

Summary
Abnormally accumulated tau protein aggregates are one of the hallmarks of neurodegenerative diseases, including Alzheimer’s disease (AD). In order to investigate proteomic alteration driven by tau aggregates, we implemented quantitative proteomics to analyze disease model mice expressing human MAPTP301S transgene (hTau-Tg) and quantified more than 9,000 proteins in total. We applied the weighted gene co-expression analysis (WGCNA) algorithm to the datasets and explored protein co-expression modules that were associated with the accumulation of tau aggregates and were preserved in proteomes of AD brains. This led us to identify four modules with functions related to neuroinflammatory responses, mitochondrial energy production processes (including the tricarboxylic acid cycle and oxidative phosphorylation), cholesterol biosynthesis, and postsynaptic density. Furthermore, a phosphoproteomics study uncovered phosphorylation sites that were highly correlated with these modules. Our datasets represent resources for understanding the molecular basis of tau-induced neurodegeneration, including AD.
Subject areas: Molecular biology, omics, proteomics
Graphical abstract
Highlights
-
•
Large-scale proteome datasets of tauopathy model mice
-
•
Protein co-expression network constructed
-
•
Four co-expression modules associated with tau-induced pathologies
-
•
Identification of phosphorylation sites correlated with the modules
Molecular biology; Omics; Proteomics.
Introduction
Alzheimer's disease (AD) is a progressive brain disorder and is the leading cause of dementia with memory and cognitive decline. The presence of extracellular β-amyloid (Aβ) deposition and intracellular accumulation of hyperphosphorylated and aggregated tau proteins remain the primary hallmarks of AD (Long and Holtzman, 2019). For decades, amyloid plaques or their major constituents, Aβ peptides, have been considered the direct cause of progressive neurodegeneration. However, more recently, aggregated tau proteins have been identified as a major cause of neurodegeneration in AD; in contrast to Aβ, the stage of tau pathology correlates well with the progression of cognitive impairment (Giannakopoulos et al., 2003; Long and Holtzman, 2019; Nelson et al., 2012). The involvement of tau protein in neurodegeneration is also supported by the fact that abnormally aggregated tau proteins are observed in other neurodegenerative diseases, which are collectively referred to as tauopathies; these include progressive supranuclear palsy (PSP), corticobasal degeneration, Pick’s disease, frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) (Didonna, 2020). Although various studies have indicated that progressive synaptic dysfunction and neuronal loss arise from a cascade of cellular derangements, including oxidative and immune-mediated injury and altered proteostasis related to aberrant transcription and translation, the precise mechanism of tau-induced neuronal injury remains to be elucidated (Didonna, 2020; Koren et al., 2019; Mangleburg et al., 2020; Wang and Mandelkow, 2016).
The enormous complexity of the central nervous system has impeded systemic exploration for decades, but recent advances in proteomics technologies using nano-scale liquid chromatography-tandem mass spectrometry (nanoLC/MS/MS) are pushing forward the frontiers of neuroscience research (Hosp and Mann, 2017). Large-scale proteomic data provide a basis for a systematic understanding of protein networks, offering unprecedented insight into complex biological processes. In particular, weighted gene co-expression network analysis (WGCNA) has often been employed in proteomic studies of AD (Johnson et al., 2020; Rayaprolu et al., 2021; Savas et al., 2017; Seyfried et al., 2017). In WGCNA, highly co-expressed proteins are organized into a module that reflects various molecular, cellular, and circuit-level phenotypes, and such modules can be correlated with biological or pathological traits, such as age, cognitive decline, and tangle deposition (Rayaprolu et al., 2021). In the largest AD proteomic study so far reported, more than 2,000 human brain tissues were profiled, and a module linked to sugar metabolism was identified as being significantly associated with AD pathology and cognitive impairment (Johnson et al., 2020). A similar approach can also be applied to the analysis of disease models. To understand the effects of Aβ peptides on the brain proteome, brain tissues dissected from a disease model with the deposition of Aβ plaques were analyzed, and this revealed a specific loss of TARPγ-2, a key α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR)-trafficking protein (Savas et al., 2017). However, fewer proteomics studies on tau pathology have been reported, and we do not yet have a proteomic dataset that is sufficiently deep for systematic analysis.
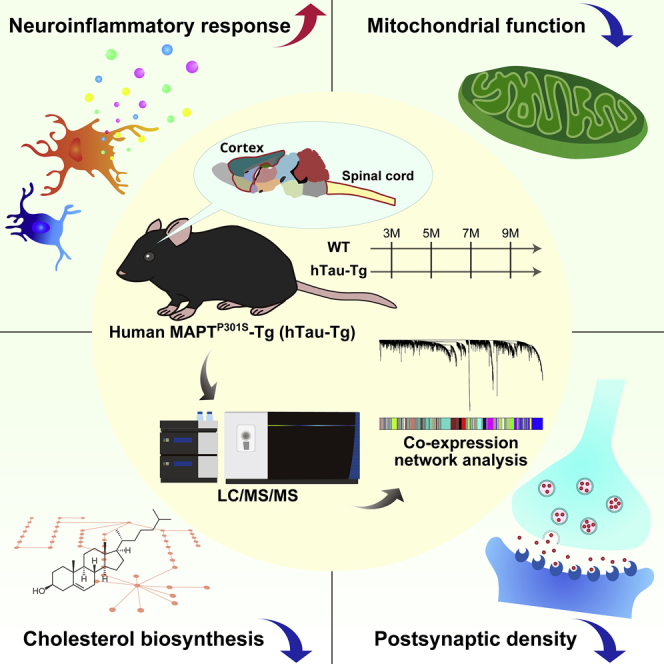
In this study, we conducted a proteomic analysis of transgenic mice expressing human tau protein with the P301S mutation under the control of the murine Thy1 promoter (line2541; hTau-Tg) in order to examine the contribution of aberrantly accumulated tau protein to the neurodegenerative processes (Allen et al., 2002). We employed a workflow consisting of sample multiplexing by tandem mass tag (TMT), peptide fractionation by high pH reversed-phase chromatography, and nanoLC/MS/MS. We applied WGCNA to our dataset and found co-expressed protein modules that were significantly correlated with tau accumulation. Importantly, some of these modules were found to be significantly preserved in the postmortem brain proteomes of patients with AD. Moreover, a phosphoproteomics study uncovered phosphorylation events related to these modules, further supporting the results of our proteomic analysis.
Results
Deep and precise proteome profiling of hTau-Tg
We investigated proteome alterations of the spinal cord and cortex dissected from wild-type (WT) and hTau-Tg mice (Allen et al., 2002) at 3 months old (12 weeks old; 3M), 5 months old (20 weeks old; 5M), 7 months old (28 weeks old; 7M), and 9 months old (36 weeks old; 9M) (Figures 1A and 1B). Six biological replicates were prepared for each condition: thus, 96 tissues were analyzed in total. For large-scale protein quantification, we employed a shotgun proteomics workflow consisting of sample multiplexing by TMT, pre-fractionation by high pH reversed-phase chromatography, and nanoLC/MS/MS (Figure 1C). For each tissue, portions of each digest were pooled, labeled with TMT-126 or TMT-131N, and spiked as internal references for bridging six TMT-10-plexes and accounting for technical variation, respectively. Our strategy resulted in the quantification of 9,039 proteins in total (Figure 1D and Table S1). There was excellent reproducibility in protein quantification, with Pearson correlation coefficients >0.997 (Figures S1A and S1B). The medians of relative standard deviation (RSD) in protein quantification were less than 1% under these conditions (Figures S1D and S1E). Thus, our strategy enabled deep and precise proteomic profiling of hTau-Tg mice.
Figure 1.

Deep and precise proteome profiling of hTau-Tg
(A–C) Graphical illustration of the samples and workflow employed for the quantitative proteomic analysis. Spinal cord (SC) and cortex (Cx) dissected from wild-type (WT) and hTau-Tg mice were analyzed at four age points. Six biological replicates were analyzed by six TMT-10-plexes for each spinal cord and cortex. Proteins were extracted using sodium dodecyl sulfate (SDS), purified by acetone precipitation, and digested with LysC and trypsin. The digests were multiplexed by TMT, fractionated by high pH reversed-phased chromatography (high pH RP), and analyzed by nanoLC/MS/MS.
(D) Numbers of quantified proteins. The numbers of proteins that were quantified in at least three samples (N≧3) and in all samples (N = 6) are shown.
Comparative analysis of tissues reveals different responses to tau mutant overexpression
Proteomic changes in hTau-Tg mice compared to WT in the spinal cord and cortex at various ages are shown as volcano plots (Figures 2A–2H). In the spinal cord, 13, 50, 462, and 989 proteins were significantly upregulated, and 4, 17, 365, and 1,114 proteins were significantly downregulated at 3M, 5M, 7M, and 9M, respectively (Figures 2A–2D). On the other hand, in the cortex, 203, 488, 353, and 103 proteins were significantly upregulated, and 157, 536, 402, and 101 proteins were significantly downregulated at the respective ages, although interestingly, the age at which the largest number of proteins was significantly regulated was 5M, not 9M, in the cortex (Figures 2E–2H). In other words, at the earlier stages, more proteins were significantly regulated in the cortex than in the spinal cord, while at the later stages, more drastic changes were observed in the spinal cord than in the cortex. Among the proteins significantly regulated at least at one point, 370 and 380 proteins, and 36 and 90 proteins showed progressive up- and downregulation in the spinal cord and cortex, respectively. Note that many of these proteins are included in the Blue module (progressively upregulated proteins) and Turquoise and Brown modules (progressively downregulated proteins) in the following co-expression network analysis.
Figure 2.

Characterization of significantly changed proteins in hTau-Tg
(A–H) Volcano plots comparing protein expression in the spinal cord (SC; A–D) and cortex (Cx; E–H) between WT and hTau-Tg at various ages. Welch’s t-tests were performed to identify significantly changed proteins (N = 6). The dashed yellow, blue, and magenta lines indicate q-value = 0.05, 0.01, and 0.001, respectively. Tau protein is highlighted in red, and proteins mentioned in the main text are highlighted in the module colors (blue, turquoise, brown, and salmon), respectively.
(I–L) GO term enrichment analysis for significantly regulated proteins in the spinal cord at 5M (I) and 9M (J) and in the cortex at 5M (K) and 9M (L). The top 5 terms are shown for significantly upregulated (blue) and downregulated proteins (orange), respectively. The underlying data and the results for 3M and 7M are shown in Table S2.
We characterized the significantly regulated proteins by gene ontology (GO) term enrichment analysis (Table S2). We show the results for 5M and 9M as representatives in Figures 2I–2L. No term was obtained for the significantly regulated proteins at 3M in the spinal cord. An interesting feature was that many terms related to mitochondrion were observed in the downregulated proteins, regardless of tissue or age. For the upregulated proteins, different terms were obtained. In the spinal cord, changes in extracellular proteins, such as exosome proteins and secretory proteins, were seen. For instance, complement proteins were upregulated only in the spinal cord, but not in the cortex (Figures 2A–2H), indicating that there was a more severe inflammatory reaction in the spinal cord than in the cortex. This is supported by the fact that glial fibrillary acidic protein (GFAP) was increased at all ages in the spinal cord, while it was not changed in the cortex (Middeldorp and Hol, 2011) (Figures 2A–2H). For the cortex, terms related to microtubule were seen. At 3M, 7M, and 9M, the top 2 significantly changed proteins were α-tubulin (TUBA1A) and β-actin (ACTB), based on the q-values (Figures 2E–2H and Table S1). Moreover, several subtypes of tubulins were included in the top 10 significantly upregulated proteins at 3M. These findings suggest changes in microtubule stability. This idea is supported by the observation of significant changes in microtubule-associated proteins, such as dynactin subunit 2 (DCTN2) (Figures 2E–2H). To explore the common changes between the tissues, we created Venn diagrams (Figures S2A–S2H). Many mitochondrial proteins, including proteins involved in the NADH dehydrogenase complex and tricarboxylic acid (TCA) cycle, were commonly downregulated, again suggesting that the downregulation of mitochondrial proteins may be the main pathology commonly observed regardless of tissue. It should be noted that these lists include mouse endogenous tau. However, we think that its apparent upregulation is an artifact, as will be discussed later.
