

Abstract
The insect gut is frequently exposed to pathogenic threats and must not only clear these potential infections, but also tolerate relatively high microbe loads. In contrast to the mechanisms that eliminate pathogens, we currently know less about the mechanisms of disease tolerance. We investigated how well-described mechanisms that prevent, signal, control or repair damage during infection contribute to the phenotype of disease tolerance. We established enteric infections with the bacterial pathogen Pseudomonas entomophila in transgenic lines of Drosophila melanogaster fruit flies affecting dcy (a major component of the peritrophic matrix), upd3 (a cytokine-like molecule), irc (a negative regulator of reactive oxygen species) and egfr1 (epithelial growth factor receptor). Flies lacking dcy experienced the highest mortality, while loss of function of either irc or upd3 reduced tolerance in both sexes. The disruption of egfr1 resulted in a severe loss in tolerance in male flies but had no substantial effect on the ability of female flies to tolerate P. entomophila infection, despite carrying greater microbe loads than males. Together, our findings provide evidence for the role of damage limitation mechanisms in disease tolerance and highlight how sexual dimorphism in these mechanisms could generate sex differences in infection outcomes.
Keywords: disease tolerance, gut epithelial immunity, tissue damage repair, oral bacterial infection, enteric infection, infection dose
1. Introduction
Many insects thrive on decomposing and decaying organic matter containing a diversity of commensal and pathogenic microorganisms. Like most animals, insects have evolved diverse responses to infection, including behavioural avoidance of infection, physical barriers to pathogen entry and a variety of humoral and cellular immune responses [1–3]. These responses have been particularly well described in the fruit fly Drosophila melanogaster, where signalling pathways such as IMD and Toll are recognized as major contributors to pathogen clearance [1,2,4–7]. In addition to mechanisms that reduce pathogen burdens, it is increasingly recognized that mechanisms promoting disease tolerance are equally important during recovery to a healthy state [8–12]. Disease tolerance is defined as the ability of hosts to maintain health despite harbouring relatively high pathogen loads [9,10,13]. Implicit in this definition is that disease tolerance cannot be measured by assessing host health or pathogen growth separately, but is instead defined by their relationship [14]. While it is possible to compare the relative health of an individual for a given pathogen burden (also known as ‘point tolerance' or the ‘per-pathogen pathogenicity' [15,16]) a more common approach is to analyse how host health (often measured as mortality) changes across a range of pathogen burdens, known as range tolerance [13,15]. Range tolerance may present as a linear decline in health with increasing pathogen burdens, where groups of hosts with steep negative slopes indicate lower tolerance to increasing pathogen numbers compared with groups with shallower slopes [13,15]. Nonlinear declines of health are also known, reflecting potential threshold dynamics in health deterioration with increasing pathogen numbers [17–19]. The phenotype of disease tolerance has been observed in several species, including insects [17,20–23] rodents [8,24,25], birds [26,27] and humans [11,18,28].
The mechanisms of pathogen clearance are well described in many animal species [29], but we currently know less about the mechanisms underlying disease tolerance. Given that tolerance reflects the ability to maintain health independently of pathogen clearance, we might expect tolerance mechanisms to be related to processes such as detoxification, reduction of inflammation, or tissue damage control and cellular renewal [9,11,30]. Genome-wide association or transcriptomic studies in Drosophila have highlighted potential candidate genes underlying phenotypic variation in disease tolerance [21], but it remains unclear how many of these genes interact with known mechanisms of immunity and recovery.
Furthermore, almost all candidate genes for disease tolerance in Drosophila arise from systemic infections, where pathogens are introduced directly into the body cavity of the fly, resulting in a septic infection [21,31,32]. This leaves a gap in our knowledge about disease tolerance during orally acquired infections, which are especially relevant in the context of the ecology of most insects, that consume decaying organic matter containing a large diversity of potentially harmful microorganisms [33–35]. The insect gut is therefore frequently exposed to pathogenic threats and must be able not only to detect and clear these potential infections, but also be able to repair the resulting damage to gut tissues in order to tolerate relatively high numbers of ingested pathogens.
Here, we aimed to specifically test how well-established mechanisms that prevent, reduce or repair tissue damage contribute to the phenotype of disease tolerance. The Drosophila gut is a compartmentalized tubular organ which is structurally and functionally similar to the vertebrate intestinal tract [2,36–38]. We can consider several stages comprising gut defence in Drosophila (electronic supplementary material, figure S1). The first involves the physical barrier of the gut epithelia and the peritrophic matrix (PM), which is a layer of chitin and glycoproteins that lines the insect midgut lumen. The PM is functionally analogous to mammalian mucus membrane in the digestive tract and acts as the first line of defence against invading pathogens [2,39]. A major component of the PM is drosocrystallin (dcy). Loss-of-function mutations in dcy increase the permeability of the peritrophic matrix to larger molecules and allow leakage of microbial cells, including pathogens, into the haemolymph. Dcy-deficient flies therefore exhibit increased susceptibility to oral bacterial infections, and this has been shown in great detail during infection with the gram-negative bactrerium Pseudomonas entomophila [2,40].
Another mode of defence during gut infections is the production of reactive oxygen species (ROS) by the gut epithelia. For example, in response to ingested Pseudomonas entomophila, ROS production is induced by two NADPH enzymes- nox (NADPH oxidase) and duox (dual oxidase), while irc (immune-reactive catalase) negatively regulates ROS production once the infection threat is controlled, which otherwise, would lead to cytotoxic effects [2,41,42]. ROS production not only targets pathogens directly, but also plays additional roles in triggering signalling pathways that lead to the production of IMD- or Toll-responsive antimicrobial peptides [42–45].
The final stage in gut defence is to repair the damage caused during the infection. Damage-signalling cytokine-like molecules upd3 are released from damaged cells which trigger the Jak/Stat-pathway, stimulating the proliferation of intestinal stem cells (ISCs) and their differentiation into enterocytes (ECs) via egfr1 (epidermal growth factor receptor) signalling [46–48]. Flies lacking Jak/Stat or Egfr are therefore highly susceptible to bacterial infections due to their inability to repair and renew damaged tissue [46,48,49].
To investigate how these mechanisms of damage prevention (dcy), signalling (upd3) control (irc) and renewal (egfr) contribute to disease tolerance during gut infections we employed oral infections in Drosophila transgenic lines with loss-of-function in each of these genes on a common genetic background (w1118). We orally challenged these flies with three infection doses of Pseudomonas entomophila and then quantified their effects on survival, pathogen loads and disease tolerance responses during the period of peak infection burden.
