascular risk. Interestingly, we found only a low correlation between progression of IMT in the CCA and the bulb. However, this is not entirely unexpected, because IMT at these two locations shows different association with risk factors and predicts different types of cardiovascular events.30, 31, 32 Shear stress, which has become recognized as a key factor in plaque development, is also markedly different in the CCA and the bulb.33
The vulnerability of atherosclerotic plaques depends on the balance between cell injury, inflammatory-driven degradation of fibrous tissue by MMPs, and the repair of this injury primarily by smooth muscle cells.1,4 The death receptors TNFR-1, TRAILR-2, and Fas are released in soluble forms following activation of the extrinsic apoptosis signal pathway, implicating them as biomarkers of ongoing apoptosis.19 Elevated levels of TRAILR-2 have been found to predict cardiovascular death independently of other major risk factors.19 We have previously shown that MMP-3 and -7 are elevated in subjects with T2D and correlate significantly with the severity of atherosclerosis.34 Interestingly, MMP-3 and -7 showed opposing associations with plaque progression in the present study, with high levels of MMP-3 being associated with regression of CCA IMT and high levels of MMP-7 with progression. Regression of bulb IMT was also associated with higher levels of the endothelial growth factor PlGF. It has previously been shown that increased circulating levels of PlGF reflect a repair response of stressed tissue and are associated with an increased risk of MI and stroke.14 Collectively, the present findings support the notion that IMT regression is associated with a more vulnerable plaque phenotype.
One of the aims of the present study was to determine whether factors that are associated with changes in carotid IMT differ between subjects with and without T2D. We identified several factors that were common; IMT progression was associated with increased levels of connective tissue cell growth factors, and regression of bulb IMT was associated with presence of large echolucent plaques at baseline. In subjects without T2D, the latter was also associated with increased circulating levels of biomarkers reflecting inflammation, cell death by apoptosis, and endothelial activation. The state of chronic, low-grade inflammation that commonly is present in subjects with T2D has been proposed as a key factor in aggravating atherosclerosis. In contrast to this notion, we found no associations between inflammatory biomarkers, such as hsCRP and IL-6, with change in carotid IMT in subjects with T2D. A possible explanation to this could be that most subjects in the present study were treated with statins, which is known to both lower hsCRP levels and plaque inflammation.35,36
In conclusion, the present study demonstrates that, in high-risk subjects receiving guide-line treatment, progression of carotid atherosclerosis mainly occurs in more echogenic, low-risk plaques and is associated with higher plasma concentrations of plaque-stabilizing growth factors. One possible interpretation of this observation is that, in subjects receiving preventive treatment, plaque growth occurs because of increased synthesis of fibrous material maintaining a stable plaque phenotype and that this process is driven by an increased concentration of pro-fibrotic growth factors in the circulation. Regression, on the other hand, mainly occurs in large plaques with a more vulnerable phenotype and is associated with increased expression of biomarkers reflecting vascular stress in plasma as well as lower concentrations of plaque-stabilizing growth factors. This is well in line with the notion that proper medical treatment is more likely to induce regression of a vulnerable plaque consisting of lipid deposits, necrotic debris, and inflammatory cells than of a stable plaque containing predominantly fibrous connective tissue.
Limitations of the study
Some limitations of the present study need to be considered. There are no standard definitions of regression and progression of carotid IMT in terms of mm change. Here, we applied the arbitrary set cutoff of ±0.05 mm change to define no change in IMT. It is possible that the use of other cutoffs to define regression and progression could have yielded different results. However, sensitivity analyses applying a cutoff of ±0.1 mm change to define no change in IMT provided almost identical findings. Most of the subjects participating in our study were high-risk individuals, and it is likely that our findings may not apply to more low-risk populations. Given that the present cohort is composed mainly of patients of European descent, the findings may not apply to individuals from other ethnicities. Also, the present findings need to be replicated in an independent cohort.