Protein co-expression network analysis identifies tau-associated protein co-expression modules
In order to gain system-level insights, we first performed WGCNA (Langfelder and Horvath, 2008) to generate protein co-expression networks in the spinal cord, in which responses to tau-induced neuronal damage were expected considering that microglia and astrocytes were activated. The amount of accumulated tau measured by staining with 1-fluoro-2,5-bis(3-carboxy-4-hydroxystyryl)benzene (FSB), a fluorescent Congo red derivative that labels filamentous tau inclusions (Velasco et al., 2008), was employed as a trait (Figure S3). Importantly, the magnitude of the proteomic alteration was consistent with the abundance of tau inclusions (Figures 2A–2D and S3), supporting the idea that the dataset does, indeed, reflect pathological changes caused by tau. We detected 19 network modules of co-expressed proteins (Figures 3A and S4A and Table S3). In WGCNA, colors were assigned as the names of different modules, and 430 proteins unassigned to any other module were collected as the Grey module (not subjected to downstream analyses). GO term enrichment analysis revealed significant enrichment for certain GO terms for all modules, encompassing diverse biological processes and cellular components (Figure 3B and Table S4). To assess the cell type dependence of each co-expression module, we performed enrichment analysis for cell-type-specific marker proteins (Sharma et al., 2015) (Figure 3C). The Turquoise, Brown, and Salmon modules showed significant enrichment for marker proteins of neurons, while the Magenta and Blue modules showed enrichment for marker proteins of glial cells. To find modules associated with the accumulation of tau, we calculated the Pearson correlation coefficients between the FSB staining intensities (Figure S3) and each module eigenprotein, which is defined as the first principal component of a given module and serves as a representative (Figures 3D–3H, S4B–S4P, and S5 and Table S3). This yielded ten significantly correlated modules: five negatively correlated modules (Turquoise, Black, Red, Brown, and Salmon) and five positively correlated modules (Blue, Yellow, Pink, Green, and Purple).
Figure 3.

Overview of the result of weighted protein co-expression analysis of spinal cord
(A–D) Summary of the WGCNA results using the spinal cord proteome dataset. (A) Module clustering dendrogram. Clustering was performed based on the eigenprotein values. (B) GO term enrichment analysis for the top 50 hub proteins (left, cellular component; right, biological process). The underlying data and the results for all module proteins are shown in Table S4. ∗, positive regulation of RNA polymerase II transcriptional preinitiation complex assembly. (C) Cell-type-specific marker protein enrichment analysis by hypergeometric test. Significant enrichment (q-value < 0.01) is highlighted with red boxes. (D) Module-trait correlations. Pearson correlation coefficients between each module eigenprotein value and the abundance of tau estimated by FSB-intensities (Figure S3), or age were calculated. The numbers in parentheses show q-values. Significant correlations (q-value < 0.01) are highlighted with red boxes.
(E–H) Eigenprotein values of Turquoise, Brown, Salmon, and Blue modules, which were the main focus, as tau-associated and AD-preserved modules, in the following analyses in this study (see Figure S6). Eigenprotein values of the remaining modules are shown in Figure S4. The top and bottom of the whisker indicate the largest value no further than 1.5 ∗ IQR and the smallest value at most 1.5 ∗ IQR, respectively, where IQR is the inter-quartile range or the distance between the first and third quartiles. Data beyond the end of the whiskers are plotted individually.
Modules preserved in human postmortem brains of Alzheimer’s disease cohorts
Alzheimer’s disease has complex pathological features that are often only partially mimicked in disease models, and therefore it is necessary to examine whether the protein co-expression modules identified in hTau-Tg are present in the brain tissues of patients with AD as well. To this end, we used module preservation statistics with a proteome dataset of dorsolateral prefrontal cortex (DLPFC) derived from 419 samples, including 91 controls, 98 asymptomatic patients with AD, and 230 patients with AD derived from multiple cohorts (3,274 proteins) (Johnson et al., 2020) (Figure S6A). Note that DLPFC is a region that is uniquely expanded in humans and is related to cognitive abilities and higher brain functions. We found that eight modules (Magenta, Blue, Brown, Salmon, Turquoise, Lightcyan, Midnightblue, and Cyan) were significantly preserved in the human AD brains with Zsummary scores >2. Notably, the Magenta and Blue modules showed Zsummary scores >10. Importantly, these eight modules included four modules (Blue, Brown, Salmon, and Turquoise) that were significantly correlated with tau accumulation (Figure 3D). Moreover, we computed synthetic module eigenproteins based on the expression in AD brains and confirmed that the eigenproteins for modules elevated in the spinal cord of hTau-Tg were elevated in AD as well, and vice versa for decreased eigenproteins (Figures 3E–3H and S6B–S6I). These results suggest that these four modules in the spinal cord of hTau-Tg reflect tau-induced pathology in AD. On the other hand, eleven modules were not significantly preserved in the AD brains. These modules may be involved in disease-model-specific events.
We also assessed whether the identified modules in the spinal cord of hTau-Tg were preserved in the cortex of hTau-Tg, and found six modules (Blue, Turquoise, Magenta, Lightcyan, Brown, and Tan) that were significantly preserved, with Zsummary scores >2 (Figures S6J–S6P). These modules included three modules that were correlated with tau accumulation (Blue, Turquoise, and Brown) (Figure 3D).
Four co-expression modules associated with tau-induced pathologies and preserved in Alzheimer’s disease brains
Based on the results of the module preservation analysis, we delved into the four modules that were correlated with the amount of tau quantified by FSB staining and that were significantly preserved in the AD brains: these were the Blue, Turquoise, Brown, and Salmon modules.
The Blue module showed the strongest positive correlation with the abundance of tau (Figure 3D), was highly preserved in AD brains (Figure S6A), and was enriched particularly for marker proteins of astrocytes and microglia (Figure 3C). Highly connected hub nodes in a module are expected to play central roles in the protein co-expression network. The top hub protein was moesin (MSN), a member of the ERM protein family consisting of ezrin (EZR), radixin (RDX), and MSN, which links cortical actin filaments to the plasma membrane to regulate membrane dynamics, cell adhesion, migration, signal transduction, and immune response (Pore and Gupta, 2015) (Figure 4A). Interestingly, EZR was also included in the hub proteins, and all of the ERM proteins were included in this module. Protein-protein interaction (PPI) analysis using STRING (Szklarczyk et al., 2019) revealed that the hub proteins, including MSN, EZR, C1qa, a key component of the complement cascade, and GFAP, belong to a PPI network with highly significant interactions (p-value = 1.0e-16) (Figure 4B). Moreover, other proteins in the classical complement cascade including C3 and C4b were also included in this module, suggesting the association of the ERM proteins with neuroinflammatory response. Notably, in the AD brains, MSN and EZR were significantly upregulated as well, although RDX was not changed (Figures S7A–S7C). Collectively, these results indicate that tau aggregates cause neuroinflammation and microglial activation involving MSN and the other ERM proteins.
Figure 4.

Four modules that were associated with tau and preserved in AD brains
(A) Top 50 hub proteins of the Blue module. The top 10 hub proteins are arranged anticlockwise from the right end in the inner circle, and the remaining hub proteins are arranged similarly in the outer circle. The size of each circle indicates the relative kWithin. The top hub protein is highlighted in red.
(B) Protein-protein interaction (PPI) analysis of the top 50 hub proteins by STRING. Edge size reflects the combined score.
(C) Top 50 hub proteins of the Turquoise module. The proteins are arranged similarly to A. Proteins with the GO term “mitochondrion” are highlighted in the shown color.
(D and E) Heatmap showing the expression ratios of the selected proteins involved in the TCA cycle (D) and OXPHOS (E).
(F) Top 50 hub proteins of the Brown module. The proteins are arranged similarly to A. Proteins with the GO term “cholesterol biosynthetic process” are highlighted in the shown color.
(G) Heatmap showing the expression ratios of the selected proteins involved in cholesterol biosynthesis.
(H) Top 50 hub proteins of the Salmon module. The proteins are arranged similarly to A. Proteins with the GO term “postsynaptic density” are highlighted in the shown color.
The Turquoise module had a significant negative correlation with tau accumulation and was enriched for neuronal marker proteins (Figure 4C). The hub proteins of this module included many mitochondrial proteins (18/50, 36%), including the top hub protein LDHB, which mediates the conversion of pyruvate and lactate. In addition, the hub proteins included proteins that are important in the TCA cycle, such as pyruvate dehydrogenase subunit beta (PDHB) and 2-oxoglutarate dehydrogenase-like protein (OGDHL), which is an isozyme of 2-oxoglutarate dehydrogenase (OGDH) expressed in the central nervous system (Bunik et al., 2008). Moreover, many other proteins in the TCA cycle pathway tended to be downregulated (Figure 4D). These results indicate that the TCA cycle is functionally degraded in hTau-Tg. Furthermore, the hub proteins of the Turquoise module also included proteins involved in oxidative phosphorylation (OXPHOS), such as cytochrome c oxidase subunit 6A1 (COX6A1) and NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 10 (NDUFA10) (Figure 4C). Many other OXPHOS proteins also tended to be downregulated in hTau-Tg (Figure 4E) as well as TCA cycle proteins. In the AD brains, PDHB, OGDHL, and NDUFA10 were significantly downregulated, while COX6A1 was not changed, and LDHB was significantly upregulated (Figures S7D–S7H), indicating that there are some differences at the individual protein level between hTau-Tg and AD. Overall, however, our results demonstrate that mitochondrial energy production was impaired by accumulated tau.
The Brown module had the highest negative correlation with the amount of tau and was enriched for neuronal marker proteins (Figures 3C, 3D, and 4F). According to the result of GO term enrichment analysis, proteins involved in cholesterol metabolism were significantly enriched among the hub proteins (Figures 3B and 4F). The top hub protein was farnesyl pyrophosphate synthase (FDPS), which is the key enzyme in isoprenoid biosynthesis, catalyzing the formation of farnesyl diphosphate, a precursor of sterols. Intriguingly, we found proteins involved in cholesterol biosynthesis were comprehensively downregulated (Figure 4G). The expression of the enzymes on the cholesterol biosynthesis pathway is tightly regulated by the master regulator, sterol regulatory element-binding protein (SREBP2) (Karasinska and Hayden, 2011). Importantly, SREBP2 also belonged to the Brown module and was significantly downregulated at 7M and 9M in hTau-Tg (Figures 2A–2D). Overall, these results indicate that cholesterol biosynthesis is reduced through the downregulation of SREBP2 in hTau-Tg. To establish whether the proteins involved in cholesterol homeostasis are also altered in AD, we investigated the AD brain proteome dataset (Figures S7I and S7J). We found that one of the downstream proteins of SREBP2, isopentenyl-diphosphate delta-isomerase 1 (IDI1), which is included in the hub proteins, was downregulated. On the other hand, some proteins in the AD brains, such as hydroxymethylglutaryl-CoA synthase 1 (HMGCS1), did not show similar changes to those in hTau-Tg, again indicating the presence of differences at the individual protein level and the complexity of AD pathology.