2. Material and methods
(a) . Fly strains
The following fly stocks were obtained from the Bloomington Stock Centre, Indiana: dcy (w1118; Mi{ET1}CrysMB08319; FB#26106) [50], irc (w1118; Mi{ET1}IrcMB11278; FB#29191) [51], (#2079), upd3 (w1118; P{XP}upd3d11639; FB#19355) [52]. These lines were subsequently isogenized by backcrossing onto the same w1118 background (VDRC stock# 60000) for at least 10 generations. The egfrt1 mutant was a kind gift from Carla Saleh (Pasteur Institute, Paris) and previously isogenized to w1118 first by replacing the chromosomes not containing the mutation using balancer chromosomes and then by backcrossing at least 10 times to the same VDRC w1118 line [53]. The VDRC w1118 line was included as the control line in all experiments. All fly lines were maintained in plastic vials (12 ml) on a standard sugar-cornmeal medium [54] at a constant temperature of 25°C (±2°C) and on a 12 h : 12 h light : dark cycle. All experimental flies were mated, 3–5-day-old males and females.
(b) . Bacterial culture preparation
To test the impact of bacterial infection on fly survival, we used the gram-negative bacteria Pseudomonas entomophila (a kind gift from Ben Longdon in 2014), that was originally isolated from a wild D. melanogaster [41] is able to establish infection in broad range of insects and other invertebrates [55]. In flies, P. entomophila infection mainly occurs in the intestinal epithelium and eventually causes death [41]. To obtain bacterial cultures for oral exposure, we inoculated frozen isogenic bacterial stock cultures stored at −80°C onto fresh 15 ml LB broth (media composition) and incubated overnight at 37°C with shaking at 120 r.p.m. (revolutions per minute). The overnight cultures were diluted 1 : 100 into 500 ml of fresh LB broth and incubated again at 30°C with shaking at 120 rpm. At the mid-log phase (OD600 = 0.75), we harvested the bacterial cells by centrifugation at 5000 r.p.m. for 15 min and re-suspended the bacterial pellet in 5% sucrose [56]. The final inoculum was adjusted to three different bacterial concentrations or infection dose OD600 = 10 (low dose), OD600 = 25 (medium dose) and OD600 = 45 (high dose).
(c) . Experimental design
Measuring tolerance as a linear decline in survival with increasing pathogen growth requires collecting matching data on survival and pathogen loads, ideally from the same individual. However, this is challenging in the fly model because quantifying microbe loads requires destructive sampling. Instead, we considered the vial as the unit of replication, and employed a split-vial design (electronic supplementary material, figure S2). In total, we set up 500 infection vials, split across 2 experimental blocks (n = 10 vials for OD600 = 10 and OD600 = 45; n = 30 vials for OD600 = 25, for each combination of sex (2) per fly line (5)—with each vial containing 27–30 flies for all doses). Following oral bacterial exposure (see below) each vial containing 25 flies of each infection treatment, sex and fly line combination were split into two vials for measuring (1) survival following infection (15 flies per combination) and (2) internal bacterial load (10 flies per combination) (electronic supplementary material, figure S2). This split-vial design allowed us to use replicate-matched data for both the proportion of flies surviving and the average bacterial load for each replicate vial to estimate the linear relationship between fly survival and internal bacterial load for each fly line.
(d) . Oral infection and survival assay
Before infecting flies we prepared infection vials by pipetting 350 µl of standard agar (1 l triple distilled H2O, 20 g agar, 84 g brown sugar, 7 ml Tegosept anti-fungal agent) onto the lids of 7 ml tubes (bijou vials) and allowed it to dry. Simultaneously, we starved the experimental flies in 12 ml agar vials (1 l triple distilled H2O, 20 g agar) for 4–5 h. Once the agar in the bijou lids dried, we placed a filter disc (Whattmann-10) in the lid and pipetted 80 µl of bacterial culture directly onto the filter disc. For control (mock) infections, we replaced bacterial culture with a 5% sucrose solution. We then orally exposed flies inside the bijou vials for 18-hours and then transferred the flies onto fresh vials containing standard sugar-cornmeal medium [56]. No flies died during the exposure period. Following this exposure period, flies were checked for survival every 3–6 h (days 1–3) and then every 12 h until 13 days following the exposure period. During the survival assay, live flies were tipped into vials with fresh medium every 3 days.
(e) . Bacterial load measurement
To test whether variation in mortality of experimental flies after P. entomophila infection is explained by the ability to clear infection, we measured bacterial load using 3–5-day-old flies (w1118 and transgenic flies) following enteric infection with either OD600 = 10 (low dose), OD600 = 25 (medium dose) or OD600 = 45 (high dose) of P. entomophila. For OD600 = 25 (medium dose), we measured bacterial load at three timepoints (immediately after oral exposure 0–15 min, 24 h and 96 h) following P. entomophila infection, while other doses CFUs were only measured at 24 h following the infection period. To confirm oral bacterial infection, we thoroughly surface-sterilized flies (group of 3) with 70% ethanol for 30–60 s and then rinsed twice with sterile distilled water. We plated the second wash on LB agar plates and incubated overnight at 30°C to confirm that surface bacteria were successfully removed after alcohol sterilization. We transferred flies onto 1.5 ml micro centrifuge tubes and homogenized using a motorized pestle for approximately 30–60 s in 100 µl LB broth (n = 30 homogenates per sex per infection treatment per fly line). We performed serial dilution of each homogenate up to 10−6-fold and added 4 µl aliquot on an LB agar plate. After this, we incubated the plate overnight for 18-h at 30°C and counted the resultant bacterial colonies manually. We note that mock-infected control fly homogenates did not produce any colonies on LB agar plates [56].
3. Statistics
(a) . Survival following oral infection
We analysed survival data using a mixed effects Cox model using the R package ‘coxme' [57]. We specified the model as: survival∼fly line × treatment × sex + (1|vials/block), with ‘fly line', ‘treatment’ and ‘sex' and their interactions as fixed effects, and ‘vials' nested in ‘block' as a random effect for w1118 and flies deficient of damage prevention and repair mechanisms.
(b) . Internal bacterial load
We found that residuals of bacterial load data were non-normally distributed when tested using Shapiro–Wilks's test. Hence, we first log-transformed the data and then confirmed that the log-transformed residuals were still non-normally distributed. We analysed the log-transformed data, using a generalized linear model best fitted to gamma distribution, with ‘fly line' and ‘sex' as a fixed effect and ‘vials' as random effect. Subsequently, for pairwise contrasts (i.e. comparing the changes in bacterial load for each fly line relative to the w1118 across males and females following oral P. entomophila infection) we used a Kruskal–Wallis test (non-parametric pairwise comparisons using Wilcoxon method).
