

Abstract
Phosphatidylserine (PS) is an anionic phospholipid in the eukaryotic membrane and is abundant in the brain. Accumulated studies have revealed that PS is involved in the multiple functions of the brain, such as activation of membrane signaling pathways, neuroinflammation, neurotransmission, and synaptic refinement. Those functions of PS are related to central nervous system (CNS) diseases. In this review, we discuss the metabolism of PS, the anti-inflammation function of PS in the brain; the alterations of PS in different CNS diseases, and the possibility of PS to serve as a therapeutic agent for diseases. Clinical studies have showed that PS has no side effects and is well tolerated. Therefore, PS and PS liposome could be a promising supplementation for these neurodegenerative and neurodevelopmental diseases.
Keywords: phosphatidylserine, phosphatidylserine liposomes, neurotransmission, synaptic refinement, central nervous system disease
Introduction
Phosphatidylserine (PS) is a structural component of the eukaryotic membrane and is accounted for 5–10% of the total lipid of cells (van Meer et al., 2008; Vance, 2015). Given its unique physical and biochemical properties as an anionic phospholipid, PS binds to various proteins and is involved in many biological processes, including enzyme activation, apoptosis, neurotransmission, and synaptic refinement (Fadok et al., 1992; Zhang et al., 2009; Huang et al., 2011; Scott-Hewitt et al., 2020). Therefore, the dysregulation on the metabolism of PS is associated with different CNS diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), major depressive disorder (MDD), stroke, and autism spectrum disease (ASD) (Enseleit et al., 1984; Fabelo et al., 2011; El-Ansary et al., 2016; Tokuoka et al., 2019; Homorogan et al., 2021). In addition, PS supplementation is proved to benefit the patients with AD, MDD, PD, or ADHD (Funfgeld et al., 1989; Maggioni et al., 1990; Crook et al., 1992; Hirayama et al., 2014). Chronic neuroinflammation is implicated in these CNS diseases, PS supplementation can inhibit excessive neuroinflammation to play a neuroprotective role. Moreover, PS supplementation can improve the cognitive function of the brain. In this review, we summarized the role of PS in the brain and its role in several related CNS diseases.
The biosynthesis, distribution, asymmetry, and degradation of phosphatidylserine
As an important glycerophospholipid, PS was first identified in the whole-brain lipid extracts in the 1940s (Folch, 1948). Its glycerol moiety contains two acyl chains at the sn-1 and sn-2 positions and a polar-head group at position sn-3, in which the neutral amino acid serine locates (Leventis and Grinstein, 2010). As shown in Figure 1, PS is produced by exchanging headgroups in mammalian cells by PS synthases; for example, PS synthase 1 is responsible for exchanging headgroup choline from PC (phosphatidylcholine), and PS synthase 2 is responsible for exchanging headgroup ethanolamine from PE (phosphatidylethanolamine). Because PS synthase 1 and 2 are uniquely expressed in the mitochondrial-associated membranes (MAMs) of the endoplasmic reticulum, PS is produced in the endoplasmic reticulum and transferred to the mitochondria or the Golgi through MAMs (Stone and Vance, 2000). In the mitochondria, a part of PS is catalyzed to PE by PS decarboxylase in the inner leaflet of mitochondria, while the other part of PS is incorporated into the mitochondrial membrane (Camici and Corazzi, 1995). Some newly synthetic PS is transferred from the endoplasmic reticulum to the Golgi intermediate compartment and the Golgi cisternae via the secretory pathway (Voelker, 2000), then PS is secreted to the plasma membrane or is delivered to the endosome and the lysosome. PS in the endosome, especially recycling endosomes, is slowly recycled to the plasma membrane (Voelker, 2000). In the normal conditions, PS is located exclusively in the cytoplasmic leaflet of the plasma membrane, endoplasmic reticulum lumen, Golgi, mitochondria, and endosomes to maintain the normal function of organelles (Yeung et al., 2008; Leventis and Grinstein, 2010; Kay and Fairn, 2019). The detailed biological events of PS and the percentage of PS in total phospholipid in different organelles were summarized and listed as in Table 1.
FIGURE 1.

The biosynthesis, distribution, and degradation of phosphatidylserine. PS is produced in ER (MAM), PSS1 catalyzes PE to PS, PSS2 catalyzes PC to PS. Some new PS is transported to mitochondria, and PS is decarboxylased and forms PE in mitochondria. PS is also transferred to the plasma membrane and other oranges by the Golgi via traditional vesicle-mediated trafficking. PS in the endosome is recycled to the plasma membrane. PS in lysosomes is derived from the Golgi apparatus or endosome. PS can be hydrolyzed by phospholipase A1 (PLA1) and phospholipase A2 (PLA2), the productions are 2-acyl-1-lyso -PS and 1-acyl-2-lyso-PS, respectively. Created with BioRender.com.
TABLE 1.
The biological events of phosphatidylserine (PS) and the percentage of PS in total phospholipid of different organelles.
| Organelles membrane | PS% | The key protein | Events of PS distribution and metabolism |
| Plasma membrane | 12 | Flippase Floppase Scramblase | Flippase transports PS from the extracellular to the cytosolic side, floppase transports PS from the cytosolic to the extracellular side, scramblases transports PS bidirectionally. |
| Endoplasmic reticulum | 3–5 | PSS1 and PSS2 scramblases | Produce PS by PSS1 and PSS2 in MAMs, scramblases translocate PS synthesized on the cytosolic side to the internal leaflet. |
| Golgi complex | 5 | P4-ATPase | Keep PS asymmetry, transport PS to plasma membrane, divert PS to the prelysosomal endocytic compartment. |
| Early endosome | 8.5 | ATP8A1, ATP8A2, ATP9A, EHD1 | ATP8A1, ATP8A2 and ATP9A are PS flippases, EHD1 is a PS effector, all of them are essential for endosomal traffic through recycling endosomes. |
| Late endosome | 2.5–3.9 | ||
| mitochondria | 1 | PS decarboxylase | Decarboxylate PS to PE on the outer leaflet of the mitochondrial inner membrane |
EHD1, Eps15 homology domain-containing protein 1.
Degradation of PS is carried out via two enzymes: PS decarboxylases and phospholipases (as shown in Figure 1). As previously described (Camici and Corazzi, 1995), PS decarboxylases catalyze PS to form PE in the mitochondria. There are two: PS-specific phospholipases A1 and A2. Both phospholipases catalyze a reaction to produce Lyso-phosphatidylserine (2-acyl-1-lyso-PS and 1-acyl-2-lyso-PS). PS-specific phospholipase A1 (PS-PLA1) hydrolyzes the sn-1 acyl chain of PS exposed on the surface of cells such as apoptotic cells or activated platelets, and generates 2-acyl-1-lyso-PS which is a mediator for the activation of mast cells, T cells and neural cells (Wen et al., 2001). PS-specific phospholipase A2 (PS-PLA2) is also essential to inflammation and the immune response. It hydrolyzes the sn-2 acyl of PS to produce 1-acyl-2-lyso-PS and further to form many bioactive lipid mediators in many biological processes (Funk, 2001). Therefore, lyso-phosphatidylserine is involved in a series of biological process such as apoptosis and T cell activation (Bellini and Bruni, 1993). For example, when PS is exposed during apoptosis, PS-PLA1 hydrolyzed PS on the cell surface and produces 1-acyl-2-lyso-PS, stimulates histamine release from mast cells in the presence of FcupvarepsilonRI cross-linker, and induces inflammation and cell death (Hosono et al., 2001). In addition, Lyso-phosphatidylserine can also enhance nerve growth factor-induced neural differentiation, and may play a neuroprotective role to improve tissue restoration after brain damage occurs (Lourenssen and Blennerhassett, 1998).
To maintain normal cellular function, PS is distributed in the inner leaflet of the lipid bilayers of the membrane; otherwise, cells are induced to apoptosis as mentioned above when PS is exposed on the outer leaflet of the lipid bilayers (Chua et al., 2019). How is the distribution asymmetry of PS regulated in the cellular lipid bilayers? Flippase, floppases, and scramblases are three lipid transporter enzymes in the membranes that dictate the fate of PS distribution. The P4 subfamily of P-type ATPases (P4-ATPases) is identified as flippase which transport PS and other lipids from the extracellular to the cytosolic side of the membrane in an ATP-independent manner. All P4-ATPases are critical to minimize PS exposure. Total fourteen P4-ATPases are identified in the human genome; some of them are located in the plasma membrane (such as ATP11A and ATP11C), while some is located in the endosome membrane (such as ATP8A1, ATP8A2, and ATP9A) (Nagata et al., 2020). Most P4-ATPases require CDC50A (TMEM30A) as a functional subunit for target localization (Coleman and Molday, 2011). Deletion of the chaperone CDC50A in the cell promotes PS exposure and cellular engulfment by macrophage (Segawa et al., 2014). Opposite to flippase, floppase transports lipid from the cytosolic to the extracellular side of the membrane. Lipid floppases are identified as members of the ATP-binding cassette (ABC) transporter superfamily. Floppase ABCA1 is responsible for PS and cholesterol transportation and has been found to have a critical role in lipid efflux and plasma membrane remodeling (Gulshan et al., 2013). Scramblases are also important lipid transporters that transport PS bidirectionally in an ATP-independent manner (Leventis and Grinstein, 2010). Two family members TMEM16 and Xk-related (XKR) protein are identified to have scramblases activity (Suzuki et al., 2013; Kalienkova et al., 2021). Among the two families, TMEM16F and XKR 8 are well-documented scramblases. As shown in Figure 2, TMEM16F is a Ca2+-dependent scramblase, while XKR8 responds to the caspase signal (Hankins et al., 2015). However, these transporters may be interacted or crosstalk between them may regulate PS metabolism. For example, in the apoptotic cells or other biological processes, the flippases are disrupted by caspase or inhibited by Ca2+; at the same time, either TMEM16F or XKR8 is activated to expose PS and participate these biological processes (Nagata et al., 2020).
FIGURE 2.
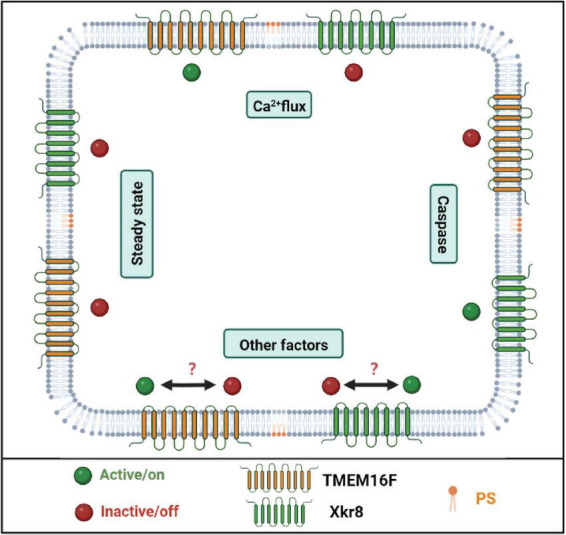
The scrambles are responsible for phosphatidylserine (PS) exposure. PS scrambles are responsible for PS translocation during cell apoptosis as well as other biological processes. TMEM16F and XKr8 are well-documented PS scrambles. When the intercellular Ca2+ concentration is upregulated, TMEM16F binds Ca2+ and transports PS to the outer leaflet of the plasma membrane. When the cell undergoes apoptosis, caspase cleaves and activates XKr8, and then XKr8 exposes PS to the outside of the cell and releases the “eat me” signal. But in other biological processes, it remains unknown which scramble is activated or both of them are activated. Created with BioRender.com.
The functions of endogenous phosphatidylserine
As the protein docking sites on the cell membrane, phosphatidylserine participates in the activation of many signaling pathways
PS is the major acidic phospholipid in the human brain, accounting for 11.4–14.4% in the cerebral cortex, and 16.0–21.1% in white matter and myelin (Svennerholm, 1968; Horrocks, 1973). Hence, PS is an essential nutrient for the brain and is implicated in the normal functions of the brain. As the structural lipid in the cell membrane, many signaling molecules or proteins interact with PS through their C2 domain or gamma-carboxyglutamic acid domain in the presence of Ca2+; PS is located inside the cells and servers as to recruit and activate signal pathways, including PKC family, phosphoinositide-3-kinase (PI3K)/AKT and Ras/Raf. For example, protein kinase C (PKC) family members bind PS through their C2 domains, while growth arrest-specific 6 (Gas6) binds PS via gamma-carboxyglutamic acid domain (Rajotte et al., 2008). In addition, due to the negatively charged headgroup of PS, some proteins bind PS in a non-specific charge-based manner, such as protein kinase Src, Rac1, and K-Ras, and activate these kinase and their downstream signals (Stace and Ktistakis, 2006). These signal pathways have been well described to support neuronal cell survival, differentiation, and proliferation (Kim et al., 2014; Glade and Smith, 2015; Kay and Fairn, 2019).
