

Summary
The RET kinase receptor is a target of mutations in neural crest tumors, including pheochromocytomas, and of oncogenic fusions in epithelial cancers. We report a RET::GRB2 fusion in a pheochromocytoma in which RET, functioning as the upstream partner, retains its kinase domain but loses critical C-terminal motifs and is fused to GRB2, a physiological RET interacting protein. RET::GRB2 is an oncogenic driver that leads to constitutive, ligand-independent RET signaling; has transforming capability dependent on RET catalytic function; and is sensitive to RET inhibitors. These observations highlight a new driver event in pheochromocytomas potentially amenable for RET-driven therapy.
Keywords: RET mutation, RET fusion, oncogenic fusion, pheochromocytoma, paraganglioma, tyrosine kinase inhibitor, GRB2, driver genetic events, selpercatinib, pralsetinib
Graphical abstract
Highlights
-
•
A RET::GRB2 fusion was found in a pheochromocytoma
-
•
RET retains its kinase domain, swaps the C tail with GRB2, a normal RET binding partner
-
•
RET::GRB2 relies on RET kinase function for transformation
-
•
RET::GRB2 renders cells sensitive to RET inhibitors
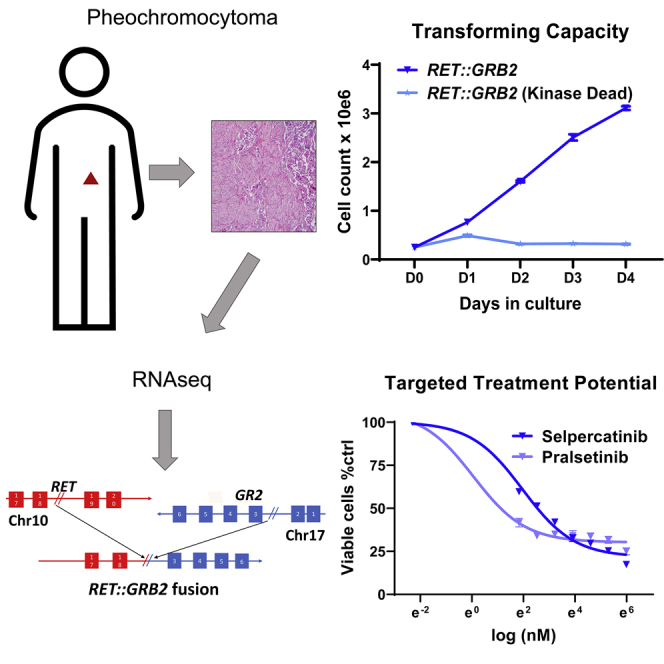
Estrada-Zuniga et al. describe a RET::GRB2 fusion in a pheochromocytoma. In this rearrangement, RET is uniquely positioned upstream to the partner gene GRB2, which encodes a natural RET interacting protein. RET::GRB2 is transforming and sensitive to RET inhibitors, supporting its driver role and highlighting opportunities for targeted therapy in pheochromocytomas.
Introduction
RET is a tyrosine kinase receptor with restricted tissue expression and relevance to cancer.1 Oncogenic somatic RET missense mutations occur in 40% of medullary thyroid carcinoma (MTC) and 5%–7% of pheochromocytomas.2 These neural-crest-derived tumors are also components of multiple endocrine neoplasia type 2A (MEN 2A) or MEN 2B hereditary syndromes, which are caused by germline RET mutations.1,2 These mutations lead to constitutive ligand-independent RET dimerization and signaling activation or change substrate specificity.1,3
Besides mutations, RET can be aberrantly activated by gene fusions in 1%–2% of lung adenocarcinomas and 10%–20% of papillary thyroid carcinoma3, 4, 5 and more rarely in leukemia, colon, prostate, and breast cancer.1 Shared elements of these RET fusions involve an intact RET tyrosine kinase domain fused to an upstream partner, often comprising proteins with coiled-coil motifs that provide dimerization domains. This fusion configuration leads to constitutive activation of RET and its effectors and endows target cells with oncogenic phenotypes.3,6
Highly selective RET inhibitors selpercatinib and pralsetinib have recently been approved for clinical use and have led to high response rates both in mutant- and fusion-related RET-driven cancers.7, 8, 9 Therefore, identification of novel forms of RET disruptions in cancer can have immediate clinical applicability. Recently, a RET::SEPTIN9 fusion was reported in a metastatic pheochromocytoma. In this instance, RET was positioned as the upstream partner of the fusion instead of at the conventional 3′ position. Notably, the patient displayed a marked clinical response to selpercatinib.10
In this report, we describe RET::GRB2, a distinct fusion in a sporadic pheochromocytoma, in which RET functions as the upstream component of the hybrid protein, in a similar configuration to the RET::SEPTIN9 fusion, although with a distinct RET breakpoint. Unlike other RET fusions, the C-terminal partner of this fusion, GRB2, is a known RET adaptor protein and signaling mediator. We show that this fusion transforms cells to growth factor independence, confirming its oncogenic driver role. Further, we demonstrate that both the kinase activity of RET and GRB2’s positioning downstream of RET are necessary for the fusion’s transforming potential and that the growth of RET::GRB2-expressing cells is readily inhibited by selpercatinib and pralsetinib.
Results
To discover novel driver events, we performed whole-transcriptome sequencing (RNA sequencing [RNA-seq]) of 30 pheochromocytomas and related tumors, paragangliomas (PPGLs), and focused on identification of chimeric oncogenic fusions (STAR Methods). Table S1 describes the key features of the cohort (Table S1). In PPGLs without a known genetic driver event, we detected 21 fusions with a minimum of three reads supporting the putative junction and at least one read spanning two genes (Table S2). Three tumors shared the same fusion transcript, UBTF-MAML3 (Figure S1), previously reported in PPGLs.11 Another pheochromocytoma was predicted to carry a fusion involving the RET and GRB2 genes. The putative RET::GRB2 fusion was supported by 492 junction reads and 18 reads spanning both genes (Figures 1A and 1B) and included the first 18 exons of RET, shared by the two main RET transcripts (NCBI: NM_020975.6 and NM_020630.6), fused in frame with exons 3–6 of the longest GRB2 transcript (Ensembl: ENSG00000177885.13, NCIB: NM_002086.5).
Figure 1.

RET::GRB2 fusion is detected as a somatic event in a pheochromocytoma
(A) Representation of the region spanning the RET exon 18 (left panel) and GRB2 exon 3 (right panel) in a pheochromocytoma predicted to carry the RET::GRB2 fusion, indicating the number of spanning reads (n = 18) and junction reads (n = 492) detected by RNA-seq. Chromosome (chr) location is indicated for each gene.
(B) Chromosomes (chrs) plotted as ideograms around the outside of the circle (CIRCOS) plot depicting the putative recombination between RET (chr10) and GRB2 (chr 17). The thickness of the connecting line represents the depth of read coverage.
(C) Hematoxylin and eosin (H&E) staining of the primary composite tumor comprised of a predominant pattern of pheochromocytoma (right) combined with a focal area of ganglioneuroma (left); magnification 100×, scale bar, 100 μm (inset, 400×, scale bar, 20 μm).
(D) Agarose gel of PCR products spanning the RET::GRB2 fusion transcript in tumor but not matched blood (leukocyte) cDNA; two other paired blood/tumor cDNA samples from patients with a RET mutation and a SDHB germline mutation, respectively, show no product (top); housekeeping gene TBP is shown (bottom). These results were repeated three times.
(E) Sanger sequencing of the product shown in (D), displaying RET sequence (exon 18) merged with GRB2 (exon 3). This experiment was repeated three times.