The Salmon module was negatively correlated with tau accumulation and was preserved in AD brains, but not in the cortex of hTau-Tg (Figures 3C and 3D, and S6A and S6J). The hub proteins were significantly enriched for neuronal marker proteins (Figure 3C), with microtubule-associated serine/threonine-protein kinase 1 (MAST1) as the top hub protein (Figure 4H). GO term enrichment analysis suggested the involvement of postsynaptic density (Figure 4H and Table S4). Postsynaptic density is a protein-dense specialization attached to the postsynaptic membrane and is essential for excitatory signaling and plasticity in mammalian neurons (Dosemeci et al., 2016; Ziff, 1997). We found seven hub proteins with the GO term “postsynaptic density” (Figure 4H). To further address functional changes in the postsynaptic density, we broadly evaluated the expression of postsynaptic density proteins, regardless of module. Notably, some of the proteins critical for the function of postsynaptic density, the subunits of AMPAR, GluA2 and GluA3, and postsynaptic density protein 95 (PSD-95), were significantly reduced at 7 and 9M (Figures 2A–2D). It is noteworthy that some of these postsynaptic density proteins were significantly reduced in the AD brains as well (Figures S7K–S7N). Taken together, these results indicate that tau accumulation is associated with a functional decline in the postsynaptic density.
Time course of changes in the tau-associated modules and causal influences
In order to obtain insight into the time course of changes in the four tau-associated modules of interest (Blue/neuroinflammatory response, Turquoise/mitochondrion, Brown/cholesterol biosynthesis, and Salmon/postsynaptic density), we investigated the effects of aging and tau mutant overexpression on the level of eigenproteins (Figure 5A). Only the Blue/neuroinflammatory response module showed a significant difference at 3M between WT and hTau-Tg, suggesting that it is involved in the earliest event(s). In addition, significant age-related changes were observed within WT samples only in this module, indicating that the Blue/neuroinflammatory response module is associated with general aging. The Brown/cholesterol biosynthesis, Turquoise/mitochondrial, and Salmon/postsynaptic density modules showed significant differences between WT and hTau-Tg by 5M, 7M, and 9M, respectively. Thus, the alteration of these four modules appears to take place in the following order: Blue/neuroinflammatory response, Brown/cholesterol biosynthesis, Turquoise/mitochondrial, and Salmon/postsynaptic density. This conclusion was supported by the result of Bayesian network analysis (Figure 5B). The Blue/neuroinflammatory response module was likely the cause of changes in the Turquoise/mitochondrial module and Brown/cholesterol biosynthesis modules, which independently led to the Salmon/postsynaptic density module. In other words, aberrant inflammatory responses may cause abnormalities in mitochondrial and cholesterol homeostasis, leading to a decline in neuronal functions.
Figure 5.

Estimation of the time course of changes in the tau-associated modules and causal influences
(A) For the Blue/neuroinflammatory response, Turquoise/mitochondrial, Brown/cholesterol biosynthesis, and Salmon/postsynaptic density modules, the effects of age and transgenic tau protein on the level of module eigenproteins were examined by two-way analysis of variance (two-way ANOVA) with the Sidak post hoc test. The bars indicate mean value ± standard deviations. ∗, p-value < 0.05; ∗∗, p-value < 0.01; ∗∗∗, p-value < 0.001.
(B) Bayesian network analysis to derive causal influences among the Blue/neuroinflammatory response, Turquoise/mitochondrial, Brown/cholesterol biosynthesis, and Salmon/postsynaptic density modules.
Phosphoproteomics identifies changed phosphorylation sites associated with tau accumulation
Aberrant phosphorylation, such as the hyperphosphorylation of tau protein, plays a key role in AD pathology (Bai et al., 2020; Morshed et al., 2020; Perluigi et al., 2016; Zhang et al., 2019). Thus, in order to support the insights obtained in our WGCNA, we subsequently conducted a phosphoproteomics study of the spinal cord, which presented the most severe tau-induced neuronal damage. The digests of the same samples used for proteomic analysis were subjected to phosphopeptide enrichment and analyzed by nanoLC/MS/MS (Figure 6A). This approach allowed us to quantify 8,453 confidently localized (class I, localization probability >0.75) phosphorylation sites mapped on 2,916 proteins (Table S5). In agreement with the other brain phosphoproteomics datasets (Liu et al., 2018; Lundby et al., 2012), the major phosphorylation site was serine (87.5%), followed by threonine (11.9%), while phosphotyrosines accounted for less than 1% of quantified phosphorylation sites in our dataset (Figure 6B). There was good reproducibility in phosphoproteomics quantification, as well as in global proteomics, with Pearson correlation coefficients >0.993 (Figure S1C). A total of 28 phosphorylation sites on the tau protein were included in the results, many of them being localized in the proline-rich region (Figure 6C). Volcano plots showed that many phosphorylation sites on the tau protein were prominently detected in hTau-Tg (Figure S8), supporting the view that the present results reflect the pathology associated with hyperphosphorylated tau.
Figure 6.

Phosphoproteomics of spinal cord identifies changed phosphorylation sites related to the tau-associated and AD-preserved modules
(A) Workflow of phosphoproteomics. Phosphopeptides were enriched from the TMT-labeled spinal cord samples (Figure 1C), fractionated by strong cation exchange (SCX) chromatography at low pH, and analyzed by nanoLC/MS/MS.
(B) Content of quantified phosphorylation sites.
(C) Mapping detected tau phosphorylation sites. The ratios (hTau-Tg/WT) are shown as a heatmap. Note that the positions are based on 2N4R tau. N1/N2, N-terminal domains; R1-R4, four major microtubule-binding repeats.
(D) Number of phosphorylation sites that showed a significant correlation with the module eigenproteins (q-value < 0.001).
(E and F) Overrepresented motifs surrounding the phosphorylation sites that showed significant correlation with the module eigenproteins. The top five motifs according to the Z scores are shown for each module.
(G) 1D-Linear motif enrichment analysis. The ratios of phosphorylation sites with a given motif were compared with the ratios of all phosphorylation sites by a two-sided Wilcoxon-Mann-Whitney test. A score near 1 indicates that the phosphorylation sites with the motif are strongly concentrated at the high end of the ratio distribution, while a score near −1 indicates that the phosphorylation sites with the motif are strongly concentrated at the low end of the ratio distribution.
Sites, where phosphorylation changes are correlated with the tau-associated protein co-expression modules, may have roles in tau-induced pathology related to those modules. To widely explore such phosphorylation sites, we comprehensively computed Pearson correlations between the phosphorylation levels of quantified sites and module eigenproteins (Figure 6D and Table S5). We here focus on the four modules of interest. Approximately 2,000 phosphorylation sites were found to be significantly correlated (p-value < 0.001; 0.46< |R|) with each of the Blue/neuroinflammatory response, Turquoise/mitochondrial, and Brown/cholesterol biosynthesis modules, whereas less than 1,000 phosphorylation sites were found to be highly correlated with the Salmon/postsynaptic density module, indicating relatively less participation of phosphorylation in Salmon-module-related events. In order to investigate the mechanisms underlying tau-induced pathologies at the phosphoproteome level, overrepresented motifs in these tau-pathology-associated phosphorylation sites were extracted (Figures 6E and 6F). In the upregulated phosphorylation sites, many motifs with basic residues were observed (Figure 6E). Interestingly, the Blue/neuroinflammatory response, Turquoise/mitochondrial, and Brown/cholesterol biosynthesis modules showed similar motifs, including a known substrate motif “R-R-X-pS” of protein kinase A (PKA). The Blue/neuroinflammatory response and Brown/cholesterol biosynthesis modules showed the “L-X-R-X-X-pS” motif of AMP-activated protein kinase (AMPK). On the other hand, the Salmon/postsynaptic density module showed unique motifs. Motifs with proline were predominantly observed in downregulated phosphorylation sites of all the modules (Figure 6F). Phosphorylation of such sites is often mediated by cyclin-dependent kinases (CDKs) and mitogen-activated protein kinases (MAPKs). These results were supported by motif 1D-enrichment analysis of all phosphorylation sites (Figure 6G). The phosphorylation sites with motifs of basophilic kinases, such as PKA and protein kinase C (PKC), were significantly upregulated, while the phosphorylation sites with motifs of proline-directed kinases such as MAPKs and CDKs were significantly downregulated. Intriguingly, the detected phosphorylation sites of tau included those known to be mediated by PKA (S214) in monkey (Carlyle et al., 2014) and by AMPK (T231, S262, S356, S396) in mouse (Domise et al., 2016) in pathological contexts. We also calculated correlations between phosphorylation and FSB intensities (Figure S3) and found that phosphorylation levels at 1,287 sites showed a significant correlation (p-value < 0.001) with FSB intensity (Table S5). The second and third most significantly correlated sites were S214 and S262 on tau, further suggesting the importance of PKA and AMPK in the tau-induced pathologies.
Discussion
By comparing the significantly regulated proteins in the spinal cord and cortex, we found that these tissues exhibit different proteomic changes in hTau-Tg. Interestingly, in contrast to the spinal cord, neuroinflammatory proteins such as complement proteins and GFAP were not significantly upregulated in the cortex, indicating that neuronal loss might be smaller in the cortex than in the spinal cord, despite the high expression of transgenic tau mutant in the cortex. This idea is also supported by the fact that the Salmon/postsynaptic density module was not preserved in the cortex. We anticipate that deeper pursuit of the cortex proteome changes may lead to the discovery of a mechanism involved in resistance to the lesions led by tau and thus benefit the development of treatments for tau-induced pathologies. On the other hand, the commonly regulated proteins between the tissues included many mitochondrial proteins. This finding was validated by identifying the Turquoise/mitochondrial module as the second most highly preserved module in the cortex, indicating the importance of mitochondrial impairment in relation to AD pathology.
In accord with the fact that genetic and histological studies have identified microglia and astrocytes as the primary mediators of neuroinflammation in tauopathies (Didonna, 2020; Paulus et al., 1993; Serrano-Pozo et al., 2011), the Blue/neuroinflammatory response module was highly enriched with marker proteins of these cells, and appeared to be involved in the earliest event(s) among the four modules we focused on in this study. Neuroinflammation is commonly observed in neurodegenerative diseases (Didonna, 2020; Kwon and Koh, 2020). Given that microglia become activated upon interaction with tau aggregates (Morales et al., 2013) and are able to internalize them (Bolós et al., 2017), their main function at these pathological sites is likely the clearance of misfolded tau (Didonna, 2020). We found that complement proteins were elevated in hTau-Tg. Complement C3 protein has a pivotal role in the type I interferon pathway leading to synapse elimination, and inhibition of this pathway has been shown to improve the pathological condition in vivo (Roy et al., 2020). Thus, our results suggest the importance of the Blue/neuroinflammatory response module for understanding tau-induced synaptic dysfunction via neuroinflammation. The importance of complement proteins for AD pathology was also suggested at the transcription network level; a study investigating an AD model mouse with human tau mutant has shown that C1qa, C1qb, and C1qc are hub genes of an immune-associated module (Castanho et al., 2020). Interestingly, MSN, the top hub protein of the Blue/neuroinflammatory response module, has also been identified as one of the hub proteins in two previous independent studies involving WGCNA of AD brain proteomes (Johnson et al., 2020; Zhang et al., 2018), indicating that MSN is a robust hub protein of the protein network in AD brains. Recently, it was shown that MSN expression in cells located in the vicinity of Aβ plaques, probably microglia, is elevated in AD brain (Rayaprolu et al., 2020). In addition, a recent preprint reported that aberrant activation of MSN in neurons by tau acts through the actin cytoskeleton to dysregulate the cellular program that maintains neuronal identity, leading to neurotoxicity (Beckmann et al., 2021). Furthermore, the National Institute on Aging’s Accelerating Medicines Partnership, Alzheimer’s Disease consortium, has nominated MSN as a drug target for Alzheimer’s disease (https://agora.ampadportal.org/genes/(genes-router:gene-details/ENSG00000147065)) (Beckmann et al., 2021). Our findings support and strengthen the idea that the Blue/neuroinflammatory response module, particularly MSN, could be a therapeutic target for AD in terms of tau pathology. However, the precise role of MSN in neuroinflammatory responses and in the relationship between microglia and tau aggregates still remains unclear.