(c) . Measuring disease tolerance
Finally, to understand how damage-signalling and repair mechanisms affect disease tolerance in males and females during oral P. entomophila infection, we analysed the linear relationship between fly survival against bacterial load (measured at 24 h, as this was the peak microbe load) by fitting linear models [8,10,17,19,22,58]. We assessed differences in disease tolerance (fly survival with increasing bacterial load) by fitting ‘fly line' and ‘sex' as categorical fixed effects, ‘average bacterial load (log10)' as a continuous covariate, and their interactions as fixed effects. Significant interaction effects between fly line and bacterial load would indicate that the slope of the relationship between fly survival and load varies between fly lines, that is, the tolerance response differs between lines. Because our interest was to quantify the effect of damage prevention and repair mechanisms on disease tolerance, we compared the slope estimates of each of the transgenic lines with the slope of w1118 line using a pairwise comparison (F-test).
4. Results
(a) . Flies lacking dcy are more susceptible to oral Pseudomonas entomophila infections than those lacking components that minimize, signal or repair damage
Following oral infection with three different doses of Pseudomonas entomophila, flies with disrupted components of damage prevention (dcy), signalling (upd3), renewal (egfr1) and regulation (irc), were all significantly more susceptible to oral P. entomophila infections compared to w1118 flies (figure 1; electronic supplementary material, table S1; figure 1a for infection dose OD600 = 25; figure 1b for infection dose OD600 = 10; figure 1c for infection dose OD600 = 45). Among these lines, dcy knockouts were particularly susceptible to infection, even at the lowest dose of OD600 = 10 (figure 1; see electronic supplementary material, figure S3 and table S2 for hazard ratios). The effect of each gene disruption on the survival of flies following infection was similar in males and females (fly line × sex × treatment interaction = non-significant; electronic supplementary material, table S1), compared to control w1118 flies at all doses (figure 1; electronic supplementary material, table S1).
Figure 1.

Kaplan–Meier curves for females and males of w1118 and transgenic flies, exposed to oral P. entomophila of infection dose. (a) OD600 = 10, n = 10 vials per treatment per sex per fly line. (b) OD600 = 25, n = 30 vials per treatment per sex per fly line. (c) OD600 = 45. n = 10 vials per treatment per sex per fly line. For all doses, 15–17 flies per replicate vial.
(b) . Both w1118 and flies with disrupted tissue damage prevention and repair mechanisms show sex differences in bacterial load during oral infections
The higher susceptibility of all transgenic flies to oral bacterial exposure could either be caused by their inability to supress the bacterial growth or due to their inability to tolerate the damage inflicted during oral infection. To distinguish between these mechanisms, we first quantified internal bacterial loads at 15 min, 24 h and 96 h following the overnight oral exposure period to OD600 = 25 of P. entomophila. We observed a peak in microbe loads at 24 h post-infection in all fly lines (see electronic supplementary material, figure S4 and table S3) at this dose, and all fly lines showed sex differences at this timepoint (figure 2; electronic supplementary material, figure S4 and table S3), though by 96 h following oral infection this sex difference was no longer present in flies lacking upd3 or egfr expression (electronic supplementary material, figure S4 and table S3). However, it is important to note that by 96 h following the exposure, a considerable fraction of flies had experienced mortality (figure 1), and bacterial loads were necessarily only measured in flies able to mount a successful immune response.
Figure 2.

Bacterial load measured as colony-forming units (CFUs) using infection dose. (a) OD600 = 10 (low dose), (b) OD600 = 25 (medium dose) and (c) OD600 = 45 (high dose) after 24 h following the end of the oral bacterial exposure with P. entomophila for w1118 flies and transgenic lines (n = 30 vials of which 12–15 flies per treatment per sex per fly line for bacterial load measurement). Significantly different transgenic lines from control w1118 flies are denoted with asterisks (*) analysed using pairwise comparisons between w1118 and for males and females, respectively.
Focusing only on the peak microbe load at 24 h following the end of the exposure period, we observed sex differences in microbe load in low OD600 = 10 and medium dose OD600 = 25, but not at the higher dose of OD600 = 45 (figure 2; electronic supplementary material, table S4). However, while the magnitude of sex differences was similar for all lines at OD600 = 10, at OD600 = 25 the magnitude of the sex differences in microbe loads depended on the fly line (electronic supplementary material, table S4; line-by-sex interaction p < 0.001). It was also notable that some w1118 flies and some with disrupted irc exposed to OD600 = 10 showed complete clearance of infection after 24 h (figure 2). Analysing microbe load at OD600 = 10 data including or removing these flies did not yield qualitatively different results (electronic supplementary material, table S5). Flies lacking irc expression exhibited levels of bacterial load similar to the w1118 flies when infected with OD10, but showed lower bacterial load relative to w1118 flies at OD600 = 25 and OD600 = 45 (figure 2; electronic supplementary material, table S3).
(c) . Damage repair mechanisms mediate sex differences in disease tolerance during oral bacterial Pseudomonas entomophila infections
While some of the variation in survival between fly lines (figure 1) may be explained by variation in resistance—that is, their ability to clear infection (figure 2)—some of that variation may also arise due to differences in tolerance. We were therefore interested in measuring disease tolerance, where the slope of the linear relationship of survival relative to peak bacterial loads (measured at 24 h post-infection) reflects the degree of tolerance: steep negative slopes indicate a rapid mortality with increases in pathogen loads (low tolerance), while less steep or flat slopes reflect relatively more tolerant host [8,17,58,59]. While we carried out this analysis for all infection doses (electronic supplementary material, figure S5 and table S6), here we focus on flies infected with the intermediate dose (OD600 = 25), as we had 30 replicate-paired measurements of survival and microbe loads, and therefore greater power to estimate tolerance slopes relative to the extreme doses, where only 10 replicates were performed for each line/sex/infection combination.
At OD600 = 25, we found that the disruption of damage prevention, signalling and repair genes resulted in reduced tolerance, measured as the rate at which fly survival changed with bacterial load relative to w1118 flies (figure 3; tables 1 and 2). The rate at the rate at which survival declined with increasing microbe loads (tolerance), depended on the mechanisms of damage repair that was disrupted, and there were also sex differences in these effects (figure 3b and table 1; significant ‘line-by-sex-bacterial load' interaction). For example, both males and females lacking the major component of the peritrophic matrix dcy showed significantly reduced survival, but did not show marked decrease in tolerance with increasing microbe loads (figures 1 and 3). By contrast, flies lacking the damage renewal mechanisms egfr1 showed sex differences in disease tolerance (figure 3 and table 2; electronic supplementary material, table S7), as the disruption of egfr1 resulted in males, but not females becoming less tolerant of P. entomophila, showing a much steeper decline in survival with increasing microbe loads compared with females (figure 3). Notably, egfr1 males were less tolerant than w1118 males despite harbouring comparable microbe loads (figure 2).
Figure 3.
