

Summary
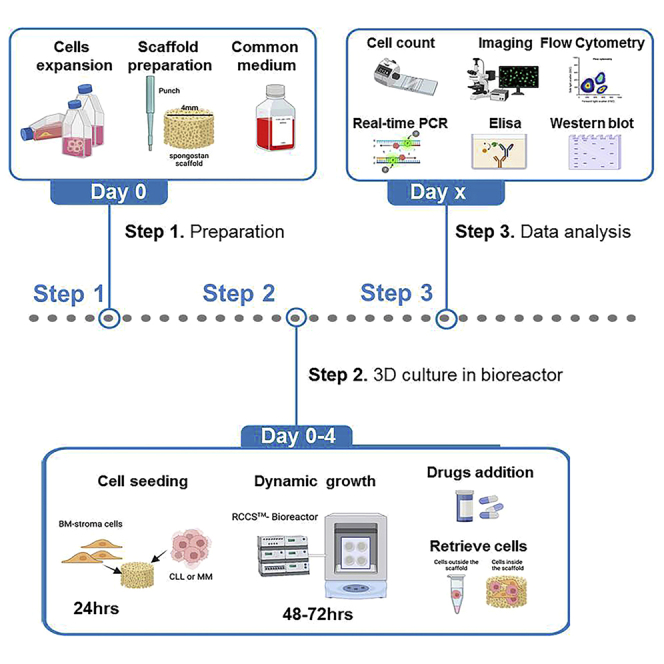
In this protocol, we describe how to generate 3D culture surrogates of chronic lymphocytic leukemia (CLL) and multiple myeloma (MM) bone marrow microenvironments. We detail the use of culturing scaffolds populated with BM stromal cells and tumor cells in the RCCS™ bioreactor. This 3D culture can efficiently recapitulate tumor-stroma crosstalk and allows the testing of drugs such as ibrutinib and bortezomib. Moreover, this protocol can be used for the generation of other and more complex tumor microenvironments.
For complete details on the use and execution of this protocol, please refer to Belloni et al. (2018) and Barbaglio et al. (2021).
Subject areas: Cell Biology, Cell culture, Cancer, Immunology, Biotechnology and bioengineering
Graphical abstract
Highlights
-
•
Step-by-step generation of a 3D bone marrow microenvironment for CLL and MM
-
•
Set up of a dynamic 3D culture in an RCCS™ bioreactor
-
•
Integrative analysis of the 3D scaffolds in response to drug treatment
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
In this protocol, we describe how to generate 3D culture surrogates of chronic lymphocytic leukemia (CLL) and multiple myeloma (MM) bone marrow microenvironments. We detail the use of culturing scaffolds populated with BM stromal cells and tumor cells in the RCCS™ bioreactor. This 3D culture can efficiently recapitulate tumor-stroma crosstalk and allows the testing of drugs such as ibrutinib and bortezomib. Moreover, this protocol can be used for the generation of other and more complex tumor microenvironments.
Before you begin
Cell lines culture and expansion
-
1.Culturing and expansion of HS-5 bone marrow stromal cell (BMSC) line.
-
a.HS-5 BMSCs are cultured in DMEM supplemented with 10% FBS and Penicillin/Streptomycin (Pen/Strep) 1% in T75 ventilated flasks.
-
b.When confluency is reached, HS-5 are washed with cold PBS and detached by Trypsin treatment (1 min at 37°C).
-
c.Cells are then retrieved and centrifuged at 285 × g for 5 min at 4°C.
-
d.To obtain the desired cell concentration, viable cells are counted by trypan blue exclusion method and re-suspended in Complete Tissue Culture Medium (CTM).
-
a.
-
2.Culture and expansion of tumor cell lines:
-
a.MEC-1 chronic lymphocytic leukemia (CLL) cell line and MM.1S multiple myeloma (MM) cell line are cultured in RPMI supplemented with 10% FBS and Pen/Strep 1%.
-
b.The cells are kept in culture in suspension at the concentration of 500 × 103 cells/mL.
-
c.CTM is changed every 2 days, centrifuging cells at 410 × g for 5 min at room temperature (RT), supernatant (SN) is discarded and cells are re-suspended at desired concentration with CTM.