STAR★Methods
Key resources table
| RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical commercial assays | ||
| OLINK CVD 1 panel | OLINK Biosciences | No longer on the market |
| Software and algorithms | ||
| SPSS version 27 | IBM | www.ibm.com/support/pages/ibm-spss-statistics-27-documentation |
| Other | ||
| Endo-PAT | Itamar Medical | www.itamar-medical.com/professionals/endopat/ |
| SphygmoCor | Actor Medical | atcormedical.com/technology/sphygmocor/ |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact Jan Nilsson (jan.nilsson@med.lu.se).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Study population
The SUMMIT VIP baseline study investigation included 985 subjects with T2D (out of which 485 had prevalent CVD) and 515 subjects without T2D (out of which 245 had prevalent CVD).16 Subjects were recruited from existing population cohorts and hospital registers at the university hospitals in Malmö (Sweden), Pisa (Italy), Dundee and Exeter (UK) between November 2010 and June 2013. T2D was defined based on contemporary or historical evidence of hyperglycemia (according to WHO 1998 criteria; fasting plasma glucose >7.0 mmol/L or 2-h plasma glucose >11.1 mmol/L, or both), or by current medication with anti-diabetic drugs. Classification of CVD included non-fatal acute MI, hospitalized unstable angina, resuscitated cardiac arrest, any coronary revascularization procedure, non-fatal stroke, transient ischemic attack confirmed by a specialist, lower extremity artery disease defined as ankle brachial pressure index (ABPI) < 0.9 with intermittent claudication or prior corrective surgery, angioplasty or above ankle amputation.16 Subjects were matched at each center for gender, age (±5 years) and duration of T2D (±5 years, when appropriate) using the T2D with CVD as reference group. Demographics, clinical characteristics including physical and laboratory examinations were obtained according to a pre-defined study protocol at all 4 participating centers. Information about medication was obtained through questionnaires or health registers.
After 36 months, 58% (n = 870) of the baseline study subjects accepted to participate in a follow-up re-examination. Information of incident cardiovascular events (same as above) during the study period was obtained through a questionnaire. Five subjects were excluded because of carotid surgery procedure during the follow-up period (Figure S1 and Table 1).
Ethics approval
The study was approved by the local ethics review boards and carried out in accordance with the principles of the Declaration of Helsinki. All study subjects provided written informed consent.
Method details
Vascular assessments
IMT in the right and left CCA and the carotid bulb was measured by ultrasound. Plaques were defined as focal thickening (≥0.8 mm) of the artery wall and the gray scale median (GSM) of the largest plaque measured using the Artery Measurement System as previously described.37,38 Endothelial function was measured using an EndoPat device (Itamar Medical, Caesarea Ind. Park, Israel) to estimate the endothelium-dependent, post-ischemic reactive hyperemia index (RHI) in response to 5-minutes of upper arm arterial occlusion. Arterial stiffness was assessed by calculating the carotid-femoral pulse wave velocity (PWV) using a SphygmoCor device (Atcor Medical, Australia). Valid PWV and RHI data were obtained from 412 and 440 subjects, respectively. Before the start of the study, staff at the participating centers completed a joint carotid ultrasound training program to minimize intra-observer variability resulting in an intra-observer variability of <5% in the CCA and <10% in the carotid bulb. Detailed information about the methods used for vascular assessments, as well as data regarding intra- and inter-observer variability and calibration between centres, has been published previously.16 The average of the left and right carotids was used to calculate change in mm in mean and maximal (max) IMT in the CCA (n = 566) and the bulb (n = 522) between the baseline and follow-up investigation. Where IMT data was available from one side only that was used as the mean value.
Biomarker analysis
Plasma levels of biomarkers reflecting inflammation (interleukin-6 (IL-6), monocyte chemoattractant protein-1 (MCP-1) and macrophage inflammatory protein-1α (MIP-1α)), degradation of extracellular matrix (matrix metalloproteinase (MMP)-3 -7 and −12), cell death by apoptosis (soluble tumor necrosis factor receptor-1 (TNFR-1), soluble TNF-related apoptosis-inducing ligand receptor-2 (TRAILR-2) and soluble Fas), and tissue-stabilizing growth factors (platelet-derived growth factor (PDGF), epidermal growth factor (EGF) and heparin-binding EGF (HBEGF)) were analyzed by the Proximity Extension Assay (PEA) technique using the Proseek Multiplex CVD96x96 reagents kit (Olink Bioscience, Uppsala, Sweden) at the Clinical Biomarkers Facility, Science for Life Laboratory, Uppsala as previously described.34 All samples were analyzed in the same run. Data analysis was performed by a preprocessing normalization procedure using Olink Wizard for GenEx (Multid Analyses, Sweden). Values are presented as arbitrary units (AU). Data regarding intra- and inter-assays variations as well as general calibrator curves to calculate the approximate concentrations are available on the OLINK homepage (http://www.olink.com). hsCRP was measured by an immune-turbidimetric method at the department of Clinical Chemistry at Skåne University Hospital, Malmö.