(F) RET (left) and GRB2 (right) mRNA expression obtained from high-throughput sequencing (HT-seq) counts of RNA-seq from 30 pheochromocytomas/paragangliomas (PPGLs; dots), depicting an exon within the fusion (x axis) and one outside the fusion (y axis). RET::GRB2-positive tumor is indicated in blue.
(G) DNA-based quantitative real-time PCR assay for copy-number assessment of the RET and GRB2 genes using primers targeting the area within (x axis) or outside the fusion (y axis) of a set of 14 PPGLs of various genotypes (dots). This experiment was repeated three times in duplicate samples.
This tumor derived from a 52-year-old African American male, who had been diagnosed with a 3.5 cm right composite pheochromocytoma, with a focal ganglioneuroma component (Figure 1C). The patient had elevated plasma and urine metanephrines (5- to 7-fold increase) and normetanephrines (1.5- to 3-fold increase), no clinical features of MEN 2A or MEN 2B, no prior radiation exposure, and no family history of PPGL. Nearly 3 years after the pheochromocytoma resection, there was no evidence of recurrence or metastasis. No mutations of known PPGL genes or other Tier 1 cancer genes were identified in germline DNA, tumor DNA, or tumor RNA from this patient.
We confirmed the RET::GRB2 fusion transcript by RT-PCR (Figure 1D) and Sanger sequencing of the tumor cDNA (Figure 1E) but not leukocyte cDNA, establishing its somatic status (Figure 1D). We extended this analysis to four other PPGLs with composite features but did not detect the fusion transcript (Figures 1D and S2A). A putative reciprocal GRB2::RET fusion transcript was not detected in the tumor with RET::GRB2 fusion. We were unable to unequivocally determine whether the fusion originated from the pheochromocytoma, the focal ganglioneuroma component, or both, as the frozen tumor sample used for RNA-seq was not obtained from the same area reflected in Figure 1C containing both histologies. Our attempts to obtain RNA from these distinct histological components in archival sections did not yield any usable sample.
The tumor carrying the RET::GRB2 fusion showed imbalanced expression between exons spanned by the fusion when compared with exons not involved in the rearrangement, as determined by analysis of our RNA-seq cohort (Figure 1F) or another set of PPGLs (Figure S2B). Moreover, combined data from the RNA-seq cohort and the TCGA pheochromocytoma-PPGL tumor set11 showed that the RET::GRB2 fusion-positive tumor displayed RET transcription levels within the range of RET mutant PPGLs and higher than those of RET wild-type tumors (Figure S2C). In addition, the RET::GRB2 fusion-expressing tumor was the top expressor of GRB2 mRNA among the 208 tumors in both cohorts (Figure S2C). To determine if the fusion was associated with copy-number changes, a DNA-based, quantitative real-time PCR assay was developed and revealed no evidence of amplification of RET or GRB2 genes within the area spanning the fusion (Figure 1G), suggesting that the overexpression of the RET::GRB2 transcript was not driven by accompanying genomic amplification of the two partner genes and thus differed from PPGLs with UBTF-MAML3 fusions or some cases of RET mutations, in which copy gain is observed.11
The putative RET::GRB2 fusion is predicted to produce a protein with 1,204 amino acids that retains the RET extracellular, transmembrane, and entire kinase domains until arginine 1,013 but lacks amino acids of the C-terminal tail (Figure 2A). The RET C terminus encodes several functionally relevant tyrosine residues and diverges after glycine 1,063 into its two main functional isoforms, RET9 and RET51 (Figure 2A).1 The RET::GRB2 fusion occurs upstream of the RET branching in its two isoforms (Figure 2A). The downstream component of the fusion encompasses an in-frame GRB2 sequence, except for its first 26 amino acids, including half of the proximal SH3, the entire SH2 domain, and the full distal SH3 domain (Figure 2A). Using protein lysates from PPGLs and an antibody directed at RET’s extracellular domain, which is preserved in the fusion, we detected a product of ∼180 kDa in the tumor carrying the RET::GRB2 fusion, compatible with the predicted size for the chimeric product (Figure 2B). This size is similar to glycosylated RET, the main detectable form of this protein.1,12 RET levels in the fusion tumor were comparable to PPGLs with RET mutations and higher than tumors with an intact RET sequence (Figure 2B). In contrast, an antibody recognizing the RET C terminus detected high levels of RET expression in PPGLs with a RET mutation, while the RET::GRB2 fusion sample had virtually undetectable levels (Figure 2B), suggesting that the main source of RET in this tumor is the fusion product rather than the wild-type RET allele. All PPGLs expressed the endogenous, 28 kDa GRB2 protein (Figure 2B). However, the tumor carrying the RET::GRB2 fusion displayed an additional GRB2 band of the expected fusion size (Figure 2B). These data confirmed the expression of the RET::GRB2 fusion protein in this pheochromocytoma. Moreover, RET phosphorylation at tyrosine 905 as well as phosphorylation of AKT and ERK were detected in the tumor carrying the RET::GRB2 fusion, similar to or at higher levels than two RET mutant pheochromocytomas (Figure 2B). Taken together, these findings suggest that the RET::GRB2 protein product is expressed and is associated with RET activation, supporting its driver role in this pheochromocytoma.
Figure 2.

Validation of the RET::GRB2 fusion protein in the pheochromocytoma and in vitro
(A) Diagram of wild-type (WT) RET, displaying relevant domains as indicated (CadLD, cadherin-like domains; CRD, cysteine-rich domain; TM, transmembrane domain; TKD, tyrosine kinase domain). The two main RET isoforms diverge at amino acid (aa) 1,063 (∗) with either 51 (RET51) or 9 (RET9) distinctive aa at the C terminus; WT GRB2 contains two SH3 domains flanking one SH2 domain. The RET (R1013) and GRB2 (V27) breakpoint sites are indicated; full-length RET::GRB2 fusion spans 1,204 aa.
(B) Western blot of protein lysates from PPGLs carrying mutations in RET, SDHB, VHL, and EPAS1 genes, and the RET::GRB2 fusion, probed with phosphorylated (P) RET (Y905) and two distinct total (T) RET antibodies directed at the extracellular, N-terminal (N-term) region around D320 and C-terminal (C-term) region beyond aa 1,100. GRB2 shows both the WT and fusion product, P- and T- AKT; ERK1/2, β-actin is a loading control; two technical replicates were performed.
(C) SH-SY5Y cells stably expressing RET9, RET51, RET::GRB2, and a control vector, probed with RET, GRB2, and α-tubulin as a loading control; three biological replicates were performed.
(D) SH-SY5Y cells expressing RET::GRB2 or RET51 were starved of serum for 3 h and exposed to 100 ng/mL GDNF (+) or vehicle (−) for 10 min, and lysates were probed with P-RET Y905 or T-RET; α-tubulin is a loading control; experiments were repeated three times.
(E) SH-SY5Y cells expressing RET::GRB2, RET 9, or RET51 constructs were treated with GDNF as in (D). Lysates were probed with GRB2 and T- and P- AKT, ERK1/2, T RET, and RET phosphorylated at Y1062, a region excluded from the RET::GRB2 fusion; α-tubulin is a loading control; experiments were repeated three times.
(F) Confocal microscopy of HEK293T cells expressing WT, mutant (C634R) RET, or RET::GRB2 fusion, labeled with a tag antibody in green (MYC for WT and C634R RET or hemagglutinin [HA] for RET::GRB2 fusion) and a membrane marker, Na/K ATPase (red). Nuclei are stained with DAPI (blue). A merged image is shown in the right panels. Scale bar: 10 μm.
(G) Quantification of the colocalized signals between RET and the Na/K ATPase using ImageJ from multiple independent images (n = 10–13 cells/genotype, two biological replicates). One way ANOVA, p = 0.048; ∗p = 0.02, two-tailed t test; ∗∗p = 0.0042, two-tailed t test.