-
a.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC, anti-human CD73, REAfinity™ (REA804) | Miltenyi Biotec | Cat# 130-111-909 RRID: AB_2659167 |
| PC7, anti-human CD19 (J3-119) | Beckman Coulter | Cat# IM3628 RRID: AB_10638575 |
| PE, anti-human CD38 (HIT2) | Thermo Fisher Scientific | Cat# MHCD3804 RRID: AB_1472940 |
| PC7, anti-human CD38 (HIT2) | BD Biosciences | Cat# 560677 RRID: AB_1727473 |
| FITC, anti-human CD138 (MI15) | BD Biosciences | Cat# 552723 RRID: AB_394443 |
| Chemicals, peptides, and recombinant proteins | ||
| Phalloidin-ATTO 565 | MERCK, Sigma Aldrich | Cat# 94072 |
| Hoechst 33342, Trihydrochloride, Trihydrate - 10 mg/mL Solution in Water | Thermo Fisher Scientific | Cat# H3570 |
| Trypan blue solution | Merck, Sigma-Aldrich | Cat# T8154 |
| Triton ™ X100 | Merck, Sigma-Aldrich | Cat# T9284 |
| RPMI 1640 with L-glutamine | Euroclone S.p.A | Cat# ECB2000 |
| DMEM with L-glutamine and High Glucose | Euroclone S.p.A | Cat# ECM0103L |
| Fetal Bovine Serum (FBS) | Euroclone S.p.A | Cat# ECS5000L |
| Penicillin-Streptomycin Solution 100× | Euroclone S.p.A | Cat#ECB3001D |
| Dulbecco’s PBS w/o Calcium & Magnesium (PBS) | Euroclone S.p.A | Cat# ECB4004L |
| PBS with Ca++/Mg++ | Merck, Sigma-Aldrich | Cat# D8662-500ML |
| Trypsin–EDTA 1× solution (0.5 g/L Trypsin-0.021 g/L EDTA | Euroclone S.p.A | Cat# ECM0920D |
| LiberaseTM TM Research Grade | Roche, Merck | Cat# 5401119001 |
| Paraformaldehyde (PFA) 4% | of your choice | N/A |
| Ibrutinib | Selleck Chemicals | Cat# PCI-32765 |
| Bortezomib (Btz) (Velcade®) | Millennium Pharmaceuticals | N/A |
| Bovine Serum Albumin (BSA) | Merck, Sigma-Aldrich | Cat# A7906 |
| cOmpleteTM: protease inhibitor cocktail | Roche, Merck | Cat# 11697498001 |
| NP40 cell lysis buffer | Thermo Fisher Scientific | Cat# FNN0021 |
| TRIzol® Reagent | Ambion®, Inc by Life Technologies | Cat# 15596018 |
| Critical commercial assays | ||
| AnnexinV-FITC/Propidium Iodide (PI) Apoptosis Detection kit | Thermo Fisher Scientific | Cat# BMS500FI-300 |
| Vybrant® CFDA SE Cell tracer Kit | Thermo Fisher Scientific | Cat# V12883 |
| Experimental models: Cell lines | ||
| HS-5: human HPV-16 E6/E7 transformed BMSCs | American Type Cell Culture (ATCC) | Cat# CRL-11882 |
| MEC-1 cells | DSMZ | Cat# ACC497 |
| MM1.S cells | American Type Cell Culture (ATCC) | Cat# CRL-2974 |
| Software and algorithms | ||
| FCS Express Flow Cytometry software | De Novo Software | https://denovosoftware.com/ |
| Other | ||
| Gelatin sponge, SpongostanTM | Ethicon, Inc. USA | Cat# MS0005/J&J |
| Rotary Cell Culture System (RCCSTM) bioreactor | Synthecon, Inc | Cat# RCCS-4DQ https://synthecon.com/pages/disposable_vessel_system_rccs4dq_synthecon_20.asp |
| 4 Pack individually wrapped gamma sterilized disposable 10 mL vessels (High Aspect Ratio Vessel-HARV) | Synthecon, Inc, USA | Cat# D-410 |
| 96-well Suspension Culture Plate, U bottom | Greiner Bio-One, CELLSTAR | Cat# 650185 |
| Forceps | of your choice | N/A |
| Biopsy punch (4 mm diameter) | of your choice | N/A |
| Sterile scalpels | of your choice | N/A |
| 5 mL syringes | of your choice | N/A |
| 2 mL syringes | of your choice | N/A |
| 1 mL syringes | of your choice | N/A |
| Micro-pipette | of your choice | N/A |
| Tips for micropipette | of your choice | N/A |
| Disposable plastic-ware for cell culture | of your choice | N/A |
| 6-well plate | of your choice | N/A |
| 48-well plate | of your choice | N/A |
| Confocal Microscope | of your choice | N/A |
| Multi-color Flow cytometer | of your choice | N/A |
| Sterile class II biosafety cabinet | of your choice | N/A |
| Incubator with humidified atmosphere, at 37°C and 5% CO2 | of your choice | N/A |
| Refrigerated centrifuge | of your choice | N/A |
| Hemocytometer, Burker chamber | of your choice | N/A |
| Water bath with controlled temperature | of your choice | N/A |
| FACS tubes with 35 μm cell strainer caps | Falcon | Cat##352235 |
Materials and equipment
| Complete tissue culture medium (CTM) | Final concentration | Amount/volume |
|---|---|---|
| RPMI1640 with L-glutamine or DMEM with 4.5 g/L glucose | 89% | 445 mL |
| Heat Inactivated FBS | 10% | 50 mL |
| Penicillin/Streptomycin solution | 1% | 5 mL |
| Total | 100% | 500 mL |
Storage notes: Prepare CTM under sterile conditions using a biosafety cabinet, store at 4°C and pre-warm at 37°C before use. CTM can be stored at 4°C for 1 month.
| Liberase™ TM research grade solution | Final concentration | Amount/volume |
|---|---|---|
| Liberase™ 100× stock solution (2,5 mg/mL) | 25 μg/mL | 100 μL |
| PBS with Ca++/Mg++ | 99% | 9.9 mL |
| Total | 10 mL |
Storage notes: Prepare Liberase™ under sterile conditions using a biosafety cabinet.
Liberase™ is dissolved in PBS with Ca++/Mg++ to obtain a 100× stock solution (2.5 mg/mL). Aliquot and store the stock solution at −20°C.