Quantification and statistcal analysis
Statistics
Values are presented as mean and standard deviation for continuous variables with normal distribution and as median and interquartile rage (IQR) for skewed variables. Biomarker values were normalized by log transformation before used in statistical analyses. Missing data were <5% for all variables except pulse wave velocity were 16% lacked data. Subjects with missing data were excluded from that analysis. Differences between means of normally distributed continuous variables were assessed with independent sample t tests and between skewed variables with the Mann-Whitney U-test. χ2 test was used for categorical variables. Correlations were calculated as Spearman’s rank correlation coefficients. Differences between IMT change categories (regression, no change and progression) were calculated with one-way ANOVA and significance of differences versus no change with the conservative Scheffe’s post hoc test. P for trend for difference in medication between IMT change categories was calculated by Chi-square linear-by-linear association. Logistic regression models were used to identity independent association between measured variables and IMT change categories. In these analyses, subjects with regression were compared with the combined no change and progression groups and vice versa for those with progression. Linear regressions models were used to identify independent associations between measured variables and carotid plaque GSM and IMT change. Bonferroni correction was used to adjust for multiple testing. Calculations were done using SPSS statistics version 22. All statistical analyses were done in accordance with the original protocol of the study.
Acknowledgments
This work was supported by funding from the Innovative Medicines Initiative (the SUMMIT consortium, IMI-2008/115006, the Swedish Heart-Lung Foundation, and the NIHR Exeter Clinical Research Facility). Funders had no role in the study. Any opinions raised in this paper are those of the authors and not those of NIHR or the UK Department of Health. J.N. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Author contributions
F.K., A.C.S., H.M.C., A.N., C.P., I.G., and J.N. designed and supervised the study. G.Ö., M.P., C.K., K.A., F.C., K.M.G., D.S., S.P., E.V., M.K., F.K., H.L., F.D., and J.B. collected clinical data and performed investigations on patients. J.N. managed data and performed statistical analyses. F.K. and J.N. drafted the manuscript, and all other authors made critical revisions.
Declaration of interests
The authors declare no competing interests.
Published: July 1, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2022.100676.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
References
- 1.Hansson G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005;352:1685–1695. doi: 10.1056/nejmra043430. [DOI] [PubMed] [Google Scholar]
- 2.Bentzon J.F., Otsuka F., Virmani R., Falk E. Mechanisms of plaque formation and rupture. Circ. Res. 2014;114:1852–1866. doi: 10.1161/CIRCRESAHA.114.302721. [DOI] [PubMed] [Google Scholar]
- 3.Libby P., Buring J.E., Badimon L., Hansson G.K., Deanfield J., Bittencourt M.S., Tokgözoğlu L., Lewis E.F. Atherosclerosis. Nat. Rev. Dis. Prim. 2019;5:56. doi: 10.1038/s41572-019-0106-z. [DOI] [PubMed] [Google Scholar]
- 4.Libby P. Mechanisms of acute coronary syndromes and their implications for therapy. N. Engl. J. Med. 2013;368:2004–2013. doi: 10.1056/NEJMra1216063. [DOI] [PubMed] [Google Scholar]