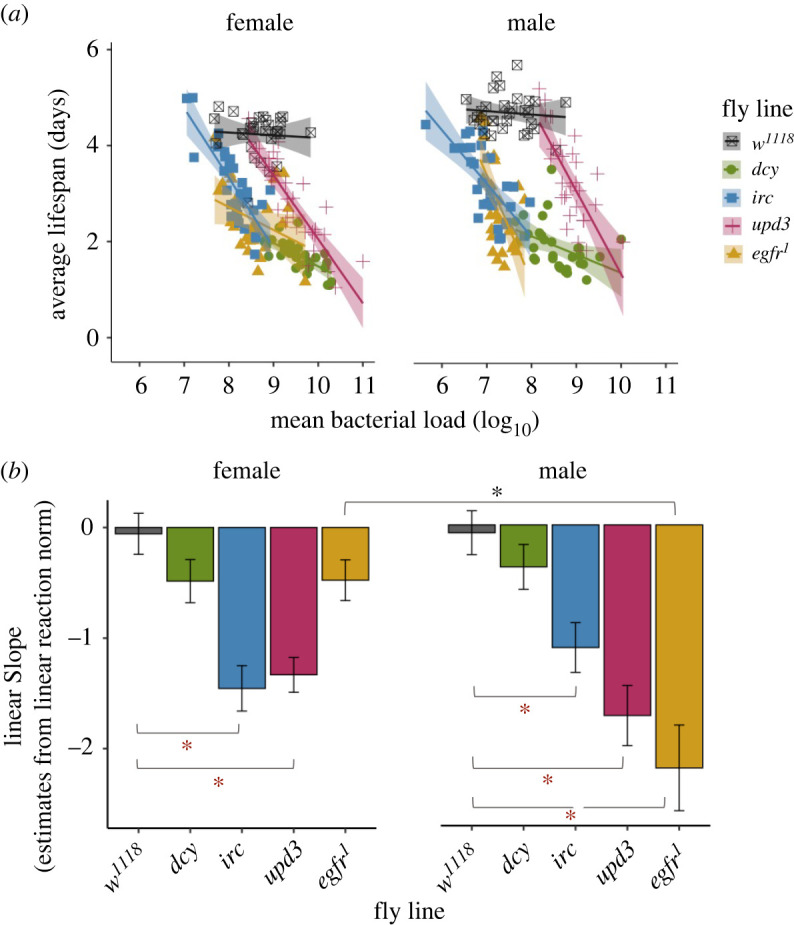
(a) The relationship between fly survival (measured as average lifespan) and peak bacterial load (CFUs measured at 24 h following the end of the exposure period), analysed using linear models for female and male flies (w1118 and flies lacking damage prevention and repair mechanisms). Each point shows data for average lifespan and mean bacterial load (CFUs) of 30 vials (with each vial containing 25 individual flies per fly line per sex combination) after 24 h post oral bacterial exposure. The data shown here are for the medium infection dose OD600 = 25. (b) The slope of the linear reaction norm extracted from the linear models. (Grey asterisk (*) indicates significant difference in tolerance between males and females (interaction between the bacterial load and the sex for each fly line measured using ANCOVA, table 1), red asterisks (*) indicate that the slopes of each fly line are significantly different from w1118, analysed using pairwise F-test from linear norm estimates; table 2).
Table 1.
Summary of ANCOVA. To assess differences in infection tolerance (fly survival with increasing bacterial burden) following oral P. entomophila infection with OD600 = 25 infection dose, after 24 h. We analysed ANCOVA and fitted ‘sex' as categorical fixed effects, ‘average bacterial load' as a continuous covariate and their interactions as fixed effects for each of the fly lines (w1118 and flies lacking damage prevention and repair mechanisms). See electronic supplementary material, table S6 for a full set of analyses on all three doses.
| dose | source | d.f. | sum of sq. | F ratio | p |
|---|---|---|---|---|---|
| OD-25 | fly line | 4 | 93.60 | 82.41 | <0.001 |
| sex | 1 | 6.006 | 21.15 | <0.001 | |
| bac. load | 1 | 47.66 | 167.8 | <0.001 | |
| fly line × sex | 4 | 10.11 | 8.904 | <0.001 | |
| fly line × bac. load | 4 | 21.18 | 18.65 | <0.001 | |
| sex × bac. load | 1 | 1.552 | 5.468 | 0.02 | |
| fly line × sex × bac. load | 4 | 5.357 | 4.716 | 0.001 |
Table 2.
Summary of pairwise comparisons (F-test) of linear slope estimates from linear reaction norm for w1118 flies and flies lacking damage prevention and repair mechanisms.
| sex | fly line | SSE | F ratio | p |
|---|---|---|---|---|
| female | dcy versus w1118 | 7.32 | 4.85 | 0.03 |
| egfr1 versus w1118 | 18.32 | 1.88 | 0.17 | |
| irc versus w1118 | 17.75 | 28.00 | <0.001 | |
| upd3 versus w1118 | 18.33 | 29.79 | <0.001 | |
| male | dcy versus w1118 | 10.66 | 2.17 | 0.14 |
| egfr1 versus w1118 | 29.07 | 21.41 | <0.001 | |
| irc versus w1118 | 16.06 | 18.72 | <0.001 | |
| upd3 versus w1118 | 24.87 | 27.01 | <0.001 |
5. Discussion
In the present work, we tested how mechanisms of damage prevention (dcy), signalling (upd3) control (irc) and renewal (egfr) contribute to disease tolerance during enteric infection. We present evidence that all these mechanisms contribute to disease tolerance during bacterial gut infection, and that some of these effects are sexually dimorphic. Previous transcriptomic, genome-wide association (GWAS) or microarrays studies have identified several candidate genes associated with disease tolerance, including—CrebA, grainyhead and debris buster, dFOXO [21,31,32,60]. However, all this work has focused on flies infected systemically by directly injecting bacteria into the fly. Here, we investigated tolerance during the natural oral route of infection, and we took a more targeted approach to specifically investigate how some of the well-described tissue damage prevention and repair mechanisms affect disease tolerance during enteric bacterial infections.
Though repairing infection-damage is crucial to fly survival, we found that flies lacking damage-preventing (dcy) are particularly susceptible to oral infections compared to those lacking components that minimize, signal or repair damage. In other words, preventing damage is clearly preferable to repairing damage from the perspective of fly survival. This result is consistent with previous work showing that loss-of-function in dcy increases the peritrophic matrix width making the gut leaky and compromising gut barrier function during oral infections with P. entomophila [2,50,61–63]. We also found increased bacterial loads relative to the w1118 control line, measured after 24 h following infection in both male and female dcy knockouts. This is likely because of the combination of leaky gut and pore-forming toxin produced by P. entomophila [2] resulting in higher bacterial growth in the fly haemolymph.