After dilution store at 4°C for 2 weeks.
| Blocking buffer | Final concentration | Amount/volume |
|---|---|---|
| PBS | 98% | 9.8 mL |
| BSA | 1% | 0.1 g |
| Heat Inactivated FBS | 1% | 100 μL |
| Total | 100% | ∼10 mL |
Storage notes: Prepare solution under sterile conditions using a biosafety cabinet and store at 4°C for 1 month.
| Hoechst 33342 | Final concentration | Amount/volume |
|---|---|---|
| Hoechst 10 mg/mL | 5 μg/mL | 5 μL |
| PBS | N/A | 9,95 mL |
| Total | 10 mL |
Storage notes: Store at 4°C for 1 week.
| Washing medium | Final concentration | Amount/volume |
|---|---|---|
| FBS | 1% | 500 μL |
| PBS | 99% | 49,5 mL |
| Total | 100% | 50 mL |
Storage notes: Prepare solution under sterile conditions using a biosafety cabinet and store at 4°C for 1 month.
| Ibrutinib | Final concentration | Amount/volume |
|---|---|---|
| Ibrutinib 10 mM | 1 μM | 1,1 μL |
| TCM | N/A | 11 mL |
| Total | ∼11 mL |
Storage note: Prepare solution under sterile conditions using a biosafety cabinet. The stock solution (10 mM) is supplied in DMSO and can be stored at −20°C in small aliquots.
Dilute it in CTM immediately before use.
| Velcade® | Final concentration | Amount/volume |
|---|---|---|
| 1st dilution (solution A) | ||
| Velcade® (6.5 mM) | 6.5 μM | 1 μL |
| CTM | N/A | 1 mL |
| 2nd dilution | ||
| Solution A | 10 nM | 16.9 μL |
| CTM | N/A | 11 |
| Total | ∼11 mL |
Storage note: The proteasome inhibitor Bortezomib (Velcade®, Millennium Pharmaceuticals, Inc., Cambridge, MA) is stored at −20°C and diluted in CTM immediately before use.
Step-by-step method details
Step 1: Static pre-seeding of gelatin scaffold with HS-5 BMSC line
Timing: 5 h 30 mintotal
This section details the method for the static pre-seeding of BMSC in the gelatin scaffold (Spongostan™).
-
1.Preparation and cutting of the gelatin scaffolds (Figure 1A).
-
a.Under sterile conditions, in a laminar flow hood, open the blister containing the gelatin sponge (Spongostan™) and place it in a Petri dish. Gelatin sponge is supplied as a cube of 10 mmx10 mmx10 mm.
-
b.Using a scalpel cut the sponge into 3 slices of about 3 mm each.
-
c.With a 4 mm diameter punch, cut four equal cylinders per slice. It is possible to obtain 12 cylinders/scaffolds per sponge with dimensions 4 mm in diameter by 3 mm in height.
-
a.
-
2.Retrieval of HS-5 BMSC by trypsin treatment. Counting and dilution of cell suspension.
-
a.Detach HS-5 BMSC cells from the culture flask by trypsin treatment and re-suspend them in CTM (DMEM+10% FBS), the volume depends on the flask (for T75 >5 mL for T25<5 mL).
-
b.Count live HS-5 BMSCs by trypan blue exclusion method.
-
c.Re-suspend the cells at the concentration of 10 × 106 cells/mL.
-
a.
-
3.Seeding of gelatin scaffolds with HS-5 BMSCs and static incubation (Figure 1B).
-
a.Place one gelatin scaffold per well in a 96-well, U-bottom, cell suspension plate.
-
b.Seed 200000 HS-5 BMSCs per scaffold (corresponding to 20 μL of cell suspension) allowing the entire volume (20 μL) to be absorbed by the sponge to homogenously distribute HS-5 BMSCs within the scaffold.
-
c.For a better distribution of the cells inside the scaffolds, slowly pipette each scaffold 3 times with a p100 micropipette equipped with a tip whose extremity is cut off (Figure 2).
-
d.Incubate the 96-well plate in a humidified incubator at 5% CO2 and 37°C for 4 h.
-
a.
CRITICAL: The gelatin scaffolds are very light and electrostatic and tend, if not well positioned, to exit the plate when located under the sterile hood. It is therefore advisable to position the scaffolds in any well of the plate with the help of forceps. To prevent evaporation of the medium in the wells containing the scaffolds, fill the empty wells surrounding the scaffolds with PBS.
Figure 1.

Preparation of the scaffolds and the RCCS™ bioreactor
(A) Preparation of scaffolds from a cube of gelatin sponge (Spongostan™).
(B) Positioning of scaffolds in the 96-well suspension culture plate and HS-5 BMSC seeding.
(C and D) The RCCS™ 4DQ bioreactor (Synthecon, Inc,): the bioreactor is composed by a working station (C) with 4 independent positions for vessels and a power supply (D) with a tachometer controlling the speed of rotation of each vessel. The rotation of vessels is counterclockwise and maintains the scaffolds in a free fall condition simulating microgravity. The dynamic culture allows optimal mass transfer, delivery of nutrients and of oxygen, avoiding cell sedimentation.
(E) High Aspect Ratio Vessel (HARV): it is composed by one Plug Fill port, two syringe ports and a silicon membrane for gas exchange.
(F) High Aspect Ratio Vessel (HARV) with the Plug Fill port opened.
Figure 2.

How to pipette each scaffold with a cut tip
Step 2: Dynamic seeding in the RCCS™ bioreactor of gelatin scaffold with HS-5 BMSC
This section describes how to complete efficiently the seeding of BMSCs in the scaffolds under dynamic conditions in the RCCS™ bioreactor (Figures 1C and 1D). This step avoids cell sedimentation, favoring the homogeneous distribution of the cells within the scaffold.
The RCCS™4DQ bioreactor is equipped with four independent vessel stations (Figure 1C) and a tachometer for rotation control (Figure 1D). The vessel rotation on a horizontal axis (clinorotation) maintains the scaffolds in a free-fall condition (mimicking microgravity), generating a constant dynamic fluid flow. This guarantees an optimal mass transfer, which results in optimal viability of the cells within the scaffolds (Ferrarini et al., 2013, 2017). Moreover, the HARV vessels are equipped with a gas exchange membrane that favors the optimal oxygenation of the scaffolds.