(H) Uniform manifold approximation and projection (UMAP) plot of RNA-seq data from pheochromocytomas/PPGLs of our cohort (n = 30, UTHSA) and TCGA (n = 178), color-coded by genotype; gray symbols are tumors with unknown mutations; RET::GRB2 fusion (red triangle).
Next, to model the specific role of the RET::GRB2 fusion in vitro, we designed lentiviral constructs containing the full-length RET::GRB2 fusion sequence and RET constructs containing the two C-terminally distinct isoforms RET51 and RET9 (Figure 2A) as controls. Expression of the RET::GRB2 fusion construct in neural-crest-derived SH-SY5Y cells yielded the expected protein products recognized by RET and GRB2 antibodies (Figure 2C). These cells have been shown to respond to the RET ligand GDNF by inducing phosphorylation and activation of RET and its downstream effectors.13 Unlike SH-SY5Y cells expressing wild-type RET51, the levels of phosphorylated RET at tyrosine 905 were similarly high at baseline and after GDNF exposure in cells expressing RET::GRB2 (Figure 2D). These results suggest that the RET::GRB2 fusion leads to constitutive, ligand-independent RET activation.
A main docking residue for RET signaling is phospho-tyrosine 1062 (Y1062), which is shared by RET9 and RET51 (Figure 2A). This is one of the key recruitment sites for multiple signaling proteins that contain an SRC homology 2 (SH2)-binding domain or phosphotyrosine binding domains and mediates activation of downstream pathways, including PI3K/AKT and ERK/MAPK.1 Y1062 is retained in typical RET fusions, where RET is the 3′ prime partner,1,3 and also in the RET::SEPTIN9 fusion described in a pheochromocytoma.10 In contrast, this site is abolished by the RET::GRB2 fusion, which contains only the first 1,013 amino acids of RET. Accordingly, we were unable to detect phosphorylated Y1062 both at baseline and after GDNF exposure, while this phosphorylation is expectably detectable in cells expressing wild-type (WT) RET9 and RET 51 (Figure 2E). Despite the absence of Y1062, RET::GRB2-expressing cells increased AKT and ERK phosphorylation, as shown in Figure 2E, suggesting that activation of RET canonical effectors is maintained in these cells and that GRB2 sequences present in the fusion might circumvent the need for the Y1062 docking site and contribute to the activation of RET downstream signaling.
Next, to gain insight into the mechanisms of RET activation by this fusion, we used confocal microscopy to examine the subcellular localization of RET::GRB2. WT RET localizes to multiple endomembrane organelles, including the endoplasmic reticulum, Golgi, endosomes, and plasma membrane (PM).1 In contrast, RET carrying a pathogenic mutation, such as RET-C634R, has enhanced PM expression relative to endomembrane.14 In HEK293 T cells, the RET::GRB2 fusion showed higher co-localization with the PM marker Na/K ATPase than both WT and RET-C634R (Figures 2F and 2G), in line with a profile of higher surface expression and constitutive RET activation.14
To investigate the effects of the RET::GRB2 fusion in an unbiased manner, we compared the transcriptome of the tumor carrying this fusion with other PPGLs from our cohort (UTHSA) and the TCGA dataset, which includes 17 RET mutants.11 Uniform manifold approximation and projection (UMAP) analysis showed clustering of the RET::GRB2 fusion tumor with PPGLs carrying RET mutations (Figure 2H), supporting their similarities. We also used gene set enrichment analysis (GSEA) of differentially expressed genes (DEGs) between the RET::GRB2 sample and the RET mutants (Table S3) to find differential pathways and found transcription regulation enrichment in the fusion sample (Table S4). Taken together, high expression of the fusion transcript and protein along with enhanced membrane surface localization suggest potential mechanisms for the constitutive RET-related signaling activation resulting from this fusion, although other mechanisms cannot be excluded.
Next, to determine if the RET::GRB2 fusion had transforming capability, we used a well-established model, Ba/F3 cells, a pro-B bone-marrow-derived cell type that requires interleukin 3 (IL-3) for growth and which becomes IL-3 independent upon oncogenic transformation.15,16 We generated Ba/F3 cells stably expressing the RET::GRB2 construct (Figure 3A) and a kinase-dead (KD) version of this fusion, RET::GRB2 KD, carrying a K758 M mutation known to render RET catalytically inactive.12,17 In addition, to evaluate whether RET alone was sufficient for transformation, we generated a construct containing a truncated version of RET spanning amino acids 1–1,013 that comprised the RET component of the fusion (Figure 3A). We also generated cells expressing the transforming RET C634R mutant as positive control, while parental Ba/F3 cells served as negative control (Figure 3A). Stable ectopic RET expression in Ba/F3 cells was confirmed at the protein (Figures 3B and S3A) and mRNA (Figure S3B) levels. We found that RET:GRB2-expressing cells and, as expected, RET C634R cells grew independently of IL-3, indicating that these variants display transforming ability and support the oncogenic role of the RET::GRB2 fusion (Figure 3C). In contrast, KD RET::GRB2 and truncated RET were unable to proliferate in the absence of IL-3, similar to parental Ba/F3 cells (Figure 3C). These findings suggest that RET kinase function is required for the oncogenic features of the fusion. In addition, the lack of IL-3-independent growth of the cells expressing truncated RET further supports GRB2 as an active contributor to the transforming properties of this fusion.
Figure 3.

RET::GRB2 fusion is oncogenic and sensitive to clinical grade RET inhibitors
(A) Constructs used to evaluate transforming activity in Ba/F3 cells: (domains are depicted as in Figure 2A); RET-C634R, pathogenic RET mutant; RET truncated, first 1,013 amino acids of RET, containing only the RET component of the fusion; RET::GRB2, contains the RET::GRB2 fusion; RET::GRB2-KD, contains the kinase-dead version of the RET::GRB2 fusion carrying a K758 M mutation in the RET catalytic domain.
(B) Western blot of lysates from Ba/F3 cells stably expressing the constructs indicated in (A), along with parental cells, probed for total RET, and α-tubulin as a loading control; these experiments were repeated 3 times.
(C) Growth rate of Ba/F3 cells stably expressing the constructs indicated in (A) and (B), and parental cells cultured in the absence of interleukin 3 (IL-3). Cells were plated in triplicate and counted daily for 4 days; experiments were repeated 3 times. ∗∗∗∗p < 0.0001, two-way ANOVA.
(D) IC50 concentration-response curves to selpercatinib at doses of 0, 6.25, 12.5, 25, 50, 100, and 400 nM for 72 h measuring inhibition of growth of Ba/F3 cells expressing the RET::GRB2 fusion (4.1 nM, 95% CI, 3.4–4.9 nM) or RET-C634R mutant (29.9 nM, 95% CI, 21.2–42.6 nM) seeded in triplicate per dose and repeated three times.
(E) Lysates from Ba/F3 cells expressing RET::GRB2 or RET-C634R treated with 25 or 100 nM selpercatinib or vehicle for 4 h were probed with P-RET (Y905), and α-tubulin as loading control; three biological replicates were performed. s.e. and l.e. indicate short and long immunoblot exposure, respectively.