Phosphatidylserine is involved in neurotransmission
Neurotransmission is a biological process by which neurons transmit information and maintain normal functions of the brain. Structurally, the neurotransmitters which are packaged in synaptic vesicles are released by presynaptic membranes via calcium-dependent exocytosis and then bind to the receptor in the postsynaptic membrane and complete the signal transmission between neurons (Kimura, 2021). PS is a component of the synaptic vesicle and involves several necessary neurotransmission steps. On the one hand, PS could directly bind to neurotransmitters, and the binding facilitated the availability of neurotransmitters for their re-uptake. For examples, PS has the highest affinity among acidic lipids to bind serotonin in brain tissues (Johnson et al., 1977); the headgroup of PS in lipid bilayer is strongly bound to dopamine or L-dopa through H-bonds (Orlowski et al., 2012). The interaction between PS and neurotransmitters may facilitate the uptake process or metabolization of neurotransmitters and exert their effects (Orlowski et al., 2012). On the other hand, synaptic vesicle exocytosis is the most important neurotransmission process. Previous studies showed that PS treatment increases synaptic vesicle number adjacent to the plasma membrane and upregulates the frequency of calcium-dependent exocytosis of synaptic vesicles (Uchiyama et al., 2007; Zhang et al., 2009). Synaptic vesicle exocytosis includes three critical steps: vesicle docking, prime, and fusion. PS may influence neurotransmitter vesicle docking and fusion at the plasma membrane. Recently a study demonstrated that PS promoted vesicle docking through interaction with α-synuclein (α-Syn). When PS levels in the vesicle membrane decrease, α-Syn inhibits the vesicle docking; however, high levels of PS can bind to α-synuclein and reverse its inhibition on the vesicle docking (Lou et al., 2017), suggesting the critical role of PS in α-Syn-mediated vesicle docking. Similarly, PS also regulates the opening and dilation of the fusion pore via interaction with synaptotagmin I (syt-1), the major calcium sensor for synaptic vesicle exocytosis (Brose et al., 1992). This process is shown in Figure 3, PS or phosphatidylinositol is able to bind the C2 domains of Syt-1 to promote the fusion of synaptic vesicle membrane with the cell membrane (Tucker et al., 2004; Gruget et al., 2020). Furthermore, the high level of PS increases the binding affinity of Syt-1 to N-ethylmaleimide-sensitive fusion protein receptor complex (SNARE) at the plasma membrane, which was the core machinery complex for exocytosis. The Syt-1/SNARE complex undergoes Ca2+- dependent oligomerized and inserts into the presynaptic membrane and facilitates the opening and dilation of the fusion pore for the neurotransmitter release (Hosono et al., 2001; Koh and Bellen, 2003; Zhang et al., 2009; Figure 3). In summary, PS is involved in the biological processes of the release and re-uptake of neurotransmitters.
FIGURE 3.

Phosphatidylserine (PS) effector protein synaptotagmin I is involved in neurotransmitter release. As the figure shows, synaptotagmin I possesses two C2 domains to bind Ca2+ and PS. N-terminus of synaptotagmin I is in synaptic vesicles and C2 domain in cytoplasmic. Synaptotagmin I binds Ca2+ when Ca2+ flows into cells from the Ca2+ channels. The Ca2+ binding further increases the binding of synaptotagmin I to SNARE complex as well as the fusion of synaptic and plasma membrane. SNARE complex includes the synaptosome-associated protein of 25 kDa (SNAP25), synataxin, and synaotobrevin. Created with BioRender.com.
Phosphatidylserine is linked to glia-mediated synaptic refinement
Synaptic refinement, as known as synaptic pruning, is a process to eliminate supernumerary synapses. Impairment of synaptic pruning is involved in the pathogenesis of ASD, MDD, AD, and other mental conditions (Brown and Neher, 2014; Vilalta and Brown, 2018). Previous studies showed that microglia and astrocytes mediate the regulation of synaptic pruning (Schuldiner and Yaron, 2015; Wilton et al., 2019). In the developmental and adult brain, glia-mediated synaptic refinement is an essential physiological process to keep a proper number of synapses to maintain functional neuronal circuits (Schuldiner and Yaron, 2015). Similar to the apoptotic process, PS externalization in synapses takes part in glia-mediated synaptic pruning process in the developing hippocampus where there are abundant synapses that need to be pruned (Scott-Hewitt et al., 2020). On the contrary, microglial synapse elimination can be prevented by blocking accessibility of exposed PS using Annexin V or TREM2 loss in microglia (Scott-Hewitt et al., 2020). In addition, mitochondrial activity reduction contributes to PS exposure on axons, which is a marker of axonal pruning (Shacham-Silverberg et al., 2018).
How PS involved in glia-mediate synaptic refinement and axonal pruning is remains unclear? A previous study showed that the complement protein C1q was also involved in the synaptic pruning process by recognizing exposed PS in synapses (Scott-Hewitt et al., 2020). Deficiency of C1q in mice reduces the microglial engulfment of synapses and results in excessive retinal innervation of lateral geniculate neurons (Scott-Hewitt et al., 2020). The S4 variant of GPR56 splicing isoforms is also found to bind PS to mediate synaptic pruning by microglia; however, the deletion of GPR56 in microglia fails to bind exposed PS and leads to excess synapses (Li et al., 2020). In addition, some PS receptors that recognize exposed PS in synapses are considered to mediate synaptic pruning. Deletion of PS flippase chaperone CDC50A induces PS exposure on neuronal somas and specifically eliminates inhibitory post-synapse through microglial PS receptor Mer (Li et al., 2021). Previous studies also showed that PS receptors TREM2 and MEFG10 contribute to synaptic refinement in microglia (Filipello et al., 2018) and in astrocytes (Lee et al., 2021), respectively. Overall, PS exposure was a marker of unwanted synapses and axons; PS receptors bind to exposed PS and trigger synaptic pruning.
Phosphatidylserine located in the outer of cell membrane is a marker of apoptosis
The increasing lines of evidence demonstrate that PS exposure in the outer leaflet of cell membrane can be caused by apoptosis including intrinsic apoptosis and extrinsic apoptosis (Kiraz et al., 2016). Intrinsic apoptosis is induced by mitochondrial stress and the release of cytochrome C from the mitochondria to the cytosol. Cytochrome C in cytosol activates apoptotic protease-activating factor 1 and caspase cascades like caspase 9 and caspases 3/7, resulting in intrinsic apoptosis (Segawa et al., 2014). Extrinsic apoptosis is mediated by death receptors including tumor necrosis factor receptor 1, the Fas receptor (CD95), and the tumor necrosis factor-related apoptosis-inducing ligand (traiL) receptors. Death receptors interact with their ligands to activate caspase 8, and activating caspase 8, in turn, activates caspase3/7. Activated caspase3/7 is a key to trigger PS exposure and cell apoptosis (Boada-Romero et al., 2020).
During apoptosis, caspases 3/7 inactivates PS flippase and activates PS scramblases to expose PS (Segawa et al., 2014). ATP11C (adenosine triphosphatase type 11C), a PS flippase family member, is essential to maintain PS asymmetry in the cell membrane (Takada et al., 2015; Segawa et al., 2018). Activated caspases 3/7 cleave ATP11C upon the recognized sites of ATP11C causing PS exposure in the outer of cell membrane (Segawa et al., 2014). PS exposure is abolished and cells are not induced to apoptosis and are not engulfed by macrophages when caspase recognized sites of ATP11C are mutated (Segawa et al., 2014). In addition, PS scramblases XKR8, which is important for PS asymmetry, is also cleaved by caspase3/7 upon the recognized site at C-terminus of XKR8 and activated (Suzuki et al., 2014). Active XKR8 forms a high-order complex with basigin and neuroplastin, two chaperone proteins of XKR8 (Suzuki et al., 2014), and then exposes PS to cell surface (Suzuki et al., 2016).
Externalization of PS serves as a marker of apoptotic cells and an “eat me” signal. Exposed PS on the cell surface is recognized by PS receptors expressed in macrophages and further initiates actin reorganization to engulf apoptotic cells by macrophages (Lemke, 2019). PS receptors in macrophages that bind PS directly or indirectly are shown in Figure 4. Several PS receptors can recognize PS directly, such as T-cell immunoglobulin and mucin domain-containing molecule-1 and -4 (TIM-1, -4), receptor for advanced glycation end products (RAGE), brain-specific angiogenesis inhibitor 1 (BAI1), triggering receptor expressed on myeloid cells 2 (TREM2), stabilin 2, and members of the CD300 family (Park et al., 2007; Paidassi et al., 2008; Park et al., 2008; He et al., 2011; Tian et al., 2014; Krasemann et al., 2017). Other PS receptors, such as integrin αvβ3 and TAM receptors, bind to PS indirectly and need bridging molecules. For example, integrin αvβ3 binds to the epidermal growth factor domain of Milk fat globule-EGF factor 8, which is a soluble PS receptor to recognize exposed PS and apoptotic cells and mediate apoptotic signal (Fuller and Van Eldik, 2008); Protein S and growth arrest specific 6 also recognize PS and bridge TAM receptors with apoptotic cell (Lemke, 2017). Most PS receptors are expressed in peripheral macrophages and microglia, the primary tissue-resident macrophages in the brain (Nazareth et al., 2021).
FIGURE 4.

Phosphatidylserine (PS) is involved in microglia-mediated neuron apoptosis. When the neuron is undergoing apoptosis, PS is exposed to the cell surface. Microglia can find and phagocyte the apoptotic neuron. Several PS effectors expressed in microglia can recognize the exposed PS directly or indirectly. TREM2, RAGE, TIM4, and BAI1 can bind PS directly. TAM receptors recognize PS indirectly through their ligands Gas6 and ProS1. Both Gas6 and ProS1 have Gla-domain to bind PS. As well as TAM receptor, intergrinαvβ3 recognize PS through MFG-E8. All of the PS effectors can trigger cytoskeletal rearrangement of microglia and engulf the dying neuron. Created with BioRender.com.
The function of exogenous phosphatidylserine
Exogenous PS could obtain from the bovine brain and krill, and also be made from soybean lecithin by the enzymatic reaction with L-serine (Glade and Smith, 2015). When exogenous PS is given, PS can be uptake and transported in the cell by PS flippase, then incorporated into the membrane system to support cell function (Glade and Smith, 2015). In addition, PS liposomes that exposed PS on the surface could mimic apoptotic cells, be recognized by PS receptors and engulfed by phagocytes, and then triggered anti-inflammatory signal pathways (Aramaki et al., 2001).
Phosphatidylserine improves the cognitive function of the brain
Increasing studies have demonstrated that supplementation of PS significantly improved the cognitive impairment caused by aging, AD, or PD (Cenacchi et al., 1993; Kim et al., 2014; More et al., 2014). In a double-blind study, the elder patients with severe cognitive decline were treated with brain cortex-derived PS (BC-PS) 300 mg/day for 6 months, compared with placebos, BC-PS administration significantly improved the storage, learning, and retrieval abilities of memory in patients. Treatment the aged patients with BC-PS (300 mg/day) for 12 weeks also resulted in significant improvement of cognitive function (Schreiber et al., 2000). These results suggested that choric PS treatment ameliorates the cognitive function of the brain. Previous literatures demonstrate that PS benefits brain functions in several ways.
First, PS may improve function of brain through PKC. Since PKC activation influences the process of cognition through regulating phosphorylation of substrates such as N-methyl-D-aspartate (NMDA) receptor, AMPA receptor, and growth-associated protein-43 (Sun and Alkon, 2010), the expressions of several PKC isoforms are down-regulated in cognition impairment conditions, including aging, PD, and AD (Mizutani et al., 1998; Alkon et al., 2007; Do Van et al., 2016). Intake of exogenous PS could promote the activity of PKC (Bell and Burns, 1991), which phosphorylated its substrates to boost cell function (Sun and Alkon, 2010). Secondly, PS may ameliorate the cognitive function through enhancing glucose mentalism, which is related to cognitive impairment. Abnormal glucose metabolism rates of AD was found in predominantly disease-affected brain regions of patients with AD and other types of dementia (Friedland et al., 1983). The administration of exogenous PS (500 mg daily for 3 weeks) increased global glucose metabolism by 14.8% in the brain cortex and significantly improved the cognition of AD patients (Heiss et al., 1991; Klinkhammer et al., 1991). Thirdly, PS may improve the cognitive function through normalization the activity of NMDA receptor. NMDA receptor-mediated excitatory transmission is essential for the cognition function of the brain (Cohen and Muller, 1992). Compared to the young brains, the NMDA receptors in the aging brains alters, such as decreasing density of NMDA receptors and enhancing affinity to L-glutamine and glycine (Cohen and Muller, 1992). Chronic treatment with PS increased the density of NMDA receptors and normalized the affinity to L-glutamine and glycine in aging mice (Cohen and Muller, 1992). Fourthly, long-term potentiation requires a persistent increase in synaptic efficacy and is a critical synaptic mechanism of cognition (Thompson, 2000). Long-term potentiation needs the activation of NMDA receptor/channel complex. Treatment with exogenous PS also elicits synaptic efficacy (Borghese et al., 1993). Furthermore, exogenous PS stimulation also increased the metabolic levels of dopamine and serotonin which is lower in the cerebrospinal fluid in Alzheimer’s presenile dementia (Argentiero and Tavolato, 1980), and a recent study reported PS also increased the release of choline, which is an important neurotransmitter and decrease in AD brains (Suzuki et al., 2001), In summary, PS can improve the cognitive function of the brain through different pathways.
Phosphatidylserine inhibits neuroinflammation in neurological diseases
Neuroinflammation is an immune response in the CNS and is mediated by microglia, astrocytes, or recruited macrophages (Woodburn et al., 2021). Neuroinflammation is involved in various conditions, including CNS injury, ischemia, infection, toxin, or autoimmunity (Woodburn et al., 2021). Neuroinflammation is a double-edged sword for the CNS:transient neuroinflammation may play a protective role during tissue repair after injury and remove the cellular debris; meanwhile, chronic neuroinflammation is related to the progression of CNS diseases such as AD, PD, stroke, and other brain injury (Catorce and Gevorkian, 2016; Leng and Edison, 2021).