In the case of upd3-knockout flies, we found reduced survival and higher bacterial loads compared to w1118 flies. Previous work has shown that in response to P. entomophila infections, excessive reactive oxygen species (ROS) produced by host cells destroy the gut epithelia and block the gut repair process [62,64,65]. The JNK and Hippo pathways are activated in damaged enterocytes, which produce upd3, in turn activating the Jak/Stat pathway in intestinal stem cells. We found that both male and female upd3 knockout flies showed reduced tolerance and this is probably because in the absence of upd3 released from damaged cells the Jak/Stat-pathway activation is reduced, which is further necessary for intestinal stem cell proliferation and differentiation into enterocytes, together renew the damaged tissues [2,36,62]. We also found that functional disruption of irc results in lower bacterial loads. This effect might be expected because irc is a negative regulator of ROS [2], and higher ROS levels would lead to improved bacterial clearance.
Regarding the effects of these damage limitation mechanisms on disease tolerance, overall, we found that both male and female w1118 flies were quite tolerant of enteric bacterial infections (reflected in their relatively flat tolerance slopes; figure 3 and table 2), while disrupting most damage prevention and repair mechanism lowered disease tolerance (decline in slopes relative to w1118). While we found reduced tolerance in all knockout lines, disrupting some components of damage limitation had particularly severe effects on disease tolerance. Significant reductions in disease tolerance were observed in flies with disrupted irc and upd3, and in these fly lines the effect was comparable in both sexes. Irc-deficient flies are unable to regulate ROS levels which would lead to increased cytotoxic effects [2,42] while upd3 cytokine molecules are important for the activation of the Jak/Stat pathway [46,49]. In the case of dcy-knockout flies, survival did not deteriorate much further, possibly because it was already too poor to worsen further (figure 3). It is important to emphasize that all tolerance analyses were carried out using data on microbe loads measured at 24 h, and therefore is necessarily biased to individuals that were able to survive past this timepoint, although survival at 24 h post-infection was still considerably high.
We observed the fastest decline in tolerance in male flies lacking egfr1, but the disease tolerance of female egfr1 knockouts appeared unaffected. This sex difference in tolerance may arise as the result of sex differences in gut physiology and repair. Recent work has demonstrated that during oral Ecc15 infection, males showed significantly lower gut intestinal stem cells in response to infection, while female had higher intestinal stem cells and were resistant to infection and other stress [66]. The differentiation and proliferation of intestinal stem cells via Jak/Stat signalling into enterocytes via egfr is indispensable for tissue damage renewal. Loss of egfr1 signalling might therefore be felt more severely in males than in females, explaining why male but not female egfr1 knockouts showed a severe decrease in disease tolerance. To date, only a small proportion of studies have compared sex differences in intestinal immunity, with the majority of work focusing on one particular sex, usually females [47,66–68].
Another possibility for the observed sex difference in damage repair process, might relate to gut-plasticity such as gut remodelling. For instance, females of mammals such as mice extensively remodel their guts, increasing both digestive and absorptive capacity depending on the nutritional demands of lactation [69]. The remodelling of the gut might be one of the possible driving factors for dimorphism in gut immunity, since males and females differ in their nutritional needs [68]. Studies using Drosophila have shown that males and females can make different diet or nutritional choices in accordance with their reproduction role and demand [70] and the Drosophila midgut plastically resizes in response to changes in dietary sugar and yeast [71]. Whether gut remodelling and nutritional choice-demand causes sex differences in damage repair process during disease tolerance remains a question for future research. This also highlights a potential limitation of the current study, as we did not measure if males and females have different feeding rates or if disruption of the genes in focus resulted in differences in feeding rates, which could affect the likelihood of pathogen acquisition and infection progression. It is possible that some of the variation in observe in tolerance could arise by small differences in pathogen intake during feeding.
6. Concluding remarks
Although host mechanisms of immune-mediated clearance are key for pathogen defence and elimination, there is an increasing appreciation that additional defence mechanisms which prevent, signal, repair or renew the extent of tissue damage are also key to infection outcomes by promoting disease tolerance [30,72]. Tissue damage repair mechanisms that promote disease tolerance are interesting from a therapeutic perspective [9,10,73]. For instance, in mice, mechanisms that prevent or repair damage have been shown to confer disease tolerance during malarial Plasmodium infection and also during co-infections by pneumonia causing bacteria (Legionella pneumophila) and influenza virus [74,75]. Understanding how tissue damage prevention and repair mechanisms contribute to disease tolerance may also help explain how other arthropods are able to vector bacterial and viral infections without substantial health loss [22,76,77]. In summary, our results show that the disruption of tissue damage repair processes resulted in severe loss of disease tolerance and highlight how sex differences in some damage repair mechanisms could generate sexual dimorphism in gut immunity.
Acknowledgements
We thank V. Mongelli and C. M. Saleh for sharing the egfr line and F. M. Waldron for backcrossing irc, upd3, dcy fly lines in the w1118 background, V. Doublet and E. Robertshaw for laboratory help, and J. C. Regan, A. B. Pedersen, D. H. Nussey, D. J. Obbard, T. S. Salminen and Ashworth fly group members for helpful discussion. Finally, we thank A. Reid, A. Fulton, L. Rowe and J. King for help with media preparation. Electronic supplementary material, figures S1 and S2 were created using Biorender.
Contributor Information
Arun Prakash, Email: arunpadmaprakash@gmail.com.
Pedro F. Vale, Email: pedro.vale@ed.ac.uk.
Data accessibility
All the raw data and analysis code can be accessed under a Creative Commons 4.0 License at https://doi.org/10.5281/zenodo.6510215 [78]. An earlier preprint is available from the biological preprint server bioRxiv at: https://www.biorxiv.org/content/10.1101/2021.10.03.462916v1 [79].
Electronic supplementary material is available online [80].
Authors' contributions
A.P.: conceptualization, data curation, formal analysis, investigation, visualization, writing—original draft, writing—review and editing; K.M.M.: investigation, methodology, writing—review and editing; P.F.V.: conceptualization, data curation, formal analysis, funding acquisition, project administration, resources, supervision, visualization, writing—original draft, writing—review and editing.
All authors gave final approval for publication and agreed to be held accountable for the work performed therein.
Conflict of interest declaration
We declare we have no competing interests.
Funding
We acknowledge funding and support to P.F.V. from Branco Weiss fellowship and Chancellor's Fellowship; A.P. was supported by the Darwin Trust PhD studentship. We thank the School of Biological Sciences, University of Edinburgh.