-
4.Preparation of HARV vessel, transfer of scaffolds from 96-well plate to HARV vessel and dynamic seeding in the RCCS™ bioreactor.
-
a.Take a new 10 mL Disposable High Aspect Ratio Vessel (HARV) and wash it by inserting 5 mL PBS through the open plug fill port (Figures 1E and 1F).
-
b.With a 5 mL pipette, through the plug fill port, remove the PBS.
-
c.With a p200 micropipette remove any residual PBS.
-
d.Through the plug fill port, add 600 μL of pre-warmed medium (DMEM+10%FBS) to the vessel.
-
e.Using a p100 pipette with a cut tip, carefully aspirate the scaffolds from the wells and one by one transfer them, through the plug fill port, in the HARV vessels (up to 3 scaffolds/vessel).
-
f.Close the plug fill port and place the vessels in the vessel station of the bioreactor, inside the incubator at 37°C and 5% CO2.
-
a.
-
5.Dynamic seeding, in the RCCS™ bioreactor, of gelatin scaffolds with HS-5 BMSC.
-
a.Set the tachometer to a rotation speed of 7.5 rpm and leave the scaffolds overnight inside the vessel.
-
a.
Step 3: Dynamic culture of CLL cells (MEC-1) or MM cells (MM.1S) inside HS-5 BMSCs populated scaffolds
This section explains how to seed the scaffolds, previously populated with stromal cells, with tumor cells and how to keep them in culture in the RCCS™ bioreactor.
-
6.Retrieval of tumor cells (MEC-1 or MM.1S) from the culture flasks and dynamic seeding in the scaffolds.
-
a.Retrieve MEC-1 cells or MM.1S cells from culture flasks, centrifuge them at 410 × g for 5 min and re-suspend them in ≅5 mL fresh pre-warmed medium (RPMI+10%FBS).
-
b.Count cells by trypan blue exclusion method.
-
c.If the vessel contains one or two scaffolds:
-
i.Re-suspend MEC-1 cells in CTM (RPMI+10%FBS) to obtain a final concentration of 10 × 106 cells/mL.OR
-
ii.Re-suspend MM1.S cells in CTM (RPMI+10%FBS) to obtain a final concentration of 2.5 × 106 cells/mL.
-
iii.To the HARV vessel add 2 × 106 MEC-1 (corresponding to 200 μL of cell suspension) or 500 × 103 MM.1S (corresponding to 200 μL of cell suspension) for each scaffold through the plug fill port to obtain inside the vessel a final volume of 800 μL for 1 scaffold or 1 mL for 2 scaffolds. (Figure 3-1).
-
i.
-
d.If the vessel contains three scaffolds:
-
i.Re-suspend MEC-1 cells in CTM (RPMI+10%FBS) to obtain a final concentration of 15 × 106 cells/mL.OR
-
ii.Re-suspend MM1.S cells in CTM (RPMI+10%FBS) to obtain a final concentration of 3.75 × 106 cells/mL.
-
iii.To the HARV vessel add 6 × 106 MEC-1 (corresponding to 400 μL of cell suspension) or 1.5 × 106 MM.1S (corresponding to 400 μL of cell suspension) through the plug fill port to obtain inside the vessel a final volume of 1 mL (Figure 3-1).
-
i.
-
e.Set rotation speed to 7.5 rpm and allow the cell seeding to proceed for 5 h in the incubator at 37°C and 5%CO2.
-
a.
-
7.Dynamic culture of populated scaffolds.
-
a.Pre-warmed CTM (RPMI+10%FBS) is added to the vessels through the syringe ports using sterile 5 mL syringes (Figure 3) using the following steps:
-
i.Insert a sterile 5 mL syringe in a syringe port with the plunger removed (syringe A in Figure 3).
-
ii.Insert another 5 mL sterile syringe (syringe B in Figure 3) to the second port.
-
iii.Fill syringe A with 5 mL of CTM (Figure 3.4).
-
iv.With syringe B, slowly draw the air out of the vessel and let the medium enter from the second syringe (Figure 3.5).
-
v.Repeat steps ii-iii to fill the vessel with a total of ≅11 mL CTM.
-
vi.Close the syringe ports and discard the syringes.
-
i.
-
b.Put the vessel in the bioreactor in the incubator at 37°C and 5%CO2.
-
c.Start the culture by setting the rotation speed initially to 7.5 rpm and then increasing it, up to 14 rpm, to prevent the sedimentation of the scaffolds and to maintain them in a free-fall condition.
-
d.Keep the scaffolds in culture in the bioreactor for 48–72 h. If 3D culture continues, change the medium.
-
a.
Note: If air bubbles are formed, these can be removed with the two syringes placed in the syringe ports.
Note: Speed rotation must be settled to maintain scaffolds in a free-fall condition: it means that the scaffolds must neither precipitate under the action of gravity, nor turn in the vessel under the action of the centrifugal force. These forces must be balanced by adjusting the rotational speed usually starting from 7.5 rpm and up to 14 rpm.
Note: It is important to monitor the color of the medium: if the color of the medium turns yellow it is important to change it. Normally when active proliferating cell lines are cultured (as MEC-1 cells), medium must be changed more frequently (after 48 h) than primary slowly or not proliferating cells.
-
8.Medium change.
-
a.Insert a sterile 5 mL syringe in a syringe port and carefully aspirate 2 mL of medium. Discard it or put it in a 15 mL tube for future analysis.