Previous studies show that exogenous PS liposomes have an anti-inflammatory effect in the CNS (Bachiller et al., 2018). Treatment with PS liposomes or PS head group phospho-L-serine, the expression of pro-inflammation cytokines (such as TNFa and IL1β) and NO synthesis induced by LPS significantly decrease, and the expression of anti-inflammation cytokines (TGFβ and PGE2) significantly increase in microglia (De et al., 2002; Huynh et al., 2002; De Simone et al., 2003; De Simone et al., 2004; Zhang et al., 2006). Those studies suggested that PS liposomes may play a neuroprotective role through modulating microglia phenotype. PS and PC liposomes significantly increase the survival retinal neurons after I/R by reducing the expression of pro-inflammatory genes in microglia, such as IL1β, IL6, and C-C Motif Chemokine Ligand 2–5 (Dvoriantchikova et al., 2009). PS and PC liposomes also inhibit the microglial activation induced by Aβ and interferon-γ through reducing the production of TNFa, NO, and superoxide (Casamenti et al., 1991). Intranasal PS liposomes prior to surgical brain injury induction significantly increases TGFβ, and decreased IL1β and TNFa in brain tissue to attenuate inflammation (De Simone et al., 2004).
It is unknown how PS or its analogs mediates the anti-inflammation effects. Previous studies show that Mer, a PS receptor belonging to TAM receptor, triggered anti-inflammation of macrophages because Mer deficiency did not reduced LPS-induced inflammation in mice (Camenisch et al., 1999; Vago et al., 2021). CD36, another PS receptor, is also involved in anti-inflammation of PS liposomes (De Simone et al., 2004). A newly identified PS receptor-phosphatidylserine-specific receptor (PSR) may be involved in anti-inflammatory effect of PS (Fadok et al., 2001). Like other PS receptors, PSR is also involved in the phagocytosis but the more important function of PSR is to inhibit excess inflammation (Fadok et al., 2001). PSR in macrophages can inhibit the phagocytosis of apoptotic cells when PSR is bound to its ligands such as PS liposomes, phospho-L-serine, or PSR antibody; but promoted the anti-inflammation induced by LPS by increasing TGFβ and decreasing TNFa (Fadok et al., 2000). It remains unknown whether other PS receptors also are involved in this process. Mechanically, the interaction of the PS liposomes and PS receptor may inhibit several important inflammation regulators, such as p38 mitogen-activated protein kinase (p38MAPK), cyclic AMP responding element-binding protein (CREB), and NFκB. PS liposomes treatment significantly inhibits p38MAPK phosphorylation induced by LPS (Aramaki et al., 2001; Ajmone-Cat et al., 2003; Ma et al., 2011). Treating microglia with PS liposomes reduces phosphorylation of CREB induced by LPS (Ajmone-Cat et al., 2003). PS liposomes also inhibited the activation of NFκB induced by LPS (Aramaki et al., 2001; Ajmone-Cat et al., 2003). In addition, treatment with PS liposomes triggers the activation of ERK in microglia as early as 5 min (Ma et al., 2011). Therefore, the interaction between PS and PS receptors not only mediated the phagocytosis of apoptotic cells but also inhibit inflammation signaling and the release of anti-inflammation cytokines to have the anti-neuroinflammation effects.
The roles of phosphatidylserine in different central nervous system diseases
As described above, a growing body of data implicate that both endogenous and exogenous PS plays critical roles in CNS diseases. Therefore, we summarized the current advances of PS in different CNS diseases, including AD, PD, MDD, ischemic stroke, ASD, and attention deficit hyperactivity disorder (ADHD).
Alzheimer’s disease
Alzheimer’s disease AD is a progressive neurodegenerative disease; the pathological features of AD are characterized by the accumulation of amyloid-β (Aβ) plaques and phosphorylated tau neurofibrillary tangles (Paasila et al., 2021). There are many hypotheses about the mechanisms of AD, including synaptic dysfunctions hypothesis, cholinergic theory, amyloid cascade hypothesis, tau cascade hypothesis, neuroinflammation, and gut-brain axis hypotheses (Craig et al., 2011; Calsolaro and Edison, 2016; Kowalski and Mulak, 2019; Ju and Tam, 2022). Among these hypotheses, abnormal lipid metabolism in the cell membrane is considered as one of the mechanisms of AD. The alteration of PS and other phospholipids changes the viscosity of cell membrane and hinders many biological processes, such as enzyme activity, signal transduction efficiency, and membrane carrier (Akyol et al., 2021). It is controversary about the alternation of PS in brain tissues of AD patients. Some studies have demonstrated PS is reduced in brains of AD patients (Corrigan et al., 1998; Pettegrew et al., 2001; Oma et al., 2012; Sabogal-Guaqueta et al., 2020; Akyol et al., 2021). Meanwhile, others studies also found PS is increased or unchanged in brains of AD patients (Wells et al., 1995; Lampl et al., 2006; Martin et al., 2010; Kim W. S. et al., 2018). The details are shown in Table 2. Similar to AD patients, the changes of PS in brains are also not consistent in different animal models of AD as shown in Table 3 (Yao et al., 2009; Gonzalez-Dominguez et al., 2014; Martinez-Gardeazabal et al., 2017; Fitzner et al., 2020; Yi et al., 2020; Dejakaisaya et al., 2021). The reasons for this inconsistency may be explained by brain regions, Braak stages, age in the AD patients, and different experimental methods in studies. Therefore, PS is not considered as a robust diagnostic marker for AD (Tokuoka et al., 2019), but PS plays an important role in the mechanisms of AD. First, PS was reported to significantly increase the spine density of hippocampal pyramidal neurons (Nunzi et al., 1989; Spires-Jones et al., 2007); the reduction of dendritic spines is related to brain cognitive impairment in AD and the aged (Spires-Jones et al., 2007). Therefore, PS may ameliorate AD symptoms by restoring dendritic spines. Secondly, PS could negatively regulate the activity of PS synthase (PSS1 and PSS2) to prevent the depletion of PC and PE, resulting in high potassium-induced acetylcholine release (Casamenti et al., 1991; Suzuki et al., 2001; Bergo et al., 2002); the increase of acetylcholine release enhances the activity of cholinergic neurons and improve the cognitive function of AD patients. Thirdly, PS reduces the production of Aβ in CHO-APP/PS1 cells and Aβ-induced toxicity to primary hippocampal neurons (Xu et al., 2021). In addition, PS could combine with others drugs or function as a drug carrier to improve AD symptoms, for example, PS combines with ferulic acid and curcumin significantly to inhibit Aβ production, phosphorylated tau, and IL1β release, and increase brain-derived neurotrophic factor and acetylcholine (Okuda et al., 2019). PS also serves as drug delivery approach for metformin and nicotinamide to ameliorate the cognitive function and inflammation (Vakilinezhad et al., 2018; Saffari et al., 2020).
TABLE 2.
The alteration of phosphatidylserine (PS) in Alzheimer’s disease (AD) patients.
| Age (years) | Gender | Tissue | Method | PS content |
| 81.33 ± 6.57 | 7M/8F | Neocortex | LC-MS/MS | Down |
| 72.9 ± 0.8 | – | Inferior parietal lobule Occipital cortex | 31P NMR | Down |
| 77.4 ± 7.2 | 3M/5F | Hippocampus | Gas chromatography | Down |
| 76.5 ± 8.1 | 7M/9F | Erythrocyte Membrane | HPLC | Down |
| 72.3 ± 10.6 | 4M/1F | Cortex | Annexin V SPECT imaging | Up |
| – | – | Hippocampus Temporal cortex | HPLC | Up |
| 70.9 ± 5.7 | 8M/6F | Blood | LC-MS/MS | NA |
| 81.2 ± 2.48 | 5M/5F | Cortex | HPTLC | NA |
| 70.1 ± 16.3 | 4M/6F | White matter | ESI-MS/MS | Up |
| Gray matter | Down | |||
| Cerebrospinal fluid | Down |
2D-HPLC, two-dimensional liquid chromatography/mass spectrometry;
HPLC, high performance liquid chromatography;
HPTLC, high performance thin layer chromatography;
31PNMR, 31P nuclear magnetic resonance.
TABLE 3.
The alteration of phosphatidylserine (PS) in Alzheimer’s disease (AD) models.
| Species | Model type | Age (month) | Tissue | Method | PS content |
| Mouse | APP/PS1 | 6 | Hippocampus cortex | GC-MS | Up |
| Mouse | ApoE KO | 20 | Corpus callosum | MS | Up |
| Mouse | Tg2576 | 6 | Cortex | LC-MS | Up |
| Rat | 192IgG-saporin induced AD | – | Whole brain | IMS | Up |
| Mouse | APP/PS1 | 9 | Brain cortex | HPLC | Down |
Although the function of PS in AD has not been well clarified, clinical studies have shown that PS benefits patients with AD (Crook et al., 1992; Heiss et al., 1994; More et al., 2014). Clinical studies have shown that PS supplementation significantly improves cognitive function and memory loss in patients with AD. In a clinical trial, treatment the AD patients with PS (100 mg, three times a day for 12 weeks) significantly improved cognitive impairment, especially in the early stages of AD (Crook et al., 1992). Co-administration of 300 mg PS and 240 mg PA in AD patients also had shown the effects on emotion and daily life quality (More et al., 2014). Combination of cognitive training twice a week and PS treatment (200 mg, twice a day) in AD patients benefited brain functions and neuropsychological symptoms at 8 and 16 weeks after the treatment (Heiss et al., 1994). These studies indicated that PS could be used as a daily brain health supplement for AD patients.
Parkinson’s disease
Parkinson’s disease is a neurodegenerative disorder, which clinically appears mainly as bradykinesia, rest tremor, and muscular rigidity (Israel and Hassin-Baer, 2005). Two pathogenesis features of PD are the loss of dopaminergic neurons and the presence of Lewy bodies in the substantia nigra and striatum. The degeneration of dopaminergic neurons leads to the lack of dopamine in the substantia nigra and striatum, while Lewy bodies contains a high concentration of α-Syn that are toxic to neurons (Witt, 2014). Recent studies have shown that abnormal lipid metabolism is also involved in the pathogenesis of PD. Phospholipid levels in peripheral blood of PD patients are higher than control subjects (Lobasso et al., 2017). It is found to have higher PS in the frontal cortex of PD at early stages (Fabelo et al., 2011; Canerina-Amaro et al., 2019). PS is also found to increased significantly in skin fibroblasts from Parkin-mutated PD patients (Li et al., 2015). Similar to PD patients, PS also increased in the brains of PD animal models (Canerina-Amaro et al., 2019). Therefore, PS is increased in the brain of PD patients and PD animals. However, the specific mechanism by which PS is involved in PD is still largely unknown and needs to be further studied.
Increasing PS was proved to promote the aggregation of α-Syn on phospholipid bilayers, which impairs the membrane permeabilization and may contribute to neuronal death in the substantia nigra in the brain of PD patients (Perrin et al., 2000; Zhao et al., 2004; Stockl et al., 2008; Lv et al., 2019; Hannestad et al., 2020). Kanamycin, an aminoglycoside antibiotic with positively charged amino groups, was reported to interfere with H-bonding between PS and α-Syn and inhibit aggregation of α-Syn on membrane (Mahapatra et al., 2019); thus, kanamycin may benefit PD patients. In addition, PS-riched exosomes also accelerated the aggregation of α-Syn and the transmission of α-Syn fibril between brain regions (Xia et al., 2019; Guo et al., 2020). Therefore, upregulated PS is related to the development of PD, and is a potential biomarker for the diagnosis of PD.
Interestingly, PS supplementation also benefits PD patients. In a double-blind study, PS administration showed a significant amelioration on some symptoms, such as motivation, anxiety, and affectivity in PD patients (Funfgeld et al., 1989). PS reversed memory impairment in reserpine-induced PD rat model (Alves et al., 2000); however, PS did not improve cognitive impairment in the classical MPTP-induced PD model (Perry et al., 2004), suggesting different mechanisms in reserpine/MPTP-induce memory impairment. Sleep disorders is a prodromal marker of PD (Tekriwal et al., 2017). In addition, PS can also serve as a drug delivery tool to elevate the bioavailability of drug, such as epigallocatechin-3-gallate and GDF5. Epigallocatechin-3-gallate, an antioxidant isolated from green tea with low bioavailability and high instability, is a potentially therapeutic for PD. Epigallocatechin-3-gallate–loaded PS liposomes reduced the production of nitric oxide, IL-1β, TNF-α, and COX induced by LPS in vivo and in vitro (Cheng et al., 2021). Furthermore, simultaneous intra-nigral injection of PS liposomes loaded with epigallocatechin-3-Gallate restored motor impairment in the rotation behavior test (Cheng et al., 2021). Similarly, PS liposomes loaded with growth differentiation factor GDF5 (a drug that can protect dopaminergic neurons from degeneration) with intranasal administration increased GDF5 concentration in the midbrain by 8 fold (Hanson et al., 2012). PS liposomes loaded with astragaloside IV and nestifin-1 facilitates the penetrating of the blood-brain barrier and reduced the expression of α-Syn (Kuo et al., 2021). Therefore, the PS liposome is a delivery tool for PD drugs.
Major depressive disorder
Major depressive disorder is a very heterogeneous mental disorder. Genetic, psychological, and environmental factors are the main causes of MDD disease (Gu et al., 2020; Nemeroff, 2020; Cao et al., 2021). Previous studies showed that the concentration of PS in peripheral blood of MDD patients increased significantly compared with healthy controls (Kim E. Y. et al., 2018; Homorogan et al., 2021). The content of PS was found to increase in the rat brains of post-traumatic stress (Chaichi et al., 2021). In addition, escitalopram, an antidepressant drug, significantly reduces the concentration of PS in the peripheral blood of patients with MDD, and also improved depressive behaviors (Pastoor and Gobburu, 2014; Homorogan et al., 2021). Interestingly, supplementation with PS significantly improves depressive symptoms in both depressive animals and MDD patients. Clinical studies have reported that treatment of elderly MDD women with PS (200–600 mg/day) for 30 days significantly improved the depressive symptoms (Maggioni et al., 1990; Brambilla et al., 1996). Chronic PS administration (300 mg/day for 1–6 months) for MDD patients also reduced apathy and sleep disturbances, and increased motivation, and interest (Palmieri et al., 1987). Combined supplementation with 100 mg PS, 119 mg docosahexaenoic acid, and 70 mg eicosapentaenoic (three times a day) for 12 weeks significantly improved depressive behaviors in MDD patients, accompanying with the correction of the base level and circadian rhythm of salivary cortisol (Komori, 2015). In animal models, PS administration reduced the immobility time in the forced swimming test in mice, implicating the significant antidepressant effect of PS (Castilho et al., 2004). Treatment post-stroke depressive mice with PS liposomes significantly reduced the immobility time in the forced swimming test and tail suspension test (Partoazar et al., 2021). In addition, intracerebroventricular injection of PS also attenuated stress-induced behaviors in chick isolation-induced stress model (Koutoku et al., 2005).