References
- 1.Buchon N, Silverman N, Cherry S. 2014. Immunity in Drosophila melanogaster— from microbial recognition to whole-organism physiology. Nat. Rev. Immunol. 14, 796-810. ( 10.1038/nri3763) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuraishi T, Hori A, Kurata S. 2013. Host-microbe interactions in the gut of Drosophila melanogaster. Front. Physiol. 4, 375. ( 10.3389/fphys.2013.00375) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vale PF, Siva-Jothy J, Morrill A, Forbes M. 2018. The influence of parasites on insect behavior. In Insect behavior: from mechanisms to ecological and evolutionary consequences (eds González-Santoyo I, Córdoba-Aguilar A, González-Tokman D), pp. 274-291. Oxford, UK: Oxford University Press. [Google Scholar]
- 4.Apidianakis Y, Rahme LG. 2011. Drosophila melanogaster as a model for human intestinal infection and pathology. Dis. Model. Mech. 4, 21-30. ( 10.1242/dmm.003970) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoffmann JA. 2003. The immune response of Drosophila. Nature 426, 33-38. ( 10.1038/nature02021) [DOI] [PubMed] [Google Scholar]
- 6.Hultmark D. 2003. Drosophila immunity: paths and patterns. Curr. Opin. Immunol. 15, 12-19. ( 10.1016/S0952-7915(02)00005-5) [DOI] [PubMed] [Google Scholar]
- 7.Lemaitre B, Hoffmann J. 2007. The host defense of Drosophila melanogaster. Annu. Rev. Immunol. 25, 697-743. ( 10.1146/annurev.immunol.25.022106.141615) [DOI] [PubMed] [Google Scholar]
- 8.Raberg L, Sim D, Read AF. 2007. Disentangling genetic variation for resistance and tolerance to infectious diseases in animals. Science 318, 812-814. ( 10.1126/science.1148526) [DOI] [PubMed] [Google Scholar]
- 9.Ayres JS, Schneider DS. 2012. Tolerance of infections. Annu. Rev. Immunol. 30, 271-294. ( 10.1146/annurev-immunol-020711-075030) [DOI] [PubMed] [Google Scholar]
- 10.Medzhitov R, Schneider DS, Soares MP. 2012. Disease tolerance as a defense strategy. Science 335, 936-941. ( 10.1126/science.1214935) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soares MP, Teixeira L, Moita LF. 2017. Disease tolerance and immunity in host protection against infection. Nat. Rev. Immunol. 17, 83-96. ( 10.1038/nri.2016.136) [DOI] [PubMed] [Google Scholar]
- 12.McCarville J, Ayres J. 2018. Disease tolerance: concept and mechanisms. Curr. Opin. Immunol. 50, 88-93. ( 10.1016/j.coi.2017.12.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Råberg L, Graham AL, Read AF. 2009. Decomposing health: tolerance and resistance to parasites in animals. Phil. Trans. R. Soc. B 364, 37-49. ( 10.1098/rstb.2008.0184) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simms EL. 2000. Defining tolerance as a norm of reaction. Evol. Ecol. 14, 563-570. ( 10.1023/A:1010956716539) [DOI] [Google Scholar]
- 15.Little TJ, Shuker DM, Colegrave N, Day T, Graham AL. 2010. The coevolution of virulence: tolerance in perspective. PLoS Pathog. 6, e1001006. ( 10.1371/journal.ppat.1001006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bertels F, et al. 2018. Dissecting HIV virulence: heritability of setpoint viral load, CD4+ T-cell decline, and per-parasite pathogenicity. Mol. Biol. Evol. 35, 27-37. ( 10.1093/molbev/msx246) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Louie A, Song KH, Hotson A, Thomas Tate A, Schneider DS. 2016. How many parameters does it take to describe disease tolerance? PLoS Biol. 14, e1002435. ( 10.1371/journal.pbio.1002435) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Regoes RR, et al. 2014. Disentangling human tolerance and resistance against HIV. PLoS Biol. 12, e1001951. ( 10.1371/journal.pbio.1001951) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupta V, Vale PF. 2017. Nonlinear disease tolerance curves reveal distinct components of host responses to viral infection. R. Soc. Open Sci. 4, 170342. ( 10.1098/rsos.170342) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Howick VM, Lazzaro BP. 2014. Genotype and diet shape resistance and tolerance across distinct phases of bacterial infection. BMC Evol. Biol. 14, 56. ( 10.1186/1471-2148-14-56) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lissner MM, Schneider DS. 2018. The physiological basis of disease tolerance in insects. Curr. Opin. Insect Sci. 29, 133-136. ( 10.1016/j.cois.2018.09.004) [DOI] [PubMed] [Google Scholar]
- 22.Oliveira JH, Bahia AC, Vale PF. 2020. How are arbovirus vectors able to tolerate infection? Dev. Comp. Immunol. 103, 103514. ( 10.1016/j.dci.2019.103514) [DOI] [PubMed] [Google Scholar]
- 23.Ayres JS, Schneider DS. 2009. The role of anorexia in resistance and tolerance to infections in Drosophila. PLoS Biol. 7, e1000150. ( 10.1371/journal.pbio.1000150) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palaferri Schieber AM, Lee YM, Chang MW, Leblanc M, Collins B, Downes M, Evans RM, Ayres JS. 2015. Disease tolerance mediated by microbiome E. coli involves inflammasome and IGF-1 signaling. Science 350, 558-563. ( 10.1126/science.aac6468) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cumnock K, Gupta AS, Lissner M, Chevee V, Davis NM, Schneider DS. 2018. Host energy source is important for disease tolerance to malaria. Curr. Biol. 28, 1635-1642. ( 10.1016/j.cub.2018.04.009) [DOI] [PubMed] [Google Scholar]
- 26.Adelman JS, Kirkpatrick L, Grodio JL, Hawley DM. 2013. House finch populations differ in early inflammatory signaling and pathogen tolerance at the peak of Mycoplasma gallisepticum infection. Am. Nat. 181, 674-689. ( 10.1086/670024) [DOI] [PubMed] [Google Scholar]
- 27.Bonneaud C, Tardy L, Giraudeau M, Hill GE, McGraw KJ, Wilson AJ. 2019. Evolution of both host resistance and tolerance to an emerging bacterial pathogen. Evol. Lett. 3, 544-554. ( 10.1002/evl3.133) [DOI] [Google Scholar]
- 28.Nahrendorf W, Ivens A, Spence PJ. 2021. Inducible mechanisms of disease tolerance provide an alternative strategy of acquired immunity to malaria. eLife 10, e63838. ( 10.7554/eLife.63838) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cooper EL. 2018. Advances in comparative immunology. Berlin, Germany: Springer International Publishing. [Google Scholar]
- 30.Soares MP, Gozzelino R, Weis S. 2014. Tissue damage control in disease tolerance. Trends Immunol. 35, 483-494. ( 10.1016/j.it.2014.08.001) [DOI] [PubMed] [Google Scholar]