The Turquoise/mitochondrial module was negatively correlated with the amount of tau aggregates and has a role in mitochondrial dysfunction, a typical phenotype of tau pathology, as shown by proteomic studies (David et al., 2005; Rhein et al., 2009). The hub proteins of this module included OGDHL and PDHB, which are members of pyruvate dehydrogenase complex (PDHC) and 2-oxoglutarate dehydrogenase complex (OGDHC), respectively, and the reactions catalyzed by these proteins are the rate-limiting steps in the complexes. OGDHC activity is reduced in various age-related neurodegenerative diseases, including AD, Parkinson's disease (PD), and PSP (Albers et al., 2000; Gibson et al., 2003, 2005; Park et al., 2001). Interestingly, Yoon et al. have demonstrated that patients with homozygous deleterious variants in OGDHL develop severe neurological features (Yoon et al., 2017). A possible pathway by which OGDHC deficit leads to neurodegeneration is that increased 2-oxoglutarate, a substrate of OGDHC, increases mTORC1 activity and impairs autophagy (Yoon et al., 2017). Functional decline of PDHC also causes the TCA cycle to fail through lack of acetyl-CoA, resulting in the accumulation of pyruvate, which is converted to lactate, eventually, leading to lactic acidosis. It is reported that patients with AD have a higher cerebrospinal fluid (CSF) level of lactate and lower levels of succinate, fumarate, and glutamine, compared to age-matched controls (Redjems-Bennani et al., 1998). Taken together, these results suggest that reduced expression of OGDHL and PDHB might be central to the tau-induced functional decline of the TCA cycle.
Our analysis showed that OXPHOS proteins, as well as TCA cycle proteins, were reduced in hTau-Tg. OXPHOS, in concert with the TCA cycle, is the main source of ATP in cells. The Turquoise/mitochondrial module was enriched with neuronal marker proteins, reflecting the fact that neurons have a high oxygen consumption rate. Impairment of the electron transport chain could cause electron leakage to molecular oxygen to generate reactive oxygen species (ROS), which cause oxidative damage in nuclear and mitochondrial DNA, lipids, and proteins (Desler et al., 2018). It is noteworthy that not only AD but also other neurodegenerative diseases including PD and Huntington's disease (HD) present mitochondrial dysfunction in common, despite their widely different genetic etiologies (Beal, 1998; Pérez et al., 2018; Wu et al., 2019). Mitochondrial dysfunction has a pivotal common role in neuronal cell death. Moreover, restoration of mitochondrial function can be a therapeutic strategy for the suppression of neurodegeneration, including that seen in PD and AD (Imai et al., 2019; Takeda et al., 2021). Our results should be helpful for understanding the molecular basis of mitochondrial dysfunction and for the development of related therapies. Furthermore, some of the OXPHOS proteins including NDUFA10, one of the hub proteins, were shown to be associated with cognitive stability in a proteomic study of human brain (Wingo et al., 2019). It is worth noting that a link between dysregulated mitochondrial function and decline of cognitive function was also suggested by the result of Bayesian network analysis, in which the Turquoise/mitochondrial module appeared as a causal module of the Salmon/postsynaptic density module. Thus, the Turquoise module proteins may be promising targets for future mechanistic studies on cognitive decline in neurodegenerative diseases.
We found that cholesterol biosynthetic enzymes were comprehensively downregulated in hTau-Tg. Abnormal cholesterol metabolism has been implicated in the pathogenesis of AD (van der Kant et al., 2019; Di Paolo and Kim, 2011). Accumulating evidence indicates that high cholesterol levels in middle age may increase the risk of developing dementia, including AD (Iwagami et al., 2021). Cholesterol accumulation is also observed in HD (Karasinska and Hayden, 2011), supporting the idea that aberrant cholesterol accumulation may lead to neurodegeneration. Importantly, abnormal cholesterol accumulation was observed in neurons affected by tau pathology in disease models (Glöckner and Ohm, 2014), although the mechanism remains unclear. Given that SREBP2 is tightly regulated by intracellular cholesterol, downregulation of SREBP2 and its downstream cholesterol biosynthetic proteins would be a likely consequence of cholesterol accumulation. Indeed, the downregulation of SREBP2 by tau aggregates in neurons of patients with AD has been reported (Wang et al., 2019). In our WGCNA, the Brown/cholesterol biosynthesis module was enriched for neuronal marker proteins, which is consistent with the report that tau causes cholesterol accumulation in neurons (Glöckner and Ohm, 2014). However, it is generally assumed that cholesterol is mainly produced by astrocytes in the central nervous system (Karasinska and Hayden, 2011). A link between the Brown/cholesterol biosynthesis module and Blue/neuroinflammatory response module was supported by the result of time series analysis combined with the Bayesian network analysis, which indicated that the change in the brown/cholesterol biosynthesis module was an earlier event possibly driven by a change in the Blue/neuroinflammatory response module. Thus, the Brown/cholesterol biosynthesis module may represent a complex relationship between neurons and glial cells, suggesting a link between the disruption of cholesterol homeostasis and neuroinflammation.
Interestingly, the Salmon/postsynaptic density module was not significantly preserved in the cortex of hTau-Tg, whereas it was significantly preserved in the AD brains. In accordance with the findings that the proteomic changes in the cortex were much smaller than those in the spinal cord, the lesions in the cortex were smaller than those in the spinal cord. The reason why the Salmon/postsynaptic density module network was not observed in the cortex of hTau-Tg might, therefore, be that the Salmon/postsynaptic density module is involved in a later stage in the cascade of tau-induced pathologies, as indicated by the results of time series estimation and Bayesian network analysis. Although there have been few studies on the function of the top hub protein MAST1, a microtubule-associated protein (Tripathy et al., 2018), the presence of autoantibodies against MAST1 in patients with AD has been reported (de Oliveira-Júnior et al., 2015). Thus, MAST1 might have an important role in AD pathology. We found that many postsynaptic density proteins in the Salmon module were decreased in hTau-Tg. The postsynaptic density mediates the functional and structural plasticity of the excitatory synapse to maintain synaptic homeostasis (Dosemeci et al., 2016; Gong and Lippa, 2010), and therefore loss of these postsynaptic proteins would likely lead to a severe decline in neuronal functions. Notably, spatiotemporal changes in the postsynaptic density protein network occur in early AD and tauopathy disease models (Dejanovic et al., 2018; Gong and Lippa, 2010). Dejanovic and colleagues investigated proteomic changes in the postsynaptic density isolated from another line of human tau transgenic mouse and reported the upregulation of EZR and C1q proteins at the postsynaptic density. They also reported that C1q tags tau-affected synapses and lead to microglial engulfment and synapse loss, indicating the association of the Salmon/postsynaptic density module with the Blue/neuroinflammatory response module (Dejanovic et al., 2018).
Our phosphoproteomic study confirmed that the expressed tau protein was highly phosphorylated. The detected tau phosphorylation sites included T231, which is particularly highly phosphorylated in AD compared with other tauopathies, and S396, which is highly phosphorylated in argyrophilic grain dementia (Samimi et al., 2021), indicating that the status of tau-phosphorylation may reflect various tauopathies rather than being specific to a particular disease. We found more than 3,000 phosphorylation sites significantly correlated with the modules associated with tau accumulation and preserved in the AD brains. Motif analysis indicated that changes in kinase activities were involved. We found upregulated levels of phosphorylation sites with a PKA substrate motif, which were correlated with the Blue/neuroinflammation module. PKA mediates the activation of microglia induced by plasminogen and gangliosides (Min et al., 2004), and might thus have a critical role in the microglial activation induced by tau aggregates. In addition, we found the upregulation of the levels of phosphorylation sites having an AMPK substrate motif, which were correlated with the Brown/cholesterol biosynthesis module. AMPK signaling is known to negatively regulate cholesterol biosynthesis in hepatic cells (Li et al., 2011; Wang et al., 2018). Thus, if this is also the case in brain, the downregulation of cholesterol biosynthesis in hTau-Tg may occur also at the phosphoproteome level. It is noteworthy that we detected phosphorylation at sites known to be targeted by PKA and AMPK in pathological contexts (Carlyle et al., 2014; Domise et al., 2016), which may indicate the participation of these kinases in tau-induced pathologies.
A critical issue in systematic analysis in proteomics is the measurement step, which is the key determinant of the quality of the generated dataset. In the past, proteomic studies investigating bulk brain tissues derived from disease models of tauopathy mainly employed 2-dimensional gel electrophoresis coupled with mass spectrometry (Chang et al., 2013; David et al., 2005; Tilleman et al., 2002). However, technologies in proteomics, particularly those utilizing nanoLC/MS/MS, have recently improved dramatically in terms of both comprehensiveness and quantitativeness (Hosp and Mann, 2017; Mann and Kelleher, 2008; McAlister et al., 2014; Navarrete-Perea et al., 2018). Here, we employed a workflow consisting of sample multiplexing using TMT, pre-fractionation by high-pH reversed-phase chromatography, and high-resolution mass spectrometry, and we precisely quantified more than 9,000 proteins in total. This is the largest proteomics dataset relevant to a tau-induced neurodegenerative disease mouse model, to our knowledge. Brain proteomes of patients with AD reflect a variety of influences including the patient’s background, aging, and Aβ and tau pathologies, which make analysis extremely complex. On the other hand, studies using hTau-Tg mice, in contrast to tissues derived from patients, represent only the downstream consequences of overexpression of mutated tau and may provide a deeper understanding of the pathology caused by tau aggregates, although disease-model-specific events need to be discriminated. In this study, we first built a protein co-expression network in hTau-Tg spinal cord by applying WGCNA and then used module preservation statistics to examine whether the detected protein co-expression modules were similarly generated in the postmortem brains of patients with AD. This approach enabled us to detect four modules that were correlated with tau accumulation and that were significantly preserved in AD brains. These results strongly support the validity of our dataset as a resource to deepen understanding of the molecular basis underlying tau-induced pathologies in AD. We wish to emphasize that this study is the first to systematically reveal in detail the proteome alterations associated with tau-induced pathologies in a mouse disease model.
In summary, we have uncovered the first detailed landscape of protein co-expression networks in a mouse model of tau-induced neurodegeneration manifesting abnormally aggregated and accumulated tau and exhibiting extensive and progressive functional changes. Moreover, we compared the findings in this disease model with the proteome datasets of patients with AD, thereby gaining a deeper insight into the complex pathologies of AD. We believe our dataset will contribute to the identification of novel treatment targets for AD and other tau-related neurodegenerative diseases.
Limitations of the study
An important limitation for understanding tau pathology using hTau-Tg mice (line 2541) is that the mice only express one isoform of human four-repeat tau (Allen et al., 2002), whereas the tau filaments observed in AD contain all six isoforms produced in the adult human brain. According to the original report on hTau-Tg, the majority of tau filaments resemble the half-twisted ribbons observed in cases of FTDP-17, with a minority of filaments resembling the paired helical filaments of AD (Allen et al., 2002). hTau-Tg mice develop a neurological phenotype characterized by general muscle weakness, tremor, and severe paraparesis, leading to a decrease in exercise capacity (Allen et al., 2002). Exercise has been generally recognized as a critical factor for maintaining brain functions (De Miguel et al., 2021; Prakash et al., 2015). For instance, physical activity is associated with cerebral glucose metabolism and influences the progression of AD (Dougherty et al., 2017). Thus, the changes in physical activity in hTau-Tg may affect the changes in the proteome.