The specific mechanism of antidepressant effects of PS is still largely unknown; A previous study demonstrated that co-injected PS with scopolamine, an antagonist of acetylcholine receptors abolished antidepressant effects of PS, indicating that muscarinic acetylcholine receptors are required for antidepressant effects of PS (Koutoku et al., 2005). Other studies have reported that PS treatment inhibits the production of ACTH, reduces the production of plasma cortisol, and then slows down the activation of the hypothalamus-pituitary-adrenal (HPA) axis in the process of MDD (Monteleone et al., 1992; Hellhammer et al., 2004; More et al., 2014). Although PS alleviated depressive behaviors in post-stroke depression mice through the reduction of pro-inflammatory cytokines such as TNF-α(Partoazar et al., 2021); the blood contents of IL1β, TNF-α, and IL6 did not alter in elderly patients with MDD after PS supplementation (Brambilla and Maggioni, 1998). Therefore, whether PS functions as an antidepressant through anti-inflammatory needs further studies or not. How PS improves depressive behaviors in MDD patients should be studied further.
Ischemic stroke
Stroke is a very common and serious central nervous system disease that remains the second-leading cause of death and the third-leading cause of death and disability (Collaborators, 2021). Stroke leads to acute brain damage and cell death, which is accompanied by a series of physiological and biochemical changes, such as the increase of reactive oxygen species, calcium-dependent excitotoxicity, the alteration of electrolyte composition, the increase of cytochrome c released by mitochondria phospholipase mediated membrane damage (Fisher and Saver, 2015; Collaborators, 2021; Feske, 2021). PS has been demonstrated to be involved in the biological process of stroke. The content of PS was found to decrease significantly in the brain after ischemic injury; however, PS still decreased and remained below to control even after the long time reperfusion, although the contents of other lipids were restored quickly (Enseleit et al., 1984; Wieloch et al., 1991; Rao et al., 2000). The main reasons for the decrease of PS are unknown and may be related to a large number of cell deaths and the degradation of membrane structure after ischemic injury. The reduction of PS influences the activity of intercellular enzymes such as PKC (Huang et al., 2011). PKC is inactive in the cytosol and active once binds to PS in the presence of Ca2+ in the cell membranes; during ischemia and reperfusion, the total activity of PKC is reduced (Huang et al., 2011). The activity decreased of PKC was partly because PKC-α was dephosphorylated, transited from dimer to trimer, and lost the activity, while PKC-β is degraded by calpain (Louis et al., 1988; Wieloch et al., 1991; Harada et al., 1999). Those studies suggest that ischemia/reperfusion destroys the membrane system and changes the lipids in the membrane, so, the activity of the membrane-bound enzyme is decreased and normal function in the brain is impaired.
Similar to other diseases, PS also has a therapeutic effect for stroke. Ischemia/reperfusion injury has been demonstrated to elicit strong inflammatory responses mediated by activated microglia/macrophages. Microglia/macrophages can be activated by exposed PS on apoptotic cells (Zhao et al., 2017); however, PS liposomes can mimic apoptotic cells to target microglia/macrophages (Hosseini et al., 2015). PS modified microbubbles could cross the blood–brain barrier and target the activated microglia/macrophages in an ischemic stroke mouse model (Zhao et al., 2018). As described above, PS liposomes treatment promoted the production of anti-inflammatory factors, and inhibited the production of pro-inflammatory in phagocytes (Zhang et al., 2006). Therefore, PS liposomes may have a neuroprotective effect through enhancing the anti-inflammatory response of microglia/macrophages in stroke.
Autism spectrum diseases
Autism spectrum disease is a neurodevelopmental disorder and is defined by communication and social deficit, coupled with repetitive and unusual sensory-motor behaviors (Mostafa et al., 2010). Genetic and environmental risk factors may contribute to ASD (Kim et al., 2019). The serum levels of PS were much lower in autistic patients than healthy subjects (El-Ansary et al., 2011). Another study found that serum PS levels decreased in autistic children with impaired sensory compared with control subjects (El-Ansary et al., 2016).
Autism spectrum disease is a heterogeneous neurodevelopment disease. Recently, the largest whole-exome sequencing study of ASD has identified 102 risk genes (Satterstrom et al., 2020). Although numerous biomarkers and risk genes have been reported, there is no robust biomarker to diagnose, prognosis, and predicted ASD. The reduction of PS in the blood of ASD patients could be a potential marker, but still need further studies to evaluate the effectivity in different subgroups.
Attention deficit hyperactivity disorder
Attention deficit hyperactivity disorder is a neurodevelopmental disorder that is characterized by impairing inattention, impulsivity, and motor hyperactivity (Polanczyk et al., 2015). ADHD is a familial disorder and its genetic factors contribute to 76% (Thapar and Cooper, 2016). Several environmental factors are also linked to ADHD, ranging from prenatal and perinatal factors, dietary factors, environmental toxins, and psychiatric social factors (Mayer et al., 2021). First-line pharmacological treatments for ADHD are CNS stimulants, for example, methylphenidate and dexamfetamine. The second-line treatment is the noradrenaline reuptake inhibitor atomoxetine (Thapar and Cooper, 2016). However, 20–30% of children has failed to respond to those drugs or cannot tolerate (Mayer et al., 2021). A clinical study reported that treatment ADHD children with PS-omega3 (250mg/day) for 30 weeks significantly improved ADHD symptoms including hyperactive-impulsive, emotionally, and behavioraly-dysregulated symptoms (Manor et al., 2012). Another clinical observation also showed that supplementation with 200 mg/day PS for 2 months resulted in significant improvements in overall symptoms and short-term auditory memory in ADHD children (Hirayama et al., 2014). However, it is also showed that treatment ADHD patients with 200–300?mg/day of PS significantly reduced inattention, while no effects on overall symptoms of ADHD and hyperactivity-impulsivity (Bruton et al., 2021). In conclusion, PS supplement serves as a no adverse effect and natural nutritional strategy for improving symptoms and ADHD patient’s quality of life (Manor et al., 2013). ADHD is a high prevalence mental disorder and affects a whole lifetime (Polanczyk et al., 2015), whether PS also works in adult ADHD patients need further studies.
Schizophrenia
Schizophrenia is a mental disorder described by impairment of cognitive, behavior, and motion. Symptoms of schizophrenia include delusions, disorganized speech, and hallucinations (Tandon et al., 2013). PS was increased in the thalamic (Schmitt et al., 2004), and not changed in hippocampus of patients with schizophrenia (Hamazaki et al., 2010), although thalamic and hippocampus both are critical brain regions for cognition, perhaps they are contributed to schizophrenia through different mechanisms. On the contrary, the level of PS is lower in fibroblasts and red blood cell from schizophrenia patients (Mahadik et al., 1994; Ozcan et al., 2008), however, alteration of PS in red blood cell membrane of schizophrenia patients is inconsistent in different studies, several studies also supports that PS is not change (Lautin et al., 1982). Abnormal membrane phosphatidylserine and other lipids of fibroblast and red blood cell may predate the onset of schizophrenia (Lautin et al., 1982), could sever as predicted biomarker. PS is also related to poor response of antipsychotics treatments in schizophrenia patients, lower level of PS is found in poor responses to risperidone, olanzapine, and quetiapine (de Almeida et al., 2020), due to the important role of PS in brain function, lower PS may be involved in worse outcomes of treatment.
Spinal cord injury
Spinal cord injury (SCI) is serious CNS disease which caused by traumatic and non-traumatic reasons, and often leads to impairments of sensory and motor function (Anjum et al., 2020). Reduction of PS in spinal cord is reported in several SCI animal models, including experimental autoimmune encephalomyelitis (Chevalier and Rosenberger, 2017), ischemia/reperfusion, and experimental traumatic injury (Lukacova et al., 1996). These studies support that SCI accompanied with the rapid degradation of PS and difficult to restore. In ischemia/reperfusion injury, PS is decreased during ischemia, and only partly region of spinal cord restore to control even after 3 hours long term reperfusion (Lukacova et al., 1996). PS is the main component of myelin sheath, and loss of myelin sheath is common accompaniment of SCI, so to increase PS may benefit SCI patients (Horrocks, 1973; Waxman, 1992; Anjum et al., 2020). Glyceryl triacetate treatment significantly increased PS in experimental autoimmune encephalomyelitis mice model, and improved the loss of myelin (Chevalier and Rosenberger, 2017).
Conclusion
Phosphatidylserine is important nutritional component in the cell membrane, especially with high proportion in the brain and has critical multiple functions involving in cellular signal transduction, cell death and survival, and inflammation. As we summarize in Table 4, alterations of PS are observed in the serum and the brains in different CNS diseases, but the specific biological effects of altered PS in different CNS diseases remain largely unknown and warrant further investigations. However, a body of evidence showed that oral PS benefits patients with different CNS diseases including AD, PD, MDD, and ADHD. In addition, clinical studies also showed that PS had no side effects and was well tolerated (Heiss et al., 1994). Therefore, PS and PS liposome could be a promising supplementation for these neurodegenerative and neurodevelopmental diseases.
TABLE 4.
The trend and function of phosphatidylserine (PS) in central nervous system (CNS) disease.