- 5.Björck L., Capewell S., O'Flaherty M., Lappas G., Bennett K., Rosengren A. Decline in coronary mortality in Sweden between 1986 and 2002: comparing contributions from primary and secondary prevention. PLoS One. 2015;10:e0124769. doi: 10.1371/journal.pone.0124769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Libby P., Pasterkamp G. Requiem for the 'vulnerable plaque. Eur. Heart J. 2015;36:2984–2987. doi: 10.1093/eurheartj/ehv349. [DOI] [PubMed] [Google Scholar]
- 7.Nilsson J. Atherosclerotic plaque vulnerability in the statin era. Eur. Heart J. 2017;38:1638–1644. doi: 10.1093/eurheartj/ehx143. [DOI] [PubMed] [Google Scholar]
- 8.Libby P. The changing landscape of atherosclerosis. Nature. 2021;592:524–533. doi: 10.1038/s41586-021-03392-8. [DOI] [PubMed] [Google Scholar]
- 9.Yeh R.W., Sidney S., Chandra M., Sorel M., Selby J.V., Go A.S. Population trends in the incidence and outcomes of acute myocardial infarction. N. Engl. J. Med. 2010;362:2155–2165. doi: 10.1056/NEJMoa0908610. [DOI] [PubMed] [Google Scholar]
- 10.Kolodgie F.D., Burke A.P., Wight T.N., Virmani R. The accumulation of specific types of proteoglycans in eroded plaques: a role in coronary thrombosis in the absence of rupture. Curr. Opin. Lipidol. 2004;15:575–582. doi: 10.1097/00041433-200410000-00012. [DOI] [PubMed] [Google Scholar]
- 11.van Lammeren G.W., den Ruijter H.M., Vrijenhoek J.E.P., van der Laan S.W., Velema E., de Vries J.P.P.M., de Kleijn D.P., Vink A., de Borst G.J., Moll F.L., et al. Time-dependent changes in atherosclerotic plaque composition in patients undergoing carotid surgery. Circulation. 2014;129:2269–2276. doi: 10.1161/CIRCULATIONAHA.113.007603. [DOI] [PubMed] [Google Scholar]
- 12.Rawshani A., Rawshani A., Franzén S., Eliasson B., Svensson A.M., Miftaraj M., McGuire D.K., Sattar N., Rosengren A., Gudbjörnsdottir S. Mortality and cardiovascular disease in type 1 and type 2 diabetes. N. Engl. J. Med. 2017;376:1407–1418. doi: 10.1056/NEJMoa1608664. [DOI] [PubMed] [Google Scholar]
- 13.Hess D.A., Verma S., Bhatt D., Bakbak E., Terenzi D.C., Puar P., Cosentino F. Vascular repair and regeneration in cardiometabolic diseases. Eur. Heart J. 2022;43:450–459. doi: 10.1093/eurheartj/ehab758. [DOI] [PubMed] [Google Scholar]
- 14.Chen Y., Nilsson A.H., Goncalves I., Edsfeldt A., Engström G., Melander O., Orho-Melander M., Rauch U., Tengryd C., Venuraju S.M., et al. Evidence for a protective role of placental growth factor in cardiovascular disease. Sci. Transl. Med. 2020;12:eabc8587. doi: 10.1126/scitranslmed.abc8587. [DOI] [PubMed] [Google Scholar]
- 15.Folkersen L., Gustafsson S., Wang Q., Hansen D.H., Hedman Å.K., Schork A., Page K., Zhernakova D.V., Wu Y., Peters J., et al. Genomic and drug target evaluation of 90 cardiovascular proteins in 30, 931 individuals. Nat. Metab. 2020;2:1135–1148. doi: 10.1038/s42255-020-00287-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shore A.C., Colhoun H.M., Natali A., Palombo C., Ostling G., Aizawa K., Kennbäck C., Kennback C., Casanova F., Persson M., et al. Measures of atherosclerotic burden are associated with clinically manifest cardiovascular disease in type 2 diabetes: a European cross-sectional study. J. Intern. Med. 2015;278:291–302. doi: 10.1111/joim.12359. [DOI] [PubMed] [Google Scholar]
- 17.Shore A.C., Colhoun H.M., Natali A., Palombo C., Khan F., Ostling G., Aizawa K., Kennbäck C., Kennback C., Casanova F., et al. Use of vascular assessments and novel biomarkers to predict cardiovascular events in type 2 diabetes: the SUMMIT VIP study. Diabetes Care. 2018;41:2212–2219. doi: 10.2337/dc18-0185. [DOI] [PubMed] [Google Scholar]