-
b.Open the second syringe port (in order to let the air enter/removing the vacuum) and with the syringe aspirate the remaining medium paying attention to not remove any scaffolds.
-
c.Insert a sterile 5 mL syringe (B) in a syringe port (Figure 3).
-
d.Insert another 5 mL sterile syringe (A), removing the plunger, in the second syringe port and fill it with CTM as explained in Step 3.7a (Figure 3).
-
e.Put the vessel in the bioreactor in the incubator and continue the culture.
-
a.
Note: It is possible to discard the medium or to store it at −20°C for further analysis.
Optional: You may recover cells in the SN medium by centrifuging it at 410 × g.
Figure 3.

How to fill vessels with medium step by step
Step 4: Recovering of the scaffolds from the RCCS™ bioreactor and subsequent analysis
This section explains how to recover scaffolds, SN and cells in SN from the vessel, and how to process them for subsequent analysis.
-
9.Recovering of scaffolds, SN and cells in SN from the vessels.
-
a.With a syringe, gently aspirate 2 mL of SN from the vessel through a syringe port and put it in a 15 mL tube.
-
b.Open the plug fill port and pour the content of the vessel into a Petri dish or a 6-well plate scaffolds and SN.
-
a.
-
10.Enzymatic digestion of scaffolds and retrieval of cells for counting and flow cytometry.
-
a.Add 500 μL of Liberase™ 25 μg/mL in a FACS tube and place one scaffold. Alternatively, add 1 mL of Liberase™ for the digestion of 2 or 3 scaffolds.
-
b.Let the tube/tubes incubate for 10 min at 37°C for the complete digestion of the scaffolds.
-
c.Vortex tube to facilitate cell disaggregation and to re-suspend cells. (Alternatively pipette digested scaffold).
-
d.Block the enzymatic digestion adding 500 μL (or 1 mL FBS if using 1 mL of Liberase™) of FBS.
-
e.Count cells by trypan blue exclusion.
-
f.Centrifuge tube/tubes at 410 × g for 5 min, wash cells with 1 mL of PBS.
-
g.Centrifuge again as in f and discard SN.
-
h.Use cells for successive staining and flow cytometry.
-
a.
Note: The ratio of Liberase™ to FBS is 1:1.
If apoptosis detection must be performed wash cells with the Binding Buffer (included in the apoptosis detection kit).
-
11.Fixation of scaffolds for confocal analysis.
-
a.Add 300 μL of PFA 4% per well of a 48-well plate.
-
b.Insert 1 scaffold per well in order to fix it and let it fix ON at room temperature (RT).
-
c.Wash scaffolds with Blocking Solution for subsequent staining and confocal analysis.
-
a.
-
12.Lysis of scaffolds for Western blot analysis.
-
a.Place one scaffold in a 1.5 mL vial.
-
b.Lyse scaffold with 100 μL/scaffold of Lysis Buffer for 30 min in ice (or 0°C).
-
a.
-
13.Lysis of scaffolds for real time PCR.
-
a.Place one scaffold in a 1.5 mL.
-
b.Lyse one scaffold with 1 mL of TRIzol at RT and freeze at −20°C.
-
a.
-
14.Recovering of cells in SN for counting and flow cytometry.
-
a.Recover SN from the Petri dish or 6-well plate and put it in a 15 mL tube.
-
b.Centrifuge tube at 410 × g for 5 min to pellet cells, remove SN and re-suspend in 5 mL of PBS and count them by trypan blue exclusion dye.
-
c.Place cells in facs tubes.
-
d.Centrifuge cells at 410 × g for 5 min and stain cells for flow cytometric analysis (as explained in Step 5.16).
-
a.
-
15.Recovering of SNs for ELISA test or metalloproteases (MMPs) evaluation by zymography.
-
a.Add SN in another 15 mL tube, eventually aliquot it in 1.5 mL vials-eppendorf and store it at −20°C.
-
b.SN can successively be analyzed by ELISA to assess concentration of soluble factors (Cytokines, chemokines, growth factors) or subjected to zymography for MMPs assessment.
-
a.
Step 5: Characterization of scaffolds
This section describes how to evaluate the cells present in the scaffold by flow cytometry and the cell distribution and organization, within the scaffold, by confocal analysis.
-
16.
Staining of cells retrieved from the scaffolds upon digestion with Liberase™ for flow cytometric analysis.
Usually, the expected number of cells retrieved from the scaffolds (Figure 4A) is:
HS-5+MEC-1 after 48 h of culture: ≅1500 × 103.
HS-5+MEC-1 after 72 h of culture: ≅2500 × 103.
HS-5+MM1.S after 24 h (reported in Belloni et al., 2018) and 48 h of culture: ≅1000 × 103.
Cells retrieved from the scaffolds populated with MEC-1 cells and BMSC are stained for anti-human CD19-PC7 (B cells marker, to detect the percentage of MEC-1 cells) and anti-human CD73-APC (MSC marker, to detect the percentage of BMSC) (Figures 4B and 4C).
Cells retrieved from the scaffolds populated with MM1.S cells and BMSC are stained for anti-human CD38-PE (alternatively it is possible to use CD138-FITC) (Plasmacells marker, to detect the percentage of MM1.S cells) and anti-human CD73-APC (MSC marker, to detect the percentage of BMSC) (Figure 4B and 4C).-
a.Incubate cells (≅ 500 × 103 cells) and antibodies for 15 min at 4°C: for CD19-PC7 add 5 μL of antibody to 200 μL of cell suspension; for CD38-PE add 0.5 μL of antibody to 200 μL of cell suspension (alternatively 10 μL of CD138-FITC); for CD73-APC add 2 μL of antibody to200 μL of cell suspension.