| Disease | Trend of PS | Function of PS | PS improves disease | |
|
|
||||
| Patient | Animal model | |||
| AD | Inconsistent | Inconsistent | Increase dendritic spine, increase acetylcholine, inhibit the microglial activation and tau hyperphosphorylation | Yes |
| PD | Up | Up | Initiate and enhance aggregation of α-Syn, function as carrier to deliver other drugs | Yes |
| MDD | Up | Up | Blunt the activation of hypothalamus pituitary adrenal axis, attenuate the cytotoxicity of corticosterone | Yes |
| Stroke | No data | Down | Reduced PS decrease the activity of PKC, PS liposomes inhibit inflammation | No data |
| ASD | down | No data | No data | No data |
| ADHD | No data | No data | No data | Yes |
| Schizophrenia | inconsistent | No data | No data | No data |
| SCI | No data | Down | No data | No data |
Author contributions
XM and XL were responsible for writing and literature review. MZ and WW were responsible for drawing the picture. BY and ZM were responsible for the conception and editing of the article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from Natural Science Foundation of Jiangsu Higher Education Institution (21KJB310020), the Science and Technology to people’s livelihood Program of Suzhou (Grant No. sys2018012), and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
- Ajmone-Cat M. A., De Simone R., Nicolini A., Minghetti L. (2003). Effects of phosphatidylserine on p38 mitogen activated protein kinase, cyclic AMP responding element binding protein and nuclear factor-kappaB activation in resting and activated microglial cells. J. Neurochem. 84 413–416. 10.1046/j.1471-4159.2003.01562.x [DOI] [PubMed] [Google Scholar]
- Akyol S., Ugur Z., Yilmaz A., Ustun I., Gorti S. K. K., Oh K., et al. (2021). Lipid Profiling of Alzheimer’s disease brain highlights enrichment in Glycerol(phospho)lipid, and sphingolipid metabolism. Cells 10:2591. 10.3390/Cells10102591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkon D. L., Sun M. K., Nelson T. J. (2007). PKC signaling deficits: A mechanistic hypothesis for the origins of Alzheimer’s disease. Trends Pharmacol. Sci. 28 51–60. 10.1016/j.tips.2006.12.002 [DOI] [PubMed] [Google Scholar]
- Alves C. S., Andreatini R., da Cunha C., Tufik S., Vital M. A. (2000). Phosphatidylserine reverses reserpine-induced amnesia. Eur. J. Pharmacol. 404 161–167. 10.1016/s0014-2999(00)00607-5 [DOI] [PubMed] [Google Scholar]
- Anjum A., Yazid M. D., Fauzi Daud M., Idris J., Ng A. M. H., Selvi Naicker A., et al. (2020). Spinal cord injury: Pathophysiology, multimolecular interactions, and underlying recovery mechanisms. Int. J. Mol. Sci. 21:7533. 10.3390/ijms21207533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramaki Y., Matsuno R., Tsuchiya S. (2001). Involvement of p38 MAP Kinase in the inhibitory effects of phosphatidylserine liposomes on nitric oxide production from macrophages stimulated with LPS. Biochem. Biophys. Res. Commun. 280 982–987. 10.1006/bbrc.2000.4204 [DOI] [PubMed] [Google Scholar]
- Argentiero V., Tavolato B. (1980). Dopamine (DA) and serotonin metabolic levels in the cerebrospinal fluid (CSF) in Alzheimer’s presenile dementia under basic conditions and after stimulation with cerebral cortex phospholipids (BC-PL). J. Neurol. 224 53–58. 10.1007/BF00313207 [DOI] [PubMed] [Google Scholar]
- Bachiller S., Jimenez-Ferrer I., Paulus A., Yang Y., Swanberg M., Deierborg T., et al. (2018). Microglia in neurological diseases: A road map to brain-disease dependent-inflammatory response. Front. Cell. Neurosci. 12:488. 10.3389/fncel.2018.00488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell R. M., Burns D. J. (1991). Lipid activation of protein kinase C. J. Biol. Chem. 266 4661–4664. [PubMed] [Google Scholar]
- Bellini F., Bruni A. (1993). Role of a serum phospholipase A1 in the phosphatidylserine-induced T cell inhibition. FEBS Lett. 316 1–4. 10.1016/0014-5793(93)81724-e [DOI] [PubMed] [Google Scholar]
- Bergo M. O., Gavino B. J., Steenbergen R., Sturbois B., Parlow A. F., Sanan D. A., et al. (2002). Defining the importance of phosphatidylserine synthase 2 in mice. J. Biol. Chem. 277 47701–47708. 10.1074/jbc.M207734200 [DOI] [PubMed] [Google Scholar]
- Boada-Romero E., Martinez J., Heckmann B. L., Green D. R. (2020). The clearance of dead cells by efferocytosis. Nat. Rev. Mol. Cell Biol. 21 398–414. 10.1038/s41580-020-0232-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghese C. M., Gomez R. A., Ramirez O. A. (1993). Phosphatidylserine increases hippocampal synaptic efficacy. Brain Res. Bull. 31 697–700. 10.1016/0361-9230(93)90143-y [DOI] [PubMed] [Google Scholar]
- Brambilla F., Maggioni M. (1998). Blood levels of cytokines in elderly patients with major depressive disorder. Acta Psychiatr. Scand. 97 309–313. 10.1111/j.1600-0447.1998.tb10005.x [DOI] [PubMed] [Google Scholar]
- Brambilla F., Maggioni M., Panerai A. E., Sacerdote P., Cenacchi T. (1996). beta-endorphin concentration in peripheral blood mononuclear cells of elderly depressed patients - Effects of phosphatidylserine therapy. Neuropsychobiology 34 18–21. 10.1159/000119285 [DOI] [PubMed] [Google Scholar]
- Brose N., Petrenko A. G., Sudhof T. C., Jahn R. (1992). Synaptotagmin: A calcium sensor on the synaptic vesicle surface. Science 256 1021–1025. 10.1126/science.1589771 [DOI] [PubMed] [Google Scholar]
- Brown G. C., Neher J. J. (2014). Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 15 209–216. 10.1038/nrn3710 [DOI] [PubMed] [Google Scholar]
- Bruton A., Nauman J., Hanes D., Gard M., Senders A. (2021). Phosphatidylserine for the treatment of pediatric attention-deficit/hyperactivity disorder: A systematic review and meta-analysis. J. Altern. Complement. Med. 27 312–322. 10.1089/acm.2020.0432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calsolaro V., Edison P. (2016). Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement. 12 719–732. 10.1016/j.jalz.2016.02.010 [DOI] [PubMed] [Google Scholar]
- Camenisch T. D., Koller B. H., Earp H. S., Matsushima G. K. (1999). A novel receptor tyrosine kinase, Mer, inhibits TNF-alpha production and lipopolysaccharide-induced endotoxic shock. J. Immunol. 162 3498–3503. [PubMed] [Google Scholar]
- Camici O., Corazzi L. (1995). Import of phosphatidylethanolamine for the assembly of rat brain mitochondrial membranes. J. Membr. Biol. 148 169–176. 10.1007/BF00207272 [DOI] [PubMed] [Google Scholar]
- Canerina-Amaro A., Pereda D., Diaz M., Rodriguez-Barreto D., Casanas-Sanchez V., Heffer M., et al. (2019). Differential aggregation and phosphorylation of alpha synuclein in membrane compartments associated with Parkinson disease. Front. Neurosci. 13:382. 10.3389/fnins.2019.00382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao P., Chen C., Liu A., Shan Q., Zhu X., Jia C., et al. (2021). Early-life inflammation promotes depressive symptoms in adolescence via microglial engulfment of dendritic spines. Neuron 109:2573. 10.1016/j.neuron.2021.06.012 [DOI] [PubMed] [Google Scholar]
- Casamenti F., Scali C., Pepeu G. (1991). Phosphatidylserine reverses the age-dependent decrease in cortical acetylcholine-release - a microdialysis study. Eur. J. Pharmacol. 194 11–16. 10.1016/0014-2999(91)90117-9 [DOI] [PubMed] [Google Scholar]
- Castilho J. C., Perry J. C., Andreatini R., Vital M. A. B. F. (2004). Phosphatidylserine: An antidepressive or a cognitive enhancer? Prog. Neuropsychopharmacol. Biol. Psychiatry 28 731–738. 10.1016/j.pnpbp.2004.05.013 [DOI] [PubMed] [Google Scholar]
- Catorce M. N., Gevorkian G. (2016). LPS-induced murine neuroinflammation model: Main features and suitability for pre-clinical assessment of nutraceuticals. Curr. Neuropharmacol. 14 155–164. 10.2174/1570159x14666151204122017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenacchi T., Bertoldin T., Farina C., Fiori M. G., Crepaldi G. (1993). Cognitive decline in the elderly: A double-blind, placebo-controlled multicenter study on efficacy of phosphatidylserine administration. Aging 5 123–133. 10.1007/BF03324139 [DOI] [PubMed] [Google Scholar]
- Chaichi A., Hasan S. M. A., Mehta N., Donnarumma F., Ebenezer P., Murray K. K., et al. (2021). Label-free lipidome study of paraventricular thalamic nucleus (PVT) of rat brain with post-traumatic stress injury by Raman imaging. Analyst 146 170–183. 10.1039/d0an01615b [DOI] [PubMed] [Google Scholar]
- Cheng C. Y., Barro L., Tsai S. T., Feng T. W., Wu X. Y., Chao C. W., et al. (2021). Epigallocatechin-3-gallate-loaded liposomes favor anti-inflammation of microglia cells and promote neuroprotection. Int. J. Mol. Sci. 22:3037. 10.3390/ijms22063037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier A. C., Rosenberger T. A. (2017). Increasing acetyl-CoA metabolism attenuates injury and alters spinal cord lipid content in mice subjected to experimental autoimmune encephalomyelitis. J. Neurochem. 141 721–737. 10.1111/jnc.14032 [DOI] [PubMed] [Google Scholar]
- Chua B. A., Ngo J. A., Situ K., Morizono K. (2019). Roles of phosphatidylserine exposed on the viral envelope and cell membrane in HIV-1 replication. Cell Commun. Signal. 17:132. 10.1186/s12964-019-0452-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S. A., Muller W. E. (1992). Age-related alterations of NMDA-receptor properties in the mouse forebrain: Partial restoration by chronic phosphatidylserine treatment. Brain Res. 584 174–180. 10.1016/0006-8993(92)90892-d [DOI] [PubMed] [Google Scholar]
- Coleman J. A., Molday R. S. (2011). Critical role of the beta-subunit CDC50A in the stable expression, assembly, subcellular localization, and lipid transport activity of the P4-ATPase ATP8A2. J. Biol. Chem. 286 17205–17216. 10.1074/jbc.M111.229419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborators G. B. D. S. (2021). Global, regional, and national burden of stroke and its risk factors, 1990-2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 20 795–820. 10.1016/S1474-4422(21)00252-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrigan F. M., Horrobin D. F., Skinner E. R., Besson J. A., Cooper M. B. (1998). Abnormal content of n-6 and n-3 long-chain unsaturated fatty acids in the phosphoglycerides and cholesterol esters of parahippocampal cortex from Alzheimer’s disease patients and its relationship to acetyl CoA content. Int. J. Biochem. Cell Biol. 30 197–207. 10.1016/s1357-2725(97)00125-8 [DOI] [PubMed] [Google Scholar]
- Craig L. A., Hong N. S., McDonald R. J. (2011). Revisiting the cholinergic hypothesis in the development of Alzheimer’s disease. Neurosci. Biobehav. Rev. 35 1397–1409. 10.1016/j.neubiorev.2011.03.001 [DOI] [PubMed] [Google Scholar]
- Crook T., Petrie W., Wells C., Massari D. C. (1992). Effects of phosphatidylserine in Alzheimer’s disease. Psychopharmacol. Bull. 28 61–66. [PubMed] [Google Scholar]
- De S. R., Ajmone-Cat M. A., Nicolini A., Minghetti L. (2002). Expression of phosphatidylserine receptor and down-regulation of pro-inflammatory molecule production by its natural ligand in rat microglial cultures. J. Neuropathol. Exp. Neurol. 61 237–244. 10.1093/jnen/61.3.237 [DOI] [PubMed] [Google Scholar]
- de Almeida V., Alexandrino G. L., Aquino A., Gomes A. F., Murgu M., Dobrowolny H., et al. (2020). Changes in the blood plasma lipidome associated with effective or poor response to atypical antipsychotic treatments in schizophrenia patients. Prog. Neuropsychopharmacol. Biol. Psychiatry 101:109945. 10.1016/j.pnpbp.2020.109945 [DOI] [PubMed] [Google Scholar]
- De Simone R., Ajmone-Cat M. A., Minghetti L. (2004). Atypical antiinflammatory activation of microglia induced by apoptotic neurons: Possible role of phosphatidylserine-phosphatidylserine receptor interaction. Mol. Neurobiol. 29 197–212. 10.1385/MN:29:2:197 [DOI] [PubMed] [Google Scholar]
- De Simone R., Ajmone-Cat M. A., Tirassa P., Minghetti L. (2003). Apoptotic PC12 cells exposing phosphatidylserine promote the production of anti-inflammatory and neuroprotective molecules by microglial cells. J. Neuropathol. Exp. Neurol. 62 208–216. 10.1093/jnen/62.2.208 [DOI] [PubMed] [Google Scholar]
- Dejakaisaya H., Harutyunyan A., Kwan P., Jones N. C. (2021). Altered metabolic pathways in a transgenic mouse model suggest mechanistic role of amyloid precursor protein overexpression in Alzheimer’s disease. Metabolomics 17:42. 10.1007/s11306-021-01793-4 [DOI] [PubMed] [Google Scholar]
- Do Van B., Gouel F., Jonneaux A., Timmerman K., Gele P., Petrault M., et al. (2016). Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 94 169–178. 10.1016/j.nbd.2016.05.011 [DOI] [PubMed] [Google Scholar]
- Dvoriantchikova G., Agudelo C., Hernandez E., Shestopalov V. I., Ivanov D. (2009). Phosphatidylserine-containing liposomes promote maximal survival of retinal neurons after ischemic injury. J. Cereb. Blood Flow Metab. 29 1755–1759. 10.1038/jcbfm.2009.95 [DOI] [PubMed] [Google Scholar]