- 18.Libby P., Tabas I., Fredman G., Fisher E.A. Inflammation and its resolution as determinants of acute coronary syndromes. Circ. Res. 2014;114:1867–1879. doi: 10.1161/CIRCRESAHA.114.302699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mattisson I.Y., Björkbacka H., Wigren M., Edsfeldt A., Melander O., Fredrikson G.N., Bengtsson E., Gonçalves I., Orho-Melander M., Engström G., et al. Elevated markers of death receptor-activated apoptosis are associated with increased risk for development of diabetes and cardiovascular disease. EBioMedicine. 2017;26:187–197. doi: 10.1016/j.ebiom.2017.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grønholdt M.L.M., Nordestgaard B.G., Wiebe B.M., Wilhjelm J.E., Sillesen H. Echo-lucency of computerized ultrasound images of carotid atherosclerotic plaques are associated with increased levels of triglyceride-rich lipoproteins as well as increased plaque lipid content. Circulation. 1998;97:34–40. doi: 10.1161/01.cir.97.1.34. [DOI] [PubMed] [Google Scholar]
- 21.Salem M.K., Bown M.J., Sayers R.D., West K., Moore D., Nicolaides A., Robinson T.G., Naylor A.R. Identification of patients with a histologically unstable carotid plaque using ultrasonic plaque image analysis. Eur. J. Vasc. Endovasc. Surg. 2014;48:118–125. doi: 10.1016/j.ejvs.2014.05.015. [DOI] [PubMed] [Google Scholar]
- 22.Grønholdt M.L.M., Nordestgaard B.G., Schroeder T.V., Vorstrup S., Sillesen H. Ultrasonic echolucent carotid plaques predict future strokes. Circulation. 2001;104:68–73. doi: 10.1161/hc2601.091704. [DOI] [PubMed] [Google Scholar]
- 23.Jung M., Parrinello C.M., Xue X., Mack W.J., Anastos K., Lazar J.M., Selzer R.H., Shircore A.M., Plankey M., Tien P., et al. Echolucency of the carotid artery intima-media complex and intima-media thickness have different cardiovascular risk factor relationships: the Women's Interagency HIV Study. J. Am. Heart Assoc. 2015;4:e001405. doi: 10.1161/JAHA.114.001405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ibrahimi P., Jashari F., Bajraktari G., Wester P., Henein M.Y. Ultrasound assessment of carotid plaque echogenicity response to statin therapy: a systematic review and meta-analysis. Int. J. Mol. Sci. 2015;16:10734–10747. doi: 10.3390/ijms160510734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nilsson J. Cytokines and smooth muscle cells in atherosclerosis. Cardiovasc. Res. 1993;27:1184–1190. doi: 10.1093/cvr/27.7.1184. [DOI] [PubMed] [Google Scholar]
- 26.Rattik S., Wigren M., Björkbacka H., Fredrikson G.N., Hedblad B., Siegbahn A., Bengtsson E., Schiopu A., Edsfeldt A., Dunér P., et al. High plasma levels of heparin-binding epidermal growth factor are associated with a more stable plaque phenotype and reduced incidence of coronary events. Arterioscler. Thromb. Vasc. Biol. 2015;35:222–228. doi: 10.1161/ATVBAHA.114.304369. [DOI] [PubMed] [Google Scholar]
- 27.Goncalves I., Stenstrom K., Skog G., Mattsson S., Nitulescu M., Nilsson J. Short communication: dating components of human atherosclerotic plaques. Circ. Res. 2010;106:1174–1177. doi: 10.1161/CIRCRESAHA.109.211201. [DOI] [PubMed] [Google Scholar]
- 28.Den Ruijter H.M., Peters S.A.E., Anderson T.J., Britton A.R., Dekker J.M., Eijkemans M.J., Engström G., Evans G.W., de Graaf J., Grobbee D.E., et al. Common carotid intima-media thickness measurements in cardiovascular risk prediction: a meta-analysis. JAMA. 2012;308:796. doi: 10.1001/jama.2012.9630. [DOI] [PubMed] [Google Scholar]