-
b.Wash cells with 1 mL of washing medium, successively centrifuge FACS tubes at 410 × g for 5 min.
-
c.Filter cells with 35 μm cell strainer caps before running FACS tubes on a cytofluorimeter in order to remove eventual cell aggregates.
-
d.Run cells on a cytofluorimeter and analyze data (Figures 4B and 4C).
-
e.Cells retrieved from the scaffolds populated with MM.1S and BMSC are stained with anti-human CD73-APC and anti-human CD38-PE.
-
f.Incubate cells and antibodies for 15 min at 4°C.
-
g.Wash cells with 1 mL of washing medium, successively centrifuge FACS tubes at 410 × g for 5 min.
-
h.Filter cells with 35 μm cell strainer caps before running the FACS tubes on a flow cytometer to remove eventual cell aggregates.
-
i.Run cells on a flow cytometer and analyze data (Figures 4B and 4C).
-
a.
-
17.Staining of scaffolds for confocal analysis (Figures 4D–4F).
-
a.After fixation with PFA 4% ON, in the 48 well, scaffolds are washed with Blocking Solution and permeabilized with 500 μL Triton X100 0.3% in Blocking Solution for 20 min RT.
-
b.Scaffolds are then washed with 500 μL of PBS.
-
c.Scaffold can be stained with primary and secondary antibodies and visualized by confocal microscope (Leica TC sp5 or Olympus Fluo view).
-
d.To evaluate cell organization and distribution inside the scaffold, scaffolds are stained with Phalloidin 565 diluted 1:50 in PBS and Hoechst 33342 (5 μg/mL) for 1 h at RT. To visualize tumor cells we stained them with 2 μM Vybrant® CFDA SE Cell tracer before dynamic seeding using the manufacturer’s instructions (Figures 4D–4F).
-
a.
Figure 4.

Characterization and organization of cells inside the scaffold
(A) number of cells (mean ± SD) usually retrieved from the scaffold after 48 h or 72 h of 3D culture in bioreactor.
(B) Characterization by flow cytometry of HS-5 cells, MEC-1 cells and MM1.S cells.
(C) Characterization of scaffolds populated with HS-5 BMSCs and MEC-1 (LEFT) or MM.1S (RIGHT).
(D) Confocal analysis of scaffold populated with HS-5 BMSC line (bar size=50 μm; Leica TC sp5).
(E) Confocal analysis of scaffold populated with HS-5 BMSC line and CLL cell line MEC (bar size=50 μm; Leica TC sp5).
(F) Confocal analysis of scaffold populated with HS-5 BMSC line and MM cell line MM.1S (bar size=50 μm; Olympus Fluo view).
Step 6: Addition of drugs to 3D cell cultures
-
18.Ibrutinib (Ibr) treatment to evaluate its effect in mobilizing CLL cells from the scaffolds.
-
a.Prepare one vessel as an untreated (Unt) condition and one vessel for Ibr treatment, both with 3 scaffolds populated with HS-5 BMSCs and CLL cells (as described in parts 1 and 2 for HS-5 and in part 3 for MEC-1 cells).
-
b.After 72 h of dynamic culture in the bioreactor, retrieve 1–2 mL of medium from the vessels with a syringe and aliquot into a 15 mL tube. During this operation let the scaffolds settle on the opposite site of the vessels in order to minimize their perturbation.
-
c.Remove the syringe, open the Plug Fill port and gently remove the remaining medium containing cells that did not enter the scaffolds.
-
d.Place the removed cells/media in the 15 mL tube from step b.
-
e.Count cells in SNs to verify that the number of cells entered in the scaffolds are homogeneously distributed between the 2 vessels (Unt and Ibr).
-
f.Clarify SNs by centrifuging tubes for 5 min at 1500 rpm RT.
-
g.Discard the cells that did not enter the scaffolds and retrieve medium.
-
h.Through the syringe ports, as explained previously, (Figure 3) gently re-add recovered SNs inside the vessels with or without the addition of Ibr 1 μM and avoid the generation of turbulence.
-
a.
-
19.Bortezomib (Btz) treatment to evaluate its action in inducing MM cells death.
-
a.Prepare one vessel for Unt condition and one vessel for Btz treatment both containing up to 3 scaffolds.
-
b.Through one syringe port with a 1 mL syringe, add Btz diluted in a little amount of medium (≅300 μL) in order to obtain a concentration of 10 nM inside the vessel.
-
a.
Step 7: Assessment of the impact of drugs on tumor cells
This section shows how to evaluate the impact of Ibr on CLL cells and of Btz on MM cells.
-
20.Assessment of the impact of Ibr on CLL cells (Barbaglio et al., 2021).
-
a.After 5 h of dynamic culture in the presence or the absence of Ibr, as previously described in Step 6.18, retrieve both the scaffolds and the SN that contains cells that are mobilized from the scaffolds. The scaffolds are then digested in FACS tubes by adding 500 μL of Liberase™ (as explained in Step 4.10) for 10 min at 37°C to retrieve the cells.
-
b.Count cells present in the SN by trypan blue exclusion method in both conditions (untreated and treated) to evaluate mobilization induced by Ibr.
-
c.As a control, count the cells inside the scaffold in both conditions (untreated and treated).
-
d.Stain CLL cells present in the SN for CD19-PC7 (as explained in Step 5.16a and b).
-
e.To evaluate viability of cells:
-
i.wash cells with 1 mL of Binding Buffer (provided in the apoptosis detection kit and prepared following the manufacturer’s instructions) and centrifuge tubes at 410 × g.