Lastly, we evaluated the sensitivity of the RET-transformed Ba/F3 cells to selpercatinib, a clinical grade RET inhibitor. We found that RET::GRB2 cells were highly sensitive to this drug, with a half-maximal inhibitory concentration (IC50) of 4.1 nM (3.4–4.9, 95% confidence interval [CI]); RET-C634R’s IC50 was 29.9 nM (21.2–42.9 nM, 95% CI; Figure 3D), similar to reported values of other responsive RET mutants.18 Consistent with the oncogenic role of active RET, growth suppression by selpercatinib led to marked inhibition of RET phosphorylation in these cells (Figure 3E). In contrast, selpercatinib was largely ineffective in the presence of IL-3 and did not inhibit the growth of parental Ba/F3 cells or cells expressing RET-truncated RET:GRB2-KD or RET-C634R (Figure S3C). Under these conditions, selpercatinib was also significantly less effective towards RET::GRB2-expressing cells, with an IC50 >20-fold higher (Figure S3C) than when these cells grew without IL-3 (Figure 3C). These results further validate the well-established specificity of selpercatinib effects18,19 and suggest that Ba/F3 cells expressing the RET::GRB2 fusion become over-reliant on RET signals for survival. In keeping with the selpercatinib response, RET::GRB2 cells were also sensitive to a second RET inhibitor, pralsetinib (Figure S3D). Also in line with these results, in a separate cell model, treatment of RET::GRB2-expressing SH-SY5Y cells with selpercatinib led to decreased RET phosphorylation (Figure S3E) and induction of cleaved PARP (Figure S3F), indicative, respectively, of RET signaling inhibition and apoptosis induction characteristic of responsive mutants.18 These results suggest that the RET::GRB2 fusion is sensitive to RET inhibitors.
Discussion
Here, we show that a RET::GRB2 gene fusion found in a pheochromocytoma constitutively activates RET downstream signaling and has transforming properties typical of oncogenic events. As pheochromocytomas carry single, mutually exclusive genetic driver events,20 our findings support this fusion as the molecular basis for this tumor’s development.
The recently reported RET::SEPTIN9 fusion in a metastatic pheochromocytoma10 is relevant to our findings. This fusion had a similar configuration to RET::GRB2 by positioning RET as the 5′ partner, and the patient showed marked clinical response to selpercatinib, in favor of the fusion playing a driver role and being therapeutically sensitive to RET inhibition, similar to our in vitro data of RET::GRB2. Despite these similarities, there are some key differences between RET::SEPTIN9 and RET::GRB2. First, the fusion partners are structurally and functionally distinct proteins, with only GRB2 being a known RET interacting partner.17 Secondly, the breakpoint site on RET is distinct: in RET::SEPTIN9, the RET breakpoint occurs after Y1062, potentially enabling recruitment of natural RET adaptors to promote signaling of canonical partner molecules, similar to conventional 3′ RET fusions.3 In contrast, in the RET::GRB2 fusion, the breakpoint occurs after R1013, and thus key docking sites of the RET C terminus are lost (Figure 3A). Yet, despite the lack of C-tail signals, RET::GRB2 confers overexpression, membrane localization, RET autophosphorylation, and activation of downstream signaling. Our results indicate that both RET catalytic activity and the presence of the GRB2 sequence are required for this fusion’s oncogenic properties. Fusion to GRB2 may preserve the ability to recruit canonical RET binding proteins to enable activation of downstream effectors.21,22 Interestingly, GRB2 has not been previously reported as a component of oncogenic fusions other than by rare in silico predictions without additional experimental verification.23
The fact that both RET::GRB2 and RET::SEPTIN9 carry RET at the 5′ prime position suggest that this anatomy might favor growth and transformation of adrenomedullary cells that give rise to pheochromocytomas. However, the true frequency of RET 5′ recombinations in cancer is unknown, as current fusion diagnostic platforms routinely focus on hot-spot RET intronic breakpoints involving introns 11 and 12,24 which would not have detected RET::GRB2 or RET::SEPTIN9 events.10 Notably, neither SEPTIN9 nor GRB2 possess coiled-coil domains, a frequent feature of the more common fusion partners of RET in lung and thyroid cancers.3 It remains to be determined whether this RET fusion configuration is exclusive to pheochromocytomas or if it can also be detected in other neural-crest-derived tumors or, even more broadly, in other cancers. Future studies should determine whether RET::GRB2, similar to other RET fusions, might also evolve as a potentially actionable resistance mechanism in cancers that develop resistance to other targeted therapies.3,6,24,25 Our findings, and those of Mweempwa et al., support RET fusion as a PPGL driver event with potential for therapeutic inhibition. These results highlight the importance of RET fusion testing in other driver-negative PPGLs.
Limitations of the study
The short-term follow up of the patient presented here limits our ability to predict the degree of aggressiveness of this tumor and determine the impact of RET::GRB2 on clinical outcome. Moreover, only upon recurrence would the tumor’s sensitivity and the clinical efficacy of RET inhibitors be verified. However, the sensitivity of RET::GRB2 to selpercatinib and pralsetinib in vitro suggests possible therapeutic opportunities for carriers of this fusion should they progress to metastatic disease, as did the patient with the RET::SEPTIN9 fusion. Our experiments support GRB2 contribution to the oncogenic features of the fusion. However, it remains to be defined whether GRB2 simply provides the structural motifs related to physiological RET downstream signaling21,22 or if the RET:GRB2 protein contributes to oncogenesis by hijacking other growth signals.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| phosphorylated RET-Y905 | Cell Signaling Technology | Cat#3221; RRID: AB_2179887 |
| phosphorylated RET-Y1062 | Abcam | Cat#ab51103; RRID: AB_870738 |
| RET E1N8X | Cell Signaling Technology | Cat#14556; RRID: AB_2798509 |
| RET | Abcam | Ab134100; RRID: AB_2920824 |
| phosphorylated AKT-S473 | Cell Signaling Technology | Cat#9271; RRID: AB_329825 |
| AKT | Cell Signaling Technology | Cat#9272; RRID: AB_329827 |
| phosphorylated ERK T202/Y204 | Cell Signaling Technology | Cat#4377; RRID: AB_331775 |
| ERK1/ERK2 | Cell Signaling Technology | Cat#4695; RRID: AB_390779 |
| GRB2 | Cell Signaling Technology | Cat#36344; RRID: AB_2920901 |
| PARP | Cell Signaling Technology | Cat#9542; RRID: AB_2160739 |
| β-actin | Cell Signaling Technology | Cat#3700; RRID: AB_2242334 |
| α-tubulin | Cell Signaling Technology | Cat#3873; RRID: AB_1904178 |
| Purified anti-HA.11 Epitope Tag Antibody | BioLegend | Cat#901501; RRID: AB_2565006 |
| Anti-Myc/c-Myc Antibody (9E10) | Santa Cruz Biotechnology | Cat#sc-40; RRID: AB_2857941 |
| Na,K-ATPase α1 (D4Y7E) Rabbit mAb | Cell Signaling Technology | Cat#23565; RRID: AB_2798866 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | Cat#A-11001; RRID: AB_2534069 |
| Alexa Fluor™ 647 Goat Anti-Rabbit SFX Kit, highly cross-adsorbed | Invitrogen | Cat#A31634 |
| Anti-Ret Antibody (8D10C9) | Santa Cruz Biotechnology | Cat#sc-101422; RRID: AB_2269605 |
| Bacterial and virus strains | ||
| ONE SHOT STBL3 COMP E COLI | Invitrogen | Cat#C737303 |
| Biological samples | ||
| Human tumor samples | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| RPMI (cell medium) | Corning | Cat#10040-CV |
| Fetal Bovine Serum | Gibco | Cat#26140079 |
| DAPI (4′,6-Diamidino-2-Phenylindole, Dilactate) | Thermo Fisher Scientific | Cat#EN62248 |
| TrypLE Select, 10× | Thermo Fisher Scientific | Cat#A1217701 |
| Hygromycin B | Invitrogen | Cat# 10687010 |
| Puromycin dihydrochloride | Gibco | Cat#A1113803 |