As described in the discussion section, the pathologies in AD are caused by multiple factors, and therefore the present results may not necessarily reflect the pathologies observed in patients with AD. In addition, even though neurodegeneration is also observed in the cortex, we focused mainly on the spinal cord in hTau-Tg (Allen et al., 2002) and utilized the AD DLPFC proteome dataset for comparison with AD pathology. The four modules we focused on were significantly preserved in the DLPFC proteome of patients with AD, indicating that these modules are preserved regardless of tissue differences.
We analyzed bulk brain tissues, and consequently, it is not possible to distinguish protein expression changes with cell-type resolution. An approach employing magnetic affinity cell sorting was used to obtain a cell-type-resolved mouse brain proteome (Sharma et al., 2015). However, such an approach is difficult to apply to multiple samples in terms of cost and throughput. We used enrichment analysis based on cell-type-specific markers (Johnson et al., 2020; Seyfried et al., 2017), which only partially overcomes this limitation.
We employed a shotgun proteomics strategy, in which proteins are digested into peptides prior to LC/MS/MS measurement. Consequently, proteins with very similar sequences cannot be correctly quantified. Furthermore, tau protein is cleaved by proteases, partially degraded, and highly phosphorylated at multiple sites, making it extremely difficult to distinguish between and correctly quantify endogenous and transgenic tau proteins. We consider that these issues may account for the apparent upregulation of mouse endogenous tau. Another possibility would be co-aggregation of the endogenous tau with the overexpressed human transgenic tau mutant.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| 16% Paraformaldehyde (w/v) | Thermo Fisher Scientific | Cat#43368 |
| 1-Fluoro-2,5-bis(3-carboxy-4-hydroxystyryl)benzene (FSB) | DOJINDO | Cat#F308 |
| 10% Sodium dodecyl sulfate (SDS) solution | Thermo Fisher Scientific | Cat# 15553027 |
| HALT protease/phosphatase inhibitor cocktail (100×) | Thermo Fisher Scientific | Cat#78444 |
| 2,2,2-Trifluoroethanol | Sigma-Aldrich | Cat#T63002 |
| Ammonium bicarbonate | FUJIFILM Wako | Cat#018-21742 |
| Tris-(2-carboxyethyl)phosphine, hydrochloride (TCEP) | Thermo Fisher Scientific | Cat#20490 |
| 2-Chloroacetamide | FUJIFILM Wako | Cat#032-09762 |
| Trypsin | Promega | Cat#V5111 |
| LysC | FUJIFILM Wako | Cat#129-02541 |
| Ammonium Dihydrogenphosphate | FUJIFILM Wako | Cat# 012-03305 |
| Iron(III) chloride | Sigma-Aldrich | Cat#157740-100 |
| 50% Hydroxylamine solution | Thermo Fisher Scientific | Cat# 90115 |
| 0.5 M Ethylenediamine-N,N,N′,N′-tetraacetic acid, disodium salt, dihydrate (EDTA) | NIPPON GENE | Cat# 311-90075 |
| Critical commercial assays | ||
| Bicinchoninic acid (BCA) assay kit | Thermo Fisher Scientific | Cat#23227 |
| InertSep RP-C18 | GL Sciences | Cat#5010-27130 |
| SDB-XC StageTip | Nikkyo Technos | Cat#NTCR-KT200-SDB-4 |
| Ni-nitrilotriacetic acid (NTA) spin columns | QIAGEN | Cat#31014 |
| TMT 10-plex | Thermo Fisher Scientific | Cat#90406 |
| Empore cation-SR membrane | GL Sciences | Cat#5010-30031 |
| Deposited data | ||
| hTau-Tg proteomics/phosphoproteomics data | This study | PXD031129 (JPST001212) |
| AD postmortem brain proteomics data | Synapse | syn20933797 |
| Experimental models: Organisms/strains | ||
| Human MAPTP301S transgenic mouse (hTau-Tg; line 2541) | MRC Laboratory of Molecular Biology | N/A (https://www2.mrc-lmb.cam.ac.uk/research/technology-transfer/recent-technology-transfer-initiatives/transgenic-mice-expressing-human-p301s-tau-protein-continued-success-since-2002/) |
| C57BL/6J mouse | The Jackson Laboratory | RRID: IMSR_JAX:000664 |
| Software and algorithms | ||
| TIBCO Spotfire | TIBCO software | https://perkinelmerinformatics.com/products/exclusive-reseller/tibco-spotfire |
| R framework v.3.6.1 | R project | https://www.r-project.org/ |
| R WGCNA package v.1.70-3 | CRAN | https://cran.r-project.org/web/packages/WGCNA/index.html |
| R limma package v.3.42.2 | Bioconductor | https://bioconductor.org/packages/release/bioc/html/limma.html |
| R bnlearn package v.4.7 | CRAN | https://cran.r-project.org/web/packages/bnlearn/index.html |
| R flushClust package v.1.01-2 | CRAN | https://cran.r-project.org/web/packages/flashClust/index.html |
| R mutoss package v.0.1-12 | CRAN | https://www.rdocumentation.org/packages/mutoss/versions/0.1-12/topics/sidak |
| PhosphositePlus Motif analysis tool v.6.6.0.4 | Hornbeck et al. (2012) | https://www.phosphosite.org/homeAction |
| STRING v.11.0 | Szklarczyk et al. (2019) | https://string-db.org/ |
| MaxQuant v.1.6.17.0 | Cox and Mann (2008) | http://www.coxdocs.org/doku.php?id=maxquant:start |
| Perseus v.1.6.14.0 | Tyanova et al. (2016) | http://www.coxdocs.org/doku.php?id=maxquant:start |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Kazuya Tsumagari (kazuya.tsumagari@riken.jp).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Homozygous male mice transgenic for human 0N4R tau with mutation (Line 2541, called as hTau-Tg in this study) (Allen et al., 2002) and C57BL/6J male (The Jackson Laboratory, Bar Harbor, ME) mice were used in this study. hTau-Tg mice were obtained and bred under license from MRC Laboratory of Molecular Biology (Cambridge, UK). The mice did not participate in any other experiment. They were housed with artificial lighting of 12 h light, 12 h dark and provided with pellet food and tap water ad libitum. Animal care and experimental procedures were performed in an animal facility accredited by the Health Science Center for Accreditation of Laboratory Animal Care and Use of the Japan Health Sciences Foundation. Experimental protocols were approved by the Institutional Animal Care and Use Committee of Eisai Co., Ltd., Tsukuba Research Laboratories.
Mice were sacrificed at 3, 5, 7 or 9 months of age and perfused with phosphate-buffered saline, then the cortices and spinal cords were collected (N = 6 in each case). For proteomics, hemispheres of the cortices or the rostral part of the spinal cords from the lumbar enlargement were immediately frozen in liquid nitrogen and stored at −80°C. For fluorescence labeling with β-sheet ligand, the caudal parts of the spinal cords from the lumbar enlargement were fixed in 4% paraformaldehyde solution overnight and stored in 20% sucrose solution.
Method details
FSB staining and fluorescence microscopy
Fixed brains or spinal cords were sectioned at 40-μm thickness using a microtome (REM-710; Yamato Koki, Saitama, Japan). Sections were mounted on glass slides, dried with air, and incubated in 20% ethanol containing 0.001% FSB solution (Dojindo, Kumamoto, Japan) at room temperature for 30 min. The samples were rinsed with 20% ethanol for 5 min, dipped into tris-buffered saline (TBS) three times for 5 min each, and mounted in Prolong Diamond Antifade Mountant (Thermo Fisher Scientific, Waltham, MA). Fluorescence images were captured using an Axioscan Z1 (Zeiss, Oberkochen, Germany). FSB-positive inclusions were detected and the fluorescence intensities of pixels in the inclusions were summed up using Zen (v.3.1; Zeiss) and TIBCO Spotfire (TIBCO Software, Palo Alto, CA).
Protein extraction, purification, and digestion
Brain tissues were freeze-crushed using a multi-beads shocker (Yasui Kikai, Osaka, Japan), and proteins were extracted with a lysis buffer consisting of 4% SDS, 100 mM Tris-HCl (pH 8.5), 10 mM tris(2-carboxyethyl)phosphine, 40 mM 2-chloroacetamide, and HALT protease/phosphatase inhibitor cocktail (Thermo Fisher Scientific). The proteins were precipitated by adding 4 volumes of cold acetone, reconstituted with 10% 2,2,2-trifluoroethanol (Sigma-Aldrich, St. Louis, MO) in 50 mM ammonium bicarbonate, and digested overnight with LysC (100:1, w/w; FUJIFILM Wako, Osaka, Japan) and trypsin (100:1, w/w; Promega, Madison, WI). The resulting peptides were desalted using InertSep RP-C18 columns (GL Sciences, Tokyo, Japan).
TMT labeling
To 100 μg peptides dissolved in 20 μL of 200 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-NaOH (pH 8.5) was added 0.5 mg of TMT reagents in 20 μL acetonitrile (ACN), and the solution was agitated at room temperature for 1 h. Excess reagents were quenched with 0.33% hydroxylamine (final concentration). The solution was acidified with 1% TFA (final concentration), and the labeled peptides were desalted using InertSep RP-C18 columns.
Peptide fractionation by high-pH reversed-phase chromatography
The chromatography system consisted of a Prominence high-performance liquid chromatography pump (Shimadzu, Kyoto, Japan), a high-pH-compatible column (XBridge BEH130C18, 3.5 μm, 4.6 × 250 mm; Waters, Milford, MA), and an auto-fraction collector FRC-40 (Shimadzu). TMT-labeled peptides were separated under basic conditions, that is, 4% B for 5 min, 4–35% B in 80 min, 35–40% B in 5 min, 40–80% B in 5 min, and 80% solvent B for 6 min (solvent A, 50 mM ammonium hydroxide; solvent B, 50 mM ammonium hydroxide in ACN) at the flow rate of 0.7 mL/min. Fractions were collected at 1 min intervals from 2 min after the start of the gradient, and 96 fractions were obtained. Then, 4 fractions, e.g., fractions 1, 25, 49, and 73, were combined into one fraction, finally affording 24 fractions. Each of these fractions was analyzed by nanoLC/MS/MS following purification by SDB-XC StageTips (Nikkyo Technos, Tokyo, Japan).
Phosphopeptide enrichment and fractionation
Phosphopeptide enrichment was performed by immobilized metal affinity chromatography (IMAC) using lab-made IMAC-StageTips (Tsai et al., 2022). IMAC-StageTip was prepared with Ni-nitrilotriacetic acid (NTA) silica resin (QIAGEN, Hilden, Germany). Ni2+ ions were removed with 50 mM ethylenediaminetetraacetic acid (EDTA) in 0.5% acetate buffer (v/v, pH 3) containing 1 M NaCl, and the tip was then activated with 100 mM FeCl3 in 6% acetate buffer (v/v, pH 3) and equilibrated with 0.5% acetate buffer. TMT-10-plexed peptides reconstituted in 0.5% acetate buffer were loaded onto the tip, followed by successive washes with 80% ACN including 1% TFA and 0.5% acetate buffer. The bound phosphopeptides were eluted with 200 mM NH4H2PO4 and purified on SDB-XC-StegeTips (Rappsilber et al., 2007). Prior to nanoLC/MS/MS analysis, the phosphopeptides were fractionated into 7 fractions by SCX under acidic conditions using SCX-StageTips (Adachi et al., 2016). Each fraction was analyzed by nanoLC/MS/MS in duplicate.