- 31.Howick VM, Lazzaro BP. 2017. The genetic architecture of defence as resistance to and tolerance of bacterial infection in Drosophila melanogaster. Mol. Ecol. 26, 1533-1546. ( 10.1111/mec.14017) [DOI] [PubMed] [Google Scholar]
- 32.Troha K, Im JH, Revah J, Lazzaro BP, Buchon N. 2018. Comparative transcriptomics reveals CrebA as a novel regulator of infection tolerance in D. melanogaster. PLoS Pathog. 14, e1006847. ( 10.1371/journal.ppat.1006847) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kapun M, et al. 2020. Genomic analysis of European Drosophila melanogaster populations reveals longitudinal structure, continent-wide selection, and previously unknown DNA viruses. Mol. Biol. Evol. 37, 2661-2678. ( 10.1093/molbev/msaa120) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wallace MA, et al. 2021. The discovery, distribution, and diversity of DNA viruses associated with Drosophila melanogaster in Europe. Virus Evol. 7, veab031. ( 10.1093/ve/veab031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chandler JA, Lang JM, Bhatnagar S, Eisen JA, Kopp A. 2011. Bacterial communities of diverse drosophila species: ecological context of a host–microbe model system. PLoS Genet. 7, e1002272. ( 10.1371/journal.pgen.1002272) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buchon N, Broderick NA, Lemaitre B. 2013. Gut homeostasis in a microbial world: insights from Drosophila melanogaster. Nat. Rev. Microbiol. 11, 615-626. ( 10.1038/nrmicro3074) [DOI] [PubMed] [Google Scholar]
- 37.Buchon N, Osman D. 2015. All for one and one for all: regionalization of the Drosophila intestine. Insect Biochem. Mol. Biol. 67, 2-8. ( 10.1016/j.ibmb.2015.05.015) [DOI] [PubMed] [Google Scholar]
- 38.Lemaitre B, Miguel-Aliaga I. 2013. The digestive tract of Drosophila melanogaster. Annu. Rev. Genet. 47, 377-404. ( 10.1146/annurev-genet-111212-133343) [DOI] [PubMed] [Google Scholar]
- 39.Hegedus D, Erlandson M, Gillott C, Toprak U. 2009. New insights into peritrophic matrix synthesis, architecture, and function. Annu. Rev. Entomol. 54, 285-302. ( 10.1146/annurev.ento.54.110807.090559) [DOI] [PubMed] [Google Scholar]
- 40.Shibata T, Maki K, Hadano J, Fujikawa T, Kitazaki K, Koshiba T, Kawabata S. 2015. Crosslinking of a peritrophic matrix protein protects gut epithelia from bacterial exotoxins. PLoS Pathog. 11, e1005244. ( 10.1371/journal.ppat.1005244) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vodovar N, Vinals M, Liehl P, Basset A, Degrouard J, Spellman P, Boccard F, Lemaitre B. 2005. Drosophila host defense after oral infection by an entomopathogenic Pseudomonas species. Proc. Natl. Acad. Sci. USA 102, 11 414-11 419. ( 10.1073/pnas.0502240102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buchon N, Broderick NA, Poidevin M, Pradervand S, Lemaitre B. 2009. Drosophila intestinal response to bacterial infection: activation of host defense and stem cell proliferation. Cell Host Microbe 5, 200-211. ( 10.1016/j.chom.2009.01.003) [DOI] [PubMed] [Google Scholar]
- 43.Myllymäki H, Valanne S, Rämet M. 2014. The Drosophila Imd signaling pathway. J. Immunol. 192, 3455-3462. ( 10.4049/jimmunol.1303309) [DOI] [PubMed] [Google Scholar]
- 44.Myllymäki H, Rämet M. 2014. JAK/STAT pathway in Drosophila immunity. Scand. J. Immunol. 79, 377-385. ( 10.1111/sji.12170) [DOI] [PubMed] [Google Scholar]
- 45.Valanne S, Wang J-H, Rämet M. 2011. The Drosophila toll signaling pathway. J. Immunol. 186, 649-656. ( 10.4049/jimmunol.1002302) [DOI] [PubMed] [Google Scholar]
- 46.Buchon N, Broderick NA, Kuraishi T, Lemaitre B. 2010. DrosophilaEGFR pathway coordinates stem cell proliferation and gut remodeling following infection. BMC Biol. 8, 152. ( 10.1186/1741-7007-8-152) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chakrabarti S, Dudzic JP, Li X, Collas EJ, Boquete J-P, Lemaitre B. 2016. Remote control of intestinal stem cell activity by haemocytes in Drosophila. PLoS Genet. 12, e1006089. ( 10.1371/journal.pgen.1006089) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang H, Grenley MO, Bravo M-J, Blumhagen RZ, Edgar BA. 2011. EGFR/Ras/MAPK signaling mediates adult midgut epithelial homeostasis and regeneration in Drosophila. Cell Stem Cell 8, 84-95. ( 10.1016/j.stem.2010.11.026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang H, Patel PH, Kohlmaier A, Grenley MO, McEwen DG, Edgar BA. 2009. Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila midgut. Cell 137, 1343-1355. ( 10.1016/j.cell.2009.05.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuraishi T, Binggeli O, Opota O, Buchon N, Lemaitre B. 2011. Genetic evidence for a protective role of the peritrophic matrix against intestinal bacterial infection in Drosophila melanogaster. Proc. Natl Acad. Sci. USA 108, 15 966-15 971. ( 10.1073/pnas.1105994108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ha E-M, Oh C-T, Ryu J-H, Bae Y-S, Kang S-W, Jang I, Brey PT, Lee W-J. 2005. An antioxidant system required for host protection against gut infection in Drosophila. Dev. Cell 8, 125-132. ( 10.1016/j.devcel.2004.11.007) [DOI] [PubMed] [Google Scholar]
- 52.Wang L, Sexton TR, Venard C, Giedt M, Guo Q, Chen Q, Harrison DA. 2014. Pleiotropy of the Drosophila JAK pathway cytokine unpaired 3 in.development and aging. Dev. Biol. 395, 218-231. ( 10.1016/j.ydbio.2014.09.015) [DOI] [PubMed] [Google Scholar]
- 53.Mongelli V, Lequime S, Kousathanas A, Gausson V, Blanc H, Nigg J, Quintana-Murci L, Elena SF, Saleh M-C. 2022. Innate immune pathways act synergistically to constrain RNA virus evolution in Drosophila melanogaster. Nat. Ecol. Evol. 6, 565-578. ( 10.1038/s41559-022-01697-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lewis E. 2014. Fly food. See http://cshprotocols.cshlp.org/content/2014/9/pdb.rec081414.full.
- 55.Dieppois G, Opota O, Lalucat J, Lemaitre B. 2015. Pseudomonas entomophila: a versatile bacterium with entomopathogenic properties. In Pseudomonas: volume 7: new aspects of Pseudomonas biology (eds Ramos J-L, Goldberg JB, Filloux A), pp. 25-49. Dordrecht, The Netherlands: Springer. [Google Scholar]
- 56.Siva-Jothy JA, Prakash A, Vasanthakrishnan RB, Monteith KM, Vale PF. 2018. Oral bacterial infection and shedding in Drosophila melanogaster. J. Vis. Exp. 135, 57676. ( 10.3791/57676) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Therneau T. 2015. Mixed effects cox models. CRAN repository.