- El-Ansary A., Hassan W. M., Qasem H., Das U. N. (2016). Identification of biomarkers of impaired sensory profiles among autistic patients. PLoS One 11:e0164153. 10.1371/journal.pone.0164153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Ansary A. K., Bacha A. G., Al-Ayahdi L. Y. (2011). Impaired plasma phospholipids and relative amounts of essential polyunsaturated fatty acids in autistic patients from Saudi Arabia. Lipids Health Dis. 10:63. 10.1186/1476-511X-10-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enseleit W. H., Domer F. R., Jarrott D. M., Baricos W. H. (1984). Cerebral phospholipid content and Na+,K+-ATPase activity during ischemia and postischemic reperfusion in the mongolian gerbil. J. Neurochem. 43 320–327. 10.1111/j.1471-4159.1984.tb00903.x [DOI] [PubMed] [Google Scholar]
- Fabelo N., Martin V., Santpere G., Marin R., Torrent L., Ferrer I., et al. (2011). Severe alterations in lipid composition of frontal cortex lipid rafts from Parkinson’s disease and incidental Parkinson’s disease. Mol. Med. 17 1107–1118. 10.2119/molmed.2011.00119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok V. A., Bratton D. L., Rose D. M., Pearson A., Ezekewitz R. A., Henson P. M. (2000). A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature 405 85–90. 10.1038/35011084 [DOI] [PubMed] [Google Scholar]
- Fadok V. A., Voelker D. R., Campbell P. A., Cohen J. J., Bratton D. L., Henson P. M. (1992). Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 148 2207–2216. [PubMed] [Google Scholar]
- Fadok V. A., Xue D., Henson P. (2001). If phosphatidylserine is the death knell, a new phosphatidylserine-specific receptor is the bellringer. Cell Death Differ. 8 582–587. 10.1038/sj.cdd.4400856 [DOI] [PubMed] [Google Scholar]
- Feske S. K. (2021). Ischemic stroke. Am. J. Med. 134 1457–1464. 10.1016/j.amjmed.2021.07.027 [DOI] [PubMed] [Google Scholar]
- Filipello F., Morini R., Corradini I., Zerbi V., Canzi A., Michalski B., et al. (2018). The microglial innate immune receptor TREM2 is required for synapse elimination and normal brain connectivity. Immunity 48 979–991.e8. 10.1016/j.immuni.2018.04.016 [DOI] [PubMed] [Google Scholar]
- Fisher M., Saver J. L. (2015). Future directions of acute ischaemic stroke therapy. Lancet Neurol. 14 758–767. 10.1016/S1474-4422(15)00054-X [DOI] [PubMed] [Google Scholar]
- Fitzner D., Bader J. M., Penkert H., Bergner C. G., Su M., Weil M. T., et al. (2020). Cell-type- and brain-region-resolved mouse brain lipidome. Cell Rep. 32:108132. 10.1016/j.celrep.2020.108132 [DOI] [PubMed] [Google Scholar]
- Folch J. (1948). The chemical structure of phospatidyl serine. J. Biol. Chem. 174 439–450. [PubMed] [Google Scholar]
- Friedland R. P., Budinger T. F., Ganz E., Yano Y., Mathis C. A., Koss B., et al. (1983). Regional cerebral metabolic alterations in dementia of the Alzheimer type: Positron emission tomography with [18F]fluorodeoxyglucose. J. Comput. Assist. Tomogr. 7 590–598. 10.1097/00004728-198308000-00003 [DOI] [PubMed] [Google Scholar]
- Fuller A. D., Van Eldik L. J. (2008). MFG-E8 regulates microglial phagocytosis of apoptotic neurons. J. Neuroimmune Pharmacol. 3 246–256. 10.1007/s11481-008-9118-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funfgeld E. W., Baggen M., Nedwidek P., Richstein B., Mistlberger G. (1989). Double-blind study with phosphatidylserine (PS) in parkinsonian patients with senile dementia of Alzheimer’s type (SDAT). Prog. Clin. Biol. Res. 317 1235–1246. [PubMed] [Google Scholar]
- Funk C. D. (2001). Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science 294 1871–1875. 10.1126/science.294.5548.1871 [DOI] [PubMed] [Google Scholar]
- Glade M. J., Smith K. (2015). Phosphatidylserine and the human brain. Nutrition 31 781–786. 10.1016/j.nut.2014.10.014 [DOI] [PubMed] [Google Scholar]
- Gonzalez-Dominguez R., Garcia-Barrera T., Vitorica J., Gomez-Ariza J. L. (2014). Region-specific metabolic alterations in the brain of the APP/PS1 transgenic mice of Alzheimer’s disease. Biochim. Biophys. Acta 1842(12 Pt A), 2395–2402. 10.1016/j.bbadis.2014.09.014 [DOI] [PubMed] [Google Scholar]
- Gruget C., Bello O., Coleman J., Krishnakumar S. S., Perez E., Rothman J. E., et al. (2020). Synaptotagmin-1 membrane binding is driven by the C2B domain and assisted cooperatively by the C2A domain. Sci. Rep. 10:18011. 10.1038/s41598-020-74923-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X. L., Guo T. J., Si Y. Q., Wang J. X., Zhang W. J., Deng F. R., et al. (2020). Association between ambient air pollution and daily hospital admissions for depression in 75 Chinese cities. Am. J. Psychiatry 177 735–743. 10.1176/appi.ajp.2020.19070748 [DOI] [PubMed] [Google Scholar]
- Gulshan K., Brubaker G., Wang S., Hazen S. L., Smith J. D. (2013). Sphingomyelin depletion impairs anionic phospholipid inward translocation and induces cholesterol efflux. J. Biol. Chem. 288 37166–37179. 10.1074/jbc.M113.512244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M., Wang J., Zhao Y., Feng Y., Han S., Dong Q., et al. (2020). Microglial exosomes facilitate alpha-synuclein transmission in Parkinson’s disease. Brain 143 1476–1497. 10.1093/brain/awaa090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamazaki K., Choi K. H., Kim H. Y. (2010). Phospholipid profile in the postmortem hippocampus of patients with schizophrenia and bipolar disorder: No changes in docosahexaenoic acid species. J. Psychiatr. Res. 44 688–693. 10.1016/j.jpsychires.2009.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hankins H. M., Baldridge R. D., Xu P., Graham T. R. (2015). Role of flippases, scramblases and transfer proteins in phosphatidylserine subcellular distribution. Traffic 16 35–47. 10.1111/tra.12233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannestad J. K., Rocha S., Agnarsson B., Zhdanov V. P., Wittung-Stafshede P., Hook F. (2020). Single-vesicle imaging reveals lipid-selective and stepwise membrane disruption by monomeric alpha-synuclein. Proc. Natl. Acad. Sci. U.S.A. 117 14178–14186. 10.1073/pnas.1914670117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson L. R., Fine J. M., Hoekman J. D., Nguyen T. M., Burns R. B., Martinez P. M., et al. (2012). Intranasal delivery of growth differentiation factor 5 to the central nervous system. Drug Deliv. 19 149–154. 10.3109/10717544.2012.657720 [DOI] [PubMed] [Google Scholar]
- Harada K., Maekawa T., Abu Shama K. M., Yamashima T., Yoshida K. (1999). Translocation and down-regulation of protein kinase C-alpha, -beta, and -gamma isoforms during ischemia-reperfusion in rat brain. J. Neurochem. 72 2556–2564. 10.1046/j.1471-4159.1999.0722556.x [DOI] [PubMed] [Google Scholar]
- He M., Kubo H., Morimoto K., Fujino N., Suzuki T., Takahasi T., et al. (2011). Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. EMBO Rep. 12 358–364. 10.1038/embor.2011.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiss W. D., Kessler J., Mielke R., Szelies B., Herholz K. (1994). Long-term effects of phosphatidylserine, pyritinol, and cognitive training in Alzheimers-Disease - a neuropsychological, eeg, and pet investigation. Dementia 5 88–98. 10.1159/000106702 [DOI] [PubMed] [Google Scholar]
- Heiss W. D., Szelies B., Kessler J., Herholz K. (1991). Abnormalities of energy metabolism in Alzheimer’s disease studied with PET. Ann. N.Y. Acad. Sci. 640 65–71. 10.1111/j.1749-6632.1991.tb00192.x [DOI] [PubMed] [Google Scholar]
- Hellhammer J., Fries E., Buss C., Engert V., Tuch A., Rutenberg D., et al. (2004). Effects of soy lecithin phosphatidic acid and phosphatidylserine complex (PAS) on the endocrine and psychological responses to mental stress. Stress 7 119–126. 10.1080/10253890410001728379 [DOI] [PubMed] [Google Scholar]
- Hirayama S., Terasawa K., Rabeler R., Hirayama T., Inoue T., Tatsumi Y., et al. (2014). The effect of phosphatidylserine administration on memory and symptoms of attention-deficit hyperactivity disorder: A randomised, double-blind, placebo-controlled clinical trial. J. Hum. Nutr. Diet. 27 (Suppl. 2), 284–291. 10.1111/jhn.12090 [DOI] [PubMed] [Google Scholar]
- Homorogan C., Nitusca D., Enatescu V., Schubart P., Moraru C., Socaciu C., et al. (2021). Untargeted plasma metabolomic profiling in patients with major depressive disorder using ultra-high performance liquid chromatography coupled with mass spectrometry. Metabolites 11:466. 10.3390/metabo11070466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horrocks L. A. (1973). Composition and metabolism of myelin phosphoglycerides during maturation and aging. Prog. Brain Res. 40 383–395. 10.1016/S0079-6123(08)60701-3 [DOI] [PubMed] [Google Scholar]
- Hosono H., Aoki J., Nagai Y., Bandoh K., Ishida M., Taguchi R., et al. (2001). Phosphatidylserine-specific phospholipase A1 stimulates histamine release from rat peritoneal mast cells through production of 2-acyl-1-lysophosphatidylserine. J. Biol. Chem. 276 29664–29670. 10.1074/jbc.M104597200 [DOI] [PubMed] [Google Scholar]
- Hosseini H., Li Y., Kanellakis P., Tay C., Cao A., Tipping P., et al. (2015). Phosphatidylserine liposomes mimic apoptotic cells to attenuate atherosclerosis by expanding polyreactive IgM producing B1a lymphocytes. Cardiovasc. Res. 106 443–452. 10.1093/cvr/cvv037 [DOI] [PubMed] [Google Scholar]
- Huang B. X., Akbar M., Kevala K., Kim H. Y. (2011). Phosphatidylserine is a critical modulator for Akt activation. J. Cell Biol. 192 979–992. 10.1083/jcb.201005100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh M. L., Fadok V. A., Henson P. M. (2002). Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J. Clin. Invest. 109 41–50. 10.1172/JCI11638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel Z., Hassin-Baer S. (2005). Subthalamic stimulation for Parkinson’s disease. Isr. Med. Assoc. J. 7 458–463. [PubMed] [Google Scholar]
- Johnson D. A., Cho T. M., Loh H. H. (1977). The binding of 5-hydroxytryptamine to acidic lipids in isobutanol. J. Neurochem. 29 1101–1103. 10.1111/j.1471-4159.1977.tb06514.x [DOI] [PubMed] [Google Scholar]
- Ju Y., Tam K. Y. (2022). Pathological mechanisms and therapeutic strategies for Alzheimer’s disease. Neural Regen. Res. 17 543–549. 10.4103/1673-5374.320970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalienkova V., Clerico Mosina V., Paulino C. (2021). The groovy TMEM16 family: Molecular mechanisms of lipid scrambling and ion conduction. J. Mol. Biol. 433:166941. 10.1016/j.jmb.2021.166941 [DOI] [PubMed] [Google Scholar]
- Kay J. G., Fairn G. D. (2019). Distribution, dynamics and functional roles of phosphatidylserine within the cell. Cell Commun. Signal. 17:126. 10.1186/s12964-019-0438-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W. S., Jary E., Pickford R., He Y., Ahmed R. M., Piguet O., et al. (2018). Lipidomics analysis of behavioral variant frontotemporal dementia: A scope for biomarker development. Front. Neurol. 9:104. 10.3389/fneur.2018.00104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E. Y., Lee J. W., Lee M. Y., Kim S. H., Mok H. J., Ha K., et al. (2018). Serum lipidomic analysis for the discovery of biomarkers for major depressive disorder in drug-free patients. Psychiatry Res. 265 174–182. 10.1016/j.psychres.2018.04.029 [DOI] [PubMed] [Google Scholar]
- Kim H. Y., Huang B. X., Spector A. A. (2014). Phosphatidylserine in the brain: Metabolism and function. Prog. Lipid Res. 56 1–18. 10.1016/j.plipres.2014.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. Y., Son M. J., Son C. Y., Radua J., Eisenhut M., Gressier F., et al. (2019). Environmental risk factors and biomarkers for autism spectrum disorder: An umbrella review of the evidence. Lancet Psychiatry 6 590–600. 10.1016/S2215-0366(19)30181-6 [DOI] [PubMed] [Google Scholar]
- Kimura H. (2021). From neurotransmission to neuronal disorders. Br. J. Pharmacol. 178 747–749. 10.1111/bph.15354 [DOI] [PubMed] [Google Scholar]
- Kiraz Y., Adan A., Kartal Yandim M., Baran Y. (2016). Major apoptotic mechanisms and genes involved in apoptosis. Tumour Biol. 37 8471–8486. 10.1007/s13277-016-5035-9 [DOI] [PubMed] [Google Scholar]
- Klinkhammer P., Szelies B., Heiss W.-D. (1991). Effect of phosphatidylserine on cerebral glucose metabolism in Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 1 197–201. [Google Scholar]
- Koh T. W., Bellen H. J. (2003). Synaptotagmin I, a Ca2+ sensor for neurotransmitter release. Trends Neurosci. 26 413–422. 10.1016/S0166-2236(03)00195-4 [DOI] [PubMed] [Google Scholar]
- Komori T. (2015). The effects of phosphatidylserine and Omega-3 fatty acid-containing supplement on late life depression. Ment. Illn. 7:5647. 10.4081/mi.2015.5647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutoku T., Takahashi H., Tomonaga S., Oikawa D., Saito S., Tachibana T., et al. (2005). Central administration of phosphatidylserine attenuates isolation stress-induced behavior in chicks. Neurochem. Int. 47 183–189. 10.1016/j.neuint.2005.03.006 [DOI] [PubMed] [Google Scholar]
- Kowalski K., Mulak A. (2019). Brain-gut-microbiota axis in Alzheimer’s disease. J. Neurogastroenterol. Motil. 25 48–60. 10.5056/jnm18087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasemann S., Madore C., Cialic R., Baufeld C., Calcagno N., El Fatimy R., et al. (2017). The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47 566–581.e9. 10.1016/j.immuni.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo Y. C., Chen I. Y., Rajesh R. (2021). Astragaloside IV- and nesfatin-1-encapsulated phosphatidylserine liposomes conjugated with wheat germ agglutinin and leptin to activate anti-apoptotic pathway and block phosphorylated tau protein expression for Parkinson’s disease treatment. Mater. Sci. Eng. C Mater. Biol. Appl. 129:112361. 10.1016/j.msec.2021.112361 [DOI] [PubMed] [Google Scholar]