Nanoscale liquid chromatography/tandem mass spectrometry
The nanoLC/MS/MS system consisted of an UltiMate 3000RSLCnano pump (Thermo Fisher Scientific) and an Orbitrap Fusion Lumos tribrid mass spectrometer (Thermo Fisher Scientific) equipped with a Dream spray electrospray ionization source (AMR Inc., Tokyo, Japan). Peptides were injected by an HTC-PAL autosampler (CTC Analytics, Zwingen, Switzerland), loaded on a 15 cm fused-silica emitter packed with 3 μm C18 beads (Nikkyo Technos), and separated by a linear gradient (5% solvent B for 1 min, 5–15% solvent B in 4 min, 15–40% solvent B in 100 min, 40–99% solvent B in 5 min, and 99% solvent B for 10 min; solvent A was 0.1% formic acid, and solvent B was 0.1% formic acid in 80% ACN) at the flow rate of 300 nL/min. Peptides were ionized at 2,000 V. Quantification of non-phosphorylated peptides was performed in the SPS-MS3 mode with the following parameters. All MS1 spectra were acquired over the range of 375–1500 m/z in the Orbitrap analyzer (resolution = 120,000, maximum injection time = 50 ms, automatic gain control = standard). For the subsequent MS/MS analysis, precursor ions were selected and isolated in top-speed mode (cycle time = 3 s, isolation window = 0.7 m/z), activated by collision-induced dissociation (CID; normalized collision energy = 35), and detected in the ion trap analyzer (turbo mode, maximum injection time = auto, automatic gain control = standard). The top 10 most intense fragment ions were subjected to TMT-reporter ion quantification by SPS-MS3 (HCD normalized collision energy = 65) (McAlister et al., 2014). Dynamic exclusion time was set to 30 s. Quantification of phosphorylated peptides was performed at the MS2 level with the following parameters. All MS1 spectra were acquired over the range of 400–1500 m/z in the Orbitrap analyzer (resolution = 120,000, maximum injection time = auto, automatic gain control = standard). For the subsequent MS/MS analysis, precursor ions were selected and isolated in top-speed mode (cycle time = 3 s, isolation window = 0.7 m/z), activated by high-energy collisional dissociation (HCD; normalized collision energy = 38), and detected in the Orbitrap analyzer (resolution = 60,000, maximum injection time = 105 ms, automatic gain control = 250%). Dynamic exclusion time was set to 60 s.
Raw LC/MS/MS data processing
LC/MS/MS raw data were processed using MaxQuant (v.1.6.17.0) (Cox and Mann, 2008). Database search was implemented against the UniProt mouse reference proteome database (March, 2019) including isoform sequences. The following parameters were applied: precursor mass tolerance of 4.5 ppm, fragment ion mass tolerance of 20 ppm, and up to two missed cleavages. TMT-126 was set to the reference channel, and the match-between-run function was enabled (Cox et al., 2020). Cysteine carbamidomethylation was set as a fixed modification, while methionine oxidation and acetylation on the protein N-terminus were allowed as variable modifications. Phosphorylation on serine, threonine, and tyrosine residues was allowed for phosphoproteomics data. False discovery rates were estimated by searching against a reversed decoy database and filtered for <1% at the peptide-spectrum match, protein, and PTM site levels. Correction for isotope impurities was done based on the manufacturer’s product data sheet of TMT reagents.
TMT-reporter intensity normalization
TMT-reporter intensity normalization among six of the 10-plexes was performed according to the internal reference scaling method (Plubell et al., 2017) by scaling the intensity of the reference channel (TMT-126) to the respective protein intensities. Then, the intensities were quantile-normalized and batch effects were corrected using the limma package (v.3.42.2) in the R framework. In the analyses of phosphorylation sites, quantification was done preferentially from mono-phosphopeptides, followed by di-phosphopeptides and tri-phosphopeptides.
Processing of AD brain proteome dataset
Processed and normalized AD brain proteome data obtained by Johnson et al. (2020) and the related trait information were obtained from the Synapse Repository (https://www.synapse.org/consensus; Synapse ID: syn20933797). Only proteins with missing values less than 50% were used for analyses. In cases where several proteins were assigned to a single UniProt accession, e.g. splicing variants, only the one with the lowest missing values was used. This processing reduced the number of proteins to 3,274.
Bioinformatics analyses
GO term enrichment analysis by Fisher exact test with subsequent calculation of q-value by the Benjamini-Hochberg method was performed using Perseus (Tyanova et al., 2016). For cell-type-specific marker protein enrichment analysis, proteins that were at least 8-fold more highly expressed in a certain cell type than in other cell types in the dataset by Sharma et al. (2015) were used as cell-type-specific marker proteins. The analysis was performed by means of the hypergeometric test, and q-values were calculated by the Benjamini-Hochberg method in the R framework. Protein-protein interaction analysis was performed using STRING (v.11.0) (Szklarczyk et al., 2019) with the default parameters and visualized using Cytoscape (v.3.8.0) (Shannon et al., 2003). Tau-pathology-associated phosphorylation sites were identified based on the Pearson correlation with the respective module eigenproteins (p-value < 0.001), and overrepresented motifs were extracted using the motif analysis tool in PhosphositePlus (Hornbeck et al., 2012) with the default parameters. 1D-Linear motif enrichment analysis was performed using Perseus (Tyanova et al., 2016) with the default parameters.
Weighted protein co-expression network analysis
A weighted protein co-expression network was constructed for the spinal cord proteome dataset matrix consisting of the 5,360 log2-transformed protein abundances × 48 samples that had no missing values and had undergone normalization and batch effect adjustment as described above, using the WGCNA package (v.1.70–3) in the R framework (Langfelder and Horvath, 2008). We confirmed that no marked outliers were included.
Expression similarities (adjacencies) were computed by the adjacency() function with the soft thresholding power of 19. The network type was set to “signed”. The adjacencies were transformed into a topological overlap matrix (TOM) by the TOMsimilarity() function, and a hierarchical clustering of proteins was performed by the flashClust() function (method = "average") in the flashClust package (v.1.01–2), based on the corresponding TOM dissimilarity (1-TOM). Modules were detected by the cutreeDynamic() function using the hybrid tree cut method (deep split = 4, minimum module size = 50, pamRespectsDendro = FALSE). Proteins not assigned to any modules were collected as the Gray module. After the initial network construction, Pearson correlations between each protein and each module eigenprotein, module memberships (kMEs) calculated by the moduleEigengenes() function, were calculated, and proteins without a significant correlation with the eigenproteins (p-value < 0.05) based on the Pearson correlation were excluded from the module and assigned to the M0 module. Then, module eigenproteins were re-calculated, and used for the downstream analyses. Module-trait relationships were calculated as Pearson correlations between the module eigenproteins and each trait (age, FSB-intensities), and q-value was calculated by the Benjamini-Hochberg method. Hub proteins of each module were identified based on the intramodular connectivity (kWithin) calculated by the intramodularConnectivity() function. Module characterization by enrichment analysis for GO terms and cell-type-specific marker proteins is described in the bioinformatic analysis section above. Default parameters were used unless otherwise stated.
Module preservation analysis
Module preservation analysis was performed using the modulePreservation() function in the WGCNA package. To assess the module preservation in human AD brains, we utilized the proteome dataset of DLPFC tissues derived from postmortem brains of 91 controls, 98 asymptomatic AD patients, 230 AD patients (total: 419 tissues) (Johnson et al., 2020), processed as described above. Human UniProt accessions were converted to mouse accessions of the corresponding orthologs based on the HGNC comparison of orthology predictions (HCOP; https://www.genenames.org/tools/hcop/). Zsummary composite preservation scores were calculated by using the spinal cord network as the template, against the hTau-Tg cortex dataset or human AD brain dataset, with 200 permutations. Random seed was set to 1 for reproducibility.
Quantification and statistical analysis
All t-tests were performed in the two-tailed mode. Welch’s t-test and subsequent q-value calculation by Benjamini-Hochberg adjustment were performed using Perseus (v.1.6.14.0) (Tyanova et al., 2016). Two-way ANOVA and the following post hoc Sidak tests were performed in the R framework with the sidak() function of the mutoss package (v.0.1-12). Note that we did not check if the data were normally distributed. Bayesian network analysis was performed based on the hill-climbing algorithm in the R framework using the hc() function (score = “bic-g”; the defaults were used for other parameters) of the bnlearn package (v.4.7). Calculation of Pearson correlations was performed in the R framework using the basic functions.
Acknowledgment
We would like to thank members of Eisai-Keio Innovation Laboratory for Dementia for fruitful discussions. KT is grateful to Koshi Imami (RIKEN) for supporting revision work at the current laboratory. This work was supported by a grant from Japan Agency for Medical Research and Development (JP17pc0101006).
Author contributions
KT designed and performed all proteomics experiments, analyzed the data, and wrote the article. AS performed immunofluorescence microscopy experiments. YS, HA, and JK supervised the study. All authors interpreted the data and reviewed the article.
Declaration of interests
YS, HA, and JK are employed by Eisai Co., Ltd.
Published: August 19, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104832.
Contributor Information
Kazuya Tsumagari, Email: kazuya.tsumagari@riken.jp.
Junro Kuromitsu, Email: j-kuromitsu@hhc.eisai.co.jp.
Supplemental information
(A) List of quantified proteins in spinal cord. (B) Result of Welch’s t-tests for proteins in spinal cord. (C) List of quantified proteins in cortex. (D) Result of Welch’s t-tests for proteins in cortex.
(A) GO term enrichment analysis of significantly upregulated proteins in the spinal cord. (B) GO term enrichment analysis of significantly downregulated proteins in the spinal cord. (C) GO term enrichment analysis of significantly upregulated proteins in the cortex. (D) GO term enrichment analysis of significantly downregulated proteins in the cortex.
(A) GO term enrichment analysis of top 50 hub proteins. (B) GO term enrichment analysis of all proteins.
(A) List of quantified phosphorylation sites. (B) Result of Welch’s t-tests. (C) Correlation between phosphorylation at quantified sites and module eigenproteins. (D) Summary of detected tau phosphorylation sites. (E) Correlation between phosphorylation at quantified sites and FSB intensities.