-
ii.Discard SN.
-
iii.Stain cells with 5 μL of Annexin V (AnnV) for 10 min at RT.
-
iv.Wash cells adding 1 mL of Binding Buffer and centrifuging at 410 × g.
-
v.Discard supernatant and resuspend cells in 500 μL of Binding Buffer.
-
vi.Add 7 μL of Propidium Iodide (PI) immediately before running the tube on a flow cytometer.
-
i.
-
f.Stain (as explained in Step 5.16a and b) cells inside the scaffold for CD73-APC, CD19-PC7 in order to evaluate the percentage of CLL cells inside the scaffold. To assess their viability, stain cells for AnnV and PI (as explained in point e).
-
g.Finally run cells on a flow cytometer in order to characterize the composition of scaffolds, of mobilized cells and to detect their viability.
-
a.
Note: Cells inside the scaffold or mobilized cells can be characterized by real time PCR and by Western blot to evaluate differences in gene expression and protein levels, respectively. For example, we demonstrated that CLL cells with inactive HS1 (a protein involved in the regulation of cytoskeleton activity), are more likely to be retained within the scaffolds despite the presence of Ibr, whose mobilizing effect is mainly exerted on those with active HS1 (Barbaglio et al., 2021).
-
21.Assessment of the impact of Btz on MM cells (Belloni et al., 2018).
-
a.After 24 h of dynamic culture in the presence or the absence of Btz, scaffolds are retrieved, as previously described in Step 5.16a and b, from the vessels and digested by means of 500 μL Liberase™.
-
b.Cells retrieved from the scaffolds are then counted by trypan blue exclusion dye in order to evaluate the percentage of dead cells (Trypan blue positive).
-
c.Stain cells for anti-human CD38-PE and AnnV or CD38-PC7 and AnnV/PI (as explained in part 5-point 1a and b for the staining with CD38-PE and CD38-PC7; as explained in Step 7.20a point 1a for staining with Ann V and PI) for flow cytometric analysis to detect the effects of the drug (Belloni et al., 2018) (Figures 5A and 5B).
-
a.
Note: The three scaffolds per vessel can be used to evaluate different outcomes or different analyses; for example, one scaffold can be enzymatically digested to detect apoptosis of MM cells after Btz treatment, one scaffold can be fixed and stained for confocal analysis and so on.
Figure 5.

Response to drugs
(A) Evaluation of the impact of Btz on scaffolds populated with HS-5 BMSC and MM.1S.
(B) Evaluation of the impact of Btz on MM.1S 2D culture and on scaffold populated with primary BMSCs and MM.1S.
Expected outcomes
Both CLL and MM are strictly dependent on their supportive microenvironment (Caligaris-Cappio et al., 2014; Burger and Gribben, 2017; Mondello et al., 2017; Hideshima et al., 2007) for both their survival and progression, so the generation of a proper milieu, that mimics interactions occurring in vivo, is crucial to understand tumor biology and to evaluate response to drugs. (Scielzo and Ghia, 2020).
This protocol allows the generation of dynamic 3D cell cultures that recapitulate tumor BM microenvironment through the integrated use of a scaffold and of a bioreactor. Here, we have detailed how to generate a simplified BM surrogate, using cell lines for both microenvironment and tumor cells, but this protocol can be implemented using primary BMSCs, patients’ derived tumor cells and by the addiction of other cell types, (including endothelial cells for the generation of vascularization) and immune cells (Belloni et al., 2018). Moreover, this protocol can also be adapted to generate other relevant tumor microenvironments by considering the use of other scaffold types.
Notably in the RCCS™ bioreactor, two distinct environments coexist: inside and outside the scaffold, so it is possible to evaluate tumor mobilization induced by specific drugs otherwise not possible in conventional 2D culture settings. It is also possible to study the phenotype of cells in the 2 compartments and we demonstrated that, for CLL cells, it mirrors the in vivo differences between tissue compartments (Barbaglio et al., 2021).
Moreover, we demonstrated that MM cells receive in our 3D reconstructed microenvironment, proper signals that confer protection against the action of drugs (Figure 5B) (Belloni et al., 2018).
Limitations
A limitation of this method is that there are only two sizes of disposable HARV vessels for our bioreactor: 10 mL and 50 mL. These volumes require a large amount of compounds or drugs, increasing the overall costs.
Moreover, such large volumes could also be responsible of the dilution of the cytokines or chemokines released in the SNs thus potentially decreasing the possibility to detect them in standard ELISA assays.
Another limitation is the limited number of conditions that can be tested; it depends on the number of positions in the vessel station of the bioreactor. Before performing 3D experiments, consider doing traditional 2D cultures to select optimal concentration/timing of drugs. Consider also using the 3 scaffolds from the same vessel for a time-course experiment retrieving each scaffold at different time points.
Troubleshooting
Problem 1
It could happen to find different number of cells among the scaffolds: Spongostan™ sponge is characterized by different size of its pores. This characteristic constitutes an advantage as it more closely mimics the different porosity present in the BM, but it also results in heterogeneity among scaffolds in terms of surface that can be populated by the cells.
Moreover, our scaffolds are generated by manual cut of the cube sponge so, the homogeneity of the scaffolds’ size, strictly depends on the manual skill of the operator.
Potential solution
To minimize this issue, scaffolds should be prepared by the same operator in a series of experiments, to decrease variability. Moreover, it is important to seed the same number of cells in each scaffold. Also having a triplicate per vessel is useful for evaluating any differences.
Furthermore, to evaluate the homogeneity of the scaffolds in the different vessels, it is advisable to count the cells not entered in the scaffold. This parameter reflects the number of cells inside the scaffolds.