| Blasticidin | Gibco | Cat#R21001 |
| Selpercatinib | ChemieTek | Cat#CT-LX292 |
| Pralsetinib | ChemieTek | Cat#CT-BLU667 |
| Human GDNF (glial-derived neurotrophic factor) | Preprotech | Cat#450-10-100UG |
| Recombinant Murine IL-3 | Preprotech | Cat#213-13 |
| Critical commercial assays | ||
| NEBNext® Ultra TM RNA Library Prep Kit for Illumina | New England BioLabs | Cat#E7760L |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat#4368814 |
| Phusion High-Fidelity PCR Master Mix with GC Buffer | Thermo Fisher Scientific | Cat#F532L |
| iQ SYBR Green Supermix | Bio-Rad | Cat#1708880 |
| Deposited data | ||
| RET::GRB2 ORF sequence | This paper | NCBI-Bankit (accession number: ON799207) |
| Experimental models: Cell lines | ||
| 293FT Cell Line | Thermofisher/Invitrogen | Cat#R70007 |
| SH-SY5Y | ATCC | Cat#CRL-2266 |
| Ba/F3 | Creative Biogene | Cat#CSC-RO0120 |
| Oligonucleotides | ||
| Primer: RET-GRB2 long isoform_F Forward: TTGCGGACATCAGCAAAG | This paper | N/A |
| Primer: RET-GRB2 long isoform_F Reverse: CAGGAGCGCTCTCACTCTCT | This paper | N/A |
| Primer: RET-GRB2 short isoform_F Forward: TGCGGACATCAGCAAAGAC | This paper | N/A |
| Primer: RET-GRB2 short isoform_R Reverse: GAACTTCACCACCCAGAGG A | This paper | N/A |
| Primer RETe4F TGGTGATGGTGCCCTTCC | This paper | N/A |
| Primer RETe5R CTGATGCAGGTACCACGTCT | This paper | N/A |
| Primer: RET_e8_cDNA_F Forward: GGCTGGAGTGTGAGGAGTGT | This paper | N/A |
| Primer: RET_e9_cDNA_R Reverse: AGGTCTTGGTGCTGGGAGA G | This paper | N/A |
| Primer: RET_e19_cDNA_F Forward: CTGGTGGACTGTAATAATGC | This paper | N/A |
| Primer: RET_e20_cDNA_R Reverse: TTGGATATCTTGGAAACCCA | This paper | N/A |
| Recombinant DNA | ||
| pLV[Exp]-Puro-EF1A>3x N-flag-RET_1-18_GRB2_2-3-4-5 | This paper | Custom order-By VectorBuilder VB210223-1137nqa |
| pLV[Exp]-Hygro- EF1A>hRET[NM_020975.6]/Myc | This paper | Custom order-By VectorBuilder VB210227-1059nqh |
| pLV[Exp]-Bsd- EF1A>hRET[NM_020630.6]/Myc | This paper | Custom order-By VectorBuilder VB210227-1062nya |
| pLV[Exp]-Puro-EF1A>[C-HA- RET_1-18-GRB2_2-3-4-5]/HA | This paper | Custom order-By VectorBuilder VB210227-1065xvg |
| pLV[Exp]-EGFP:T2A:Puro- EF1A>mCherry | VectorBuilder Inc | By VectorBuilder VB160109-10005 |
| pLV[Exp]-Puro- EF1A>[C-HA-RET_1-18-GRB2_2-3-4-5]/HA | This paper | Custom order-By VectorBuilder VB210227-1065xvg |
| psPAX2 | Addgene | Addgene #12260; RRID:Addgene_12260 |
| pMD2.G | Addgene | Addgene #12259; RRID:Addgene_12259 |
| pLV-RET51-c-Myc-C634R | This paper | N/A |
| pLV-RET51-c-Myc-K758M | This paper | N/A |
| pLV-RET9-c-Myc-D1014X | This paper | N/A |
| pLV-RETGRB2-K758M-HA | This paper | N/A |
| Software and algorithms | ||
| Spliced Transcripts Alignment to a Reference (STAR) | Dobin et al., 201326 | http://code.google.com/p/rna-star/ |
| HTSeq v.0.6.1 | Putri et al., 202127 | http://www-huber.embl.de/HTSeq |
| Gene Set Enrichment Analysis (GSEA) | Subramanian et al., 200528 | https://www.gsea-msigdb.org/gsea/index.jsp |
| ImageJ | Abramoff et al., 201229 | https://imagej.nih.gov/ij/ |
| MiSeq | Illumina | https://www.illumina.com/systems/sequencing-platforms/miseq.html |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Patricia L.M. Dahia (dahia@uthscsa.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed materials transfer agreement.
Experimental model and subject details
Human samples
Samples from 52 pheochromocytomas and paragangliomas (PPGLs), diagnosed using standard procedures and confirmed histologically, were obtained from discarded tumor material from surgery and blood or saliva from patients who provided informed consent to participate in our repository study (NCT03160274), approved by the University of Texas Health Science Center at San Antonio institutional review board (IRB). This repository is open for enrollment of patients with pheochromocytoma and paraganglioma regardless of age, gender, ethnicity, race or genotype.
Cell lines
HEK-293FT cells (obtained from Thermofisher/Invitrogen Cat#R70007) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin in a humidified 37°C incubator with 5% CO2.
SH-SY5Y neural crest derived human cell line of neuroblastoma lineage, a gift from Dr. Lois Mulligan, Dr Lois Mulligan and her lab at Queens University, Kingston, Ontario, Canada, originally obtained from ATCC (Cat#CRL-2266) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin in a humidified 37°C incubator with 5% CO2.
Ba/F3 murine pro-B bone marrow derived cell lines were obtained from Dr. Ricardo Aguiar16 (University of Texas Health Science Center at San Antonio). Cell lines were authenticated using MCLab services (Molecular Cloning Labs, San Francisco, California). Ba/F3 were cultured in RPMI, 10% FBS, pen-strep, and 10 ng/mL murine interleukin 3 (IL3).
Method details
Nucleic acid isolation
DNA from germline, frozen tumors or cell lines was isolated using standard method by Qiagen Genomic-Tip (Qiagen, Valencia, CA). All samples from human subjects underwent genetic screening for pheochromocytoma/paraganglioma susceptibility genes.30 A summary of the general features of these samples is shown on Table S1.
Total RNA was isolated using TRIzol reagent kit following the manufacturer’s protocol (Invitrogen).
RNA-seq and fusion detection analysis
Total RNA obtained from 30 pheochromocytomas or paragangliomas was processed for RNAseq at Novogene (Beijing, China). Five of the 30 tumor samples had known mutations, VHL, TMEM127, SDHB, FH and EPAS1 genes, and were included as controls, the remaining were tumors without an identified driver event. Sequencing libraries were generated using NEBNext® Ultra TM RNA Library Prep Kit for Illumina® (NEB, USA) using 1 μg of total RNA per tumor, following manufacturer’s recommendations and paired end sequenced on a NovaSeq at Novogene. Raw FASTQ files were processed through fastp; reads containing adapter and poly-N sequences and reads with low quality from raw data were removed. Paired-end clean reads were aligned to the reference genome (hg38) using the Spliced Transcripts Alignment to a Reference (STAR) software.26 HTSeq v0.6.1 was used to count the read numbers mapped for each gene, and then normalized by gene length to generate FPKM (Reads Per Kilobase per Million).27 Fusion detection was performed using the STAR-Fusion software which maps split and discordant read alignments to reference gene annotations (STAR-Fusion predict), fusion predictions were further filtered using STAR- ‘Fusion filter’ to examine features of the fusion genes in the context of the individual fusion pair, and in comparison to other fusion predictions called in that sample to remove spurious fusions.31
Reverse Transcription Polymerase Chain Reaction (RT-PCR), and cDNA-based or genomic DNA-based quantitative real time PCR
Total RNA from paired leukocyte (n = 3) and PPGLs RNA (n = 51) were converted into cDNA using the High-Capacity cDNA Reverse Transcription kit and random hexamers (Thermo Fisher) according to the manufacturer’s instructions. Primer pairs were designed to span the RET::GRB2 fusion transcript, based on the predicted breakpoint to involve the first 18 exons of RET (common to both RET transcript isoforms) in frame with the last 4 exons of GRB2. Two distinct transcripts encoding longer (NM_002086.5, which we termed ‘isoform 216AA based on the length in amino acids of the translated product) and shorter (NM_203506.3, or 176AA) isoforms of GRB2 that differ by the presence or absence of exon 4, respectively, were distinguished by two separate reverse primers. PCRs were performed using thermal cycler (Bio-Rad T100) and Platinum Taq DNA Polymerase (Thermo Fisher) with the following conditions initial denaturing at 95°C, followed by 40 cycles of 98°C for 15 s, 54°C for 15 s and 59°C for 15 s, and 40 s extension at 72°C. Results were visualized on a 2% agarose gel. Sanger sequencing was performed by the Molecular Cloning Laboratories (MCLAB, San Francisco, CA).