Data and code availability
The proteomics datasets and analysis data generated in this study are available in the jPOST repository PXD031129 (JPST001212) (Okuda et al., 2017) (https://repository.jpostdb.org/). This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Adachi J., Hashiguchi K., Nagano M., Sato M., Sato A., Fukamizu K., Ishihama Y., Tomonaga T. Improved proteome and phosphoproteome analysis on a cation exchanger by a combined acid and salt gradient. Anal. Chem. 2016;88:7899–7903. doi: 10.1021/acs.analchem.6b01232. [DOI] [PubMed] [Google Scholar]
- Albers D.S., Augood S.J., Park L.C., Browne S.E., Martin D.M., Adamson J., Hutton M., Standaert D.G., Vonsattel J.P., Gibson G.E., Beal M.F. Frontal lobe dysfunction in progressive supranuclear palsy. J. Neurochem. 2000;74:878–881. doi: 10.1046/j.1471-4159.2000.740878.x. [DOI] [PubMed] [Google Scholar]
- Allen B., Ingram E., Takao M., Smith M.J., Jakes R., Virdee K., Yoshida H., Holzer M., Craxton M., Emson P.C., et al. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J. Neurosci. 2002;22:9340–9351. doi: 10.1523/JNEUROSCI.22-21-09340.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai B., Wang X., Li Y., Chen P.-C., Yu K., Dey K.K., Yarbro J.M., Han X., Lutz B.M., Rao S., et al. Deep multilayer brain proteomics identifies molecular networks in Alzheimer’s disease progression. Neuron. 2020;105:975–991.e7. doi: 10.1016/j.neuron.2019.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal M.F. Mitochondrial dysfunction in neurodegenerative diseases. Biochim. Biophys. Acta. 1998;1366:211–223. doi: 10.1016/s0005-2728(98)00114-5. [DOI] [PubMed] [Google Scholar]
- Beckmann A., Ramirez P., Gamez M., Ray W.J., Frost B. Pathogenic tau disrupts the cellular program that maintains neuronal identity. bioRxiv. 2021 doi: 10.1101/2021.03.05.434166. Preprint at. [DOI] [Google Scholar]
- Bolós M., Llorens-Martín M., Perea J.R., Jurado-Arjona J., Rábano A., Hernández F., Avila J. Absence of CX3CR1 impairs the internalization of tau by microglia. Mol. Neurodegener. 2017;12:59. doi: 10.1186/s13024-017-0200-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunik V., Kaehne T., Degtyarev D., Shcherbakova T., Reiser G. Novel isoenzyme of 2-oxoglutarate dehydrogenase is identified in brain, but not in heart. FEBS J. 2008;275:4990–5006. doi: 10.1111/j.1742-4658.2008.06632.x. [DOI] [PubMed] [Google Scholar]
- Carlyle B.C., Nairn A.C., Wang M., Yang Y., Jin L.E., Simen A.A., Ramos B.P., Bordner K.A., Craft G.E., Davies P., et al. CAMP-PKA phosphorylation of tau confers risk for degeneration in aging association cortex. Proc. Natl. Acad. Sci. USA. 2014;111:5036–5041. doi: 10.1073/PNAS.1322360111/SUPPL_FILE/PNAS.201322360SI.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanho I., Murray T.K., Hannon E., Jeffries A., Walker E., Laing E., Baulf H., Harvey J., Bradshaw L., Randall A., et al. Transcriptional signatures of tau and amyloid neuropathology. Cell Rep. 2020;30:2040–2054.e5. doi: 10.1016/J.CELREP.2020.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S.H., Jung I.S., Han G.Y., Kim N.H., Kim H.J., Kim C.W. Proteomic profiling of brain cortex tissues in a Tau transgenic mouse model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2013;430:670–675. doi: 10.1016/j.bbrc.2012.11.093. [DOI] [PubMed] [Google Scholar]
- Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Yu S.H., Kyriakidou P., Cox J. Isobaric matching between runs and novel PSM-level normalization in maxquant strongly improve reporter ion-based quantification. J. Proteome Res. 2020;19:3945–3954. doi: 10.1021/acs.jproteome.0c00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David D.C., Hauptmann S., Scherping I., Schuessel K., Keil U., Rizzu P., Ravid R., Dröse S., Brandt U., Müller W.E., et al. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J. Biol. Chem. 2005;280:23802–23814. doi: 10.1074/jbc.M500356200. [DOI] [PubMed] [Google Scholar]
- Dejanovic B., Huntley M.A., De Mazière A., Meilandt W.J., Wu T., Srinivasan K., Jiang Z., Gandham V., Friedman B.A., Ngu H., et al. Changes in the synaptic proteome in tauopathy and rescue of tau-induced synapse loss by C1q antibodies. Neuron. 2018;100:1322–1336.e7. doi: 10.1016/j.neuron.2018.10.014. [DOI] [PubMed] [Google Scholar]
- Desler C., Lillenes M.S., Tønjum T., Rasmussen L.J. The role of mitochondrial dysfunction in the progression of Alzheimer’s disease. Curr. Med. Chem. 2018;25:5578–5587. doi: 10.2174/0929867324666170616110111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didonna A. Tau at the interface between neurodegeneration and neuroinflammation. Gene Immun. 2020;21:288–300. doi: 10.1038/s41435-020-00113-5. [DOI] [PubMed] [Google Scholar]
- Domise M., Didier S., Marinangeli C., Zhao H., Chandakkar P., Buée L., Viollet B., Davies P., Marambaud P., Vingtdeux V. AMP-activated protein kinase modulates tau phosphorylation and tau pathology in vivo. Sci. Rep. 2016;6:26758. doi: 10.1038/srep26758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosemeci A., Weinberg R.J., Reese T.S., Tao-Cheng J.-H. The postsynaptic density: there is more than meets the eye. Front. Synaptic Neurosci. 2016;8:23. doi: 10.3389/FNSYN.2016.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty R.J., Schultz S.A., Kirby T.K., Boots E.A., Oh J.M., Edwards D., Gallagher C.L., Carlsson C.M., Bendlin B.B., Asthana S., et al. Moderate physical activity is associated with cerebral glucose metabolism in adults at risk for Alzheimer’s disease. J. Alzheimers Dis. 2017;58:1089–1097. doi: 10.3233/JAD-161067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakopoulos P., Herrmann F.R., Bussière T., Bouras C., Kövari E., Perl D.P., Morrison J.H., Gold G., Hof P.R. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology. 2003;60:1495–1500. doi: 10.1212/01.WNL.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- Gibson G.E., Kingsbury A.E., Xu H., Lindsay J.G., Daniel S., Foster O.J.F., Lees A.J., Blass J.P. Deficits in a tricarboxylic acid cycle enzyme in brains from patients with Parkinson’s disease. Neurochem. Int. 2003;43:129–135. doi: 10.1016/S0197-0186(02)00225-5. [DOI] [PubMed] [Google Scholar]
- Gibson G.E., Blass J.P., Beal M.F., Bunik V. The α-ketoglutarate-dehydrogenase complex. Mol. Neurobiol. 2005;31:43–63. doi: 10.1385/MN:31:1-3:043. [DOI] [PubMed] [Google Scholar]
- Glöckner F., Ohm T.G. Tau pathology induces intraneuronal cholesterol accumulation. J. Neuropathol. Exp. Neurol. 2014;73:846–854. doi: 10.1097/NEN.0000000000000103. [DOI] [PubMed] [Google Scholar]
- Hornbeck P.V., Kornhauser J.M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., Sullivan M. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40:D261–D270. doi: 10.1093/NAR/GKR1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosp F., Mann M. A primer on concepts and applications of proteomics in neuroscience. Neuron. 2017;96:558–571. doi: 10.1016/j.neuron.2017.09.025. [DOI] [PubMed] [Google Scholar]
- Imai Y., Inoshita T., Meng H., Shiba-Fukushima K., Hara K.Y., Sawamura N., Hattori N. Light-driven activation of mitochondrial proton-motive force improves motor behaviors in a Drosophila model of Parkinson’s disease. Commun. Biol. 2019;2:424–511. doi: 10.1038/s42003-019-0674-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwagami M., Qizilbash N., Gregson J., Douglas I., Johnson M., Pearce N., Evans S., Pocock S. Blood cholesterol and risk of dementia in more than 1·8 million people over two decades: a retrospective cohort study. Lancet Healthy Longevity. 2021;2:e498–e506. doi: 10.1016/S2666-7568(21)00150-1. [DOI] [PubMed] [Google Scholar]
- Johnson E.C.B., Dammer E.B., Duong D.M., Ping L., Zhou M., Yin L., Higginbotham L.A., Guajardo A., White B., Troncoso J.C., et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 2020;26:769–780. doi: 10.1038/s41591-020-0815-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kant R., Langness V.F., Herrera C.M., Williams D.A., Fong L.K., Leestemaker Y., Steenvoorden E., Rynearson K.D., Brouwers J.F., Helms J.B., et al. Cholesterol metabolism is a druggable Axis that independently regulates tau and amyloid-β in iPSC-derived Alzheimer’s disease neurons. Cell Stem Cell. 2019;24:363–375.e9. doi: 10.1016/j.stem.2018.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasinska J.M., Hayden M.R. Cholesterol metabolism in Huntington disease. Nat. Rev. Neurol. 2011;7:561–572. doi: 10.1038/nrneurol.2011.132. [DOI] [PubMed] [Google Scholar]
- Koren S.A., Hamm M.J., Meier S.E., Weiss B.E., Nation G.K., Chishti E.A., Arango J.P., Chen J., Zhu H., Blalock E.M., Abisambra J.F. Tau drives translational selectivity by interacting with ribosomal proteins. Acta Neuropathol. 2019;137:571–583. doi: 10.1007/s00401-019-01970-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H.S., Koh S.-H. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl. Neurodegener. 2020;9:42. doi: 10.1186/S40035-020-00221-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9:559–613. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Xu S., Mihaylova M.M., Zheng B., Hou X., Jiang B., Park O., Luo Z., Lefai E., Shyy J.Y.J., et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13:376–388. doi: 10.1016/J.CMET.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.J., Sharma K., Zangrandi L., Chen C., Humphrey S.J., Chiu Y.-T., Spetea M., Liu-Chen L.-Y., Schwarzer C., Mann M. In vivo brain GPCR signaling elucidated by phosphoproteomics. Science. 2018;360:eaao4927. doi: 10.1126/science.aao4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J.M., Holtzman D.M. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. 2019;179:312–339. doi: 10.1016/j.cell.2019.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundby A., Secher A., Lage K., Nordsborg N.B., Dmytriyev A., Lundby C., Olsen J.V. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat. Commun. 2012;3:876–910. doi: 10.1038/ncomms1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangleburg C.G., Wu T., Yalamanchili H.K., Guo C., Hsieh Y.C., Duong D.M., Dammer E.B., De Jager P.L., Seyfried N.T., Liu Z., Shulman J.M. Integrated analysis of the aging brain transcriptome and proteome in tauopathy. Mol. Neurodegener. 2020;15:56. doi: 10.1186/s13024-020-00405-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann M., Kelleher N.L. Precision proteomics: the case for high resolution and high mass accuracy. Proc. Natl. Acad. Sci. USA. 2008;105:18132–18138. doi: 10.1073/pnas.0800788105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister G.C., Nusinow D.P., Jedrychowski M.P., Wühr M., Huttlin E.L., Erickson B.K., Rad R., Haas W., Gygi S.P. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 2014;86:7150–7158. doi: 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middeldorp J., Hol E.M. GFAP in health and disease. Prog. Neurobiol. 2011;93:421–443. doi: 10.1016/J.PNEUROBIO.2011.01.005. [DOI] [PubMed] [Google Scholar]
- De Miguel Z., Khoury N., Betley M.J., Lehallier B., Willoughby D., Olsson N., Yang A.C., Hahn O., Lu N., Vest R.T., et al. Exercise plasma boosts memory and dampens brain inflammation via clusterin. Nature. 2021;600:494–499. doi: 10.1038/s41586-021-04183-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min K.-J., Yang M.-S., Jou I., Joe E.h. Protein kinase A mediates microglial activation induced by plasminogen and gangliosides. Exp. Mol. Med. 2004;36:461–467. doi: 10.1038/emm.2004.58. [DOI] [PubMed] [Google Scholar]
- Morales I., Jiménez J.M., Mancilla M., Maccioni R.B. Tau oligomers and fibrils induce activation of microglial cells. J. Alzheimers Dis. 2013;37:849–856. doi: 10.3233/JAD-131843. [DOI] [PubMed] [Google Scholar]