- 29.Lorenz M.W., Gao L., Ziegelbauer K., Norata G.D., Empana J.P., Schmidtmann I., Lin H.J., McLachlan S., Bokemark L., Ronkainen K., et al. Predictive value for cardiovascular events of common carotid intima media thickness and its rate of change in individuals at high cardiovascular risk - results from the PROG-IMT collaboration. PLoS One. 2018;13:e0191172. doi: 10.1371/journal.pone.0191172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ebrahim S., Papacosta O., Whincup P., Wannamethee G., Walker M., Nicolaides A.N., Dhanjil S., Griffin M., Belcaro G., Rumley A., Lowe G.D. Carotid plaque, intima media thickness, cardiovascular risk factors, and prevalent cardiovascular disease in men and women: the British Regional Heart Study. Stroke. 1999;30:841–850. doi: 10.1161/01.str.30.4.841. [DOI] [PubMed] [Google Scholar]
- 31.Naqvi T.Z., Lee M.S. Carotid intima-media thickness and plaque in cardiovascular risk assessment. JACC Cardiovasc Imaging. 2014;7:1025–1038. doi: 10.1016/j.jcmg.2013.11.014. [DOI] [PubMed] [Google Scholar]
- 32.Rosvall M., Persson M., Ostling G., Nilsson P.M., Melander O., Hedblad B., Engström G., Engstrom G. Risk factors for the progression of carotid intima-media thickness over a 16-year follow-up period: the Malmö Diet and Cancer Study. Atherosclerosis. 2015;239:615–621. doi: 10.1016/j.atherosclerosis.2015.01.030. [DOI] [PubMed] [Google Scholar]
- 33.Brown A.J., Teng Z., Evans P.C., Gillard J.H., Samady H., Bennett M.R. Role of biomechanical forces in the natural history of coronary atherosclerosis. Nat. Rev. Cardiol. 2016;13:210–220. doi: 10.1038/nrcardio.2015.203. [DOI] [PubMed] [Google Scholar]
- 34.Goncalves I., Bengtsson E., Colhoun H.M., Shore A.C., Palombo C., Natali A., Edsfeldt A., Dunér P., Fredrikson G.N., Björkbacka H., et al. Elevated plasma levels of MMP-12 are associated with atherosclerotic burden and symptomatic cardiovascular disease in subjects with type 2 diabetes. Arterioscler. Thromb. Vasc. Biol. 2015;35:1723–1731. doi: 10.1161/ATVBAHA.115.305631. [DOI] [PubMed] [Google Scholar]
- 35.Ridker P.M., Rifai N., Pfeffer M.A., Sacks F.M., Moye L.A., Goldman S., Flaker G.C., Braunwald E. Inflammation, pravastatin, and the risk of coronary events after myocardial infarction in patients with average cholesterol levels. Circulation. 1998;98:839–844. doi: 10.1161/01.cir.98.9.839. [DOI] [PubMed] [Google Scholar]
- 36.Crisby M., Nordin-Fredriksson G., Shah P.K., Yano J., Zhu J., Nilsson J. Pravastatin treatment increases collagen content and decreases lipid content, inflammation, metalloproteinases, and cell death in human carotid plaques: implications for plaque stabilization. Circulation. 2001;103:926–933. doi: 10.1161/01.cir.103.7.926. [DOI] [PubMed] [Google Scholar]
- 37.Goncalves I., Lindholm M.W., Pedro L.M., Dias N., Fernandes e Fernandes J., Fredrikson G.N., Nilsson J., Moses J., Ares M.P. Elastin and calcium rather than collagen or lipid content are associated with echogenicity of human carotid plaques. Stroke. 2004;35:2795–2800. doi: 10.1161/01.str.0000147038.12073.59. [DOI] [PubMed] [Google Scholar]
- 38.Ostling G., Persson M., Hedblad B., Gonçalves I., Goncalves I. Comparison of grey scale median (GSM) measurement in ultrasound images of human carotid plaques using two different softwares. Clin. Physiol. Funct. Imag. 2013;33:431–435. doi: 10.1111/cpf.12049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.