For quantification of RET and GRB2 tumor mRNA, real time PCR assays were designed to measure RET and GRB2 transcription using primers spanning the region within (RET exons 8 and 9; GRB2 exons 3 and 4), and a separate region outside the fusion (RET exons 19 and 20; GRB2 exon 1) using SYBRGreen (Bio-Rad) in a QuantStudio6 instrument (Thermo Fisher). Expression levels were calculated using the delta-delta Ct method. Primer sequences are listed on STAR Methods.
We also designed a strategy to quantify RET and GRB2 copy number by quantitative real-time based PCR from tumor DNA. A pair of RET primers spanning an area included in the fusion (exon 11) and one pair spanning an exon downstream, excluded from the fusion (exon 19). Likewise, we designed primers spanning the exon 1 of GRB2 e1, not included in the fusion, and primers located in exons 4 and 5, belonging to the fusion product. Results were analyzed using by calculating the delta-delta method, as reported.32
Constructs and transductions
The following constructs were purchased from VectorBuilder Inc (Illinois, USA) using a lentiviral backbone under a EF1A promoter (pLV). Constructs were designed to encode the wild type RET 51 (NM_020975.6) and RET 9 (NM_020630.6) open reading frames carrying a MYC tag at the C-terminus, or the RET::GRB2 fusion containing RET exons 1–18 mRNA and GRB2 (NM_002086.5) exons 3-6 mRNA in frame, and containing a C-terminus HA tag, or the same fusion sequence with an N-terminus flag tag. A pLV-GFP-EF1A-mCherry was used as a control vector. We used site-directed mutagenesis with Phusion High-Fidelity DNA Polymerase (Thermo Fisher) to generate pLV-RET51-C634R, pLV-RET-1-1013AA (encoding RET truncated at amino acid 1014, the site of the fusion), pLV-RET::GRB2-K758M-HA (encoding the fusion product carrying a mutation on lysine 758, which renders RET catalytically inactive.17 A diagram depicting the various constructs is shown in Figure 3A. Virus generation was performed as we previously described.33 In brief, HEK-293 T cells were co-transfected with the pLV constructs above and the packing and envelop plasmids PAX2 (Addgene # 12260,) and pMD2.G (Addgene #12259), both gifts from Didier Trono. Parental SH-SY5Y cells were transduced with supernatant at 48 and 72 h and polybrene 8ug/mL, and Ba/F3 cells were transduced by spinoculation followed by antibiotic selection of polyclonal populations.16
Immunoblot analysis
Lysates were obtained from frozen pheochromocytomas and paragangliomas, and from the cell lines with a buffer containing 1% NP40, 50 mM Tris pH8.0,150 mM NaCl, 10% glycerol, 1 mM EDTA and Halt Protease and Phosphate Inhibitor (Thermo Fisher), mechanically disrupted, incubated on ice for 15 min and centrifuged at 4C for recovery of supernatant. Whole protein lysates were boiled in denaturing loading buffer(8% SDS, 0.4 M DTT, 0.2 M Tris-HCl, 4.3 M glycerol, 6 mM bromophenol blue), separated by SDS/polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride (PVDF) membrane at 110 V for 1-2 h at 4C.
Membranes were blocked in 5% skim milk and then probed at 4°C overnight with the following primary antibodies: phosphorylated RET-Y905 [Cell Signaling Technology (CST) 3221, 1:750 dilution], phosphorylated RET-Y1062 (Abcam ab51103, 1:750), RET E1N8X (CST 14556, 1:1500), RET (Abcam ab134100, 1:1200), phosphorylated AKT-S473 (CST 9271, 1:750), AKT (CST 9272, 1:1000), phosphorylated ERK T202/Y204(CST 4377, 1:1500), ERK1/ERK2 (CST 4695, 1:1500), GRB2 (CST 36344, 1:2000), PARP (CST 9542, 1:1000), β-actin (CST 3700, 1:2000), α-tubulin (CST 3873, 1:2000). Blots were developed with chemiluminescent detection (Millipore Kit, cat WBKLS0500). Image J (NIH) was used for quantification of scanned blots JPEG files.
TCGA data merge and clustering GSEA analysis
HTSeq raw counts from RNAseq data from 178 pheochromocytomas/paragangliomas TCGA dataset were downloaded from the GDC data portal (https://portal.gdc.cancer.gov/) and normalized to our RNAseq cohort of 30 independent pheochromocytomas/paragangliomas, including the tumor carrying the RET::GRB2 fusion were consolidated. Raw counts were processed to remove batch effect by ComBat_seq34 then normalized using trimmed mean of M values (TMM)35 and transformed to log2CPM. Data from RET gene and GRB2 gene counts were plotted for all samples, split between those with intact RET sequence (RET-WT) or RET disrupted, including 17 RET mutants and 1 RET::GRB2 fusion (RET-mutant). The entire dataset was used for uniform manifold approximation and projection (UMAP) plotting for visualization. Next, DeSeq2 was performed in data from 17 tumors carrying germline or somatic RET mutations vs. 1 RET::GRB2 to identify differentially expressed genes (DEGs) using logfold change with Padj<0.05 (Table S1). DEGs were used for a pre-ranked GSEA analysis against multiple datasets.28
Confocal microscopy
HEK293 T cells were transduced with pLV-RET-GRB2-HA (referred to as RET::GRB2), pLV-RET51-WT-MYC (referred to as RET-WT) and pLV-RET51-C634R-MYC (referred to as RET-C634R) constructs were seeded on glass coverslips and 48 h later the cells were fixed using 4% formaldehyde for 10 min after media removal and two phosphate buffered saline (PBS) washes. The cells were permeabilized using 0.1% Triton X-100 (10 min, room temperature). After 3 washes with PBS, the cells were blocked with 2% bovine serum albumin (BSA) for 30 min at room temperature. The cells were then stained for RET-GRB2 using anti-HA (Biologend, cat #901501,1:100, overnight at 4°C) or MYC (for tagged RET51-WT or RETC634R, Santa Cruz Biotechnology cat #SC-40, 1:100, 4°C) and the plasma membrane marker NA/K ATPase (Cell Signaling cat #23565, 1:50, 4°C). The cells were then washed with PBS and stained with anti-mouse Alexa488 antibody (1:100) and anti-rabbit Alexa647 antibody (1:100), along with DAPI (2.5 μg/mL) for 30 min in the dark at room temperature. All antibodies were diluted in 2% BSA. The cells were washed with PBS and mounted on glass slides with 50% glycerol in PBS. Cells were imaged using a ZEISS LSM710 confocal microscope (×40, oil immersion objective, DAPI excitation at 405 nm, Alexa488 and Alexa647 excitation at 488 and 647 nm, respectively). Quantification was performed as reported.36 In brief, plasma membrane signals from individual cells were selected by drawing a 3-pixel wide line along the Na/KATPase signal and obtaining the intensity values from Na/KATPase and HA or Myc signals using ImageJ. The average intensity values were then obtained from each channel, and a ratio of HA or MYC over Na/K ATPase was calculated from 7–13 fields containing 1-5 cells per field. ANOVA and two-tailed t-test were used to compare WT, C34R and RET::GRB2 ratios.