- Morshed N., Ralvenius W.T., Nott A., Watson L.A., Rodriguez F.H., Akay L.A., Joughin B.A., Pao P.C., Penney J., LaRocque L., et al. Phosphoproteomics identifies microglial Siglec-F inflammatory response during neurodegeneration. Mol. Syst. Biol. 2020;16:e9819. doi: 10.15252/msb.20209819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarrete-Perea J., Yu Q., Gygi S.P., Paulo J.A. Streamlined tandem mass tag (SL-TMT) protocol: an efficient strategy for quantitative (Phospho)proteome profiling using tandem mass tag-synchronous precursor selection-MS3. J. Proteome Res. 2018;17:2226–2236. doi: 10.1021/acs.jproteome.8b00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson P.T., Alafuzoff I., Bigio E.H., Bouras C., Braak H., Cairns N.J., Castellani R.J., Crain B.J., Davies P., Del Tredici K., et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol. 2012;71:362–381. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda S., Watanabe Y., Moriya Y., Kawano S., Yamamoto T., Matsumoto M., Takami T., Kobayashi D., Araki N., Yoshizawa A.C., et al. jPOSTrepo: an international standard data repository for proteomes. Nucleic Acids Res. 2017;45:D1107–D1111. doi: 10.1093/nar/gkw1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira-Júnior L.C., Araújo Santos F.d.A., Goulart L.R., Ueira-Vieira C. Epitope fingerprinting for recognition of the polyclonal serum autoantibodies of Alzheimer’s disease. BioMed Res. Int. 2015;2015:267989. doi: 10.1155/2015/267989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G., Kim T.-W. Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat. Rev. Neurosci. 2011;12:284–296. doi: 10.1038/nrn3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park L.C., Albers D.S., Xu H., Lindsay J.G., Beal M.F., Gibson G.E. Mitochondrial impairment in the cerebellum of the patients with progressive supranuclear palsy. J. Neurosci. Res. 2001;66:1028–1034. doi: 10.1002/JNR.10062. [DOI] [PubMed] [Google Scholar]
- Paulus W., Bancher C., Jellinger K. Microglial reaction in Pick’s disease. Neurosci. Lett. 1993;161:89–92. doi: 10.1016/0304-3940(93)90147-D. [DOI] [PubMed] [Google Scholar]
- Pérez M.J., Jara C., Quintanilla R.A. Contribution of tau pathology to mitochondrial impairment in neurodegeneration. Front. Neurosci. 2018;12:441. doi: 10.3389/FNINS.2018.00441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perluigi M., Barone E., Di Domenico F., Butterfield D.A. Aberrant protein phosphorylation in Alzheimer disease brain disturbs pro-survival and cell death pathways. Biochim. Biophys. Acta. 2016;1862:1871–1882. doi: 10.1016/j.bbadis.2016.07.005. [DOI] [PubMed] [Google Scholar]
- Plubell D.L., Wilmarth P.A., Zhao Y., Fenton A.M., Minnier J., Reddy A.P., Klimek J., Yang X., David L.L., Pamir N. Extended multiplexing of tandem mass tags (TMT) labeling reveals age and high fat diet specific proteome changes in mouse epididymal adipose tissue. Mol. Cell. Proteomics. 2017;16:873–890. doi: 10.1074/mcp.M116.065524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pore D., Gupta N. The ezrin-radixin-moesin family of proteins in the regulation of B-cell immune response. Crit. Rev. Immunol. 2015;35:15–31. doi: 10.1615/CritRevImmunol.2015012327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash R.S., Voss M.W., Erickson K.I., Kramer A.F. Physical activity and cognitive vitality. Annu. Rev. Psychol. 2015;66:769–797. doi: 10.1146/ANNUREV-PSYCH-010814-015249. [DOI] [PubMed] [Google Scholar]
- Rappsilber J., Mann M., Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007;2:1896–1906. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- Rayaprolu S., Higginbotham L., Bagchi P., Watson C.M., Zhang T., Levey A.I., Rangaraju S., Seyfried N.T. Systems-based proteomics to resolve the biology of Alzheimer’s disease beyond amyloid and tau. Neuropsychopharmacology. 2021;46:98–115. doi: 10.1038/s41386-020-00840-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayaprolu S., Gao T., Xiao H., Ramesha S., Weinstock L.D., Shah J., Duong D.M., Dammer E.B., Webster J.A., Lah J.J., et al. Flow-cytometric microglial sorting coupled with quantitative proteomics identifies moesin as a highly-abundant microglial protein with relevance to Alzheimer’s disease. Mol. Neurodegener. 2020;15:28. doi: 10.1186/s13024-020-00377-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redjems-Bennani N., Jeandel C., Lefebvre E., Blain H., Vidailhet M., Guéant J.L. Abnormal substrate levels that depend upon mitochondrial function in cerebrospinal fluid from alzheimer patients. Gerontology. 1998;44:300–304. doi: 10.1159/000022031. [DOI] [PubMed] [Google Scholar]
- Rhein V., Song X., Wiesner A., Ittner L.M., Baysang G., Meier F., Ozmen L., Bluethmann H., Dröse S., Brandt U., et al. Amyloid-β and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA. 2009;106:20057–20062. doi: 10.1073/pnas.0905529106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy E.R., Wang B., Wan Y.W., Chiu G., Cole A., Yin Z., Propson N.E., Xu Y., Jankowsky J.L., Liu Z., et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J. Clin. Invest. 2020;130:1912–1930. doi: 10.1172/JCI133737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samimi N., Sharma G., Kimura T., Matsubara T., Huo A., Chiba K., Saito Y., Murayama S., Akatsu H., Hashizume Y., et al. Distinct phosphorylation profiles of tau in brains of patients with different tauopathies. Neurobiol. Aging. 2021;108:72–79. doi: 10.1016/J.NEUROBIOLAGING.2021.08.011. [DOI] [PubMed] [Google Scholar]
- Savas J.N., Wang Y.Z., DeNardo L.A., Martinez-Bartolome S., McClatchy D.B., Hark T.J., Shanks N.F., Cozzolino K.A., Lavallée-Adam M., Smukowski S.N., et al. Amyloid accumulation drives proteome-wide alterations in mouse models of Alzheimer’s disease-like pathology. Cell Rep. 2017;21:2614–2627. doi: 10.1016/j.celrep.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Pozo A., Mielke M.L., Gómez-Isla T., Betensky R.A., Growdon J.H., Frosch M.P., Hyman B.T. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am. J. Pathol. 2011;179:1373–1384. doi: 10.1016/j.ajpath.2011.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyfried N.T., Dammer E.B., Swarup V., Nandakumar D., Duong D.M., Yin L., Deng Q., Nguyen T., Hales C.M., Wingo T., et al. A multi-network approach identifies protein-specific Co-expression in asymptomatic and symptomatic Alzheimer’s disease. Cell Syst. 2017;4:60–72.e4. doi: 10.1016/j.cels.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K., Schmitt S., Bergner C.G., Tyanova S., Kannaiyan N., Manrique-Hoyos N., Kongi K., Cantuti L., Hanisch U.K., Philips M.A., et al. Cell type- and brain region-resolved mouse brain proteome. Nat. Neurosci. 2015;18:1819–1831. doi: 10.1038/nn.4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D., Gable A.L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N.T., Morris J.H., Bork P., et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K., Uda A., Mitsubori M., Nagashima S., Iwasaki H., Ito N., Shiiba I., Ishido S., Matsuoka M., Inatome R., Yanagi S. Mitochondrial ubiquitin ligase alleviates Alzheimer’s disease pathology via blocking the toxic amyloid-β oligomer generation. Commun. Biol. 2021;4:192. doi: 10.1038/s42003-021-01720-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilleman K., Van den Haute C., Geerts H., van Leuven F., Esmans E.L., Moens L. Proteomics analysis of the neurodegeneration in the brain of tau transgenic mice. Proteomics. 2002;2:656–665. doi: 10.1002/1615-9861(200206)2:6<656::AID-PROT656>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Tripathy R., Leca I., van Dijk T., Weiss J., van Bon B.W., Sergaki M.C., Gstrein T., Breuss M., Tian G., Bahi-Buisson N., et al. Mutations in MAST1 cause mega-corpus-callosum syndrome with cerebellar hypoplasia and cortical malformations. Neuron. 2018;100:1354–1368.e5. doi: 10.1016/J.NEURON.2018.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai C.F., Ogata K., Sugiyama N., Ishihama Y. Motif-centric phosphoproteomics to target kinase-mediated signaling pathways. Cell Rep. Methods. 2022;2:100138. doi: 10.1016/J.CRMETH.2021.100138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- Velasco A., Fraser G., Delobel P., Ghetti B., Lavenir I., Goedert M. Detection of filamentous tau inclusions by the fluorescent Congo red derivative FSB [(trans, trans)-1-fluoro-2, 5-bis(3-hydroxycarbonyl-4-hydroxy)styrylbenzene] FEBS Lett. 2008;582:901–906. doi: 10.1016/J.FEBSLET.2008.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Mandelkow E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016;17:5–21. doi: 10.1038/nrn.2015.1. [DOI] [PubMed] [Google Scholar]
- Wang C., Zhao F., Shen K., Wang W., Siedlak S.L., Lee H.G., Phelix C.F., Perry G., Shen L., Tang B., et al. The sterol regulatory element-binding protein 2 is dysregulated by tau alterations in Alzheimer disease. Brain Pathol. 2019;29:530–543. doi: 10.1111/BPA.12691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Liu S., Zhai A., Zhang B., Tian G. AMPK-mediated regulation of lipid metabolism by phosphorylation. Biol. Pharm. Bull. 2018;41:985–993. doi: 10.1248/BPB.B17-00724. [DOI] [PubMed] [Google Scholar]
- Wingo A.P., Dammer E.B., Breen M.S., Logsdon B.A., Duong D.M., Troncosco J.C., Thambisetty M., Beach T.G., Serrano G.E., Reiman E.M., et al. Large-scale proteomic analysis of human brain identifies proteins associated with cognitive trajectory in advanced age. Nat. Commun. 2019;10:1619–1714. doi: 10.1038/s41467-019-09613-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Chen M., Jiang J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion. 2019;49:35–45. doi: 10.1016/j.mito.2019.07.003. [DOI] [PubMed] [Google Scholar]
- Yoon W.H., Sandoval H., Nagarkar-Jaiswal S., Jaiswal M., Yamamoto S., Haelterman N.A., Putluri N., Putluri V., Sreekumar A., Tos T., et al. Loss of nardilysin, a mitochondrial Co-chaperone for α-ketoglutarate dehydrogenase, promotes mTORC1 activation and neurodegeneration. Neuron. 2017;93:115–131. doi: 10.1016/J.NEURON.2016.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y., Lippa C.F. Review: disruption of the postsynaptic density in Alzheimer’s disease and other neurodegenerative dementias. Am. J. Alzheimers Dis. Other Demen. 2010;25:547–555. doi: 10.1177/1533317510382893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Ma C., Gearing M., Wang P.G., Chin L.S., Li L. Integrated proteomics and network analysis identifies protein hubs and network alterations in Alzheimer’s disease. Acta Neuropathol. Commun. 2018;6:19. doi: 10.1186/s40478-018-0524-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T., Chen D., Lee T.H. Phosphorylation signaling in APP processing in Alzheimer’s disease. Int. J. Mol. Sci. 2019;21:209. doi: 10.3390/ijms21010209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziff E.B. Enlightening the postsynaptic density. Neuron. 1997;19:1163–1174. doi: 10.1016/S0896-6273(00)80409-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) List of quantified proteins in spinal cord. (B) Result of Welch’s t-tests for proteins in spinal cord. (C) List of quantified proteins in cortex. (D) Result of Welch’s t-tests for proteins in cortex.
(A) GO term enrichment analysis of significantly upregulated proteins in the spinal cord. (B) GO term enrichment analysis of significantly downregulated proteins in the spinal cord. (C) GO term enrichment analysis of significantly upregulated proteins in the cortex. (D) GO term enrichment analysis of significantly downregulated proteins in the cortex.
(A) GO term enrichment analysis of top 50 hub proteins. (B) GO term enrichment analysis of all proteins.
(A) List of quantified phosphorylation sites. (B) Result of Welch’s t-tests. (C) Correlation between phosphorylation at quantified sites and module eigenproteins. (D) Summary of detected tau phosphorylation sites. (E) Correlation between phosphorylation at quantified sites and FSB intensities.
Data Availability Statement
The proteomics datasets and analysis data generated in this study are available in the jPOST repository PXD031129 (JPST001212) (Okuda et al., 2017) (https://repository.jpostdb.org/). This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.