- Lampl Y., Lorberboym M., Blankenberg F. G., Sadeh M., Gilad R. (2006). Annexin V SPECT imaging of phosphatidylserine expression in patients with dementia. Neurology 66 1253–1254. 10.1212/01.wnl.0000208436.75615.8c [DOI] [PubMed] [Google Scholar]
- Lautin A., Cordasco D. M., Segarnick D. J., Wood L., Mason M. F., Wolkin A., et al. (1982). Red cell phospholipids in schizophrenia. Life Sci. 31 3051–3056. 10.1016/0024-3205(82)90074-1 [DOI] [PubMed] [Google Scholar]
- Lee J. H., Kim J. Y., Noh S., Lee H., Lee S. Y., Mun J. Y., et al. (2021). Astrocytes phagocytose adult hippocampal synapses for circuit homeostasis. Nature 590 612–617. 10.1038/s41586-020-03060-3 [DOI] [PubMed] [Google Scholar]
- Lemke G. (2017). Phosphatidylserine Is the Signal for TAM Receptors and Their Ligands. Trends Biochem. Sci. 42 738–748. 10.1016/j.tibs.2017.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke G. (2019). How macrophages deal with death. Nat. Rev. Immunol. 19 539–549. 10.1038/s41577-019-0167-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng F., Edison P. (2021). Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 17 157–172. 10.1038/s41582-020-00435-y [DOI] [PubMed] [Google Scholar]
- Leventis P. A., Grinstein S. (2010). The distribution and function of phosphatidylserine in cellular membranes. Annu. Rev. Biophys. 39 407–427. 10.1146/annurev.biophys.093008.131234 [DOI] [PubMed] [Google Scholar]
- Li T., Chiou B., Gilman C. K., Luo R., Koshi T., Yu D. K., et al. (2020). A splicing isoform of GPR56 mediates microglial synaptic refinement via phosphatidylserine binding. EMBO J. 39:e104136. 10.15252/embj.2019104136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T., Yu D., Oak H. C., Zhu B., Wang L., Jiang X., et al. (2021). Phospholipid-flippase chaperone CDC50A is required for synapse maintenance by regulating phosphatidylserine exposure. EMBO J. 40:e107915. 10.15252/embj.2021107915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z. G., Zhang J. B., Sun H. R. (2015). Increased plasma levels of phospholipid in Parkinson’s disease with mild cognitive impairment. J. Clin. Neurosci. 22 1268–1271. 10.1016/j.jocn.2015.02.013 [DOI] [PubMed] [Google Scholar]
- Lobasso S., Tanzarella P., Vergara D., Maffia M., Cocco T., Corcelli A. (2017). Lipid profiling of parkin-mutant human skin fibroblasts. J. Cell. Physiol. 232 3540–3551. 10.1002/jcp.25815 [DOI] [PubMed] [Google Scholar]
- Lou X., Kim J., Hawk B. J., Shin Y. K. (2017). alpha-Synuclein may cross-bridge v-SNARE and acidic phospholipids to facilitate SNARE-dependent vesicle docking. Biochem. J. 474 2039–2049. 10.1042/BCJ20170200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis J. C., Magal E., Yavin E. (1988). Protein kinase C alterations in the fetal rat brain after global ischemia. J. Biol. Chem. 263 19282–19285. [PubMed] [Google Scholar]
- Lourenssen S., Blennerhassett M. G. (1998). Lysophosphatidylserine potentiates nerve growth factor-induced differentiation of PC12 cells. Neurosci. Lett. 248 77–80. 10.1016/s0304-3940(98)00275-4 [DOI] [PubMed] [Google Scholar]
- Lukacova N., Halat G., Chavko M., Marsala J. (1996). Ischemia-reperfusion injury in the spinal cord of rabbits strongly enhances lipid peroxidation and modifies phospholipid profiles. Neurochem. Res. 21 869–873. 10.1007/BF02532334 [DOI] [PubMed] [Google Scholar]
- Lv Z., Hashemi M., Banerjee S., Zagorski K., Rochet J. C., Lyubchenko Y. L. (2019). Assembly of alpha-synuclein aggregates on phospholipid bilayers. Biochim. Biophys. Acta Proteins Proteom. 1867 802–812. 10.1016/j.bbapap.2019.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H. M., Wu Z., Nakanishi H. (2011). Phosphatidylserine-containing liposomes suppress inflammatory bone loss by ameliorating the cytokine imbalance provoked by infiltrated macrophages. Lab. Invest. 91 921–931. 10.1038/labinvest.2011.54 [DOI] [PubMed] [Google Scholar]
- Maggioni M., Picotti G. B., Bondiolotti G. P., Panerai A., Cenacchi T., Nobile P., et al. (1990). Effects of phosphatidylserine therapy in geriatric-patients with depressive-disorders. Acta Psychiatr. Scand. 81 265–270. 10.1111/j.1600-0447.1990.tb06494.x [DOI] [PubMed] [Google Scholar]
- Mahadik S. P., Mukherjee S., Correnti E. E., Kelkar H. S., Wakade C. G., Costa R. M., et al. (1994). Plasma membrane phospholipid and cholesterol distribution of skin fibroblasts from drug-naive patients at the onset of psychosis. Schizophr. Res. 13 239–247. 10.1016/0920-9964(94)90048-5 [DOI] [PubMed] [Google Scholar]
- Mahapatra A., Sarkar S., Biswas S. C., Chattopadhyay K. (2019). An aminoglycoside antibiotic inhibits both lipid-induced and solution-phase fibrillation of alpha-synuclein in vitro. Chem. Commun. 55 11052–11055. 10.1039/c9cc04251b [DOI] [PubMed] [Google Scholar]
- Manor I., Magen A., Keidar D., Rosen S., Tasker H., Cohen T., et al. (2012). The effect of phosphatidylserine containing Omega3 fatty-acids on attention-deficit hyperactivity disorder symptoms in children: A double-blind placebo-controlled trial, followed by an open-label extension. Eur. Psychiatry 27 335–342. 10.1016/j.eurpsy.2011.05.004 [DOI] [PubMed] [Google Scholar]
- Manor I., Magen A., Keidar D., Rosen S., Tasker H., Cohen T., et al. (2013). Safety of phosphatidylserine containing omega3 fatty acids in ADHD children: A double-blind placebo-controlled trial followed by an open-label extension. Eur. Psychiatry 28 386–391. 10.1016/j.eurpsy.2012.11.001 [DOI] [PubMed] [Google Scholar]
- Martin V., Fabelo N., Santpere G., Puig B., Marin R., Ferrer I., et al. (2010). Lipid alterations in lipid rafts from Alzheimer’s disease human brain cortex. J. Alzheimers Dis. 19 489–502. 10.3233/JAD-2010-1242 [DOI] [PubMed] [Google Scholar]
- Martinez-Gardeazabal J., Gonzalez de San Roman E., Moreno-Rodriguez M., Llorente-Ovejero A., Manuel I., Rodriguez-Puertas R. (2017). Lipid mapping of the rat brain for models of disease. Biochim. Biophys. Acta Biomembr. 1859(9 Pt B), 1548–1557. 10.1016/j.bbamem.2017.02.011 [DOI] [PubMed] [Google Scholar]
- Mayer J. S., Bernhard A., Fann N., Boxhoorn S., Hartman C. A., Reif A., et al. (2021). Cognitive mechanisms underlying depressive disorders in ADHD: A systematic review. Neurosci. Biobehav. Rev. 121 307–345. 10.1016/j.neubiorev.2020.12.018 [DOI] [PubMed] [Google Scholar]
- Mizutani T., Nakashima S., Nozawa Y. (1998). Changes in the expression of protein kinase C (PKC), phospholipases C (PLC) and D (PLD) isoforms in spleen, brain and kidney of the aged rat: RT-PCR and Western blot analysis. Mech. Ageing Dev. 105 151–172. 10.1016/s0047-6374(98)00094-3 [DOI] [PubMed] [Google Scholar]
- Monteleone P., Maj M., Beinat L., Natale M., Kemali D. (1992). Blunting by chronic phosphatidylserine administration of the stress-induced activation of the hypothalamo-pituitary-adrenal axis in healthy men. Eur. J. Clin. Pharmacol. 42 385–388. 10.1007/BF00280123 [DOI] [PubMed] [Google Scholar]
- More M. I., Freitas U., Rutenberg D. (2014). Positive effects of soy lecithin-derived phosphatidylserine plus phosphatidic acid on memory, cognition, daily functioning, and mood in elderly patients with Alzheimer’s disease and dementia. Adv. Ther. 31 1247–1262. 10.1007/s12325-014-0165-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafa G. A., El-Hadidi E. S., Hewedi D. H., Abdou M. M. (2010). Oxidative stress in Egyptian children with autism: Relation to autoimmunity. J. Neuroimmunol. 219 114–118. 10.1016/j.jneuroim.2009.12.003 [DOI] [PubMed] [Google Scholar]
- Nagata S., Sakuragi T., Segawa K. (2020). Flippase and scramblase for phosphatidylserine exposure. Curr. Opin. Immunol. 62 31–38. 10.1016/j.coi.2019.11.009 [DOI] [PubMed] [Google Scholar]
- Nazareth L., St John J., Murtaza M., Ekberg J. (2021). Phagocytosis by peripheral glia: Importance for nervous system functions and implications in injury and disease. Front. Cell. Dev. Biol. 9:660259. 10.3389/fcell.2021.660259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeroff C. B. (2020). The state of our understanding of the pathophysiology and optimal treatment of depression: Glass half full or half empty? Am. J. Psychiatry 177 671–685. 10.1176/appi.ajp.2020.20060845 [DOI] [PubMed] [Google Scholar]
- Nunzi M. G., Milan F., Guidolin D., Polato P., Toffano G. (1989). Effects of phosphatidylserine administration of aged-related structural changes in the rat hippocampus and septal complex. Pharmacopsychiatry 22 (Suppl. 2), 125–128. 10.1055/s-2007-1014632 [DOI] [PubMed] [Google Scholar]
- Okuda M., Fujita Y., Sugimoto H. (2019). The additive effects of low dose intake of ferulic acid, phosphatidylserine and curcumin, not alone, improve cognitive function in APPswe/PS1dE9 transgenic mice. Biol. Pharm. Bull. 42 1694–1706. 10.1248/bpb.b19-00332 [DOI] [PubMed] [Google Scholar]
- Oma S., Mawatari S., Saito K., Wakana C., Tsuboi Y., Yamada T., et al. (2012). Changes in phospholipid composition of erythrocyte membrane in Alzheimer’s disease. Dement. Geriatr. Cogn. Dis. Extra 2 298–303. 10.1159/000341603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlowski A., Grzybek M., Bunker A., Pasenkiewicz-Gierula M., Vattulainen I., Mannisto P. T., et al. (2012). Strong preferences of dopamine and l-dopa towards lipid head group: Importance of lipid composition and implication for neurotransmitter metabolism. J. Neurochem. 122 681–690. 10.1111/j.1471-4159.2012.07813.x [DOI] [PubMed] [Google Scholar]
- Ozcan O., Ipcioglu O. M., Gultepe M., Basogglu C. (2008). Altered red cell membrane compositions related to functional vitamin B(12) deficiency manifested by elevated urine methylmalonic acid concentrations in patients with schizophrenia. Ann. Clin. Biochem. 45(Pt 1), 44–49. 10.1258/acb.2007.007057 [DOI] [PubMed] [Google Scholar]
- Paasila P. J., Aramideh J. A., Sutherland G. T., Graeber M. B. (2021). Synapses, microglia, and lipids in Alzheimer’s disease. Front. Neurosci. 15:778822. 10.3389/fnins.2021.778822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paidassi H., Tacnet-Delorme P., Garlatti V., Darnault C., Ghebrehiwet B., Gaboriaud C., et al. (2008). C1q binds phosphatidylserine and likely acts as a multiligand-bridging molecule in apoptotic cell recognition. J. Immunol. 180 2329–2338. 10.4049/jimmunol.180.4.2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri G., Palmieri R., Inzoli M. R., Lombardi G., Sottini C., Tavolato B., et al. (1987). Double-blind controlled trial of phosphatidylserine in patients with senile mental deterioration. Clin. Trials J. 24 73–83. [Google Scholar]
- Park D., Tosello-Trampont A. C., Elliott M. R., Lu M., Haney L. B., Ma Z., et al. (2007). BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature 450 430–434. 10.1038/nature06329 [DOI] [PubMed] [Google Scholar]
- Park S. Y., Kang K. B., Thapa N., Kim S. Y., Lee S. J., Kim I. S. (2008). Requirement of adaptor protein GULP during stabilin-2-mediated cell corpse engulfment. J. Biol. Chem. 283 10593–10600. 10.1074/jbc.M709105200 [DOI] [PubMed] [Google Scholar]
- Partoazar A., Seyyedian Z., Zamanian G., Saffari P. M., Muhammadnejad A., Dehpour A. R., et al. (2021). Neuroprotective phosphatidylserine liposomes alleviate depressive-like behavior related to stroke through neuroinflammation attenuation in the mouse hippocampus. Psychopharmacology 238 1531–1539. 10.1007/s00213-021-05783-1 [DOI] [PubMed] [Google Scholar]
- Pastoor D., Gobburu J. (2014). Clinical pharmacology review of escitalopram for the treatment of depression. Expert Opin. Drug Metab. Toxicol. 10 121–128. 10.1517/17425255.2014.863873 [DOI] [PubMed] [Google Scholar]
- Perrin R. J., Woods W. S., Clayton D. F., George J. M. (2000). Interaction of human alpha-Synuclein and Parkinson’s disease variants with phospholipids. Structural analysis using site-directed mutagenesis. J. Biol. Chem. 275 34393–34398. 10.1074/jbc.M004851200 [DOI] [PubMed] [Google Scholar]
- Perry J. C., Da Cunha C., Anselmo-Franci J., Andreatini R., Miyoshi E., Tufik S., et al. (2004). Behavioural and neurochemical effects of phosphatidylserine in MPTP lesion of the substantia nigra of rats. Eur. J. Pharmacol. 484 225–233. 10.1016/j.ejphar.2003.11.029 [DOI] [PubMed] [Google Scholar]
- Pettegrew J. W., Panchalingam K., Hamilton R. L., McClure R. J. (2001). Brain membrane phospholipid alterations in Alzheimer’s disease. Neurochem. Res. 26 771–782. 10.1023/a:1011603916962 [DOI] [PubMed] [Google Scholar]
- Polanczyk G. V., Salum G. A., Sugaya L. S., Caye A., Rohde L. A. (2015). Annual Research Review: A meta-analysis of the worldwide prevalence of mental disorders in children and adolescents. J. Child Psychol. Psychiatry 56 345–365. 10.1111/jcpp.12381 [DOI] [PubMed] [Google Scholar]