Alternatively, when performing experiments were determining the number of cells populating the scaffolds is critical, you can opt to use other types of spongy scaffolds characterized by constant porosity and supplied in sheets of fixed thickness (i.e., Avitene™ Ultrafoam™ Collagen Sponge, BD) (Frenquelli et al., 2020) and use flow count.
Problem 2
Scaffolds under microgravity conditions could stick together as they tend to position themselves in the same zone of the vessel.
Potential solution
Monitor the 3D culture in the bioreactor and if this occurs immediately recover scaffolds and separate them. Then continue the dynamic 3D culture. Moreover, never use more than 3 scaffolds/vessel.
Problem 3
Scaffold could become difficult to be disaggregated for further analysis over time.
Potential solution
Depending on the specific read out it is possible to increase the mechanic disaggregation (pipetting) or enzymatic incubation time. Verify that the prolonged enzymatic activity is not becoming toxic for the cells.
Problem 4
The protocol here presented is optimized for MM and CLL cells, in case of different cell types the number of cells or incubation may need to be adjusted and the proliferation rate must be evaluated for further read-outs such as drug testing.
Potential solution
Before start with the dynamic growth try to populate the scaffold in static condition with different number of cells and time points and evaluate the proliferation rate of each cell type involved.
Problem 5
Each drug can have a different mechanism of action in 3D culture vs conventional 2D cultures.
Potential solution
For each drug different from those reported in this manuscript you will have to find the working concentration and time of incubation.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Cristina Scielzo, scielzo.cristina@hsr.it.
Materials availability
All reagents are commercially available.
Acknowledgments
C.S. project is supported by Associazione Italiana per la Ricerca sul Cancro AIRC under IG 2018 - ID. C.S. acknowledges financial support from EHA Advanced Research Grant 2020. P.G. acknowledges financial support from Special Program on Metastatic Disease – 5 per mille #2119. Graphical abstract and Figure 1B have been created with BioRender.com. We acknowledge Alembic and Fractal facilities.
Author contributions
B.D. and S.C. set up the protocol, wrote the manuscript, and prepared the figures. F.M., F.E., G.V., and B.F. helped in the setup of the protocol. F.M., F.E., and G.P. revised the manuscript.
Declaration of interests
The authors declare no competing interests.
Data and code availability
No datasets or codes were generated in this study.
References
- Belloni D., Heltai S., Ponzoni M., Villa A., Vergani B., Pecciarini L., Marcatti M., Girlanda S., Tonon G., Ciceri F., et al. Modeling multiple myeloma-bone marrow interactions and response to drugs in a 3D surrogate microenvironment. Haematologica. 2018;103:707–716. doi: 10.3324/haematol.2017.167486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbaglio F., Belloni D., Scarfò L., Sbrana F.V., Ponzoni M., Bongiovanni L., Pavesi L., Zambroni D., Stamatopoulos K., Caiolfa V.R., et al. Three-dimensional co-culture model of chronic lymphocytic leukemia bone marrow microenvironment predicts patient-specific response to mobilizing agents. Haematologica. 2021;106:2334–2344. doi: 10.3324/haematol.2020.248112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger J.A., Gribben J.G. The microenvironment in chronic lymphocytic leukemia (CLL) and other B cell malignancies: insight into disease biology and new targeted therapies. Semin. Cancer Biol. 2017;24:71–81. doi: 10.1016/j.semcancer.2013.08.011. [DOI] [PubMed] [Google Scholar]
- Caligaris-Cappio F., Bertilaccio M.T.S., Scielzo C. How the microenvironment wires the natural history of chronic lymphocytic leukemia. Semin. Cancer Biol. 2014;24:43–48. doi: 10.1016/j.semcancer.2013.06.010. [DOI] [PubMed] [Google Scholar]
- Ferrarini M., Steimberg N., Boniotti J., Berenzi A., Belloni D., Mazzoleni G., Ferrero E. 3D-Dynamic culture models of multiple myeloma. Methods Mol. Biol. 2017;1612:177–190. doi: 10.1007/978-1-4939-7021-6_13. [DOI] [PubMed] [Google Scholar]
- Ferrarini M., Steimberg N., Ponzoni M., Belloni D., Berenzi A., Girlanda S., Caligaris-Cappio F., Mazzoleni G., Ferrero E. Ex-vivo dynamic 3-D culture of human tissues in the RCCSTM bioreactor allows the study of Multiple Myeloma biology and response to therapy. PLoS One. 2013;26:e71613. doi: 10.1371/journal.pone.0071613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenquelli M., Caridi N., Antonini E., Storti F., Viganò V., Gaviraghi M., Occhionorelli M., Bianchessi S., Bongiovanni L., Spinelli A., et al. The WNT receptor ROR2 drives the interaction of multiple myeloma cells with the microenvironment through AKT activation. Leukemia. 2020;34:257–270. doi: 10.1038/s41375-019-0486-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hideshima T., Mitsiades C., Tonon G., Richardson P.G., Anderson K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer. 2007;7:585–598. doi: 10.1038/nrc2189. [DOI] [PubMed] [Google Scholar]
- Mondello P., Cuzzocrea S., Navarra M., Mian M. Bone marrow micro-environment is a crucial player for myelomagenesis and disease progression. Oncotarget. 2017;8:20394–20409. doi: 10.18632/oncotarget.14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scielzo C., Ghia P. Modeling the leukemia microenviroment in vitro. Front. Oncol. 2020;10:607608. doi: 10.3389/fonc.2020.607608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No datasets or codes were generated in this study.