Transformation assays
Ba/F3 cells stably expressing RET::GRB2, RET::GRB2-K758 M (RET::GRB2-KD), RET51-C634R, RET truncated (RET 1-1013) constructs, or parental cells, were seeded at 2.5 × 105 per well in triplicate were incubated in RPMI 1640 medium (Invitrogen) with 10% FBS without IL3. For the next 4 days, cells were counted daily using an automated cell counter (Cellometer K2 Fluorescent Cell Counter, Nexcellom). Each assay was performed with three biological replicates.
Selpercatinib and pralsetinib growth and RET phosphorylation inhibition
Ba/F3 cells expressing RET::GRB2 or RET51-C634R were plated at 400,000 cells per well in triplicate in RPMI 1640 medium (Invitrogen) with 10% FBS without IL3 alone or containing the following doses of selpercatinib(Chemietek, Indianapolis) 6.25, 12.5, 25, 50, 100 and 200 nM for 72 h.7 Cell count was determined as above. Similar experiments and dose range were performed with pralsetinib (Chemietek, Indianapolis). Half-maximal inhibitory concentration (IC50) for proliferation curves was calculated for each drug.
Ba/F3 cells expressing RET::GRB2, RET::GRB2-K758 M (RET::GRB2-KD), RET51-C634R, RET truncated (RET 1–1013) were plated at 400,000 cells per well in triplicate in RPMI 1640 medium (Invitrogen) with 10% FBS without IL3 alone or containing the following doses of selpercatinib (Chemietek, Indianapolis) 6.25, 12.5, 25, 50, 100 and 200 nM for 72 h.7 Cell count was determined as above. Similar experiments and dose range were performed with pralsetinib (Chemietek, Indianapolis). Half-maximal inhibitory concentration (IC50) for proliferation curves was calculated for each drug.
Ba/F3 cells stably expressing RET::GRB2 or RET-C634% were seeded in equal number and maintained in IL3 minus media containing 25 nM or 100 nM selpercatinib or vehicle control for 4 h. Cells were harvested and lysates prepared as above and evaluated for phosphorylated RET (Y905).
SH-SY5Y cells stably expressing RET::GRB2, were seeded in equal number and maintained in full-media containing 50 nM selpercatinib. Cells were harvested and lysates prepared as above and evaluated for phosphorylated RET, total RET (4 h treatment) and PARP (24 h treatment) to evaluate RET activity and apoptosis, respectively.
Quantification and statistical analysis
Data analyses were performed with the GraphPad Prism software (version 9.2.0, GraphPad Software Inc) and Excel (Microsoft), with Student’s two-tailed t test or one-way ANOVA. p < 0.05 was considered significant. These analyses were applied to confocal microscopy measurements and cell growth curve.
IC50 for proliferation curves was calculated for each drug using a 4- parameter logistic nonlinear regression model in GraphPad.
Acknowledgments
We are grateful to the patients who donated samples to the study. We acknowledge the UTHSCSA Genomics and Optical Imaging Core Facilities for services, supported by NIH-P30-CA54174, and the South Texas Research Laboratory (STRL) Histology-Immunohistochemistry Laboratory at UTHSA and Ms. Dominie Cerda for processing histological samples. P.L.M.D. is a recipient of funds from the NIH (GM114102 and CA264248) and the Neuroendocrine Tumor Research Foundation and is the holder of the Robert Tucker Hayes Distinguished Chair in Oncology. This work was supported by funds from UT System Star Awards (to P.L.M.D.). R.C.T.A. acknowledges funding support from NIH R01ES031522, NIH R01GM140456, I01BX001882 (Veterans Administration Merit Award), and RP190043 (CPRIT).
Author contributions
C.M.E.-Z. conducted experiments and wrote manuscript draft; Z.-M.C., P.E., Q.G., H.G.-C., H.L., B.N.L., Y.D., and X.W. conducted experiments; E.A., A.Z., and N.A. performed clinical activities and collected data; R.C.T.A designed and supervised experiments and wrote the paper; and P.L.M.D. conceived the study, designed experiments, supervised the study, and wrote the paper.
Declaration of interests
The authors have no conflicts of interest to declare.
Inclusion and diversity
We worked to ensure gender balance, ethnic or other types of diversity in the recruitment of human subjects. We worked to ensure diversity in experimental samples through the selection of the genomic datasets. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science.
Published: July 19, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2022.100686.
Supplemental information
column 1 contains gene ID, column 2 contains the class difference weight
Data and code availability
-
•
RNA sequence data have been deposited at BankIt NCBI (accession number: ON799207).
-
•
This paper does not report original code
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Mulligan L.M. RET revisited: expanding the oncogenic portfolio. Nat. Rev. Cancer. 2014;14:173–186. doi: 10.1038/nrc3680. [DOI] [PubMed] [Google Scholar]
- 2.Dahia P.L.M. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat. Rev. Cancer. 2014;14:108–119. doi: 10.1038/nrc3648. [DOI] [PubMed] [Google Scholar]
- 3.Santoro M., Moccia M., Federico G., Carlomagno F. RET gene fusions in malignancies of the thyroid and other tissues. Genes. 2020;11:424. doi: 10.3390/genes11040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grieco M., Santoro M., Berlingieri M.T., Melillo R.M., Donghi R., Bongarzone I., Pierotti M.A., Della Ports G., Fusco A., Vecchiot G. PTC is a novel rearranged form of the ret proto-oncogene and is frequently expressed in vivo in human papillary thyroid carcinomas. Cell. 1990;60:557–563. doi: 10.1016/0092-8674(90)90659-3. [DOI] [PubMed] [Google Scholar]
- 5.Kohno T., Ichikawa H., Totoki Y., Yasuda K., Hiramoto M., Nammo T., Sakamoto H., Tsuta K., Furuta K., Shimada Y., et al. KIF5B-RET fusions in lung adenocarcinoma. Nat. Med. 2012;18:375–377. doi: 10.1038/nm.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ou S.-H.I., Zhu V.W. Catalog of 5′ fusion partners in RET+ NSCLC Circa 2020. JTO Clin. Res. Rep. 2020;1:100037. doi: 10.1016/j.jtocrr.2020.100037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thein K.Z., Velcheti V., Mooers B.H.M., Wu J., Subbiah V. Precision therapy for RET-altered cancers with RET inhibitors. Trends Cancer. 2021;12:1074–1088. doi: 10.1016/j.trecan.2021.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Subbiah V., Gainor J.F., Rahal R., Brubaker J.D., Kim J.L., Maynard M., Hu W., Cao Q., Sheets M.P., Wilson D., et al. Precision targeted therapy with BLU-667 for RET-driven cancers. Cancer Discov. 2018;8:836–849. doi: 10.1158/2159-8290.CD-18-0338. [DOI] [PubMed] [Google Scholar]
- 9.Wirth L.J., Sherman E., Robinson B., Solomon B., Kang H., Lorch J., Worden F., Brose M., Patel J., Leboulleux S., et al. Efficacy of selpercatinib in RET-altered thyroid cancers. N. Engl. J. Med. 2020;383:825–835. doi: 10.1056/NEJMoa2005651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mweempwa A., Xu H., Vissers J.H.A., Tothill R.W., Pattison A.D., Fellowes A.P., Thomas D.M., Richardson G., Hicks R.J., Grimmond S.M., et al. Novel RET fusion RET-SEPTIN9 predicts response to selective RET inhibition with selpercatinib in malignant pheochromocytoma. JCO Precis. Oncol. 2021;5:1160–1165. doi: 10.1200/po.21.00127. [DOI] [PubMed] [Google Scholar]