- Rajotte I., Hasanbasic I., Blostein M. (2008). Gas6-mediated signaling is dependent on the engagement of its gamma-carboxyglutamic acid domain with phosphatidylserine. Biochem. Biophys. Res. Commun. 376 70–73. 10.1016/j.bbrc.2008.08.083 [DOI] [PubMed] [Google Scholar]
- Rao A. M., Hatcher J. F., Dempsey R. J. (2000). Lipid alterations in transient forebrain ischemia: Possible new mechanisms of CDP-choline neuroprotection. J. Neurochem. 75 2528–2535. 10.1046/j.1471-4159.2000.0752528.x [DOI] [PubMed] [Google Scholar]
- Sabogal-Guaqueta A. M., Arias-Londono J. D., Gutierrez-Vargas J., Sepulveda-Falla D., Glatzel M., Villegas-Lanau A., et al. (2020). Common disbalance in the brain parenchyma of dementias: Phospholipid profile analysis between CADASIL and sporadic Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 1866:165797. 10.1016/j.bbadis.2020.165797 [DOI] [PubMed] [Google Scholar]
- Saffari P. M., Alijanpour S., Takzaree N., Sahebgharani M., Etemad-Moghadam S., Noorbakhsh F., et al. (2020). Metformin loaded phosphatidylserine nanoliposomes improve memory deficit and reduce neuroinflammation in streptozotocin-induced Alzheimer’s disease model. Life Sci. 255:117861. 10.1016/j.lfs.2020.117861 [DOI] [PubMed] [Google Scholar]
- Satterstrom F. K., Kosmicki J. A., Wang J., Breen M. S., De Rubeis S., An J. Y., et al. (2020). Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180 568–584.e23. 10.1016/j.cell.2019.12.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt A., Wilczek K., Blennow K., Maras A., Jatzko A., Petroianu G., et al. (2004). Altered thalamic membrane phospholipids in schizophrenia: A postmortem study. Biol. Psychiatry 56 41–45. 10.1016/j.biopsych.2004.03.019 [DOI] [PubMed] [Google Scholar]
- Schreiber S., Kampf-Sherf O., Gorfine M., Kelly D., Oppenheim Y., Lerer B. (2000). An open trial of plant-source derived phosphatydilserine for treatment of age-related cognitive decline. Isr. J. Psychiatry Relat. Sci. 37 302–307. [PubMed] [Google Scholar]
- Schuldiner O., Yaron A. (2015). Mechanisms of developmental neurite pruning. Cell. Mol. Life Sci. 72 101–119. 10.1007/s00018-014-1729-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott-Hewitt N., Perrucci F., Morini R., Erreni M., Mahoney M., Witkowska A., et al. (2020). Local externalization of phosphatidylserine mediates developmental synaptic pruning by microglia. EMBO J. 39:e105380. 10.15252/embj.2020105380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segawa K., Kurata S., Yanagihashi Y., Brummelkamp T. R., Matsuda F., Nagata S. (2014). Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 344 1164–1168. 10.1126/science.1252809 [DOI] [PubMed] [Google Scholar]
- Segawa K., Yanagihashi Y., Yamada K., Suzuki C., Uchiyama Y., Nagata S. (2018). Phospholipid flippases enable precursor B cells to flee engulfment by macrophages. Proc. Natl. Acad. Sci. U.S.A. 115 12212–12217. 10.1073/pnas.1814323115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shacham-Silverberg V., Sar Shalom H., Goldner R., Golan-Vaishenker Y., Gurwicz N., Gokhman I., et al. (2018). Phosphatidylserine is a marker for axonal debris engulfment but its exposure can be decoupled from degeneration. Cell Death Dis. 9:1116. 10.1038/s41419-018-1155-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires-Jones T. L., Meyer-Luehmann M., Osetek J. D., Jones P. B., Stern E. A., Bacskai B. J., et al. (2007). Impaired spine stability underlies plaque-related spine loss in an Alzheimer’s disease mouse model. Am. J. Pathol. 171 1304–1311. 10.2353/ajpath.2007.070055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stace C. L., Ktistakis N. T. (2006). Phosphatidic acid- and phosphatidylserine-binding proteins. Biochim. Biophys. Acta 1761 913–926. 10.1016/j.bbalip.2006.03.006 [DOI] [PubMed] [Google Scholar]
- Stockl M., Fischer P., Wanker E., Herrmann A. (2008). alpha-Synuclein selectively binds to anionic phospholipids embedded in liquid-disordered domains. J. Mol. Biol. 375 1394–1404. 10.1016/j.jmb.2007.11.051 [DOI] [PubMed] [Google Scholar]
- Stone S. J., Vance J. E. (2000). Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J. Biol. Chem. 275 34534–34540. 10.1074/jbc.M002865200 [DOI] [PubMed] [Google Scholar]
- Sun M. K., Alkon D. L. (2010). Pharmacology of protein kinase C activators: Cognition-enhancing and antidementic therapeutics. Pharmacol. Ther. 127 66–77. 10.1016/j.pharmthera.2010.03.001 [DOI] [PubMed] [Google Scholar]
- Suzuki J., Denning D. P., Imanishi E., Horvitz H. R., Nagata S. (2013). Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 341 403–406. 10.1126/science.1236758 [DOI] [PubMed] [Google Scholar]
- Suzuki J., Imanishi E., Nagata S. (2014). Exposure of phosphatidylserine by Xk-related protein family members during apoptosis. J. Biol. Chem. 289 30257–30267. 10.1074/jbc.M114.583419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki J., Imanishi E., Nagata S. (2016). Xkr8 phospholipid scrambling complex in apoptotic phosphatidylserine exposure. Proc. Natl. Acad. Sci. U.S.A. 113 9509–9514. 10.1073/pnas.1610403113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S., Yamatoya H., Sakai M., Kataoka A., Furushiro M., Kudo S. (2001). Oral administration of soybean lecithin transphosphatidylated phosphatidylserine improves memory impairment in aged rats. J. Nutr. 131 2951–2956. 10.1093/jn/131.11.2951 [DOI] [PubMed] [Google Scholar]
- Svennerholm L. (1968). Distribution and fatty acid composition of phosphoglycerides in normal human brain. J. Lipid Res. 9 570–579. [PubMed] [Google Scholar]
- Takada N., Takatsu H., Miyano R., Nakayama K., Shin H. W. (2015). ATP11C mutation is responsible for the defect in phosphatidylserine uptake in UPS-1 cells. J. Lipid Res. 56 2151–2157. 10.1194/jlr.M062547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandon R., Gaebel W., Barch D. M., Bustillo J., Gur R. E., Heckers S., et al. (2013). Definition and description of schizophrenia in the DSM-5. Schizophr. Res. 150 3–10. 10.1016/j.schres.2013.05.028 [DOI] [PubMed] [Google Scholar]
- Tekriwal A., Kern D. S., Tsai J., Ince N. F., Wu J., Thompson J. A., et al. (2017). REM sleep behaviour disorder: Prodromal and mechanistic insights for Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 88 445–451. 10.1136/jnnp-2016-314471 [DOI] [PubMed] [Google Scholar]
- Thapar A., Cooper M. (2016). Attention deficit hyperactivity disorder. Lancet 387 1240–1250. 10.1016/S0140-6736(15)00238-X [DOI] [PubMed] [Google Scholar]
- Thompson S. M. (2000). Cognition: Long-term potentiation. Am. J. Psychiatry 157:492. 10.1176/appi.ajp.157.4.492 [DOI] [PubMed] [Google Scholar]
- Tian L., Choi S. C., Murakami Y., Allen J., Morse H. C., III, Qi C. F., et al. (2014). p85alpha recruitment by the CD300f phosphatidylserine receptor mediates apoptotic cell clearance required for autoimmunity suppression. Nat. Commun. 5:3146. 10.1038/ncomms4146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuoka S. M., Kita Y., Shimizu T., Oda Y. (2019). Isobaric mass tagging and triple quadrupole mass spectrometry to determine lipid biomarker candidates for Alzheimer’s disease. PLoS One 14:e0226073. 10.1371/journal.pone.0226073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker W. C., Weber T., Chapman E. R. (2004). Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science 304 435–438. 10.1126/science.1097196 [DOI] [PubMed] [Google Scholar]
- Uchiyama Y., Maxson M. M., Sawada T., Nakano A., Ewing A. G. (2007). Phospholipid mediated plasticity in exocytosis observed in PC12 cells. Brain Res. 1151 46–54. 10.1016/j.brainres.2007.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vago J. P., Amaral F. A., van de Loo F. A. J. (2021). Resolving inflammation by TAM receptor activation. Pharmacol. Ther. 227:107893. 10.1016/j.pharmthera.2021.107893 [DOI] [PubMed] [Google Scholar]
- Vakilinezhad M. A., Amini A., Akbari Javar H., Baha’addini Beigi Zarandi B. F., Montaseri H., Dinarvand R. (2018). Nicotinamide loaded functionalized solid lipid nanoparticles improves cognition in Alzheimer’s disease animal model by reducing Tau hyperphosphorylation. Daru 26 165–177. 10.1007/s40199-018-0221-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G., Voelker D. R., Feigenson G. W. (2008). Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9 112–124. 10.1038/nrm2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance J. E. (2015). Phospholipid synthesis and transport in mammalian cells. Traffic 16 1–18. 10.1111/tra.12230 [DOI] [PubMed] [Google Scholar]
- Vilalta A., Brown G. C. (2018). Neurophagy, the phagocytosis of live neurons and synapses by glia, contributes to brain development and disease. FEBS J. 285 3566–3575. 10.1111/febs.14323 [DOI] [PubMed] [Google Scholar]
- Voelker D. R. (2000). Interorganelle transport of aminoglycerophospholipids. Biochim. Biophys. Acta 1486 97–107. 10.1016/s1388-1981(00)00051-2 [DOI] [PubMed] [Google Scholar]
- Waxman S. G. (1992). Demyelination in spinal cord injury and multiple sclerosis: What can we do to enhance functional recovery? J. Neurotrauma 9 (Suppl. 1), S105–S117. [PubMed] [Google Scholar]
- Wells K., Farooqui A. A., Liss L., Horrocks L. A. (1995). Neural membrane phospholipids in Alzheimer disease. Neurochem. Res. 20 1329–1333. 10.1007/BF00992508 [DOI] [PubMed] [Google Scholar]
- Wen X. Y., Stewart A. K., Skaug J., Wei E., Tsui L. C. (2001). Murine phosphatidylserine-specific phospholipase A1 (Ps-pla1) maps to chromosome 16 but is distinct from the lpd (lipid defect) locus. Mamm. Genome 12 129–132. 10.1007/s003350010256 [DOI] [PubMed] [Google Scholar]
- Wieloch T., Cardell M., Bingren H., Zivin J., Saitoh T. (1991). Changes in the activity of protein kinase C and the differential subcellular redistribution of its isozymes in the rat striatum during and following transient forebrain ischemia. J. Neurochem. 56 1227–1235. 10.1111/j.1471-4159.1991.tb11415.x [DOI] [PubMed] [Google Scholar]
- Wilton D. K., Dissing-Olesen L., Stevens B. (2019). Neuron-glia signaling in synapse elimination. Annu. Rev. Neurosci. 42 107–127. 10.1146/annurev-neuro-070918-050306 [DOI] [PubMed] [Google Scholar]
- Witt S. N. (2014). Lipid disequilibrium in biological membranes, a possible pathway to neurodegeneration. Commun. Integr. Biol. 7:e993266. 10.4161/19420889.2014.993266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodburn S. C., Bollinger J. L., Wohleb E. S. (2021). The semantics of microglia activation: Neuroinflammation, homeostasis, and stress. J. Neuroinflammation 18:258. 10.1186/s12974-021-02309-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y., Zhang G., Han C., Ma K., Guo X., Wan F., et al. (2019). Microglia as modulators of exosomal alpha-synuclein transmission. Cell Death Dis. 10:174. 10.1038/s41419-019-1404-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z. J., Li Q., Ding L., Shi H. H., Xue C. H., Mao X. Z., et al. (2021). A comparative study of the effects of phosphatidylserine rich in DHA and EPA on Abeta-induced Alzheimer’s disease using cell models. Food Funct. 12 4411–4423. 10.1039/d1fo00286d [DOI] [PubMed] [Google Scholar]
- Yao J. K., Wengenack T. M., Curran G. L., Poduslo J. F. (2009). Reduced membrane lipids in the cortex of Alzheimer’s disease transgenic mice. Neurochem. Res. 34 102–108. 10.1007/s11064-008-9673-1 [DOI] [PubMed] [Google Scholar]
- Yeung T., Gilbert G. E., Shi J., Silvius J., Kapus A., Grinstein S. (2008). Membrane phosphatidylserine regulates surface charge and protein localization. Science 319 210–213. 10.1126/science.1152066 [DOI] [PubMed] [Google Scholar]
- Yi M., Zhang C., Zhang Z., Yi P., Xu P., Huang J., et al. (2020). Integrated metabolomic and lipidomic analysis reveals the neuroprotective mechanisms of Bushen Tiansui formula in an abeta1-42-induced rat model of Alzheimer’s disease. Oxid. Med. Cell. Longev. 2020:5243453. 10.1155/2020/5243453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Fujii S., Wu Z., Hashioka S., Tanaka Y., Shiratsuchi A., et al. (2006). Involvement of COX-1 and up-regulated prostaglandin E synthases in phosphatidylserine liposome-induced prostaglandin E2 production by microglia. J. Neuroimmunol. 172 112–120. 10.1016/j.jneuroim.2005.11.008 [DOI] [PubMed] [Google Scholar]
- Zhang Z., Hui E., Chapman E. R., Jackson M. B. (2009). Phosphatidylserine regulation of Ca2+-triggered exocytosis and fusion pores in PC12 cells. Mol. Biol. Cell 20 5086–5095. 10.1091/mbc.E09-08-0691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H., Tuominen E. K., Kinnunen P. K. (2004). Formation of amyloid fibers triggered by phosphatidylserine-containing membranes. Biochemistry 43 10302–10307. 10.1021/bi049002c [DOI] [PubMed] [Google Scholar]
- Zhao R., Jiang J., Li H., Chen M., Liu R., Sun S., et al. (2018). Phosphatidylserine-microbubble targeting-activated microglia/macrophage in inflammation combined with ultrasound for breaking through the blood-brain barrier. J. Neuroinflammation 15:334. 10.1186/s12974-018-1368-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S. C., Ma L. S., Chu Z. H., Xu H., Wu W. Q., Liu F. (2017). Regulation of microglial activation in stroke. Acta Pharmacol. Sin. 38 445–458. 10.1038/aps.2016.162 [DOI] [PMC free article] [PubMed] [Google Scholar]