- 58.Schneider DS, Ayres JS. 2008. Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat. Rev. Immunol. 8, 889-895. ( 10.1038/nri2432) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kutzer MAM, Armitage SAO. 2016. Maximising fitness in the face of parasites: a review of host tolerance. Zoology 119, 281-289. ( 10.1016/j.zool.2016.05.011) [DOI] [PubMed] [Google Scholar]
- 60.Dionne MS, Pham LN, Shirasu-Hiza M, Schneider DS. 2006. Akt and foxo dysregulation contribute to infection-induced wasting in Drosophila. Curr. Biol. 16, 1977-1985. ( 10.1016/j.cub.2006.08.052) [DOI] [PubMed] [Google Scholar]
- 61.Opota O, Vallet-Gély I, Vincentelli R, Kellenberger C, Iacovache I, Gonzalez MR, Roussel A, van der Goot F-G, Lemaitre B. 2011. Monalysin, a novel ß-pore-forming toxin from the Drosophila pathogen Pseudomonas entomophila, contributes to host intestinal damage and lethality. PLoS Pathog. 7, e1002259. ( 10.1371/journal.ppat.1002259) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chakrabarti S, Liehl P, Buchon N, Lemaitre B. 2012. Infection-induced host translational blockage inhibits immune responses and epithelial renewal in the Drosophila gut. Cell Host Microbe 12, 60-70. ( 10.1016/j.chom.2012.06.001) [DOI] [PubMed] [Google Scholar]
- 63.Blemont M, Vincentelli R, Kellenberger C, Opota O, Lemaitre B, Roussel A, Leone P. 2013. Crystallization and preliminary X-ray analysis of monalysin, a novel β-pore-forming toxin from the entomopathogen Pseudomonas entomophila. Acta Crystallograph. Sect. F Struct. Biol. Cryst. Commun. 69, 930-933. ( 10.1107/S174430911301885X) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Han J, Ulevitch RJ. 2005. Limiting inflammatory responses during activation of innate immunity. Nat. Immunol. 6, 1198-1205. ( 10.1038/ni1274) [DOI] [PubMed] [Google Scholar]
- 65.Lambeth JD. 2007. Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radic. Biol. Med. 43, 332-347. ( 10.1016/j.freeradbiomed.2007.03.027) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Regan JC, Khericha M, Dobson AJ, Bolukbasi E, Rattanavirotkul N, Partridge L. 2016. Sex difference in pathology of the ageing gut mediates the greater response of female lifespan to dietary restriction. eLife 5, e10956. ( 10.7554/eLife.10956) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ayyaz A, Li H, Jasper H. 2015. Haemocytes control stem cell activity in the Drosophila intestine. Nat. Cell Biol. 17, 736-748. ( 10.1038/ncb3174) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Belmonte RL, Corbally M-K, Duneau DF, Regan JC. 2020. Sexual dimorphisms in innate immunity and responses to infection in Drosophila melanogaster. Front. Immunol. 10, 3075. ( 10.3389/fimmu.2019.03075) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Speakman JR. 2008. The physiological costs of reproduction in small mammals. Phil. Trans. R. Soc. B 363, 375-398. ( 10.1098/rstb.2007.2145) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Camus MF, Piper MD, Reuter M. 2019. Sex-specific transcriptomic responses to changes in the nutritional environment. eLife 8, e47262. ( 10.7554/eLife.47262) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bonfini A, Dobson AJ, Duneau D, Revah J, Liu X, Houtz P, Buchon N. 2021. Multiscale analysis reveals that diet-dependent midgut plasticity emerges from alterations in both stem cell niche coupling and enterocyte size. eLife 10, e64125. ( 10.7554/eLife.64125) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vale PF, Fenton A, Brown SP. 2014. Limiting damage during infection: lessons from infection tolerance for novel therapeutics. PLoS Biol. 12, e1001769. ( 10.1371/journal.pbio.1001769) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vale PF, McNally L, Doeschl-Wilson A, King KC, Popat R, Domingo-Sananes MR, Allen JE, Soares MP, Kümmerli R. 2016. Beyond killing: can we find new ways to manage infection? Evol. Med. Public Health 2016, 148-157. ( 10.1093/emph/eow012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ferreira A, et al. 2011. Sickle hemoglobin confers tolerance to plasmodium infection. Cell 145, 398-409. ( 10.1016/j.cell.2011.03.049) [DOI] [PubMed] [Google Scholar]
- 75.Jamieson AM, Pasman L, Yu S, Gamradt P, Homer RJ, Decker T, Medzhitov R. 2013. Role of tissue protection in lethal respiratory viral-bacterial coinfection. Science 340, 1230-1234. ( 10.1126/science.1233632) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Taracena ML, et al. 2018. Regulation of midgut cell proliferation impacts Aedes aegypti susceptibility to dengue virus. PLoS Negl. Trop. Dis. 12, e0006498. ( 10.1371/journal.pntd.0006498) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lambrechts L, Saleh M-C. 2019. Manipulating mosquito tolerance for arbovirus control. Cell Host Microbe 26, 309-313. ( 10.1016/j.chom.2019.08.005) [DOI] [PubMed] [Google Scholar]
- 78.Vale P, Prakash A. 2021. Data and code for mechanisms of damage prevention, signalling, and repair impact the ability of Drosophila to tolerate enteric bacterial infection. Zenodo ( 10.5281/zenodo.5564919) [DOI]
- 79.Prakash A, Monteith KM, Vale PF. 2021. Mechanisms of damage prevention, signalling, and repair impact the ability of Drosophila to tolerate enteric bacterial infection. bioRxiv, 2021.10.03.462916. ( 10.5281/zenodo.5564919) [DOI]
- 80.Prakash A, Monteith KM, Vale PF. 2022. Mechanisms of damage prevention, signalling and repair impact disease tolerance. Figshare. ( 10.6084/m9.figshare.c.6135651) [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Vale P, Prakash A. 2021. Data and code for mechanisms of damage prevention, signalling, and repair impact the ability of Drosophila to tolerate enteric bacterial infection. Zenodo ( 10.5281/zenodo.5564919) [DOI]
- Prakash A, Monteith KM, Vale PF. 2022. Mechanisms of damage prevention, signalling and repair impact disease tolerance. Figshare. ( 10.6084/m9.figshare.c.6135651) [DOI] [PMC free article] [PubMed]
Data Availability Statement
All the raw data and analysis code can be accessed under a Creative Commons 4.0 License at https://doi.org/10.5281/zenodo.6510215 [78]. An earlier preprint is available from the biological preprint server bioRxiv at: https://www.biorxiv.org/content/10.1101/2021.10.03.462916v1 [79].
Electronic supplementary material is available online [80].