- 11.Fishbein L., Leshchiner I., Walter V., Danilova L., Robertson A.G., Johnson A.R., Lichtenberg T.M., Murray B.A., Ghayee H.K., Else T., et al. Cancer Genome Atlas Research Network Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell. 2017;31:181–193. doi: 10.1016/j.ccell.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hyndman B.D., Gujral T.S., Krieger J.R., Cockburn J.G., Mulligan L.M. Multiple functional effects of RET kinase domain sequence variants in hirschsprung disease. Hum. Mutat. 2013;34:132–142. doi: 10.1002/humu.22170. [DOI] [PubMed] [Google Scholar]
- 13.Hyndman B.D., Crupi M.J.F., Peng S., Bone L.N., Rekab A.N., Lian E.Y., Wagner S.M., Antonescu C.N., Mulligan L.M. Differential recruitment of E3 ubiquitin ligase complexes regulates RET isoform internalization. J. Cell Sci. 2017;130:3282–3296. doi: 10.1242/jcs.203885. [DOI] [PubMed] [Google Scholar]
- 14.Asai N., Iwashita T., Matsuyama M., Takahashi M. Mechanism of activation of the ret proto-oncogene by multiple endocrine neoplasia 2A mutations. Mol. Cell Biol. 1995;15:1613–1619. doi: 10.1128/mcb.15.3.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warmuth M., Kim S., Gu X-j, Xia G., Adrián F. Ba/F3 cells and their use in kinase drug discovery. Curr. Opin. Oncol. 2007;19:55–60. doi: 10.1097/cco.0b013e328011a25f. [DOI] [PubMed] [Google Scholar]
- 16.Bouamar H., Jiang D., Wang L., Lin A.P., Ortega M., Aguiar R.C.T. MicroRNA 155 control of p53 activity is context dependent and mediated by Aicda and Socs1. Mol. Cell Biol. 2015;35:1329–1340. doi: 10.1128/MCB.01446-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scott R.P., Eketjäll S., Aineskog H., Ibáñez C.F. Distinct turnover of alternatively spliced isoforms of the RET kinase receptor mediated by differential recruitment of the Cbl ubiquitin ligase. J. Biol. Chem. 2005;280:13442–13449. doi: 10.1074/jbc.M500507200. [DOI] [PubMed] [Google Scholar]
- 18.Subbiah V., Shen T., Terzyan S.S., Liu X., Hu X., Patel K.P., Hu M., Cabanillas M., Behrang A., Meric-Bernstam F., et al. Structural basis of acquired resistance to selpercatinib and pralsetinib mediated by non-gatekeeper RET mutations. Ann. Oncol. 2021;32:261–268. doi: 10.1016/j.annonc.2020.10.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Subbiah V., Shen T., Tetzlaff M., Weissferdt A., Byers L.A., Cascone T., Behrang A., Meric-Bernstam F., Mooers B.H.M., Rothenberg S.M., et al. Patient-driven discovery and post-clinical validation of NTRK3 fusion as an acquired resistance mechanism to selpercatinib in RET fusion-positive lung cancer. Ann. Oncol. 2021;32:817–819. doi: 10.1016/j.annonc.2021.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dahia P.L.M. Pheochromocytomas and paragangliomas, genetically diverse and minimalist, all at once. Cancer Cell. 2017;31:159–161. doi: 10.1016/j.ccell.2017.01.009. [DOI] [PubMed] [Google Scholar]
- 21.Besset V., Scott R.P., Ibáñez C.F. Signaling complexes and protein-protein interactions involved in the activation of the Ras and phosphatidylinositol 3-kinase pathways by the c-ret receptor tyrosine kinase. J. Biol. Chem. 2000;275:39159–39166. doi: 10.1074/jbc.M006908200. [DOI] [PubMed] [Google Scholar]
- 22.Alberti L., Borrello M.G., Ghizzoni S., Torriti F., Rizzetti M.G., Pierotti M.A. Grb2 binding to the different isoforms of Ret tyrosine kinase. Oncogene. 1998;17:1079–1087. doi: 10.1038/sj.onc.1202046. [DOI] [PubMed] [Google Scholar]
- 23.Gao Q., Liang W.W., Foltz S.M., Mutharasu G., Jayasinghe R.G., Cao S., Chen K., Lazar A.J., Fields R.C., Wendl M.C., et al. Driver fusions and their implications in the development and treatment of human cancers. Cell Rep. 2018;23:227–238.e3. doi: 10.1016/j.celrep.2018.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang S.R., Aypar U., Rosen E.Y., Mata D.A., Benayed R., Mullaney K., Jayakumaran G., Zhang Y., Frosina D., Drilon A., et al. A performance comparison of commonly used assays to detect RET fusions. Clin. Cancer Res. 2021;27:1316–1328. doi: 10.1158/1078-0432.CCR-20-3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li B., Qu H., Zhang J., Pan W., Liu M., Yan X., Huang X., He X., Lin D., Liu S., et al. Genomic characterization and outcome evaluation of kinome fusions in lung cancer revealed novel druggable fusions. npj Precis. Oncol. 2021;5:81. doi: 10.1038/s41698-021-00221-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Putri G.H., Anders S., Pyl P.T., Pimanda J.E., Zanini F. Analysing high-throughput sequencing data in Python with HTSeq 2.0. Bioinformatics. 2022;38:2943–2945. doi: 10.1093/bioinformatics/btac166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abràmoff M.D., Magalhães P.J., Ram S.J. Image processing with ImageJ. Biophot. Int. 2004;11:36–42. [Google Scholar]
- 30.Flores S.K., Deng Y., Cheng Z., Zhang X., Tao S., Saliba A., Chu I., Burnichon N., Gimenez-Roqueplo A.P., Wang E., et al. Functional characterization of TMEM127 variants reveals novel insights into its membrane topology and trafficking. J. Clin. Endocrinol. Metab. 2020;105:e3156. doi: 10.1210/clinem/dgaa396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haas B.J., Dobin A., Stransky N., Li B., Yang X., Tickle T., Bankapur A., Ganote C., Doak T.G., Pochet N., Sun J. STAR-fusion: fast and accurate fusion transcript detection from RNA-seq. bioRxiv. 2017 doi: 10.1101/120295. Preprint at. [DOI] [Google Scholar]
- 32.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 33.Srikantan S., Deng Y., Cheng Z.M., Luo A., Qin Y., Gao Q., Sande-Docor G.M., Tao S., Zhang X., Harper N., et al. The tumor suppressor TMEM127 regulates insulin sensitivity in a tissue-specific manner. Nat. Commun. 2019;10:4720. doi: 10.1038/s41467-019-12661-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y., Parmigiani G., Johnson W.E. ComBat-seq: batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform. 2020;2:lqaa078. doi: 10.1093/nargab/lqaa078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robinson M.D., Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Richardson D.S., Rodrigues D.M., Hyndman B.D., Crupi M.J.F., Nicolescu A.C., Mulligan L.M. Alternative splicing results in RET isoforms with distinct trafficking properties. Mol. Biol. Cell. 2012;23:3838–3850. doi: 10.1091/mbc.E12-02-0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
column 1 contains gene ID, column 2 contains the class difference weight
Data Availability Statement
-
•
RNA sequence data have been deposited at BankIt NCBI (accession number: ON799207).
-
•
This paper does not report original code
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.